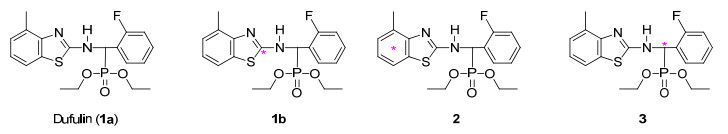
毒氟磷(Dufulin, 1a , 图 1 [1 .近年来, 对于毒氟磷的研究主要集中于其作用机理、结构优化、田间药效和毒性等方面的研究, 对其在土壤和水体中的环境毒理学研究鲜见报道[2 6 .迄今, 对毒氟磷的代谢、残留和环境行为等仍缺乏深入的研究[7 .放射性同位素示踪技术具有痕量精准、溯源追踪、直观简便等独特优势, 是其他任何技术所不可替代的, 在国际上被广泛应用于农医药等化学品的代谢途径、作用机理、环境行为与归宿等研究[8 17 .为满足国际农药安全评判试验标准和登记要求(与我国2017年11月份新实施的标准和要求一致), 必须借助于放射性同位素示踪技术开展毒氟磷在生物(植物、哺乳动物和家禽)体内的代谢、残留及其在土壤中的环境行为等研究, 旨在为毒氟磷安全评判提供更丰富的试验数据, 进而推动毒氟磷走向国际市场[7 .
图 1
在前期研究中, 笔者以[14 C]碳酸钡为同位素原料, 制备了高比活度[噻唑基-2-14 C]毒氟磷(1b )[7 .毒氟磷(1a )含3个芳环片段, 从化学稳定性、代谢稳定性和毒理学重要性的角度考虑, 还需进一步对1a 中2个苯环进行标记.对1a 中含氟苯环片段而言, 苯环碳-14标记合成路线长, 费用高; 考虑到农医药等化学品分子中与芳环直接相连的碳原子在示踪过程中通常比较稳定而不易脱落.因而, 本研究以苄基碳标记(3 )代替对苯环的标记, 以降低成本.本文报道了作为示踪剂使用的两种放射性同位素碳-14标记毒氟磷2 , 3 的合成(Schemes 1 2
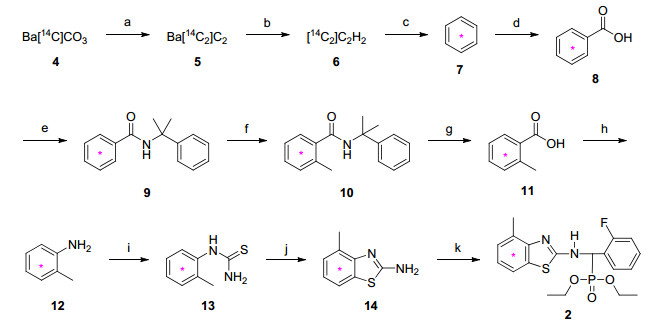
图式 1
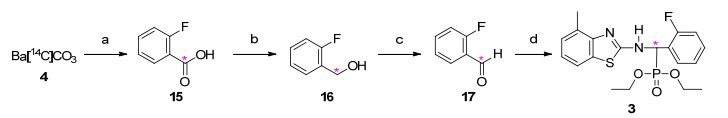
图式 2
1.
结果与讨论
在反应方法筛选、合成工艺优化和探索性放射性合成基础上, 以[14 C]碳酸钡为放射性同位素原料, 通过乙炔环三聚、苯羧基化和甲基化、环化、胺化、亲核取代、格氏、还原、氧化等反应, 分别经11步、5步反应制备了2种放射性同位素碳-14标记毒氟磷[N -[2-(4-甲基[苯基-U-14 C6 ]苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基甲基膦酸酯(2 , 20.6 mCi)和N -[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基[14 C]甲基膦酸酯(3 , 32.4 mCi)], 反应总放化收率/化学收率分别为31%和67%, 其结构经1 H NMR和在线放射性高效液相色谱-二极管阵列检测器/质谱联用(HPLC-FSA/PDA/MS, 简称HFPM)分析确认.放射性薄层层析-同位素成像分析(TLC-IIA, Isotope Imaging Analysis)、离线放射性高效液相色谱(HPLC-LSC, Liquid Scintillation Counter)、HFPM和LSC分析表明, 2 和3 的放化纯度和化学纯度均大于98%, 其比活度(SA, Specific Activity)分别为25.5, 55.5 mCi/ mmol.两种标记物的质量指标完全满足放射性同位素示踪试验的要求, 可作为示踪剂, 用于毒氟磷的代谢、残留和环境行为等研究.
在农医药等化学品同位素标记合成中, 首先要考虑标记核素(碳-14, 氚等)和标记位置的选择.氚标记物易制备, 氚单一位置标记物的理论最大比活度为28.77 Ci/mmol, 但氚标记物储存超过半年会因衰变和辐射自分解而引起放化纯度和比活度的显著降低.前期研究经验表明, 氚在示踪过程中易与研究对象介质中氢原子发生一定程度的交换而脱落, 造成研究对象介质污染, 最终导致对代谢物的追踪溯源困难而易造成误判, 也难以精确定量代谢物的含量[12 15 .相比较而言, 碳是农医药等化学品的主要构成元素; 碳-14半衰期长, 其单一位置标记物的理论最大比活度为62.41 mCi/mmol, 这足以满足示踪试验的要求.因而, 在农医药等化学品的标记中优先考虑以碳-14作为标记核素[9 12 .通常选择化学品分子中具备结构和代谢稳定性的骨架碳原子进行标记, 其优点是核素标记牢固, 不易脱落.因而, 本研究以碳-14对毒氟磷分子中苯并噻唑片段中的苯环(2 )和苄基碳(3 )进行标记.
在放射合成中, 含碳-14合成砌块国际市场价格昂贵且种类极少.本研究所需原料[U-14 C6 ]苯(7 )和2-氟[羰基-14 C]苯甲酸(15 )用量大, 定制价格高, 供货期长, 这成为本研究的瓶颈.故笔者从易得的[14 C]碳酸钡出发, 在高温下将其转化为[14 C2 ]碳化钡, 水解获得了[14 C2 ]乙炔, 经环三聚反应制备了高比活度含碳-14合成砌块[U-14 C6 ]苯(7 , SA>300 mCi/mmol).该砌块的成功制备使本研究摆脱了对国际市场原料供应的限制, 也为后续开展高比活度含[U-14 C6 ]苯环片段标记物的合成奠定了基础.为降低成本, 本研究在满足示踪试验要求的前提下, 将其SA调至约25.5 mCi/mmol而开展后续工作.
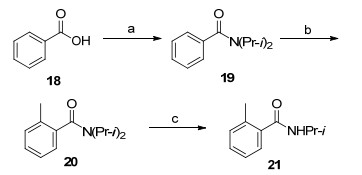
为在[苯基-U-14 C6 ]苯甲酸(8 )的羧基邻位引入甲基, 本研究将8 转化为酰胺9 , 利用酰胺基对苯环锂化的导向作用实现羧基邻位锂化, 通过碘甲烷(MeI)亲电进攻苯环而引入甲基, 脱去α -甲基苯乙烯而获得邻甲[苯基-U-14 C6 ]苯甲酸(11 ).冷实验表明, MeI务必在尽可能短时间内完成加料, 否则体系会产生非放10 苄基位甲基化产物、苄基和氮同时甲基化产物, 导致非放10 收率急剧降低.在冷反应中, 笔者曾尝试以二异丙基胺将苯甲酸转化为N , N -二异丙基苯甲酰胺(19 ), 利用s -BuLi、四甲基乙二胺(TMEDA)和MeI在19 苯环羰基的邻位引入甲基, 高收率获得了N , N -二异丙基邻甲苯甲酰胺(20 , Scheme 3 [18 19 .尽管以19 为模型底物可高效地脱去二异丙氨基而获得苯甲酸, 但经过多种条件尝试发现20 易脱去一个异丙基而获得21 , 难以脱去第二个异丙基, 因而放弃该路线.
图式 3
叠氮磷酸二苯酯(DPPA)将11 转化为2-甲基[苯基-U-14 C6 ]苯甲酰基叠氮, 在加热、避光条件下转为2-异氰酸甲[苯基-U-14 C6 ]苯酯, 与叔丁醇反应产生叔丁基(邻甲基[苯基-U-14 C6 ]苯基)氨基甲酸酯, 水解转化为2-甲基[苯基-U-14 C6 ]苯胺(12 ).在冷反应中, 曾尝试从苯出发, 经硝化、还原、乙酰化、乙酰氨基邻位碘化[20 和甲基化反应[21 获得邻甲基苯胺(Scheme 4
图式 4
在工业合成中, 非放14 与2-氟苯甲醛缩合得亚胺, 进而与亚磷酸二乙酯亲核加成得到毒氟磷, 反应副产物水用甲苯蒸馏带出而促进反应完全[2 .在放射合成中, 反应为微量反应, 因而改用预处理的无水硫酸钠除水以促进反应彻底, 其优点是吸水能量大且对标记物吸附量少, 分别以83%和74%收率获得碳-14标记毒氟磷2 和3 .
在2-氟[羰基-14 C]苯甲酸(15 )制备中, 反应中微量的二氧化碳会引起[14 C]CO2 比活度急剧降低, 导致标记合成失败.本研究利用自制的集成式微型放射性二氧化碳反应系统高收率获得15 , 这表明该系统密封性好, 能确保其中二氧化碳被彻底排出, 又能防止空气进入而有效地防止产品SA降低, 促进[14 C]CO2 被高效地转化.
通常碳-14标记物的1 H NMR数据与对应非标记物的一致性, 但两者的质谱有所不同.标记物2 是苯环上6个碳原子均标记, 其比活度为25.5 mCi/mmol, 远低于苯环均标记物的理论最大比活度, 因而2 属于低比活度标记物.除加速器质谱之外, 常规质谱难以检测到2 中14 C信号特征[22 .根据HFPM分析中毒氟磷对应HPLC洗脱组分的放射性来间接确定2 中14 C的存在, 这与笔者前期鉴定低比活度碳-14标记丙酯草醚结构的方法相同[9 10 .标记物3 均为单碳定位标记, 其比活度为55.5 mCi/mmol, 接近单碳定位标记物的理论最大比活度, 因而3 属于高比活度标记物, 常规质谱易检测到14 C信号特征(标记物相对分子量比毒氟磷大2)[7 .
放射性同位素碳-14标记物可靠的质量指标是示踪实验成败的关键, 尤其是放化纯度.本研究中采用TLC-IIA, HPLC-LSC和HPLC-FSA (在线放射性高效液相色谱)三种方法测定了2种标记物的放化纯度, 其结果相互补充印证[7 9 12 .分析表明, 2 和3 的放化纯度均大于98%.为了减少放射性物质不必要的损失, 通常高比活度碳-14标记物(SA≥50 mCi/mmol)的化学纯度根据HPLC-UV色谱图以面积归一法计算; 而对于低比活度标记物(SA: 1~10 mCi/mmol), 可考虑用外标法或内标法测定其化学纯度[7 . HPLC-UV分析表明, 2 和3 的化学纯度分别为98.7%和99.1%.
2.
结论
以[14 C]碳酸钡为放射性同位素原料, 通过乙炔环三聚、苯羧基化和甲基化、环化、亲核取代、格氏、还原、氧化等反应, 分别通过11步、5步反应分别制备2种放射性同位素碳-14标记毒氟磷[N -[2-(4-甲基[苯基-U-14 C6 ]苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基甲基膦酸酯(2 )和N -[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基[14 C]甲基膦酸酯(3 )], 标记物的质量指标满足放射性同位素示踪研究的要求, 可作为放射性示踪剂, 用于毒氟磷的代谢、残留和环境行为等研究.
3.
实验部分
3.1
仪器与试剂
放射性[14 C]碳酸钡(SA: 58.0 mCi/mmol; 放化纯度: 99.9%;化学纯度>99.8%)购自美国ARC公司; 闪烁体2, 5-二苯基噁唑(HPLC>99%)和1, 4-双(5-苯基-2-噁唑)苯(HPLC>98%)均购自日本TCI公司, LSC用闪烁液自行配置[9 ; FSA用PerkinElmer Optiphase HiSafe 3闪烁液; HPLC和HPLC-MS所用Fisher公司的甲醇、乙腈分别为色谱级和质谱级; 毒氟磷标样用工业品经多次重结晶自行制备, 由上海启甄环境科技有限公司代谢安评中心GLP实验室分析认可; 其它试剂均为市售分析纯; 溶剂按照文献[23
上海仝科TCGC-1700高温水平管式电炉; Agilent 7890B GC-5977B MS气质联用系统; Waters Alliance e2695 HPLC-2489 UV/Acquity QDa MS液质联用系统; Waters Alliance e2695 HPLC-AIM ν. ARC FSA/Waters 2998 PDA/Acquity QDa MS多检测器联用分析系统(FSA与PDA并联; MS串联于PDA后); Waters 2545 HPLC-2998 PDA制备液相色谱系统(配备fraction collector Ⅲ); Waters Acquity Arc-Xevo G2-XS QTof高分辨质谱; Varian 400 MHz核磁共振仪; PerkinElmer Tri-Carb 4910TR液体闪烁测量仪; Bioscan TLC薄层放射性扫描仪; GE Typhoon FLA9500 IP多功能激光成像仪; Milli-Q Reference S. Kit (18.2 MΩ/cm, 25 ℃)超纯水制备系统; Sartorius BSA22 4S-CW (1 mg)和BT25S (0.01 mg)电子天平.以上与定量分析相关仪器均通过3Q认证和上海市计量测试技术研究院校准.
3.2
放射性同位素标记物的合成
3.2.1
[U-14 C6 ]苯(7 )的合成
以[14 C]碳酸钡(4 , 1393 mg, 406.0 mCi, 58.0 mCi/mmol)为放射性同位素原料, 在高温下将其转化为[14 C2 ]碳化钡(5 , 542.9 mg, 381.6 mCi, 94%), 水解变为[14 C2 ]乙炔(6 , 97 mg, 374.0 mCi, 98%), 利用自行设计的乙炔环三聚装置制备了[U-14 C6 ]苯(7 , 81 mg, 332.9 mCi, 89%)[24 28 .采用GC-MS和LSC跟踪反应.用无水苯将7 的SA调低至约25.5 mCi/mmol后备用. 1 H NMR (400 MHz, CDCl3 ) δ : 7.36 (s, 6H). HFPM分析显示, 标记物7 的HPLC-UV色谱保留时间与HPLC-FSA色谱中保留时间一致, 且两者与相同色谱条件下苯标样的保留时间一致. HFPM分析条件: Diamonsil C18柱, 5 μm, 4.6 mm×150 mm(美国迪马公司), 流速0.80 mL/min, 波长254 nm, 进样量10 μL; 梯度洗脱(min/% A)控制: 0/10, 5/10, 25/100, 30/100; A为甲醇, B为水+0.1%甲酸; HPLC柱后洗脱液分别进入FSA和PDA的分流比为1/1;进入FSA检测器中洗脱液与Optiphase HiSafe 3闪烁液的流速比为1/8.下文如无说明均使用此条件.
3.2.2
[苯基-U-14 C6 ]苯甲酸(8 )的合成
在室温和CO氛围下, 将三氟乙酸(9 mL)加入过硫酸钾(1737 mg, 6435 μmol)和醋酸钯(289 mg, 1287 μmol)的混合物中, 剧烈搅拌, 然后加入[U-14 C6 ]苯(7 , 203 mg, 65.6 mCi, 25.5 mCi/mmol).在常压下继续剧烈搅拌10 h[29 . HFPM监测显示7 完全转化.浓缩反应液至干, 加水(50 mL), 用NaOH水溶液(2 mol•L-1 )调节pH至13, 水相用甲基叔丁基醚(MTBE)萃取(50 mL×4)除去杂质, 用浓盐酸调pH至1, 乙酸乙酯萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 浓缩得白色固体8 (275 mg, 57.1 mCi, 87%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 12.96 (s, 1H), 7.97~7.92 (m, 2H), 7.65~7.59 (m, 1H), 7.53~7.46 (m, 2H); ESI-MS m /z : 123 [M+H]+ .
3.2.3
N -(2-苯基-2-丙基)[苯基-U-14 C6 ]苯甲酰胺(9 )的合成
在氩气保护下, 将[苯基-U-14 C6 ]苯甲酸(8 , 274 mg, 56.9 mCi)加入二氯亚砜(5 mL)中, 搅拌, 回流2 h, 降至室温, 减压蒸掉二氯亚砜得[苯基-U-14 C6 ]苯甲酰氯, 直接用于下一步反应.
在氩气保护下, 将无水α , α -二甲基苄胺(308 mg, 2278 μmol)、无水三乙胺(518 mg, 4466 μmol)溶于干燥二氯甲烷(10 mL)中, 冷却至0 ℃; 将该溶液缓慢滴入搅拌的[苯基-U-14 C6 ]苯甲酰氯干燥二氯甲烷溶液(5 mL)中, 保持温度低于10 ℃.滴加完毕后, 升温至25 ℃, 搅拌4 h. HFPM监测显示体系仅有1个放射性物质9 .向反应体系加水(80 mL), 二氯甲烷萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 过滤, 浓缩, 快速柱层析[V (二氯甲烷):V (石油醚)=1:1~2:1]得白色固体9 (507 mg, 54.1 mCi, 95%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 8.44 (s, 1H), 7.84 (d, J =7.3 Hz, 2H), 7.55~7.34 (m, 6H), 7.28 (t, J =7.5 Hz, 2H), 7.20~7.13 (m, 1H), 1.67 (s, 6H); ESI-MS m /z : 262 [M+Na]+ .
3.2.4
N -(2-苯基-2-丙基)-2-甲基[苯基-U-14 C6 ]苯甲酰胺(10 )的合成
在氩气保护下, 将干燥TMEDA (785 mg, 6765 μmol)加入到N -(2-苯基-2-丙基)[苯基-U-14 C6 ]苯甲酰胺(9 , 505 mg, 53.9 mCi)的无水四氢呋喃溶液(7 mL)中, 搅拌, 降温至-78 ℃, 缓慢滴加s -BuLi (6.8 mL, 1.0 mol•L-1 ), 控制温度低于-65 ℃.滴加完毕搅拌10 min后, 在-40 ℃下搅拌1 h.反应再次降温至-78 ℃, 将MeI (660 mg, 4651 μmol)快速(<5 s)加入反应体系, 反应因放热而升温至-55 ℃, 在此温度下搅拌0.5 h. HFPM监测显示9 完全转化.将反应液转入冰-水中, 乙酸乙酯萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 浓缩, 快速柱层析[V (乙酸乙酯):V (石油醚)=1:8~1:4]得白色固体10 (465 mg, 46.9 mCi, 87%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 8.48 (s, 1H), 7.46~7.40 (m, 2H), 7.36~7.16 (m, 7H), 2.29 (s, 3H), 1.63 (s, 6H); ESI-MS m /z : 276 [M+Na]+ .
3.2.5
2-甲基[苯基-U-14 C6 ]苯甲酸(11 )的合成
在氩气保护下, 将N -(2-苯基-2-丙基)-2-甲基[苯基-U-14 C6 ]苯甲酰胺(10 , 464 mg, 46.8 mCi)溶于三氟乙酸(TFA) (8 mL), 升温至30 ℃, 搅拌1.5 h. HFPM监测显示10 彻底消耗.浓缩得2-甲基[苯基-U-14 C6 ]苯甲酰胺粗品, 用油泵抽真空以除去副产物α -甲基苯乙烯.
在氩气保护下, 将2-甲基[苯基-U-14 C6 ]苯甲酰胺溶于TFA (8 mL)中, 降温至-10 ℃, 在3 min内分4批加入NaNO2 (253 mg, 3672 μmol), 搅拌1 h, 反应液呈黑色; 升温至20 ℃, 搅拌2 h. HFPM监测显示体系仅有1个放射性物质11 .反应液浓缩至干, 用NaOH水溶液(2 mol•L-1 )调制pH至13, 水相用MTBE萃取(50 mL×4)除去杂质, 用浓盐酸调水相pH至1, 乙酸乙酯萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 浓缩得白色固体11 (227 mg, 42.6 mCi, 91%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 12.80 (s, 1H), 7.80 (d, J =7.6 Hz, 1H), 7.49~7.37 (m, 1H), 7.34~7.21 (m, 2H), 2.50 (s, 3H); ESI-MS m /z : 137 [M+H]+ .
3.2.6
2-甲基[苯基-U-14 C6 ]苯胺(12 )的合成
在氩气保护下, 将DPPA (530 mg, 1830 μmol)滴入2-甲基[苯基-U-14 C6 ]苯甲酸(11 , 226 mg, 42.4 mCi)、无水三乙胺(185 mg, 1830 μmol)的无水叔丁醇(3 mL)溶液中, 加热至85 ℃, 搅拌8 h. HFPM监测显示11 彻底消耗.反应降至室温, 加水(80 mL), 乙酸乙酯萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 过滤浓缩得中间体叔丁基-N -(2-甲基[苯基-U-14 C6 ]苯基)氨基甲酸酯粗品.向得到粗品中加入MeOH (7 mL)和浓盐酸(1 mL), 回流2 h. HFPM监测显示放射性中间体完全转化.将反应液冷至0 ℃, 用NaOH溶液(4 mol•L-1 )中和盐酸, 二氯甲烷萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 浓缩, 快速柱层析[V (二氯甲烷):V (石油醚)=1:2~3:1]得无色透明液体12 (160 mg, 38.2 mCi, 90%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 6.94~6.80 (m, 2H), 6.63~6.39 (m, 2H), 4.78 (s, 2H), 2.03 (s, 3H). HFPM分析显示, 12 的HPLC-UV色谱保留时间与HPLC-FSA色谱中保留时间一致, 且两者与相同色谱条件下邻甲苯胺标样的保留时间一致.
3.2.7
2-甲基[苯基-U-14 C6 ]苯基硫脲(13 )的合成
在氩气保护和避光条件下, 将苯甲酰基异硫氰酸酯(268 mg, 1645 μmol)缓慢滴入搅拌的2-甲基[苯基-U-14 C6 ]苯胺(12 , 160 mg, 38.1 mCi)无水四氢呋喃溶液(8 mL)中, 室温搅拌1.5 h[30 . HFPM监测显示12 完全消失.反应液浓缩至干, 加入甲醇(8 mL)、水(4 mL)和碳酸钾(620 mg, 4493 μmol), 升温至80 ℃, 搅拌12 h. HFPM监测显示中间体1-苯甲酰基-3-(2-甲基[苯基-U-14 C6 ]苯基)硫脲完全水解.浓缩除去反应液中大部分甲醇, 倒入冰-水(80 mL)中, 乙酸乙酯萃取(50 mL×4), 合并有机相, 干燥, 浓缩, 快速柱层析[V (乙酸乙酯):V (石油醚)=1:2~2:1]得2-甲基[苯基-U-14 C6 ]苯基硫脲(13 , 241 mg, 37.0 mCi, 97%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 9.22 (s, 1H), 7.71~6.72 (brs, 6H), 2.18 (s, 3H); ESI-MS m /z : 167 [M+H]+ .
3.2.8
2-氨基-4-甲基[苯基-U-14 C6 ]苯并噻唑(14 )的合成
在氩气保护下, 将二氯亚砜(5 mL)加入2-甲基[苯基-U-14 C6 ]苯基硫脲(13 , 174 mg, 36.8 mCi)中, 加热至50 ℃, 搅拌1.5 h. HFPM监测显示13 完全消失.反应降至室温, 将反应液倒入饱和碳酸氢钠水溶液(80 mL, 0 ℃)中, 乙酸乙酯萃取(50 mL×4), 合并有机相, 饱和食盐水洗涤, 干燥, 浓缩, 快速柱层析[V (乙酸乙酯):V (石油醚)=2:3~1:1]得白色固体14 (160 mg, 24.9 mCi, 93%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 7.53~7.39 (m, 3H), 7.06~7.00 (m, 1H), 6.94~6.87 (m, 1H), 2.41 (s, 3H); ESI-MS m /z : 165 [M+H]+ .
3.2.9
N -[2-(4-甲基[苯基-U-14 C6 ]苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基甲基膦酸酯(2 , [苯基-U-14 C6 ]毒氟磷)的合成
在氩气保护下, 将2-氨基-4-甲基[苯基-U-14 C6 ]苯并噻唑(14 , 160 mg, 24.8 mCi)无水甲苯(8 mL)溶液、2-氟苯甲醛(124 mg, 992 μmol)和亚磷酸二乙酯(205 mg, 1488 μmol)依次加入无水对甲苯磺酸(8 mg, 48.6 μmol), 无水硫酸钠(775 mg, 5456 μmol, 在350 ℃预干燥2 h后使用)和无水甲苯(8 mL)的混合物中, 氩气置换5次, 搅拌, 回流8 h[7 . HFPM监测显示14 完全消失.反应降至室温, 过滤, 浓缩, 制备型HPLC纯化得白色固体(2 , 329 mg, 20.6 mCi, 83%).色谱条件: xBridge Prep C18柱(10 μm, 150 mm×19 mm; Waters Co., MA, USA), 流速9.00 mL/min, 波长254 nm, 进样量600 μL; 梯度洗脱(min/% A)控制: 0/60, 5/60, 10/100, 15/100, 20/60, 25/60; A为乙腈, B为水.收集保留时间为11.48~13.68 min组分. 1 H NMR (400 MHz, CDCl3 ) δ : 7.64~7.59 (m, 1H), 7.38 (d, J =7.8 Hz, 1H), 7.32~7.26 (m, 1H), 7.15~7.07 (m, 3H), 7.00 (t, J =7.6 Hz, 1H), 6.59 (s, 1H), 5.80 (d, J =22.7 Hz, 1H), 4.30~4.19 (m, 2H), 4.07~4.00 (m, 1H), 3.89~3.81 (m, 1H), 2.53 (s, 3H), 1.33 (t, J =7.1 Hz, 3H), 1.14 (t, J =7.1 Hz, 3H; ESI-MS m /z : 409 [M+H]+ , 431 [M+Na]+ . HRMS calcd for C19 H23 FN2 O3 PS [M+H]+ 409.1151, found 409.1143.
3.2.10
2-氟[羰基-14 C]苯甲酸(15 )的合成
在氩气保护下, 将预干燥的自制微型集成式放射性二氧化碳反应系统(包含二氧化碳发生器、气体压力缓冲器、微型干燥器、二氧化碳反应器、微型气体循环泵、玻璃旋塞和连接管线等功能单元)固定在铁架上.向二氧化碳发生器中加入[14 C]碳酸钡(199 mg, 57.8 mCi, 58.0 mCi/mmol)和现烘碳酸钡(20 mg, 0.1 mmol), 密封所有管口.热风枪加热(250 ℃), 氩气置换5次; 继续加热, 油泵抽真空至系统内压力(0.07 Pa)稳定, 关闭系统的所有旋塞, 使系统处于高真空状态, 自然冷却至室温.
向二氧化碳反应器中加入邻甲苯基溴化镁四氢呋喃溶液(1 mol•L-1 , 1.5 mL)和脱除CO2 的无水四氢呋喃(14 mL).将二氧化碳发生器和二氧化碳反应器冷至-10~-5 ℃, 搅拌, 向二氧化碳发生器中缓慢滴加脱除CO2 的浓硫酸(5 mL), 产生的[14 C]CO2 经微型干燥器(P2 O5 )干燥后进入二氧化碳反应器与格氏试剂反应.经30 min后二氧化碳反应器液面无明显气泡溢出.接通二氧化碳反应器和二氧化碳发生器之间气体回路上的旋塞, 启动微型气体循环泵, 将二氧化碳发生器升温至50 ℃; 向系统补充适量的干燥氩气, 以氩气运载系统中残余[14 C]CO2 与格氏试剂反应.经GC-MS和气体放射性检测, 氩气循环2 h后[14 C]CO2 彻底消耗.以NaOH溶液(0.5 mol•L-1 )淬灭反应, 调溶液pH至13, 在45 ℃减压蒸除四氢呋喃, 余液加水至80 mL, 用MTBE (60 mL)萃取除去杂质; 以稀盐酸调水相pH至2~3, 乙酸乙酯萃取(60 mL×6), 合并有机相, 饱和食盐水洗涤, 干燥, 过滤, 浓缩得白色固体15 (142 mg, 55.4 mCi, 两步收率为96%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 13.24 (brs, 1H), 7.91~7.83 (m, 1H), 7.69~7.60 (m, 1H), 7.37~7.23 (m, 2H); ESI-MS m /z (%): 143 [M+H]+ , 125 (100), 142 (M+ , 71), 95 (42), 75 (22), 50 (8), 124 (8), 69 (6), 76 (6).
3.2.11
(2-氟苯基)[14 C]甲醇(16 )的合成
在氩气保护下, 将2-氟[羰基-14 C]苯甲酸(15 , 136 mg, 53.1 mCi)无水四氢呋喃(10 mL)溶液冷至0 ℃, 缓慢滴入BH3 四氢呋喃溶液(4.5 mL, 4.5 mmol, 1 mol•L-1 ), 防止产生气体引起冲料.滴加完毕, 加热至65 ℃, 搅拌3 h. HFPM监测显示15 完全消失.反应冷至室温, 滴加MeOH (10 mL)淬灭反应, 浓缩, 快速柱层析[V (乙酸乙酯):V (石油醚)=1:25~1:20)得无色油状物16 (110 mg, 47.8 mCi, 90%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 7.52~7.43 (m, 1H), 7.35~7.24 (m, 1H), 7.23~7.07 (m, 2H), 5.30 (t, J =5.7 Hz, 1H), 4.56 (d, J =5.7 Hz, 2H). ESI-MS m /z (%): 129 [M+H]+ , 128 (M+ , 49), 127 (26), 126 (63), 107 (49), 105 (47), 97 (100), 95 (31), 77 (62), 51 (25).
3.2.12
2-氟[羰基-14 C]苯甲醛(17 )的合成
在氩气保护下, 将氯铬酸吡啶(PCC) (1.209 g, 2.5 mmol)、硅胶(2.4 g, 100~200目)和无水二氯甲烷(10 mL)混合, 搅拌, 滴入(2-氟苯基)[14 C]甲醇(16 , 110 mg, 47.6 mCi)无水二氯甲烷(5 mL)溶液, 室温搅拌12 h. HFPM监测显示16 完全消失.过滤反应液, 滤饼用无水二氯甲烷(20 mL×5)洗至无放射性为止, 合并滤液, 低温浓缩得无色油状物17 (100 mg, 43.8 mCi, 92%). 1 H NMR (400 MHz, CDCl3 ) δ : 10.36 (s, 1H), 7.86 (s, 1H), 7.61 (s, 1H), 7.26 (s, 1H), 7.17 (t, J =9.1 Hz, 1H); EI-MS m /z (%): 126 (M+ , 81), 125 (100), 95 (50), 75 (21), 96 (13), 50 (12), 70 (8), 127 (7), 74 (6).
3.2.13
N -[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟苯基- O , O -二乙基[14 C]甲基膦酸酯(3 , [苄基-14 C]毒氟磷)的合成
以17 (99 mg, 43.7 mCi)为放射性同位素原料, 按照第3.2.9节中方法反应12 h, 同法处理获得了N -[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟苯基-O , O -二乙基[14 C]甲基膦酸酯(3 , 394 mg, 32.4 mCi, 74%). 1 H NMR (400 MHz, DMSO-d 6 ) δ : 9.13~9.07 (m, 1H), 7.66~7.59 (m, 1H), 7.49 (d, J =7.7 Hz, 1H), 7.40~7.33 (m, 1H), 7.28~7.20 (m, 2H), 7.04 (d, J =7.3 Hz, 1H), 6.94 (t, J =7.6 Hz, 1H), 6.06~5.97 (m, 1H), 4.15~4.04 (m, 2H), 3.98~3.90 (m, 1H), 3.87~3.79 (m, 1H), 2.41 (s, 3H), 1.19 (t, J =7.0 Hz, 3H), 1.04 (t, J =7.0 Hz, 3H); ESI-MS m /z : 411 [M+H], + 433 [M+Na]+ ; HRMS calcd for C18 14 CH23 FN2 O3 PS [M+H]+ 411.1183, found 411.1194.
3.3
放射性同位素标记物的质量指标分析
按照前期研究中高比活度[噻唑基-2-14 C]毒氟磷(1b )的定量分析条件和方法[7 , 标记物2 , 3 的放化纯度分别以TLC-IIA, HPLC-LSC和HPLC-FSA法测定, 化学纯度以HPLC-PDA外标法测定, 比活度以LSC法测定[9 12 .
辅助材料(Supporting Information) 放射性同位素标记物2 、3 的1 H NMR, ESI-HRMS, HPLC-FSA, HPLC-UV谱图、标记物的质量指标分析条件和方法.这些材料可以免费从本刊网站(http://sioc-journal.cn/ )上下载.

 下载:
下载:

 下载:
下载:






