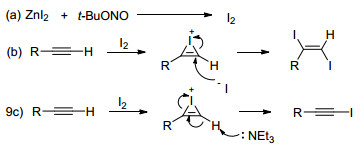
Scheme 1.
Previous methods for the synthesis of iodoalkynes and diiodoalkenes and this work
Switchable Synthesis of Iodoalkynes and Diiodoalkenes from Terminal Alkynes
Suo Chen , Xiaowei Zhang , Hui Zhao , Xiaohong Guo , Xiangguo Hu

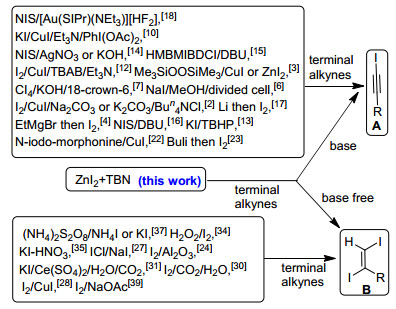
Iodo-substituted compounds are valuable intermediates used extensively in organic synthesis for carbon-carbon, carbon-oxygen and carbon-nitrogen bond formation. Among them, iodoalkynes and diiodoalkenes continue to be important building blocks because of their ability of participating in different coupling reactions and heterocyclic ring formations. Although different methods have been developed for the synthesis of these compounds, the direct iodination of terminal alkynes is the most appealing strategy owing to the commercial availability of these compounds.[1] As shown in Scheme 1, iodoalkyne[2~24] and diiodoalkenes[25~44] can be prepared from an I+ (e.g. NIS, I2 etc.) under various conditions. Alternatively, they can also be synthesized from I- reagents (e.g. NaI, KI, NH4I, CuI, ZnI2) together with an oxidant. Despite the obvious similarities that different methods of the latter approach share, no reaction system can be used for the synthesis of both iodoalkynes and diiodoalkenes. Herein, we report a novel and switchable synthesis of iodoalkynes and diiodoalkenes from terminal alkynes with zinc iodide (ZnI2) and tert-butylnitrite (TBN).
At the onset, we aimed to develop a new procedure for iodoalkynes. tert-Butyl nitrite was chosen as the oxidant based on its good reactivity towards different alkynes.[41~43] 1a and triethylamine were selected as the model compound and the base, respectively. Then different I- reagents and solvents were screened, and it was found that ZnI2 in CHCl3 gave the highest yield (Entries 1~7, Table 1). Other bases, such as 1, 5-diazabicylo[5, 4, 0]undec-5-ene (DBU) and sodium carbonate afforded diminished yields (Entries 8, 9, Table 1). Finally, through the fine tuning the reaction parameters (Entries 10~15, Table 1). The optimized conditions were identified as following: ZnI2 (1.1 equiv.), tert-butyl nitrite (TBN, 3 equiv.), NEt3 (0.5 equiv.) in chloroform at room temperature (Entry 15, Table 1).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | I- reagent (equiv.) |
Base (equiv.) |
Solvent | Yielda/% |
| 1b | NaI (1.5) | Et3N (2.0) | Toluene | Trace |
| 2b | KI (1.5) | Et3N (2.0) | Toluene | Trace |
| 3b | CuI (1.5) | Et3N (2.0) | Toluene | Trace |
| 4b | ZnI2 (1.5) | Et3N (2.0) | Toluene | 35 |
| 5b | ZnI2 (1.5) | Et3N (2.0) | CH3CN | Trace |
| 6b | ZnI2 (1.5) | Et3N (2.0) | THF | Trace |
| 7b | ZnI2 (1.5) | Et3N (2.0) | CHCl3 | 38 |
| 8b | ZnI2 (1.5) | Na2CO3 (2.0) | CHCl3 | Trace |
| 9b | ZnI2 (1.5) | DBU (2.0) | CHCl3 | 34 |
| 10b | ZnI2 (1.5) | Et3N (1.5) | CHCl3 | 62 |
| 11b | ZnI2 (1.5) | Et3N (1.0) | CHCl3 | 75 |
| 12b | ZnI2 (1.5) | Et3N (0.5) | CHCl3 | 90 |
| 13c | ZnI2 (1.5) | Et3N (0.5) | CHCl3 | 86 |
| 14d | ZnI2 (1.1) | Et3N (0.5) | CHCl3 | 91 |
| 15e | ZnI2 (1.1) | Et3N (0.5) | CHCl3 | 93 |
| a Isolated yield; b 1.5 equiv. TBN and 50 ℃; c 1.5 equiv. TBN and room temperature; d 2.0 equiv. TBN and room temperature; e 3.0 equiv. TBN and room temperature. | ||||
With the optimized conditions in hand, we next proceeded to investigate the substrate scope. As shown in Table 2, the reaction conditions were mild enough to tolerate different functional groups. Alkyl (2b, 2e), alkyloxy (2c, 2d), halo (2g~2i), cyano (2j), nitro (2k), ester (2l) substituted substrates all underwent the reaction successfully to furnish the desired products in 61%~95% yields. The reaction was not sensitive to the electron density of the substrates, as similar yields were obtained for both electron-rich compounds (2c, 2d) and the electron-poor substrates (2j, 2k). Because only weak base (triethylamine) is used, this method is well complementary to those in which strong bases (e.g., EtMgBr, [4] BuLi, [23] and DBU[16]) are needed.
Much efforts to tune the aforementioned conditions to furnish the diiodoalkene proved to be unfruitful. To our surprise, diiodoalkene 3a was obtained in 73% yield with the same reagent system (ZnI2/TBN) in the absence of triethylamine. It is interesting to note that the reaction is very sensitive to the electron property of the substrates. For electron-rich substrates (e.g., 3c, 3d), the desired transformation occurred at room temperature smoothly. For the remaining compounds shown in Scheme 3 (e.g., 3e~3i), heating at 100 ℃ was required to get the reaction work. No products could be obtained with Substrates bearing electron-withdrawing substituents such as ester and nitro groups. In all cases, iodoalkynes were obtained as the minor products and thus complicated the column purification, which resulted in the relative moderate yields for the desired diiodo-compounds.
下载:
导出CSV
 |
 |
| a Isolated yields of diiodoalkenes; b isolated yields of iodoalkynes. |
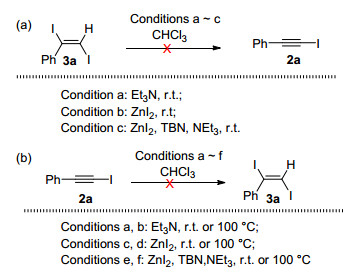
In order to gain some insight into the aforementioned two types of reactions, we have run the following control experiments (Scheme 2). It is reasonable to expect that iodoalkynes and diiodoalkenes are interchangeable under the reaction conditions. However, we found that triethylamine alone, or together with TBN and ZnI2, could not convert diiodoalkene 3a to iodoalkyne 2a (Scheme 2a). Similarly, iodoalkyne 2a could not be transformed to diiodoalkene 3a under conditions shown in Scheme 2b.
Based on the control experiment and literature precedents, [27] We postulate that the two reactions proceed through the reaction pathways shown in Scheme 3. ZnI2 serves as an iodine source[3] and TBN as an oxidant[45] (Scheme 3a). The diiodoalkene is formed by reaction of the terminal alkyne with iodine to form a iodonium intermediate, followed by ring opening with an iodide (Scheme 3b).[27] The mechanism corroborates well with the experimental results (only E-diiodoalkenes were formed). One the other hand, the iodoalkyne is possibly formed by deprotonation of the iodonium intermediate with triethylamine, followed by the elimination to yield the terminal alkyne (Scheme 3c). As the iodonium intermediate is generated from the terminal alkyne, the mechanism proposed also explains why the diiodoalkenes 2 and the alkynes 3 were not interchangeable, which was proved by the control experiments (Scheme 2).
In conclusion, we have developed a novel and switchable protocol for the synthesis of iodoalkynes and diiodoalkenes, which were formed from terminal alkynes using the same reagent system of ZnI2 and TBN in the presence or absence of triethylamine, respectively. The iodoalkyne formation is operationally simple, mild (weak base and room temperature), and tolerant of a large range of functional groups. As only weak base (triethylamine) is used, this method is well complementary to other approaches in which strong bases (e.g., EtMgBr, [4] BuLi, [23] and DBU[16]) are required. However, the diiodoalkene transformation shows interesting dependence on the electron property, and only electron-rich and neutral compounds are viable substrates. The control experiments performed suggest that iodoalkynes and diiodoalkenes are not interchangeable under the reaction conditions, which supports both of the reaction involve a iodonium intermediate formed directly from a terminal alkyne.
The alkyne (0.2 mmol) was added to a solution of zinc iodide (0.22 mmol), triethylamine (0.1mmol) and TBN (0.6 mmol) in dry CHCl3 (5 mL). The reaction mixture was stirred at room temperature under N2 atmosphere. After completion, the reaction was quenched with aqueous 10% Na2S2O3 solution. The reaction mixture was then diluted with CH2Cl2 (20mL), washed with brine (5 mL×2), and dried with anhydrous Na2SO4. After removal of the solvent under vacuum, the residue was purified by column chromatography.
(Iodoethynyl)benzene (2a):[10] Yellow oil, 93% yield. 1H NMR (400 MHz, CDCl3)δ: 7.48~7.40 (m, 2H), 7.36~7.28 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 132.4, 128.9, 128.3, 123.5, 94.2, 6.2.
1-(Iodoethynyl)-4-methylbenzene (2b):[10] Colorless oil, 74% yield. 1H NMR (400 MHz, CDCl3) δ: 7.33 (d, J=7.9 Hz, 2H), 7.12 (d, J=7.8 Hz, 2H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 139.1, 132.3, 129.1, 120.5, 94.4, 21.6, 4.9.
1-(Iodoethynyl)-4-methoxybenzene (2c): Yellow solid, 85% yield. m.p. 59~61 ℃ (Lit.[10] m.p. 61~62 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.46~7.33 (m, 2H), 6.89~6.78 (m, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 133.9, 115.6, 113.9, 94.0, 55.4, 3.9.
1-(Iodoethynyl)-2-methoxybenzene (2d):[18] Yellow oil, 87% yield. 1H NMR (400 MHz, CDCl3) δ: 7.40 (dd, J=7.6, 1.7 Hz, 1H), 7.34~7.24 (m, 1H), 6.95~6.82 (m, 2H), 3.88 (s, 3H); 13C NMR (100MHz, CDCl3) δ: 161.1, 134.5, 130.3, 120.4, 112.6, 110.7, 90.5, 55.9, 9.3.
1-(Iodoethynyl)-4-pentylbenzene (2e): Pale-yellow oil, 95% yield. 1H NMR (400 MHz, CDCl3) δ: 7.40~7.32 (m, 2H), 7.13 (d, J=8.1 Hz, 2H), 2.66~2.55 (m, 2H), 1.61 (q, J=7.6 Hz, 2H), 1.32 (tq, J=10.4, 4.3, 3.2 Hz, 4H), 0.90 (t, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 144.2, 132.3, 128.4, 120.7, 94.4, 36.0, 31.56, 31.0, 22.6, 14.1, 5.0; IR ν: 3027, 2956, 2166, 1605, 1410, 1378, 824 cm-1. HRMS (ESI) calcd for C13H16I [M+H]+ 299.02912, found 299.0288.
1-(Iodoethynyl)-4-(trifluoromethyl)benzene (2f): White solid, 61% yield. m.p. 117~119 ℃ (Lit.[48a] m.p. 119~121 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J=8.3 Hz, 2H), 7.53 (d, J=8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 132.8, 130.6 (q, J=33 Hz), 127.2, 125.4 (q, J=3.8 Hz), 122.6, 93.0, 10.2.
1-Chloro-4-(iodoethynyl)benzene (2g): Pale-yellow solid, 71% yield. m.p. 78~80 ℃ (Lit.[48a] m.p. 83~84 ℃), 7.28 (d, J=8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 135.0, 133.7, 128.8, 120.0, 93.1, 7.8.
1-Chloro-2-(iodoethynyl)benzene (2):[48b] Yellow oil, 86% yield. 1H NMR (400 MHz, CDCl3) δ: 7.50~7.44 (m, 1H), 7.42~7.36 (m, 1H), 7.29~7.17 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 136.8, 134.3, 129.9, 129.3, 126.5, 123.3, 91.0, 12.4.
1-Bromo-4-(iodoethynyl)benzene (2i): Pale-yellow solid, 79% yield. m.p. 92~93 ℃ (Lit.[10] m.p. 92~94 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.45 (d, J=8.5 Hz, 2H), 7.30 (d, J=8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 133.8, 131.7, 123.3, 122.4, 93.2, 8.1.
4-(Iodoethynyl)benzonitrile (2j): White solid, 87% yield. m.p. 166~168 ℃ (Lit.[48a]: m.p. 169~170 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.60 (d, J=8.8 Hz, 2H), 7.50 (d, J=8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 133.0, 132.1, 128.2, 118.3, 112.3, 92.7, 13.2.
1-(Iodoethynyl)-4-nitrobenzene (2k): Pale-yellow solid, 88% yield. m.p. 169~170 ℃ (Lit.[48a] m.p. 67~168 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.23~8.14 (m, 2H), 7.61~7.54 (m, 2H); 13C NMR (100MHz, CDCl3) δ: 147.5, 133.3, 130.1, 123.7, 92.5, 14.2.
Methyl 4-(iodoethynyl)benzoate (2l): White solid, 89% yield. m.p. 133~135 ℃ (Lit.[48c] m.p. 135~137 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.04~7.94 (m, 2H), 7.53~7.44 (m, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.4, 132.4, 130.1, 129.5, 128.0, 93.5, 52.4, 10.7.
The alkyne (0.2 mmol) was added to a solution of zinc iodide (0.22 mmol) and TBN (0.6 mmol) in dry CHCl3 (5 mL). The reaction mixture was stirred at room temperature under N2 atmosphere. After completion, the reaction was quenched with aqueous 10% Na2S2O3 solution. The reaction mixture was then diluted with CH2Cl2 (20 mL), washed with brine (5 mL×2), and dried with anhydrous Na2SO4. After removal of the solvent under vacuum, the residue was purified by column chromatography.
(E)-(1, 2-Diiodovinyl)benzene (3a): White solid, 73% yield. m.p. 78~79 ℃ (Lit.[38] m.p. 73~75 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.40~7.32 (m, 5H), 7.27 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 143.2, 129.1, 128.6, 128.5, 96.3, 80.9.
(E)-1-(1, 2-Diiodovinyl)-4-methylbenzene (3b):[38] Orange oil, 61% yield. 1H NMR (400 MHz, CDCl3) δ: 7.27 (d, J=7.9 Hz, 2H), 7.23 (s, 1H), 7.17 (d, J=7.9 Hz, 2H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 140.3, 139.2, 129.2, 128.6, 96.7, 80.3, 21.5.
(E)-1-(1, 2-Diiodovinyl)-4-methoxybenzene (3c):[38] Yellow oil, 78% yield. 1H NMR (400 MHz, CDCl3) δ: 7.41~7.28 (m, 2H), 7.20 (s, 1H), 6.96~6.80 (m, 2H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.9, 135.4, 130.3, 113.8, 96.7, 80.0, 55.4.
(E)-1-(1, 2-Diiodovinyl)-2-methoxybenzene (3d): Yellow oil, 59% yield. 1H NMR (400 MHz, CDCl3) δ: 7.35 (ddd, J=8.3, 7.4, 1.1 Hz, 1H), 7.27 (s, 1H), 7.15 (dd, J=7.6, 1.8 Hz, 1H), 6.98 (td, J=7.5, 1.1 Hz, 1H), 6.91 (dd, J=8.4, 1.1 Hz, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 155.4, 132.2, 130.7, 129.6, 120.8, 111.8, 92.7, 83.2, 55.9; IR ν: 3066, 2923, 1296, 1180, 840, 748 cm-1; HRMS (ESI) calcd for C9H9I2O [M+H]+ 386.8737, found 386.8731.
(E)-1-(1, 2-Diiodovinyl)-4-pentylbenzene (3e):[38] Yellow oil, 60% yield. 1H NMR (400 MHz, CDCl3) δ: 7.29 (d, J=8.2 Hz, 2H), 7.22 (s, 1H), 7.16 (d, J=8.2 Hz, 2H), 2.60 (t, J=7.7 Hz, 2H), 1.70~1.57 (m, 2H), 1.41~1.29 (m, 4H), 0.90 (t, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 144.2, 140.3, 128.7, 128.5, 96.9, 80.2, 35.9, 31.7, 30.9, 22.7, 14.2.
(E)-1-(1, 2-Diiodovinyl)-4-(trifluoromethyl)benzene (3f):[48d] Orange oil, 50% yield. 1H NMR (400 MHz, CDCl3) δ: 7.68~7.60 (m, 2H), 7.51~7.43 (m, 2H), 7.36 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 146.8, 131.1, 129.1, 126.7 (q, J=271 Hz), 125.7 (q, J=3.8 Hz), 122.5, 93.8, 82.5.
(E)-1-(1, 2-Diiodovinyl)-4-fluorobenzene (3g):[38] Orange oil, 55% yield. 1H NMR (400 MHz, CDCl3) δ: 7.39~7.31 (m, 2H), 7.27 (s, 1H), 7.09~7.01 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 162.6 (d, JC—F=250 Hz, 1C), 139.2 (d, JC—F=3.6 Hz, 1C), 130.7 (d, JC—F=8.6 Hz, 2C), 115.6 (d, JC—F=22.4Hz, 2C), 94.9, 81.6.
(E)-1-Chloro-4-(1, 2-diiodovinyl)benzene (3h):[38] Yellow oil, 47% yield. 1H NMR (400 MHz, CDCl3) δ: 7.37~7.33 (m, 2H), 7.32~7.27 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 141.5, 134.8, 129.9, 128.7, 94.5, 81.6.
(E)-1-Bromo-4-(1, 2-diiodovinyl)benzene (3i): Yellow solid, 60% yield. m.p. 63~65 ℃ (Lit.[38] m.p. 62~64 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.55~7.47 (m, 2H), 7.29 (s, 1H), 7.26~7.20 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 142.1, 131.8, 130.3, 123.2, 94.6, 81.8.
Supporting Information The Supporting Information containing the 1H NMR and 13C NMR spectra of this paper is available free of charge via the Internet at http://sioc-journal.cn.
Wu, W. Q.; Jiang, H. F. Acc. Chem. Res. 2014, 47, 2483. doi: 10.1021/ar5001499
Jeffery, T. J. Chem. Soc., Chem. Commun. 1988, 909.
Casarini, A.; Dembech, P.; Reginato, G.; Ricci, A.; Seconi, G. Tetrahedron Lett. 1991, 32, 2169. doi: 10.1016/S0040-4039(00)71266-4
Rao, M. L. N.; Periasamy, M. Synth. Commun. 1995, 25, 2295. doi: 10.1080/00397919508011785
Monenschein, H.; Sourkouni-Argirusi, G.; Schubothe, K. M.; O'Hare, T.; Kirschning, A. Org. Lett. 1999, 1, 2101. doi: 10.1021/ol991149m
Nishiguchi, I.; Kanbe, O.; Itoh, K.; Maekawa, H. Synlett 2000, 89.
Abele, E.; Fleisher, M.; Rubina, K.; Abele, R.; Lukevics, E. J. Mol. Catal. A:Chem. 2001 165, 121. doi: 10.1016/S1381-1169(00)00451-9
Lahrache, H.; Robin, S.; Rousseau, G. Tetrahedron Lett. 2005, 46, 1635. doi: 10.1016/j.tetlet.2005.01.089
Stefani, H. A.; Cella, R.; D rr, F. A.; Pereira, C. M. P.; Gomes, F. P.; Zeni, G. Tetrahedron Lett. 2005, 46, 2001. doi: 10.1016/j.tetlet.2005.01.161
Yan, J.; Li, J. H.; Cheng, D. P. Synlett 2007, 2442. http://downloads.hindawi.com/archive/2013/406049.xml
Meng, L. G.; Cai, P. J.; Guo, Q. X.; Xue, S. Synth. Commun. 2008, 38, 225. doi: 10.1080/00397910701749724
Chen, S. N.; Lin, T. C.; Hung, T. T.; Tsai, F. Y. J. Chin. Chem. Soc. 2009, 56, 1078. doi: 10.1002/jccs.v56.5
Reddy, K. R.; Venkateshwar, M.; Maheswari, C. U.; Kumar, P. S. Tetrahedron Lett. 2010, 51, 2170. doi: 10.1016/j.tetlet.2010.02.074
Nosel, P.; Lauterbach, T.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Chem. Eur. J. 2013, 19, 8634. doi: 10.1002/chem.v19.26
Nouzarian, M.; Hosseinzadeh, R.; Golchoubian, H. Synth. Commun. 2013, 43, 2913. doi: 10.1080/00397911.2012.749991
Li, M. R.; Li, Y. J.; Zhao, B. Z.; Liang, F. S.; Jin, L. Y. RSC Adv. 2014, 4, 30046. doi: 10.1039/C4RA04736B
Chowdhury, R. M.; Wilden, J. D. Org. Biomol. Chem. 2015, 13, 5859. doi: 10.1039/C5OB00494B
Gómez-Herrera, A.; Nahra, F.; Brill, M.; Nolan, S. P.; Cazin, C. S. J. ChemCatChem 2016, 8, 3381. doi: 10.1002/cctc.v8.21
Karpov, G. V.; Popik, V. V. J. Am. Chem. Soc. 2007, 129, 3792. doi: 10.1021/ja064470q
Barsoum, D. N.; Okashah, N.; Zhang, X. G.; Zhu, L. J. Org. Chem. 2015, 80, 9542. doi: 10.1021/acs.joc.5b01536
Chen, W. W.; Zhang, J. L.; Wang, B.; Zhao, Z. X.; Wang, X. Y.; Hu, Y. F. J. Org. Chem. 2015, 80, 2413. doi: 10.1021/jo502634h
Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018. doi: 10.1002/anie.v48:43
Wilkins, L. C.; Lawson, J. R.; Wieneke, P.; Rominger, F.; Hashmi, A. S. K.; Hansmann, M. M.; Melen, R. L. Chem. Eur. J. 2016, 22, 14618. doi: 10.1002/chem.201602719
贺干武, 刘凤伟, 许新华, 有机化学, 2007, 27, 663. doi: 10.3321/j.issn:0253-2786.2007.05.018He, G. W.; Liu, F. W.; Xu, X. H. Chin. J. Org. Chem. 2007, 27, 663(in Chinese). doi: 10.3321/j.issn:0253-2786.2007.05.018
Larson, S.; Luidhardt, T.; Kabalka, G. W.; Pagni, R. M. Tetrahedron Lett. 1988, 29, 35. doi: 10.1016/0040-4039(88)80009-1
Hondrogiannis, G.; Lee, L. C.; Kabalka, G. W.; Pagni, R. M. Tetrahedron Lett. 1989, 30, 2069. doi: 10.1016/S0040-4039(01)93713-X
Barluenga, J.; Rodriguez, M. A.; Campos, P. J. J. Org. Chem. 1990, 55, 3104. doi: 10.1021/jo00297a027
Henaff, N.; Stewart, S. K.; Whiting, A. Tetrahedron Lett. 1997, 38, 4525. doi: 10.1016/S0040-4039(97)00920-9
Duan, J. X.; Dolbier, W. R.; Jr.; Chen, Q. Y. J. Org. Chem. 1998, 63, 9486. doi: 10.1021/jo9816663
Hénaff, N.; Whiting, A. J. Chem. Soc., Perkin Trans. 1 2000, 395. https://es.scribd.com/document/81091798/Energetic-Materials
Li, J. H.; Xie, Y. X.; Yin, D. L. Green Chem. 2002, 4, 505. doi: 10.1039/B206536C
Li, J. H.; Xie, Y. X.; Yin, D. L.; Jiang, H. F. Chin. J. Chem. 2003, 21, 714. https://www.sciencedirect.com/science/article/pii/S0012821X14005810
Jereb, M.; Zupan, M.; Stavber, S. Chem. Commun. 2004, 2614.
Pavlinac, J.; Zupan, M.; Stavber, S. Org. Biomol. Chem. 2007, 5, 699. doi: 10.1039/B614819K
Terent'ev, A. O.; Borisov, D. A.; Krylov, I. B.; Nikishin, G. I. Synth. Commun. 2007, 37, 3151. doi: 10.1080/00397910701545171
Tveryakova, E. N.; Miroshnichenko, Y. Y.; Perederina, I. A.; Yusubov, M. S. Russ. J. Org. Chem. 2007, 43, 152. doi: 10.1134/S1070428007010216
Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Adv. Synth. Catal. 2008, 350, 2921. doi: 10.1002/adsc.v350:18
Guo, C. C.; Jiang, Q.; Wang, J. Y. Synthesis 2015, 47, 2081. doi: 10.1055/s-00000084
Lin, Y. M.; Lu, G. P.; Cai, C.; Yi, W. B. Org. Lett. 2015, 17, 3310. doi: 10.1021/acs.orglett.5b01488
Tang, S.; Liu, K.; Long, Y.; Qi, X. T.; Lan, Y.; Lei, A. W. Chem. Commun. 2015, 51, 8769. doi: 10.1039/C5CC01825K
Wu, Y. M.; Zhang, J. M.; Guo, Y. W.; Wang, X. J.; Zhu, Z. T. Synlett 2016, 27, 2259. doi: 10.1055/s-0035-1562115
李金恒, 谢叶香, 尹笃林, 有机化学, 2002, 22, 894. doi: 10.3321/j.issn:0253-2786.2002.11.014Li, J.H.; Xie, J. X; Yin, D. L. Chin. J. Org. Chem. 2002, 22, 894(in Chinese). doi: 10.3321/j.issn:0253-2786.2002.11.014
余开辉, 张小丽, 胡乔生, 刘良先, 有机化学, 2010, 30, 266.Yu, K.; Zhang, X.; Hu, Q.; Liu, L. Chin. J. Org. Chem. 2010, 30, 266(in Chinese).
For selected example of synthesis of diiodoalkynes from intermal alkynes, see: (a) Su, L. ; Lei, C. -Y. ; Fan, W. -Y. ; Liu, L. -X. Synth. Commun. 2011, 41, 1200.
(b) Liu, Y. ; Huang, D. ; Huang, J. ; Maruoka, K. J. Org. Chem. 2017, 82, 11865.
Liu, Y. Synlett 2011, 1184.
Dutta, U.; Maity, S.; Kancherla, R.; Maiti, D. Org. Lett. 2014, 16, 6302. doi: 10.1021/ol503025n
Dutta, U.; Lupton, D. W.; Maiti, D. Org. Lett. 2016, 18, 860. doi: 10.1021/acs.orglett.6b00147
(a) Oliver, D. ; Dino, W. ; Nils, T. ; Nancy, G. ; Francois, D. Org. Lett. 2014, 16, 4722.
(b) Pelletier, G. ; Lie, S. ; Mousseau, J. J. ; Charette, A. B. Org. Lett. 2012, 14, 5464.
(c) Dan, L. ; Joaquin, M. A. ; Emil, B. L. ; William, R. D. Chem. Eur. J. 2015, 21, 18122.
(d) Madabhushi, S. ; Jillella, R. ; R. Mallu, K. K. ; Godala, K. R. ; Vangipuram, V. S. Tetrahedron Lett. 2013, 54, 3993.
(e) Abe, H. ; Suzuki, H. Bull. Chem. Soc. Jpn. 1999, 72, 787.

Scheme 1 Previous methods for the synthesis of iodoalkynes and diiodoalkenes and this work
Table 1. Optimization of direct iodination of terminal alkyne
|
||||
| Entry | I- reagent (equiv.) |
Base (equiv.) |
Solvent | Yielda/% |
| 1b | NaI (1.5) | Et3N (2.0) | Toluene | Trace |
| 2b | KI (1.5) | Et3N (2.0) | Toluene | Trace |
| 3b | CuI (1.5) | Et3N (2.0) | Toluene | Trace |
| 4b | ZnI2 (1.5) | Et3N (2.0) | Toluene | 35 |
| 5b | ZnI2 (1.5) | Et3N (2.0) | CH3CN | Trace |
| 6b | ZnI2 (1.5) | Et3N (2.0) | THF | Trace |
| 7b | ZnI2 (1.5) | Et3N (2.0) | CHCl3 | 38 |
| 8b | ZnI2 (1.5) | Na2CO3 (2.0) | CHCl3 | Trace |
| 9b | ZnI2 (1.5) | DBU (2.0) | CHCl3 | 34 |
| 10b | ZnI2 (1.5) | Et3N (1.5) | CHCl3 | 62 |
| 11b | ZnI2 (1.5) | Et3N (1.0) | CHCl3 | 75 |
| 12b | ZnI2 (1.5) | Et3N (0.5) | CHCl3 | 90 |
| 13c | ZnI2 (1.5) | Et3N (0.5) | CHCl3 | 86 |
| 14d | ZnI2 (1.1) | Et3N (0.5) | CHCl3 | 91 |
| 15e | ZnI2 (1.1) | Et3N (0.5) | CHCl3 | 93 |
| a Isolated yield; b 1.5 equiv. TBN and 50 ℃; c 1.5 equiv. TBN and room temperature; d 2.0 equiv. TBN and room temperature; e 3.0 equiv. TBN and room temperature. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 3. Synthesis of diiodoalkenes
|
|
| a Isolated yields of diiodoalkenes; b isolated yields of iodoalkynes. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们