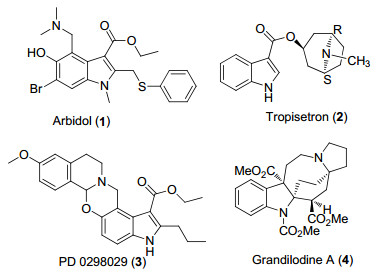
图 1.
3-吲哚羧酸酯类药物分子和天然产物
Figure 1.
Drug molecules and natural products containing 3-indolecarboxylates

在天然产物和药物分子中, 吲哚环是最广泛存在的杂环结构单元之一[1].大部分含有吲哚环的化合物具有一定的生物活性, 因此吲哚环被认为是药物分子设计的“优势结构”.其中, 3-吲哚羧酸酯类化合物是广泛存在于天然产物及药物分子中的重要的结构单元[2], 如药物分子盐酸阿比多尔(1, 抗流感病毒药)[3]、托烷司琼(2, 选择性5-羟色胺受体拮抗剂)[4]、PD 0298029 (3, 毒蕈碱乙酰胆碱受体M4的选择性拮抗剂)[5]、天然产物Grandilodine A (4, 用于逆转VCR KB细胞中的多药耐药性)[6]等均为此类化合物(图 1).
鉴于吲哚尤其是3-吲哚羧酸酯类化合物的重要性, 吲哚环的构建以及吲哚的选择性官能团化受到了化学家们广泛的关注, 许多简便、高效的合成方法逐渐被开发出来[7].金属催化的以及非金属条件下的合环反应用来构建吲哚中的吡咯环是近年来研究的热点, 而在吲哚C-3位直接进行官能团化合成酯基官能团也显现出较高的合成效率.本文在本课题组相关研究的基础上, 按照上述分类方法, 综述了近年来3-吲哚羧酸酯类化合物的合成研究进展.通过合环反应构建吲哚环以及直接在吲哚C-3位进行官能团化的方法均以所使用的金属催化剂或其它试剂来分类描述.
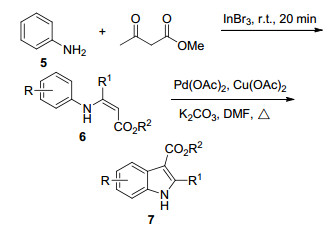
2008年, Glorius等[8]报道了由市售的苯胺类化合物通过Pd催化的分子内的C—H氧化偶联来合成3-吲哚羧酸酯类化合物的方法(Scheme 1).在三溴化铟催化作用下, 苯胺(5)与乙酰乙酸甲酯于室温搅拌可快速得到相应的烯胺羧酸酯6, 然后再以醋酸钯为催化剂、醋酸铜为氧化剂进行环化反应得到相应的3-吲哚羧酸酯类化合物7, 并提出了可能的反应机理.该方法产率较高, 应用广泛.对某些底物, 提高反应温度后, 反应时间会相应缩短, 产率也会随之变高.
同年, Sole等[9]报道了零价钯催化的β-(2-碘苯胺基)酯8的分子内α-芳基化反应合成3-吲哚羧酸酯类化合物9的新方法(Eq. 1).对芳环上连有不同取代基的底物均作了尝试, 发现在芳环上连有吸电子基团(氯原子、氟原子、甲酯等)时比在芳环上连有给电子(甲氧基)的或电中性(氢原子或甲基)基团时反应的产率要低.通常情况下, 在3-单取代的化合物中, 不会得到中间体二氢吲哚, 反应直接得到相应的吲哚衍生物; 而在少数情况下, 会生成二氢吲哚, 但其迅速被氧化为吲哚.
|
|
(1) |
同年, Soderberg等[10]首先由2-(2-硝基苯基)乙酸甲酯与1, 3, 5-三噁烷反应制得2-(2-硝基苯基)丙烯酸甲酯(10)(收率为83%), 然后在醋酸钯和三苯基膦作用下, 以一氧化碳为还原剂制备了3-吲哚甲酸甲酯(11)(收率为91%, Eq. 2).
|
|
(2) |
2015年, 张辉和曹卫国等[11]由苯胺12和炔烃13进行分子内的交叉脱氢偶联反应(CDC)制备2-全氟烷基- 3-吲哚甲酸甲酯14 (Eq. 3).该方法条件温和高效, “一锅法”制备, 在钯催化的CDC反应中只需要氧气作为最终的氧化剂.
|
|
(3) |
2011年, Alper等[12]发现了一种以一氧化碳为还原剂, 以离子二胺铑配合物为催化剂的2-乙烯基硝基芳烃15的N-环化反应来合成吲哚衍生物16的方法(Eq. 4).这一催化体系可以直接用来合成3-吲哚羧酸酯类化合物.该方法具有产率高、底物适用范围广等优点.
|
|
(4) |
2016年, 林爱俊和姚和权等[13]由N-(2-嘧啶基)苯胺17和2-重氮基乙酰乙酸酯18合成出了一系列的1-嘧啶基-吲哚-3-羧酸酯19 (Eq. 5).在这一催化体系中, 对于区域选择性的合成出吲哚-3-羧酸酯, 嘧啶基起着至关重要的作用.在所得产物中, 再次利用嘧啶基的导向作用, 可以进行吲哚C(7)位的官能化反应.另外, 在温和的反应条件下, 可以很容易的脱除掉导向基团嘧啶基得到游离的N-H吲哚羧酸酯.
|
|
(5) |
同年, 王柏全等[14]几乎同时报道了合成嘧啶基-3-吲哚羧酸酯19 (Eq. 5)的方法, 所用原料同为N-(2-嘧啶基)苯胺17和2-重氮基乙酰乙酸酯18, 不同的是, 王柏全小组直接用Cp*Rh(MeCN)3](SbF6)2作为催化剂, 不需再加入额外的AgSbF6, 其催化效率相当, 所得产物的产率也基本相同.两个小组均用氘作为标记, 考察了反应的决速步骤和反应过程, 推测了可能的反应机理.
2017年, Mutoh和Saito等[15]报道了一种钌催化的炔烃分子内环化反应, 这也是首次关于过渡金属催化的2-炔基苯胺20通过1, 2-碳迁移合成3-取代吲哚21的报道(Eq. 6).该反应条件温和, 收率较高, 具有较好的底物普适性.文中提出了可能的反应机理, 关于反应过程中1, 2-碳迁移的具体机理还在进一步研究中.
|
|
(6) |
2017年, 万小兵等[16]以N, N-二甲基甲酰胺和乙醇的混合溶液为溶剂, 偶氮二氰基戊酸为引发剂, 由芳胺衍生物22和重氮化合物23在四水合醋酸锰催化作用下发生自由基-卡宾偶联反应(RCC反应)合成出3-吲哚羧酸酯类化合物24 (Eq. 7).该方法条件温和、操作简便、所使用的催化剂价格较低.
2015年, 张小祥等[17]在阳离子金催化剂t-BuX-PhosAuNTf2的催化条件下, 成功实现了2-炔基叠氮25与羧酸合成3-吲哚酯26的串联反应(Eq. 8).该反应条件温和、底物普适性广、收率良好.并推测了可能的反应过程, 反应涉及金催化2-炔基叠氮产生α-亚胺金卡宾与羧酸的串联反应.
|
|
(7) |
|
|
(8) |
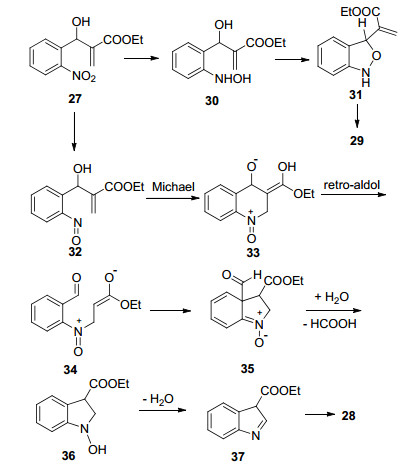
2007年, Kim等[18]以Baylis-Hillman的产物27为原料, 利用SnCl2为还原试剂, 1, 4-二氧六环为溶剂, 在加热回流条件下, 完成了化合物28的合成(Eq. 9).但该反应的产率不高, 产率为40%~50%, 会生成副产物29.通过改变反应的溶剂、温度以及SnCl2的用量, 产率都没有得到提高.并推测了反应机理.
|
|
(9) |
其可能的反应机理为(Scheme 2):首先SnCl2将27中的硝基还原为羟胺30, 再发生苄位的取代合环得到中间体31, 双键重排即可生产副产物29.
原料27还原为亚硝基中间体32, 再经Michael加成、逆aldol过程得到活泼中间体34, 34中的烯醇负离子在亚硝酰阳离子的吸电子作用下与芳环发生合环反应得到中间体35, 再经分子内的氧化还原、脱羧得到化合物36, 脱水、双键转移即可得到目标产物28.
2010年, Fournier等[19]由2-氨基苯甲醛38和重氮基乙酸乙酯39在路易斯酸作用下, 完成了3-吲哚甲酸乙酯40的制备(Eq. 10).该反应条件温和, 产率高, 反应底物易得.该反应中, 苯胺中氮原子上的保护基可以为苄基、对甲氧基苄基和甲基等烷基保护基.当芳环上有吸电子基团时反应产率较高.此反应芳环上取代基为溴和碘时, 可以通过交叉偶联反应进行官能团取代来得到更多的重要化合物.
|
|
(10) |
1995年, Mahavir等[20]首先通过烯胺41的官能团化, 然后再通过氢化, 合成了一系列3位取代的吲哚衍生物43 (Scheme 3).这一方法是Leimgruber-Batcho合成方法的新应用.
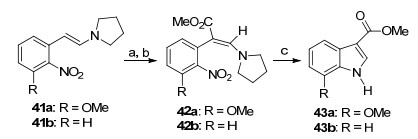
Reagents and conditions: (a) COCl2, Et3N, toluene, -20 ℃ to r.t.; (b) MeOH, -10 ℃ to r.t.; (c) H2, 10% Pd/C, MeOH, EtOAc, CH3-CO2H, r.t.
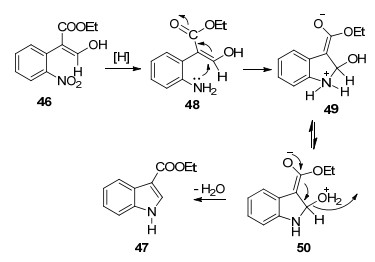
2006年, Hossain等[21]由易得的邻硝基苯甲醛44通过两步反应合成了吲哚-3-羧酸乙酯47 (Scheme 4).该方法中涉及的成环反应与传统的Reissert吲哚合成方法相似.该方法高效、简单、区域选择性好, 并且收率较高.
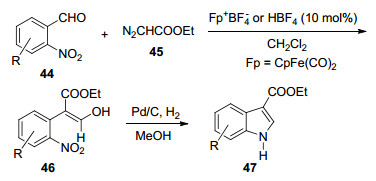
提出了第二步的反应机理(Scheme 5):硝基化合物46经催化氢化还原为苯胺48, 经Michael加成得到吡咯环中间体49, 分子内氢转移再经脱水即可得到吲哚- 3-羧酸乙酯47.
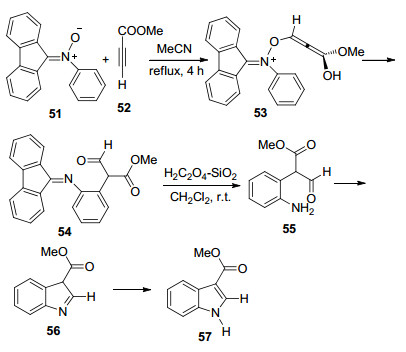
2015年, Radhamani和Prathapan等[22a]由易得的硝酮51和缺电子的乙炔52制备了3-取代吲哚57 (Scheme 6).具体的反应过程为:化合物51与化合物52首先发生加成反应生成化合物53, 再经迁移重排生成化合物54, 化合物54在酸催化下亚胺水解得到化合物55, 再发生分子内的胺-羰基脱水缩合生成化合物56, 化合物56经重排反应得到吲哚C(3)位酯化的产物57.该方法底物拓展性较好, 当化合物51中与氮原子相连的苯环上或者化合物52的炔基端碳有取代基时, 可以在吲哚环的其他位置引入取代基.
2017年, 雷爱文等[22b]开发了电催化N-芳基烯胺58的分子内脱氢环化反应合成3-吲哚羧酸酯59的新方法(Eq. 11).反应过程中由碘化钾作为电解质, 官能团耐受性较好.该方法属于环境友好型的合成方法, 在无化学氧化剂、无过渡金属存在下, 产物的收率依然很高.
|
|
(11) |
3-吲哚羧酸酯类化合物最传统的合成方法是由吲哚-3-羧酸或者吲哚-3-酰氯与醇反应来制备[23].近年来, 金属催化或非金属条件下的吲哚C(3)—H活化成为更加高效的合成3-吲哚羧酸酯的方法, 该类方法以一氧化碳、四溴化碳、氯甲酸酯等为酯羰基的C来源.
2011年, 夏春谷和李福伟等[24]以双(1, 5-环辛二烯氯化铑)为催化剂, 以过硫酸钾为氧化剂, 由吲哚60与一氧化碳、醇在甲苯中反应生成3-吲哚羧酸酯类化合物61 (Eq. 12).对于多种取代的吲哚, 直链或是环状醇均能够在此条件下反应.该方法反应条件温和, 具有很高的区域选择性.并提出了可能的反应机理, 催化剂双(1, 5-环辛二烯氯化铑)首先被氧化为三价铑, 然后在吲哚3位进行亲电子金属化反应, 形成C—Rh键, 随后, CO插入到C—Rh键中, 再与醇配位; 最后经过还原消除得到产物61, 并再生了催化剂.
|
|
(12) |
夏春谷和李福伟等[24, 25]发现, 上述方法(Eq. 12)具有一定的缺点, 例如, 铑催化剂价格昂贵、N-未保护的吲哚底物生成3-吲哚羧酸酯类化合物的收率较低. 2012年, 该小组[25]又报道了以醋酸钯为催化剂, 在碘/碳酸钾存在下, 由吲哚及其衍生物60与一氧化碳、醇在N, N-二甲基甲酰胺(DMF)溶液中反应得到相应的3-吲哚羧酸酯类化合物61 (Eq. 13).该方法底物拓展性好、产率较高.利用该方法可以由简单易得的吲哚和托品来合成具有生物活性的托烷司琼及其衍生物.
|
|
(13) |
2011年, 雷爱文小组[26]以二(三苯基膦)二氯化钯、三苯基膦和醋酸铜为催化剂, 甲苯和二甲基亚砜为混合溶剂, 以空气为氧化剂, N-甲基吲哚及取代的吲哚62与脂肪族伯醇或仲醇、一氧化碳发生反应制备相应的吲哚-3-羧酸酯衍生物63 (Eq. 14).当吲哚的N—H未经取代时, 在此反应条件下, 得到的是相应的氨基羧酸酯的产物.该方法区域选择性好, 且氧化剂空气属于环境友好型氧化剂.
|
|
(14) |
2015年, Ceroni等[27]以四溴化碳65和甲醇为溶剂, 由可见光诱导直接进行位点选择性的吲哚的C—H官能团化反应(Eq. 15).该反应所需条件温和, 7 W蓝色LED光照射下室温数小时即可完成反应.
|
|
(15) |
2016年, Tanaka和Hattori等[28]以甲苯和正己烷为混合溶剂, 在二甲基氯化铝存在下, 由取代的吲哚67与氯甲酸乙酯反应得到了一系列3-吲哚甲酸乙酯的衍生物68 (Eq. 16).吲哚环氮原子的保护基可以为甲基、三异丙基硅烷基和苄基, 该方法对于吲哚环的2位和5位有取代基的底物收率较高.
|
|
(16) |
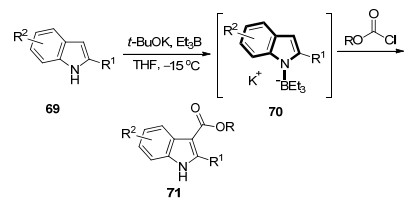
2016年, 本课题组[29]以吲哚69为原料, 通过碱性条件下与三乙基硼反应制得活泼的吲哚N-B中间体70, 再与亲电试剂氯甲酸酯反应可快速得到一系列3-吲哚羧酸酯类化合物71 (Scheme 7).该反应收率最高可达93%, 底物拓展性好, 氯甲酸甲酯、氯甲酸烯丙酯、氯甲酸苄酯与吲哚及取代的吲哚均能够在温和的条件下反应.更重要的是, N-B中间体70是非常活泼的中间体, 极大地提高了吲哚C-3位的反应活性和选择性, N—B键的存在还可以作为氮原子临时的保护基, 不仅避免了氮原子上可能的竞争反应, 反应结束后常规的后处理就可以得到氮原子上无取代的吲哚羧酸酯类化合物, 显示出了较高的合成效率.
综上所述, 现代合成技术的发展尤其是金属催化的C—H官能团化的迅速发展, 使得合成3-吲哚羧酸酯类化合物更加的简便、高效.过渡金属Pd、Rh、Ru、Mn、Au以及路易斯酸等催化的C—H活化/合环反应均被成功地应用到了重要的3-吲哚羧酸酯类化合物的合成中.而直接在吲哚C-3位进行金属催化或非金属条件下的官能团化来合成此类活性分子也有其合成优势.合成方法的多样性将极大地促进相关天然产物、药物分子以及活性分子的合成效率.但在某些方法中, 仍然存在原料或催化剂价格昂贵、反应条件苛刻、反应收率较低以及官能团兼容性差等不利因素.这将促使化学家们努力探索更加高效、绿色、经济的合成方法, 以促进天然产物及药物分子的合成效率.
Somei, M.; Yamada, F. Nat. Prod. Rep. 2003, 34, 761. https://www.sciencedirect.com/science/article/pii/S0304416516305153
Zhao, C.; Zhao, Y.; Chai, H.; Gong, P. Bioorg. Med. Chem. 2006, 14, 2552. doi: 10.1016/j.bmc.2005.11.033
Boriskin, Y. S.; Leneva, I. A.; Pecheur, E. I.; Polyak, S. J. Curr. Med. Chem. 2008, 15, 997. doi: 10.2174/092986708784049658
Sheridan, R. P.; Korzekwa, K. R.; Torres, R. A.; Walker, M. J. J. Med. Chem. 2007, 50, 3173. doi: 10.1021/jm0613471
Augelli-Szafran, C. E.; Jaen, J. C.; Morelang, D. W.; Nelson, C. B.; Penvose-Yi, J. R.; Schwarz, R. D. Bioorg. Med. Chem. Lett. 1998, 8, 1991. doi: 10.1016/S0960-894X(98)00351-5
Schultz, E. E.; Pujanauski, B. G.; Sarpong, R. Org. Lett. 2012, 14, 648. doi: 10.1021/ol203302f
Taber, D. F.; Tirunahari, P. K. Tetrahedron 2011, 67, 7195. doi: 10.1016/j.tet.2011.06.040
Wurtz, S.; Rakshit, S.; Neumann, J. J.; Droge, T.; Glorius, F. Angew. Chem., Int. Ed. 2008, 47, 7230. doi: 10.1002/anie.v47:38
Sole, D.; Serrano, O. J. Org. Chem. 2008, 73, 2476. doi: 10.1021/jo800034m
Söderberg, B. C. G.; Banini, S. R.; Turner, M. R.; Minter, A. R.; Arrington, S. K. Synthesis 2008, 903.
Shen, D.; Han, J.; Chen, J.; Deng, H.; Shao, M.; Zhang, H.; Cao, W. Org. Lett. 2015, 17, 3283. doi: 10.1021/acs.orglett.5b01479
Okuro, K.; Gurnham, J.; Alper, H. J. Org. Chem. 2011, 76, 4715. doi: 10.1021/jo200320k
Jiang, H.; Gao, S.; Xu, J.; Wu, X.; Lin, A.; Yao, H. Adv. Synth. Catal. 2016, 358, 188. doi: 10.1002/adsc.201500769
Yu, K.; Liang, Y.; Li, B.; Wang, B. Adv. Synth. Catal. 2016, 358, 661. doi: 10.1002/adsc.201500954
Watanabe, T.; Mutoh, Y.; Saito, S. J. Am. Chem. Soc. 2017, 139, 7749. doi: 10.1021/jacs.7b04564
Li, H.; Zhao, Y.; Ma, L.; Ma, M.; Jiang, J.; Wan, X. Chem. Commun. 2017, 53, 5993. doi: 10.1039/C7CC02440A
张小祥, 孙小萍, 张海飞, 崔杏丽, 马猛涛, 有机化学, 2015, 35, 1469. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201507008&dbname=CJFD&dbcode=CJFQZhang, X.-X.; Sun, X.-P.; Zhang, H.-F.; Cui, X.-L.; Ma, M.-T. Chin. J. Org. Chem. 2015, 35, 1469(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201507008&dbname=CJFD&dbcode=CJFQ
Lee, K.-Y.; Lee, H.-S.; Kim, J.-N. Bull. Korean Chem. Soc. 2007, 28, 333. doi: 10.5012/bkcs.2007.28.2.333
Levesque, P.; Fournier, P. A. J. Org. Chem. 2010, 75, 7033. doi: 10.1021/jo1016713
Mahavir, P.; Vecchia, L. L.; Kapa, P.; Oljan, R. Synth. Commun. 1995, 25, 95. doi: 10.1080/00397919508010793
Islam, M. S.; Brennan, C.; Wang, Q.; Hossain, M.-M. J. Org. Chem. 2006, 71, 4675. doi: 10.1021/jo0601821
(a) Natarajan, R. ; Rappai, J. ; Unnikrishnan, P. ; Radhamani, S. ; Prathapan, S. Synlett 2015, 26, 2467.
(b) Tang, S. ; Gao, X. ; Lei, A. Chem. Commum. 2017, 53, 3354.
Peterson, P.; Iii, J. W.; Niemann, C. J. Org. Chem. 1958, 23, 303. doi: 10.1021/jo01096a606
Lang, R.; Wu, J.; Shi, L.; Xia, C.; Li, F. Chem. Commun. 2011, 47, 12553. doi: 10.1039/c1cc15143f
Lang, R.; Shi, L.; Li, D.; Xia, C.; Li, F. Org. Lett. 2012, 14, 4130. doi: 10.1021/ol3017726
Zhang, H.; Liu, D.; Chen, C.; Liu, C.; Lei, A. Chem.-Eur. J. 2011, 17, 9581. doi: 10.1002/chem.v17.35
Yang, Q.-Q.; Marchini, M.; Xiao, W.-J.; Ceroni, P.; Bandini, M. Chem.-Eur. J. 2015, 21, 18052. doi: 10.1002/chem.201503787
Nemoto, K.; Tanaka, S.; Konno, M.; Onozawa, S. Tetrahedron 2016, 72, 734. doi: 10.1016/j.tet.2015.12.028
Zhang, Z.-W.; Xue, H.; Li, H.; Kang, H.; Feng, J.; Lin, A. Org. Lett. 2016, 18, 3918. doi: 10.1021/acs.orglett.6b01970

图 1 3-吲哚羧酸酯类药物分子和天然产物
Figure 1 Drug molecules and natural products containing 3-indolecarboxylates

Scheme 1 烯胺羧酸酯6用来合成3-吲哚羧酸酯7
Scheme 1 Synthesis of 3-indolecarboxylate 7 from enamine ester 6

Scheme 3 Leimgruber-Batcho合成方法的新应用
Scheme 3 New application of Leimgruber-Batcho synthesis
Reagents and conditions: (a) COCl2, Et3N, toluene, -20 ℃ to r.t.; (b) MeOH, -10 ℃ to r.t.; (c) H2, 10% Pd/C, MeOH, EtOAc, CH3-CO2H, r.t.

Scheme 4 由邻硝基苯甲醛合成吲哚-3-羧酸乙酯
Scheme 4 Synthesis of indole-3-carboxylic acid ethyl ester from o-nitrobenzaldehyde

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
















 下载:
下载:



