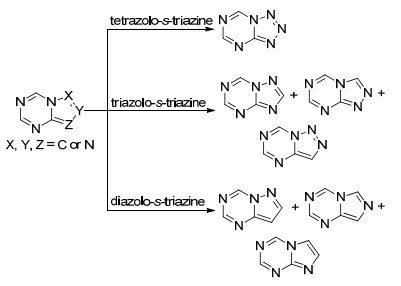
Scheme 1.
氮唑并均三嗪类化合物的分类
Scheme 1.
Classification of nitrogen zole s-triazine compounds

均三嗪类化合物在工业领域[1, 2]、医药领域[3, 4]等都有重要应用价值从而被广泛研究.不仅如此, 在均三嗪的稠合物里面, 氮唑并均三嗪类化合物表现出良好的生物活性.如四氮唑并均三嗪具有抗增殖、抗氧化[5]等活性; 三氮唑并均三嗪具有抗增殖、抗氧化[5]、抗血管增生[6], 以及抑制腺苷受体[7]和抑制胸苷磷酸化酶[8]等活性; 咪唑并均三嗪在抗病毒[9]和遗传学[10]等方面都有应用, 吡唑并均三嗪具有抗癌[11]、抗增殖[12]、抑制酶的活性[13]等性质, 经过修饰还可以作为核苷和脱氧核苷类似物[14].这些化合物都是由氮唑环与均三嗪稠合而成, 它们不仅在结构上相似, 而且在部分生物活性上也表现出了类似的性质.正是由于这些化合物具有良好的生物活性和广阔的应用前景, 所以引起了很多药物化学家及相关领域研究工作者的浓厚兴趣.
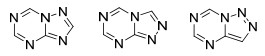
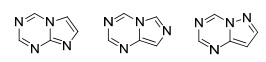
本文根据氮唑并均三嗪类化合物中氮唑的种类不同将该类化合物分为四氮唑并均三嗪, 三氮唑并均三嗪, 二氮唑并均三嗪三部分, 并从这三个方面出发综述了这类化合物的合成方法.其中三氮唑并均三嗪还可以分为1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪、1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪和1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪三种结构; 二氮唑并均三嗪还可以分为咪唑并[1, 2-a]-1, 3, 5-三嗪、咪唑并[1, 5-a]-1, 3, 5-三嗪和吡唑并[1, 5-a]-1, 3, 5-三嗪这三种结构(Scheme 1).
合成四氮唑并均三嗪类化合物的方法主要包括两方面:一方面是从5-氨基四氮唑出发通过构建三嗪环来合成目标化合物; 另一方面就是在三嗪环上引入叠氮基, 进一步发生环合来合成目标化合物. 1987年, Hafez等[15]首次报道了关于合成四氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物的反应.该反应是以5-氨基-四氮唑(1)与肉桂酰异硫氰酸酯(2)为原料在丙酮中加热回流3 h生成7-苯乙烯基-5-硫代四氮唑并[1, 5-a]-1, 3, 5-三嗪(3) (Eq. 1).
2005年, Bekircan等[5]利用5-氨基四氮唑(1)和N-羰基亚氨酸乙酯(4)在165~170 ℃的条件下反应2 h生成5, 7-二芳基四氮唑并[1, 5-a]-1, 3, 5-三嗪(5) (Eq. 2).
|
|
(1) |
|
|
(2) |
2001年, Schulz等[16]报道了2, 4, 6-三叠氮基-1, 3, 5-三嗪(6)与等量的三苯基膦在无水乙醚中室温反应2 h生成5-叠氮基-7-三苯基膦亚氨四氮唑并[1, 5-a]-1, 3, 5-三嗪(7) (Eq. 3), 化合物的结构经过X射线单晶衍射得到了确认. 2016年, Chapyshev等[17]还报道了化合物7再与一分子三苯基膦加成生成5, 7-双三苯基膦亚氨四氮唑并[1, 5-a]-1, 3, 5-三嗪的反应.
|
|
(3) |
2005年, Bakharev课题组[18]报道了2-羟基-4, 6-二(三硝基甲基)三嗪四甲基铵盐(8)与叠氮化钠在丙酮和水的混合溶剂中于20~25 ℃条件下反应2.5~3 h生成5-三硝基甲基-7-氧代四氮唑并[1, 5-a]-1, 3, 5-三嗪四甲基铵盐(9)[R=C(NO2)3]的反应.在该反应中4, 6-二(三硝基甲基)均三嗪和产物5-三硝基甲基四氮唑并均三嗪都有爆炸的危险, 在后处理和保存的过程中都需要谨慎处理. 2006年, 该课题组[19]直接利用4-叠氮基-2-氧代- 1, 3, 5-三嗪类化合物(10)在氢氧化钠的作用下于20~25 ℃条件下反应生成5-取代-7-氧代四氮唑并[1, 5-a]-1, 3, 5-三嗪钠盐(9), 其结构经X射线单晶衍射得到了确认.该化合物在酸性条件下又发生开环又生成原料10 (Scheme 2).四氮唑并[1, 5-a]-1, 3, 5-三嗪钠盐是水溶性的物质, 在有机溶剂中的溶解度很差.
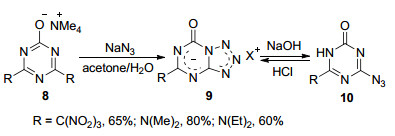
2014年, Parfenov等[20]报道了4-氨基-6-氯-2-氧代- 1, 3, 5-三嗪(11)与2 equiv.的叠氮化钠在DMF/丙酮混合溶剂中反应生成5-氨基-7-氧代-四氮唑并[1, 5-a]-1, 3, 5-三嗪钠盐(12)的反应.化合物12与氯化叔丁铵在水溶液中发生离子交换生成5-氨基-7-氧代四氮唑并[1, 5-a]-1, 3, 5-三嗪叔丁铵盐(13) (Scheme 3), 其结构经X射线单晶衍射得到了确认.化合物13相对于化合物12来说溶解度得到了改善, 可以溶于多种极性或非极性溶剂.
三氮唑并均三嗪类化合物从三氮唑的结构及其与均三嗪的稠合方式上可以分为以下三类(图 1): 1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪、1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪和1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪.
脒基取代的1, 2, 4-三氮唑是合成三氮唑并均三嗪类化合物的重要原料或中间体. 1970年, Bokaldere等[21]以1-脒基-5-氨基-1, 2, 4-三氮唑(14)为原料, 在原甲酸三乙酯中加热回流生成2-取代-7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物15.当他们以甲酸代替原甲酸三乙酯参与反应也能得到目标化合物. 2007年, Dolzhenko课题组[22]同样以1-脒基-5-氨基-1, 2, 4-三氮唑为原料, 在哌啶作用下与醛或酮在乙醇中加热回流得到生成7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物(16) (Scheme 4).

当脒基上的亚氨基被氧原子或硫原子取代时, 三氮唑并均三嗪类化合物的7号位就分别引入了氧代和硫代两种重要官能团. 1965年, Taylor等[23]首次报道了1-酰胺-5-氨基-1, 2, 4-三氮唑(17)与原甲酸三乙酯在100 ℃条件下反应18 h, 生成7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(18)的方法(Eq. 4).
|
|
(4) |
1985年, Evers等[24]报道了1-硫代酰胺-5-氨基- 1, 2, 4-三氮唑衍生物(19)在回流条件下与取代的原甲酸三乙酯发生成环反应, 生成2-甲硫基-7-硫代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物20的方法(Eq. 5).
|
|
(5) |
1989年, Dorokhov等[25]报道了以3-脒基-1, 2, 4-三氮唑盐酸盐(21)和原甲酸三乙酯为原料在甲醇钠甲醇溶液中加热回流3~5 h生成5-芳基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪22的反应(Eq. 6).
|
|
(6) |
1991年, Dorokhov等[26]同样是以3-脒基取代的1, 2, 4-三氮唑盐酸盐(21)为原料先在甲醇钠作用下生成游离碱, 然后与三氯乙腈在二甲苯中加热回流生成7-三氯甲基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物(23)(Eq. 7).化合物23中的三氯甲基活性很高, 可以与伯胺或仲胺反应生成一系列7号位被胺取代的三氮唑并均三嗪衍生物[27], 这为合成该类化合物提供了新的方法.
|
|
(7) |
2012年, Dolzhenko课题组[28]也报道了一种关于三氯甲基取代的三氮唑并均三嗪类化合物的合成方法(Scheme 5).该方法以5-氨基-1, 2, 4-三氮唑(24)为原料, 先与三氯乙腈在甲苯中加热回流得到脒基取代的1, 2, 4-三氮唑中间体25, 然后在原甲酸三乙酯中加热回流8 h生成目标产物5-三氯甲基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(26).
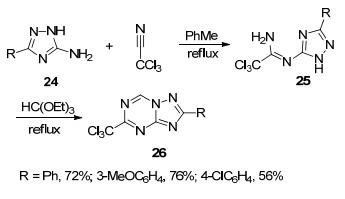
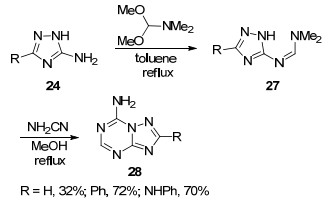
2013年, Dolzhenko课题组[29]报道了5-氨基-1, 2, 4-三氮唑(24)与N, N-二甲基甲酰胺二甲缩醛为原料合成了一种新的中间体N, N-二甲基甲脒取代的1, 2, 4-三氮唑(27)的反应, 该中间体与氨基腈在甲醇中加热回流生成7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物28 (Scheme 6).
胍基取代的三氮唑与脒基取代的三氮唑在结构上相似, 也是合成三氮唑并均三嗪类化合物的重要原料. 1998年, Zohdi等[30]报道了3-三氟甲基-5-胍基-1, 2, 4-三氮唑衍生物29与碳酸二乙酯(30)加热回流5 h生成6-苯基-2-三氟甲基-5-苯胺基-7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(31)的反应(Eq. 8).
|
|
(8) |
2007年, Dolzhenko课题组[22]以3-苯基-5-胍基- 1, 2, 4-三氮唑(32)为原料在哌啶作用下与醛或酮在乙醇中加热回流生成5-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物(33) (Eq. 9). 2008年, 该课题组[31]以3-吡啶基- 5-胍基-1, 2, 4-三氮唑和氟代苯甲醛为原料, 在相同条件下合成出了一系列氟代的7-芳基-2-吡啶基-5-氨基- 1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物.
2008年, 该课题组[27a]同样是以3-苯基-5-胍基- 1, 2, 4-三氮唑(32)为原料与三氯乙腈在甲苯中加热回流7 h, 生成2-苯基-7-三氯甲基-5-氨基-1, 2, 4-三氮唑并[1, 5-a]- 1, 3, 5-三嗪(34).当该反应在乙醇中回流反应时, 却生成2-苯基-5, 7-二氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(Eq. 10).实验结果表明, 胍基取代的三氮唑与三氯乙腈的反应具有化学选择性, 这种选择性与溶剂有关.
|
|
(9) |
|
|
(10) |
3(5)-氨基-1, 2, 4-三氮唑在合成三氮唑并均三嗪类化合物上具有广泛的应用性. 1980年, Lalezari等[32]报道了3-取代-5-氨基-1, 2, 4-三氮唑(24)与N-氰基亚氨酸乙酯在甲醇中加热回流1 h生成2-取代-7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物35的反应(Eq. 11).
|
|
(11) |
1998年, Naito等[33]利用5-氨基-1, 2, 4-三氮唑(24)和N-氰亚胺基-S, S-二硫代碳酸二甲酯(36)为原料在吡啶中回流2.5 h, 生成2-取代-7-氨基-5-甲硫基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(37); 当该反应在吡啶中于50 ℃条件下反应36 h, 生成2-取代-5-氨基-7-甲硫基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(38)(Eq. 12). 2002年, Bereczm等[34]报道了3-取代-5-氨基1, 2, 4-三氮唑和异硫脲衍生物反应生成两种互为异构体的5, 7-二氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的反应.
|
|
(12) |
2005年, Bekircan等[5]报道了5-氨基-1, 2, 4-三氮唑(39)与N-羰基亚氨酸乙酯(40)在165~170 ℃条件下反应2 h得到1, 2, 4-三氮并[1, 5-a]-1, 3, 5-三嗪类化合物(41)的反应(Eq. 13).
|
|
(13) |
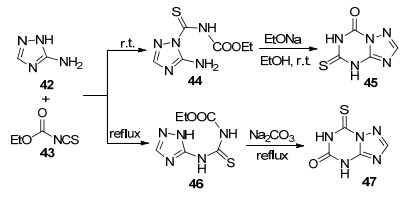
异(硫)氰酸酯类化合物在合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物中发挥了重要作用, 很多三氮唑并均三嗪类化合物中的重要官能团(如硫代, 氧代等)都是通过异(硫)氰酸酯引入的, 而且这类化合物一般都具有良好的生物活性[8]. 1972年, Driscoll课题组[35]报道了5-氨基-1, 2, 4-三氮唑(42)与N-乙氧羰基异硫氰酸酯(43)在室温条件下反应生成硫代酰胺中间体44, 然后在乙醇钠的乙醇溶液中室温反应2 h生成5-氧代-7-硫代- 1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(45).从相同的原料出发通过加热回流可以得到硫脲中间体46, 接着在碳酸钠的作用下环合生成7-氧代-5-硫代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(47)(Scheme 7). 1973年, 该课题组[36]又利用N-乙氧羰基异氰酸酯与5-氨基-1, 2, 4-三氮唑反应生成了5, 7-二氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪.这种合成方法在三嗪环上引入了氧代和硫代这两种重要官能团, 为合成该类化合物提供了新的方法.
1, 3-二烯季铵盐类化合物在参与合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物中也有其应用性. 1994年, Okide[37]报道了1, 3-二烯季铵盐类化合物48与3-氨基-1, 2, 4-三氮唑(49)在乙腈中回流反应1 h生成5-苯基- 7-二甲氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪高氯酸盐(50)的反应(Eq. 14).
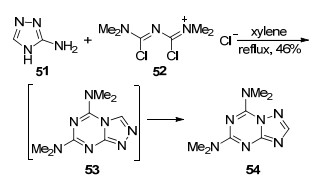
2001年, Kaddachi等[38]利用3-氨基-1, 2, 4-三氮唑(51)和氮菁(52)为原料, 在二甲苯中加热回流25 h得到5, 7-二(二甲氨基)-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(54)(Scheme 8).在该反应中3-氨基-1, 2, 4-三氮唑先和氮菁反应生成中间体1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪(53), 该中间体很快发生重排转化成其异构体1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(54).另外, 该反应操作严格, 要求在干燥且需要有惰性气体保护的环境中进行.
|
|
(14) |
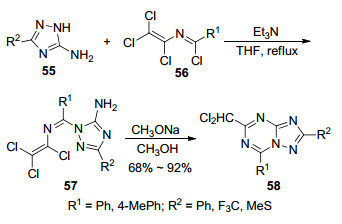
2007年, Drach等[39]报道了5-氨基-1, 2, 4-三氮唑(55)与2-氮杂-1, 3-丁二烯类化合物56反应生成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物58的方法.在该反应中三氮唑与1, 3-丁二烯在三乙胺作用下于四氢呋喃中长时间回流(84 h)脱去一分子氯化氢生成缩合中间体57, 再发生分子内环合生成目标化合物(Scheme 9).
2013年, Zamigailo等[40]利用5-氨基-1, 2, 4-三氮唑(42)和4-氧代-1, 3-苯并噁嗪类化合物59为原料, 在DMF中持续回流8 h生成7-甲基-5-芳基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪(60)(Eq. 15).
|
|
(15) |
2006年, Fronabarger等[41]利用3, 4, 5-三氨基-1, 2, 4-三氮唑(61)与毒性较大的溴化氰为原料, DMF作溶剂, 于80 ℃条件下一锅法合成含有1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪母核的三元环化合物62 (Eq. 16).
|
|
(16) |
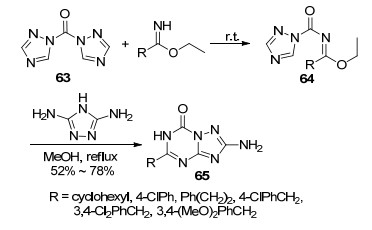
2010年, Geffken等[42]报道了一种高效的方法合成一系列5-取代-2-氨基-7-氧代-1, 2, 4-三氮唑并[1, 5-a]- 1, 3, 5-三嗪衍生物65 (Scheme 10).该方法首先以取代的亚氨酸乙酯与N, N'-羰基二(1, 2, 4-三氮唑)(CDT) (63)为起始原料, 室温反应1 h得到中间体N-羰基三氮唑亚氨酸乙酯衍生物64, 然后与3, 5-二氨基-1, 2, 4-三氮唑在无水甲醇中加热回流0.5 h生成目标化合物.其结构经过X射线单晶衍射得到了确认.该反应具有区域选择性高, 产物结构单一的优点.
2013年, Dolzhenko等[43]首次报道了在微波促进下三组分一锅法合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物67的方法(Eq. 17).该方法是以5-氨基-1, 2, 4-三氮唑(66)、原甲酸三乙酯和氨基腈为原料, 通过优化反应条件确定以甲醇作溶剂, 在150 ℃条件下反应20 min时能够取得最高收率.通过对不同底物的反应适用性考察表明, 大部分底物都能取得中等到良好收率.这种方法具有效率高、节约成本和环境友好等优点.
|
|
(17) |
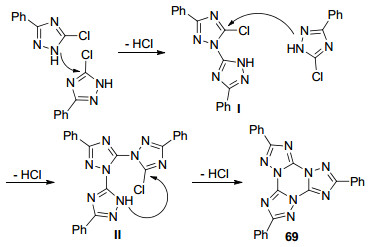
2005年, Tartakovsky等[44]报道了3-取代5-氯-1, 2, 4-三氮唑(68)在高温条件下环合生成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物69的反应(Eq. 18), 这类物质是由三个1, 2, 4-三氮唑环以[1, 5-a]的方式与1, 3, 5-三嗪环稠合而成.当R=Ph时, 反应需要在270 ℃条件下完成; 当R=Cl时, 反应需要在环丁砜作溶剂240~270 ℃条件下完成.
|
|
(18) |
作者推测反应机理如Scheme 11所示. 5-氯-1, 2, 4-三氮唑中的NH作为亲核试剂进攻另一分子三氮唑中的5号碳原子, 发生加成消除反应并脱去1分子HCl生成中间体Ⅰ, 接着与第三分子三氮唑经历同样的过程生成中间体Ⅱ, 最后发生分子内的加成消除反应得到目标产物69, 整个反应过程脱去三分子HCl.
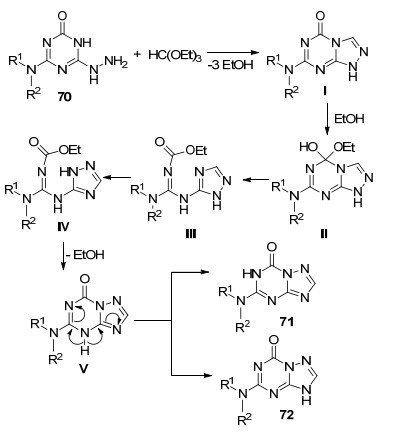
除了从1, 2, 4-三氮唑出发合成三氮唑并均三嗪类化合物之外, 从1, 3, 5-三嗪出发合成目标化合物也有文献报道, 其中的代表就是肼基取代的1, 3, 5-三嗪.这类三嗪在参与合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物的过程中一般是通过加成环合反应先生成其异构体1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪, 然后发生重排得到目标化合物. 2014年, Parfenov课题组[45]报道了4-烷胺基取代的6-肼基-2-氧代-1, 3, 5-三嗪(70)与超过量的原甲酸三乙酯加热回流6~14 h, 生成两种5-烷胺基取代的7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物的反应(Eq. 19).两种化合物的结构都经过X射线单晶衍射确认.对三嗪底物适用性考察表明, 大多数底物都能取得良好的收率, 其中当NR1R2=NHi-Pr时, 化合物71和72同时存在.
|
|
(19) |
作者提出了如Scheme 12所示的反应机理.首先6-肼基-1, 3, 5-三嗪衍生物(70)与原甲酸三乙酯加热回流脱去三分子乙醇得到5-氧代-1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物Ⅰ, 再与反应中生成的乙醇发生加成得到化合物Ⅱ.化合物Ⅱ中1, 3, 5-三嗪环开环转化为化合物Ⅲ.接着发生重排得到化合物Ⅳ, 然后脱掉一分子乙醇闭环就得到1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物Ⅴ.化合物Ⅴ中4号N原子上的H迁移到3号N原子或6号N原子上, 就得到了两种互为同分异构体的化合物71和72.
2015年, 该课题组[46]用苯甲醛类化合物代替原甲酸三乙酯, 与4-烷胺基取代的6-肼基-2-氧代-1, 3, 5-三嗪反应合成出了多个1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物(Scheme 13).化合物的结构经过X射线单晶衍射确认.在该反应中, 1, 3, 5-三嗪(73)和苯甲醛(74)在室温条件下反应先生成中间体腙75, 然后在醋酸铅作用下发生氧化环化得到目标产物1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物76.该反应的整体收率较低, 而且使用Pb(OAc)4这种重金属对环境污染大.
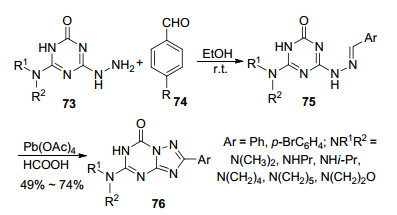
苯甲醛类化合物参与合成三氮唑并均三嗪类化合物的反应机理如Scheme 14所示.首先化合物Ⅰ在醋酸铅作用下生成中间体1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪衍生物Ⅱ, 醋酸与中间体Ⅱ的羰基发生加成生成中间体Ⅲ, 接着三嗪环开环形成中间体Ⅳ, 中间体Ⅳ很容易转化成中间体B, 最后脱去一分子羧酸就得到了重排产物7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物Ⅵ.
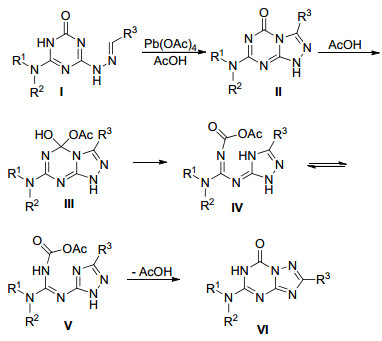
同年, Bakharev等[47]同样是以4-烷胺基取代的6-肼基-2-氧代-1, 3, 5-三嗪73为原料, 在纯甲酸中加热回流40~48 h生成了7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物77和副产物3-胍基-1, 2, 4-三氮唑衍生物78 (Eq. 20).从考察甲酸浓度对反应结果的影响来看, 甲酸浓度越大, 主产物77收率越高; 当甲酸浓度低于70%时, 主产物收率仅为5%或反应无法进行.
|
|
(20) |
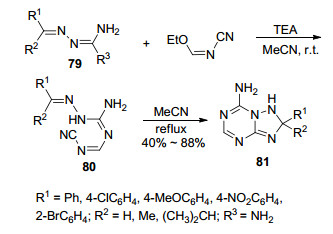
在合成三氮唑并均三嗪类化合物的方法中, 多数是以三氮唑环为基础来构建均三嗪环或以均三嗪环为基础来构建三氮唑环; 同时构建三氮唑环和均三嗪环来合成目标化合物也有文献报道. 1990年, Miyamoto课题组[48]研究了在不同条件下以腙类化合物79与N-氰基亚氨酸乙酯为起始原料, 合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物的两种方法, 即中间体转化法和直接合成法(Scheme 15).中间体转化法是从起始原料出发, 在三乙胺作用下室温反应生成链状中间体80, 然后发生分子内环合生成目标产物81; 直接合成法也是从起始原料出发在回流条件下直接生成目标产物81.除了个别化合物以外, 直接合成法与中间体转化法效果相当, 而且部分底物的收率会更高.
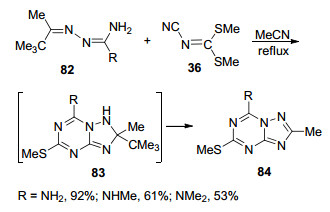
2000年, 该课题组[49]又以叔丁基取代的腙类化合物82与N-氰亚胺基-S, S-二硫代碳酸二甲酯36为原料, 在乙腈中加热回流1 h生成全共轭的1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物84 (Scheme 16).该反应先生成2号位同时具有甲基和叔丁基取代的三氮唑并三嗪的中间体83, 由于叔丁基的位阻太大, 所以很快离去生成全共轭的产物84.
1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物在热力学上的稳定性较差, 在加热或碱的作用下转化成更稳定的同分异构体1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪类化合物.这类化合物的合成方法主要是以肼基取代的1, 3, 5-三嗪为原料或者合成出该类中间体, 通过构建三氮唑环来合成目标化合物.
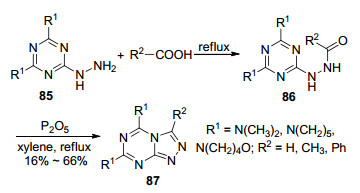
1974年, Deshpande等[50]利用取代的2-肼基-1, 3, 5-三嗪(85)与羧酸类化合物为原料来合成1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪衍生物(87) (Scheme 17).该反应首先在回流条件下生成中间体酰肼86, 最后在五氧化二磷作用下于无水二甲苯中回流得到目标化合物87.
2002年, Koppes等[51]报道了2, 4-二氨基-6-肼基- 1, 3, 5-三嗪(88)在1 mol/L盐酸中与溴化腈于室温条件下合成3, 5, 7-三胺基-1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪(89)的方法(Eq. 21).该反应具有条件温和, 后处理简单的优点; 且目标化合物可以用作医药, 工业等领域的原料.
|
|
(21) |
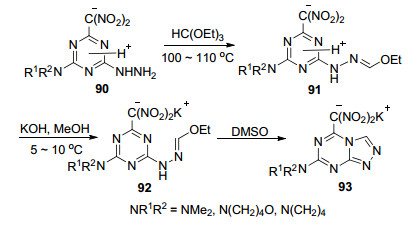
2013年, Golovina等[52]首次报道了5-二硝基甲基- 1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪钾盐(90)的合成及其结构鉴定.反应路线如Scheme 18所示。首先4-肼基-1, 3, 5-三嗪衍生物与原甲酸三乙酯反应得到中间体91, 接着在氢氧化钾甲醇溶液中碱化得其钾92, 最后发生区域选择性环化得到目标化合物93。化合物的结构经X射线单晶衍射得到了确认.
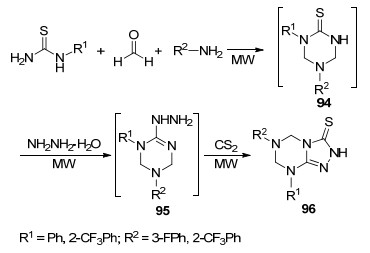
2004年, Dandia等[53]报道了在微波促进下一锅法合成1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物96的方法(Scheme 19).该方法是以芳基硫脲、甲醛和芳胺衍生物为起始原料, 首先得到2-硫代-1, 3, 5-三嗪中间体94, 然后与水合肼反应生成2-肼基-1, 3, 5-三嗪中间体95, 最后与二硫化碳环合得到目标产物96, 整个反应过程都是在微波辐射下完成的.一锅法提高了反应效率, 节约了反应成本.另外微波法还具有环境友好等优点.
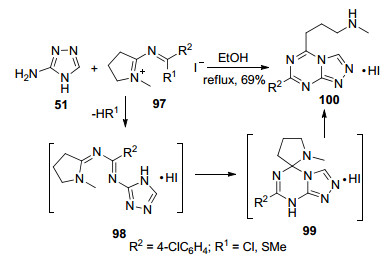
与上述方法不同的是, 2003年, Liebscher等[54]利用3-氨基-1, 2, 4-三氮唑(51)与吡咯衍生物97在乙醇中加热回流, 一步生成1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物100 (Scheme 20).在该反应中, 首先三氮唑中的氨基作为亲核试剂进攻与R1相连的碳原子, R1被三氮唑环取代形成中间体98, 然后发生分子内环化反应形成螺环化合物99, 最后在芳构化作用下四氢吡咯环开环形成目标化合物.整个反应经历了闭环和开环两个过程的相互转化.
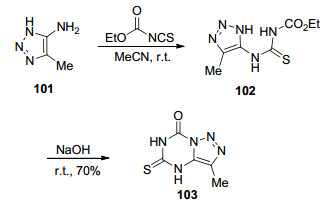
1976年, Fox等[55]首次报道了这种杂环体系, 并且提出了这类化合物的合成方法.该方法是以4-甲基-5-氨基-1, 2, 3-三氮唑101为原料, 先与乙氧羰基异硫氰酸酯在乙腈中室温反应0.5 h得到硫脲中间体102, 然后在氢氧化钠溶液中发生分子内环化生成目标化合物4-甲基-8-氧代-6-硫代-1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪(103) (Scheme 21).
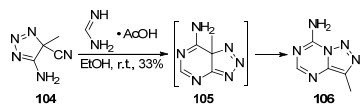
1985年, Leonard等[56]报道了以4-甲基-4-氰基-5-氨基-1, 2, 3-三氮唑(104)和甲脒醋酸盐为原料在无水乙醇中室温反应9 h生成4-甲基-8-氨基-1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪(106)的反应(Scheme 22).该反应可能先生成三氮唑并嘧啶中间体105, 然后迅速发生迁移重排得到目标化合物.
二氮唑并均三嗪类化合物从二氮唑的结构及其与均三嗪的稠合方式上可以分为以下三类(图 2):脒唑并[1, 2-a]-1, 3, 5-三嗪、脒唑并[1, 5-a]-1, 3, 5-三嗪和吡唑并[1, 5-a]-1, 3, 5-三嗪.
2-胍基苯并咪唑类化合物是合成苯并咪唑[1, 2-a]-1, 3, 5-三嗪衍生物的重要原料.其中, 1980年, Lalezari等[57]利用2-胍基苯并咪唑107和原甲酸三乙酯为原料, 加热回流3 h得到2-氨基苯并咪唑[1, 2-a]-1, 3, 5-三嗪108 (Eq. 22).原甲酸三乙酯在该反应中提供环加成反应的碳源, 另外, 还有文献报道偶氮二羧酸二乙酯[58]、二硫化碳、乙氧基亚甲基丙二酸二乙酯[59]、异氰酸苯酯[60]、4-氧代-1, 3-苯并恶嗪[61]等均可作为碳源供体参与反应.
|
|
(22) |
2005年, Bekircan等[5]利用2-氨基苯并咪唑(109)与N-羰基亚氨酸乙酯为原料在无溶剂条件下反应得到苯并咪唑[1, 2-a]-1, 3, 5-三嗪类化合物110 (Eq. 23).
|
|
(23) |
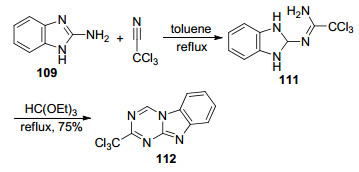
2012年, Dolzhenko等[28]利用了2-氨基苯并咪唑(109)与三氯乙腈在甲苯中加热回流5 h得到脒基取代的苯并咪唑中间体111, 然后在原甲酸三乙酯中加热回流3 h得到目标化合物2-三氯甲基苯并咪唑[1, 2-a]-1, 3, 5-三嗪(112) (Scheme 23).
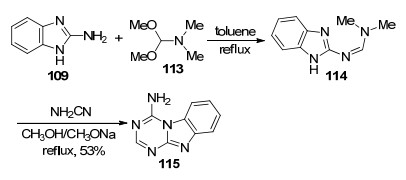
2013年, Dolzhenko等[29]以2-氨基苯并咪唑(109)与N, N-二甲基甲酰胺二甲缩醛(113)为原料, 先在甲苯中回流10 min得到脒基取代的苯并咪唑中间体114, 然后在甲醇钠甲醇溶液中与氨基腈加热回流24 h得到目标化合物4-氨基苯并咪唑[1, 2-a]-1, 3, 5-三嗪115 (Scheme 24).
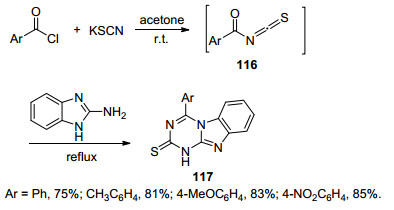
2016年, Neshati等[62]利用硫氰化钾, 芳基酰氯和2-氨基苯并咪唑为原料一锅法反应生成4-芳基-2-硫代苯并咪唑[1, 2-a]-1, 3, 5-三嗪(117) (Scheme 25).该反应中硫氰化钾和芳基酰氯先生成芳甲酰基异硫氰酸酯(116), 再与2-氨基苯并咪唑回流得到目标化合物117.
咪唑并[1, 2-a]-1, 3, 5-三嗪类化合物中除了苯并咪唑并三嗪这种三环类化合物外, 还有咪唑并[1, 2-a]-1, 3, 5-三嗪这种双环类化合物. 1968年, Tisler等[63]首次报道了这种新的杂环体系. 1991年, Nair等[65]利用2-氨基-4-氧代-1, 3, 5-三嗪(118)与氯乙醛为原料, 水作溶剂, 在45 ℃条件下反应5 d得到4-氧代咪唑并[1, 2-a]-1, 3, 5-三嗪(119) (Eq. 24). 2013年, Chen等[66]从2, 4-二氨基-1, 3, 5-三嗪出发与氯乙醛在DMSO中于120 ℃条件下反应生成咪唑并[1, 2-a]-1, 3, 5-三嗪类化合物. 2015年, 该课题组[64]用相同的策略合成出了具有抗肿瘤活性的粘附斑激酶抑制剂咪唑并[1, 2-a]-1, 3, 5-三嗪衍生物.
|
|
(24) |
1993年, Benner等[67]报道了2-氨基咪唑(120)与异硫氰酸基甲酸苯硫酯(X=S) (121)在碳酸钠作用下于乙腈中室温反应1 h得到2-氧代-4-硫代咪唑并[1, 2-a]-1, 3, 5-三嗪(122) (Eq. 25).之后他们[68]又报道了2-氨基咪唑类化合物与异氰酸基甲酸苯酯(X=O)在无水二氧六环中室温反应8 h得到2, 4-二氧代咪唑并[1, 2-a]-1, 3, 5-三嗪类化合物.
|
|
(25) |
我们课题组多年来致力于三嗪类化合物及其稠环化合物的研究与开发. 2017年, 我们[69]报道了以20 mol%氯化铜为催化剂, 在2 equiv.碘的促进下以2-氨基- 1, 3, 5-三嗪(123)与查耳酮(124)为原料高效合成咪唑并[1, 2-a]-1, 3, 5-三嗪类化合物125的方法(Eq. 26).通过对不同底物的适用性考察表明, 大部分底物都能取得良好收率.这种合成方法减少了反应步骤, 提高了反应效率, 节约了反应成本; 并且底物适用范围广, 收率高, 为合成咪唑并三嗪类化合物提供了新的思路.
|
|
(26) |
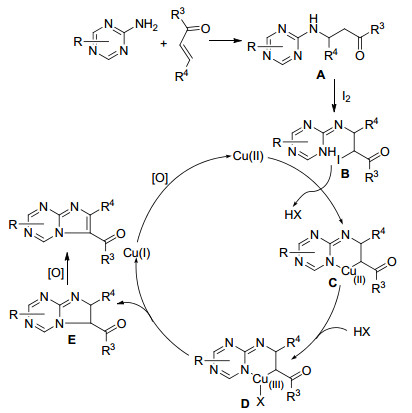
我们提出了Scheme 26所示的反应机理.首先, 2-氨基-1, 3, 5-三嗪与查尔酮发生Michael加成生成中间体A.后者与碘作用生成中间体B, 接着Cu盐很快结合上氮原子, 形成有机铜中间体C, 中间体C上的铜与卤素配位形成中间体D, 再发生还原消除得到稠环化合物E, 最后被氧化成目标化合物咪唑并[1, 2-a]-1, 3, 5-三嗪衍生物, 一价铜被氧气氧化成二价铜进入下一步循环.在该反应中铜离子的价态经历了二价、三价和一价的循环.
有关咪唑并[1, 5-a]-1, 3, 5-三嗪类化合物的合成虽有报道, 但是对于能够提供多取代基的咪唑并三嗪类化合物的合成方法, 相关文献报道还不多.
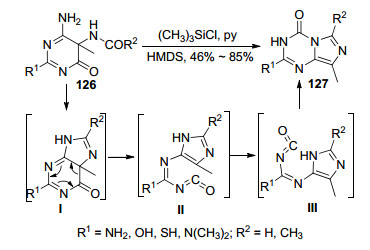
1979年, Leonard等[70]报道了嘧啶衍生物发生环化重排生成咪唑并[1, 5-a]-1, 3, 5-三嗪衍生物127的反应(Scheme 27).该反应是以为嘧啶衍生物126为原料, 先与无水吡啶和三甲基氯硅烷在室温条件下反应30 min, 然后加入六甲基二硅氮烷并回流10 min得到目标化合物2, 6-二取代-8-甲基-4-氧代咪唑并[1, 5-a]-1, 3, 5-三嗪(127).在该反应中嘧啶衍生物首先发生分子内加成消除反应生成咪唑并嘧啶中间体Ⅰ, 然后发生电子转移碳碳键断裂生成异氰酸酯中间体Ⅱ, 中间体Ⅱ中的单键旋转得到中间体Ⅲ, 最后咪唑环上的氨基与异氰酸基中的羰基加成生成目标化合物.
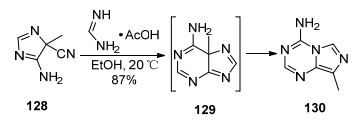
1982年, 他们[71]又报道了以4-甲基-4-氰基-5-氨基咪唑(128)和甲脒醋酸盐为原料合成8-甲基-4-氨基咪唑并[1, 5-a]-1, 3, 5-三嗪(130)的反应(Scheme 28).该反应以无水乙醇作溶剂, 先是在N2环境中于20 ℃条件下反应1 h, 最后加入乙醇钠在空气环境中继续反应7 h得到目标化合物.该反应可能先生成中间体5-甲基腺嘌呤129, 然后迅速发生迁移重排得到目标化合物. 1983年, 该课题组[72]报道了类似的迁移重排反应, 生成了多取代的咪唑并[1, 5-a]-1, 3, 5-三嗪衍生物.
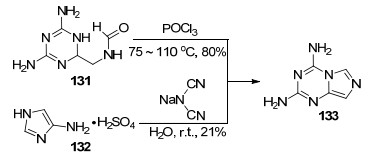
2003年, Wang等[73]报道了两种合成2, 4-二氨基咪唑并[1, 5-a]-1, 3, 5-三嗪(133)的方法(Scheme 29).方法一是以1, 3, 5-三嗪衍生物131为原料在三氯氧磷中于75 ℃条件下反应2 h, 再回流3 h得到目标化合物; 方法二是以4-氨基咪唑硫酸盐132和二氰胺钠为原料在水中室温反应48 h得到目标化合物.
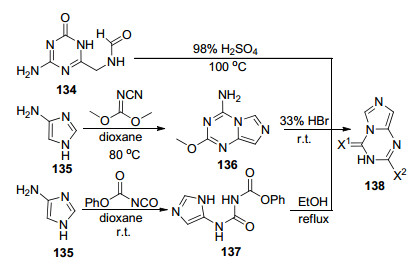
2004年, Han等[74]报道了四种咪唑并[1, 5-a]-1, 3, 5-三嗪类化合物的合成方法(Scheme 30).首先甲酰胺取代的均三嗪衍生物134在98%浓硫酸的作用下发生分子内环化得到2-氨基-4-氧代咪唑并[1, 5-a]-1, 3, 5-三嗪(138)(X1=O, X2=NH2). 2005年, Wagner等[75]用相同的方法合成了2-氨基-4-氧代咪唑并[1, 5-a]-1, 3, 5-三嗪.其次, 以4-氨基咪唑(135)与N-氰基羰亚胺二甲酯为原料在二氧六环中于80 ℃条件下反应3 h得到4-氨基-2-甲氧基咪唑并[1, 5-a]-1, 3, 5-三嗪(136), 然后在33%氢溴酸醋酸溶液中室温反应12 h得到4-氨基-2-氧代咪唑并[1, 5-a]-1, 3, 5-三嗪(X1=NH2, X2=O).最后, 以4-氨基咪唑与异氰酸基甲酰苯酯为原料在二氧六环中室温反应2 h得到咪唑中间体137, 137在乙醇中加热回流3 h得到化合物2, 4-二氧代咪唑并[1, 5-a]-1, 3, 5-三嗪(57)(X1= X2=O).
1974年, Novinson等[76]报道了合成一系列不对称2, 4-二烷基吡唑并[1, 5-a]-1, 3, 5-三嗪类化合物140的反应(Eq. 27).这类反应都是以甲脒取代的吡唑衍生物139为原料, 通过环加成反应来合成目标化合物.二硫化碳、溴化腈与原甲酸三乙酯一样都是作为碳源供体参与到环加成反应中.还有文献报道[77]二乙氧羰基、N, N'-羰基二咪唑(CDI)在与脒基取代的吡唑衍生物的反应中起到了相同的作用.
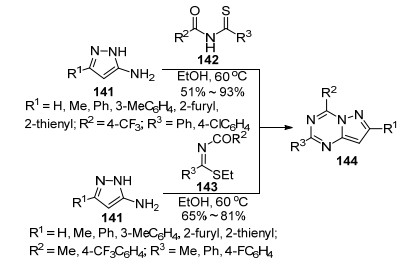
脒基或胍基取代的吡唑是合成吡唑并均三嗪类化合物的重要原料或中间体, 而以5(3)-氨基吡唑为原料来合成吡唑并均三嗪类化合物更是常用的方法. 1985年, Lin等[78]报道了5-氨基吡唑(141)与酰胺衍生物142在乙醇中于60 ℃条件下反应24 h生成2, 4, 7-三取代吡唑并[1, 5-a]-1, 3, 5-三嗪类化合物144.另外化合物141与碘乙烷反应生成N-羰基硫代亚胺酸乙酯143, 化合物143与5-氨基吡唑在同样条件下反应也能得到吡唑并三嗪类化合物(Scheme 31).
2005年, Bekircan等[5]利用5-氨基吡唑(145)与N-羰基亚氨酸乙酯(4)为原料在165~170 ℃条件下反应2 h得到2, 4-二芳基吡唑并[1, 5-a]-1, 3, 5-三嗪(146)(Eq. 28).
|
|
(28) |
2006年, Insuasty等[79]报道了区域选择性反应合成4-取代-7-甲基-2-乙硫基吡唑并[1, 5-a]-1, 3, 5-三嗪(149)的方法, 该反应是以5-氨基吡唑(147)和N-羰基硫代亚氨酸乙酯(148)为原料在DMF中加热回流0.5~1.5 h得到目标化合物(Eq. 29).这种合成方法具有反应时间短, 收率高的优点.
|
|
(29) |
2007年, Demidchuk等[39]报道了3-取代-5-氨基吡唑(150)与2-氮杂-1, 3-丁二烯类化合物56为原料在三乙胺四氢呋喃溶液中回流84 h生成2, 7-二取代-4-二氯甲基吡唑并[1, 5-a]-1, 3, 5-三嗪(151)的反应(Eq. 30).该方法需要长时间回流, 且收率较低.
|
|
(30) |
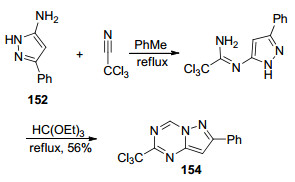
2012年, Dolzhenko等[28]报道了3-苯基-5-氨基吡唑(152)与三氯乙腈在甲苯中加热回流2 h得到脒基取代的吡唑中间体153, 然后在原甲酸三乙酯中加热回流6 h生成7-苯基-2-三氯甲基吡唑并[1, 5-a]-1, 3, 5-三嗪(154)的反应(Scheme 32).三氯甲基作为活性基团与胺类化合物反应可以直接在原位引入胺基, 是合成胺基取代的吡唑并三嗪类化合物的重要方法.
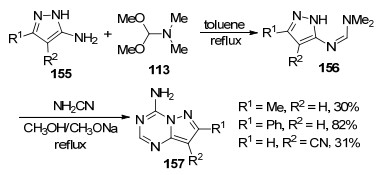
2013年, 他们[29]又利用3, 4-二取代-5-氨基吡唑(155)与N, N-二甲基甲酰胺二甲缩醛(113)为原料在甲苯中加热回流1~7 h得到脒基取代的吡唑中间体156, 然后在甲醇钠甲醇溶液中与氨基腈加热回流24 h生成7, 8-二取代-4-氨基吡唑并[1, 5-a]-1, 3, 5-三嗪(157) (Scheme 33).
2014年, Dolzhenko等[80]又报道了在微波促进下三组分一锅法合成吡唑并[1, 5-a]-1, 3, 5-三嗪类化合物157的方法(Eq. 31).该方法是以3-苯基-5-氨基吡唑155原甲酸三乙酯和氨基腈为原料, 在微波辐射下完成的, 而用传统的加热回流方法不能获得较高收率.通过对底物适用性考察表明, 大部分底物都能取得中等到良好收率.
|
|
(31) |
2013年, Zamigailo等[40]利用4-氧代-1, 3-苯并噁嗪类化合物158与等量的3-甲基-5-氨基吡唑(147)为原料在甲醇钠甲醇溶液中加热回流2.5 h生成4, 7-二甲基-2-芳基吡唑并[1, 5-a]-1, 3, 5-三嗪(159) (Eq. 32).
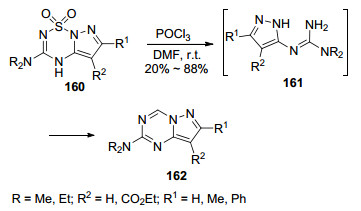
2016年, Norman等[81]报道了吡唑[1, 5-b][1,2.4,6]-恶三嗪-1, 1-二酮(160)与维尔斯迈尔试剂在室温条件下反应生成吡唑并[1, 5-a]-1, 3, 5-三嗪类化合物162的方法(Scheme 34).在反应过程中噁三嗪衍生物在酸性条件下发生氮硫键断裂释放出SO2气体形成5-胍基吡唑中间体161, 然后与维尔斯迈尔试剂反应生成最终产物7, 8-二取代-2-氨基吡唑并[1, 5-a]-1, 3, 5-三嗪.
|
|
(32) |
整体看来, 在氮唑并均三嗪类化合物当中, 对于报道合成1, 2, 4-三氮唑并[1, 5-a]均三嗪和吡唑并均三嗪这两类化合物的方法较多, 这些合成方法条件相对温和且大部分都不需要金属催化, 符合绿色化学发展理念; 在合成步骤上还有从多步法到一锅法转变的趋势.另外, 随着合成方法的日新月异, 对这两类化合物的活性研究也在深入.对于四氮唑并均三嗪, 部分三氮唑并均三嗪以及咪唑并均三嗪这三类化合物来说, 无论是合成方法还是到生物活性研究相关报道都很少, 而且现有的合成方法适用范围窄.所以我们期待未来要解决的问题是完善氮唑并均三嗪类化合物的合成方法, 并深入研究这类化合物的生物活性, 随着这个研究方向不断向前发展, 我们也坚信这些问题能够得到解决.
王光荣, 曾和平, 有机化学, 2009, 29, 1115. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu200907014&dbname=CJFD&dbcode=CJFQWang, G.-R.; Zeng, H.-P. Chin. J. Org. Chem. 2009, 29, 1115(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu200907014&dbname=CJFD&dbcode=CJFQ
Charoensirisomboon, P.; Saito, H.; Inoue, T.; Oishi, Y.; Mori, K. Polymer 1998, 39, 2089. doi: 10.1016/S0032-3861(97)00530-2
(a) Liu, Y. -N. ; Sun, X. -N. ; Gao, R. ; Li, C. -Y. ; Wang, J. ; Li, Y. -Z. ; Zhang, C. -L. Chin. J. Org. Chem. 2017, 37, 2057(in Chinese).
(刘亚宁, 孙晓娜, 高然, 李传银, 王静, 李益政, 张成路, 有机化学, 2017, 37, 2057. )
(b) Sączewski, F. ; Bułakowska, A. Eur. J. Med. Chem. 2006, 41, 611.
(c) Zheng, M. F. ; Xu, C. H. ; Ma, J. W. ; Sun, Y. ; Du, F. F. ; Liu, H. ; Lin, L. P. ; Li, C. ; Ding, J. ; Chen, K. X. ; Jiang, H. L. Bioorg. Med. Chem. 2007, 15, 1858.
El, F. A.; Soliman, S. M.; Ghabbour, H. A.; Elnakady, Y. A.; Mohaya, T. A.; Siddiqui, M. R. H.; Albericio, F. J. Mol. Struct. 2016, 1125, 121. doi: 10.1016/j.molstruc.2016.06.061
Bekircan, O.; Küxük, M.; Kahveci, B.; Kolaylı, S. Arch. Pharm. Chem. Life Sci. 2005, 338, 365. doi: 10.1002/(ISSN)1521-4184
Bera, H.; Tan, B. J.; Sun, L. Y.; Dolzhenko, A. V.; Chui, W. K.; Chiu, G. N. C. Eur. J. Med. Chem. 2013, 67, 325. doi: 10.1016/j.ejmech.2013.06.051
(a) Federico, S. ; Paoletta, S. ; Cheong, S. L. ; Pastorin, G. ; Cacciari, B. ; Stragliotto, S. ; Klotz, K. N. ; Siegel, J. ; Gao, Z. G. ; Jacobson, K. A. ; Moro, S. ; Spalluto, G. J. Med. Chem. 2011, 54, 877.
(b) Pastorin, G. ; Federico, S. ; Paoletta, S. ; Corradino, M. ; Cateni, F. ; Cacciari, B. ; Klotz, K. N. ; Gao, Z. G. ; Jacobson, K. A. ; Spalluto, G. ; Moro, S. Bioorg. Med. Chem. 2010, 18, 2524.
(c) Jörg, M. ; May, L. T. ; Mak, F. S. ; Lee, K. C. K. ; Miller, N. D. ; Scammells, P. J. ; Capuano, B. J. Med. Chem. 2015, 58, 718.
(d) Jörg, M. ; Shonberg, J. ; Mak, F. S. ; Miller, N. D. ; Elizabeth, Y. ; Scammells, P. J. ; Capuano, B. Bioorg. Med. Chem. 2013, 23, 3427.
(e) Federico, S. ; Antonella, C. ; Porta, N. ; Redenti, S. ; Pastorin, G. ; Cacciari, B. ; Klotz, K. N. ; Moro, S. ; Spalluto, G. Eur. J. Med. Chem. 2016, 108, 529.
(a) Bera, H. ; Chui, W. K. ; Gupta, S. D. ; Dolzhenko, A. V. ; Sun, L. Y. Med. Chem. Res. 2013, 22, 6010.
(b) Bera, H. ; Lee, M. H. ; Sun, L. Y. ; Dolzhenko, A. V. ; Chui, W. K. Bioorg. Chem. 2013, 50, 34.
Golankiewicz, B.; Januszczyk, P.; Ikeda, S.; Balzarini, J.; Clercq, E. D. J. Med. Chem. 1995, 38, 3558. doi: 10.1021/jm00018a015
(a) Matsuura, M. F. ; Winiger, C. B. ; Shaw, R. W. ; Kim, M. J. ; Kim, M. S. ; Daugherty, A. B. ; Chen, F. ; Moussatche, P. ; Moses, J. D. ; Lutz, S. ; Benner, S. A. ACS Synth. Biol. 2017, 6, 388.
(b) Winiger, C. B. ; Shaw, R. W. ; Kim, M. J. ; Moses, J. D. ; Matsuura, M. F. ; Benner, S. A. ; ACS Synth. Biol. 2017, 6, 194.
Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Lu, J.; Averill, A.; Almassy, R.; Chu, S. S. Bioorg. Med. Chem. Lett. 2008, 18, 619. doi: 10.1016/j.bmcl.2007.11.074
Popowycz, F.; Schneider, C.; DeBonis, S.; Skoufias, D. A.; Kozielski, F.; Galmarini, C. M.; Joseph, B. Bioorg. Med. Chem. 2009, 17, 3471. doi: 10.1016/j.bmc.2009.03.007
(a) Raboisson, P. ; Schultz, D. ; Muller, C. ; Reimund, J. Marie. ; Pinna, G. ; Mathieu, R. ; Bernard, P. ; Do, Q. T. ; DesJarlais. R. L. ; Justiano, H. ; Lugnier, C. ; Bourguignon, J. J. Eur. J. Med. Chem. 2008, 43, 816.
(b) Nie, Z. ; Perretta, C. ; Erickson, P. ; Margosiak, S. ; Almassy, R. ; Lu, J. ; Averill, A. ; Yagera, K. M. ; Chua, S. S. Bioorg. Med. Chem. Lett. 2007, 17, 4191.
(c) Sun, L. Y. ; Bera, H. ; Chui, W. K. Eur. J. Med. Chem. 2013, 65, 1.
(d) Laufer, R. ; Li, S. Wan. ; Liu, Y. ; Ng, G. ; Lang, Y. H. ; Feher, M. ; Brokx, R. ; Beletskaya, I. ; Hodgson, R. ; Mao, G. D. ; Plotnikova, O. ; Awrey, D. E. ; Mason, J. M. ; Wei, X. ; Lin, D. C. C. ; Che, Y. ; Kiarash, R. ; Madeira, B. ; Fletcher, G. C. ; Mak, T. W. ; Bray, M. R. ; Pauls, H. W. Bioorg. Med. Chem. Lett. 2016, 26, 3562.
(e) Senga, K. ; O'Brien, D. E. ; Scholten, M. B. ; Novinson, T. ; Miller, J. P. ; Robins, R. K. J. Med. Chem. 1982, 25, 243.
(a) Raboisson, P. ; Baurand, A. ; Cazenave, J. P. ; Gachet, C. ; Schultz, D. ; Spiess, B. ; Bourguignon, J. J. J. Org. Chem. 2002, 67, 8063.
(b) Lefoix, M. ; Mathis, G. ; Kleinmann, T. ; Truffert, J. C. ; Asseline, U. J. Org. Chem. 2014, 79, 3221.
Hafez, E. A. A.; Elmoghayar, M. R. H.; Ramiz, M. M. M. Liebigs Ann. Chem. 1987, 65. doi: 10.1007/bf00809296
Kessenich, E.; Polborn, K.; Schulz, A. Inorg. Chem. 2001, 40, 1102. doi: 10.1021/ic000526k
Chapyshev, S. V.; Chernyak, A. V.; Yakushchenko, I. K. J. Heterocycl. Chem. 2016, 53, 970. doi: 10.1002/jhet.v53.3
Fedorov, B. S.; Fadeev, M. A.; Gidaspov, A. A.; Kosareva, E. A.; Bakharev, V. V. Chem. Heterocycl. Compd. 2005, 41, 228. doi: 10.1007/s10593-005-0132-5
Bakharev, V. V.; Ghidaspov, A. A.; Krivolapov, D. B.; Mironova, E. V.; Litvinov, I. A. Chem. Heterocycl. Compd. 2006, 42, 1051. doi: 10.1007/s10593-006-0203-2
Parfenov, V. E.; Bakharev, V. V.; Zavodskaya, A. V.; Selezneva, E. V.; Gidaspov, A. A.; Suponitsky, K. Y. Tetrahedron Lett. 2014, 55, 7072. doi: 10.1016/j.tetlet.2014.10.143
Bokaldere, R. P.; Grinshtein, V. Y. Khim. Geterotsikl. Soedin. 1970, 6, 563. 10.1007/bf01172865
Dolzhenko, A. V.; Dolzhenko, A. V.; Chui, W. K. Tetrahedron 2007, 63, 12888. doi: 10.1016/j.tet.2007.10.046
Taylor, E. C.; Hendess, R. W. J. Am. Chem. Soc. 1965, 87, 1980. doi: 10.1021/ja01087a022
Evers, R.; Fischer, E. J. Prakt. Chem. 1985, 327, 609. doi: 10.1002/(ISSN)1521-3897
Dorokhov, V. A.; Amamchyan, A. R.; Bogdanov, V. S. Izv. Akad. Nauk SSSR, Ser. Khim. 1989, 2386.
Dorokhov, V. A.; Amamchyan, A. R.; Bogdanov, V. S.; Ugrak, B. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1991, 241.
(a) Dolzhenko, A. V. ; Pastorin, G. ; Dolzhenko, A. V. ; Chui, W. K. Tetrahedron Lett. 2008, 49, 7180.
(b) Dolzhenko, A. V. ; Tan, B. J. ; Chiu, G. N. C. ; Chui, W. K. ; Dolzhenko, A. V. J. Fluorine Chem. 2015, 175, 68.
Kalinin, D. V.; Kalinina, S. A.; Dolzhenko, A. V. Heterocycles 2012, 85, 2515. doi: 10.3987/COM-12-12542
Kalinin, D. V.; Kalinina, S. A.; Dolzhenko, A. V. Heterocycles 2013, 87, 147. doi: 10.3987/COM-12-12601
Zohdi, H. F. J. Chem. Res., Synop. 1998, 536. https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=30004SI7.TXT
Dolzhenko, A. V.; Tan, B. J.; Dolzhenko, A. V.; Chiu, G. N. C.; Chui, W. K. J. Fluorine Chem. 2008, 129, 429. doi: 10.1016/j.jfluchem.2008.02.007
Lalezari, I.; Nabahi, S. J. Heterocycl. Chem. 1980, 17, 1121. doi: 10.1002/jhet.v17:5
Akahoshi, F.; Takeda, S.; Okada, T.; Kajii, M.; Nishimura, H.; Sugiura, M.; Inoue, Y.; Fukaya, C.; Naito, Y.; Imagawa, T.; Nakamura, N. J. Med. Chem. 1998, 41, 2985. doi: 10.1021/jm970759u
Bereczm, G.; Pongó, L.; Kövesdi, I.; Reiter, J. J. Heterocycl. Chem. 2002, 39, 327. doi: 10.1002/jhet.5570090116
Hirata, T.; Twanmoh, L. M.; Wood, H. R.; Jr.; Goldin, A.; Driscoll, J. S. J. Heterocycl. Chem. 1972, 9, 99. doi: 10.1002/jhet.5570100210
Hirata, T.; Wood, H. B.; Driscoll, J. S. J. Chem. Soc., Perkin Trans. 1 1973, 1209. https://es.scribd.com/document/135298421/Alternative-Anode-Materials-for-Methane-Oxidation-in-Solid-Oxide-Fuel-Cells
Okide, G. B. J. Heterocycl. Chem. 1994, 31, 535. doi: 10.1002/jhet.v31:2
Kaddachi, M. T.; Zouari, S.; Benammar, H.; Cossy, J.; Kahn, P. J. Soc. Chim. Tunis. 2001, 4, 1171. http://www.documentation.ird.fr/hor/fdi:010062961
Demidchuk, B. A.; Brovarets, V. S.; Chernega, A. N.; Howard, J. A. K.; Vasilenko, A. N.; Turov, A. V.; Drach, B. S. Russ. J. Gen. Chem. 2007, 77, 474. doi: 10.1134/S107036320703022X
Zamigailo, L. L.; Petrova, O. N.; Shirobokova, M. G.; Lipson, V. V. Russ. J. Org. Chem. 2013, 49, 288. doi: 10.1134/S1070428013020188
Fronabarger, J. W.; Chapman, R. D.; Gilardi, R. D. Tetrahedron Lett. 2006, 47, 7707. doi: 10.1016/j.tetlet.2006.08.106
Khankischpur, M.; Hansen, F. K.; Geffken, D. Synthesis 2010, 1645. https://www.frontiersin.org/research-topics/3876/epub
Dolzhenko, A. V.; Kalinina, S. A.; Kalinin, D. V. RSC Adv. 2013, 3, 15850. doi: 10.1039/c3ra41932k
Tartakovsky, V. A.; Frumkin, A. E.; Churakov, A. M.; Strelenko, Y. A. Russ. Chem. Bull. 2005, 54, 719. doi: 10.1007/s11172-005-0310-8
Bakharev, V. V.; Parfenov, V. E.; Ul'yankin, I. V.; Zavodskaya, A. V.; Selezneva, E. V.; Gidaspov, A. A.; Eltsov, O. S.; Slepukhin, P. A. Tetrahedron 2014, 70, 6825. doi: 10.1016/j.tet.2014.07.058
Zavodskaya, A. V.; Bakharev, V. V.; Parfenov, V. E.; Gidaspov, A. A.; Slepukhin, P. A.; Isenov, M. L.; Eltsov, O. S. Tetrahedron Lett. 2015, 56, 1103. doi: 10.1016/j.tetlet.2015.01.151
Bakharev, V. V.; Parfenov, V. E.; Ul'yankina, I. V.; Zavodskaya, A. V.; Gidaspov, A. A.; Slepukhin, P. A.; Chem. Heterocycl. Compd. 2015, 51, 1014. doi: 10.1007/s10593-016-1812-z
Miyamoto, Y.; Yamazaki, C.; Matzui, M. J. Heterocycl. Chem. 1990, 27, 1553. doi: 10.1002/jhet.v27:6
Miyamoto, Y. J. Heterocycl. Chem. 2000, 37, 1587. doi: 10.1002/jhet.v37:6
Deshpande, R. J.; Roa, A. V. R. Synthesis 1974, 863. http://www.academia.edu/29719564/Chitinolytic_enzymes_classification_and_applications_An_Overview
Koppes, W. M.; Sitzmann, M. E. US 6423844, 2002. doi: 10.1108/9781786350633
Golovina, O. V.; Bakharev, V. V.; Golovin, E. V.; Parfenov, V. E.; Slepukhin, P. A. Tetrahedron Lett. 2013, 54, 3858. doi: 10.1016/j.tetlet.2013.05.052
Dandia, A.; Arya, K.; Sati, M. Synth. Commun. 2004, 34, 1141. doi: 10.1081/SCC-120028646
Karanik, M.; P tzel, M.; Liebscher, J. Synthesis 2003, 1201.
Klein, R. S.; De, L. H.; Federico, G.; Tam, S. Y. K.; Wempen, I.; Fox, J. J. J. Heterocycl. Chem. 1976, 13, 589. doi: 10.1002/jhet.5570130333
Oakes, F. T.; Leonard, N. J. J. Org. Chem. 1985, 50, 4986. doi: 10.1021/jo00224a074
Lalezari, I.; Nabahi, S. J. Heterocycl. Chem. 1980, 17, 1121. doi: 10.1002/jhet.v17:5
Furukawa, M.; Kawanabe, K.; Yoshimi, A.; Okawara, T.; Noguchi, Y. Chem. Pharm. Bull. 1983, 31, 2473. doi: 10.1248/cpb.31.2473
Badawey, E. A. M.; Kappe, T. Arch. Pharm. Pharm. Med. Chem. 1997, 330, 59. doi: 10.1002/(ISSN)1521-4184
Dolzhenko, A. V.; Chui, W. K.; Dolzhenko, A. V. Synthesis 2006, 597.
Suvorova, E. Y.; Vikrishchuk, N. I.; Popov, L. D.; Starikova, Z. A.; Vikrishchuk, A. D.; Zhdanov, Y. A. Russ. J. Org. Chem. 2007, 43, 1553. doi: 10.1134/S1070428007100259
Esmaeili, N.; Neshati, J.; Yavari, I. Res. Chem. Intermed. 2016, 42, 5339. doi: 10.1007/s11164-015-2369-7
Kobe, J.; Stanovnik, B.; Tisler, M. Chem. Commun. (London) 1968, 1456.
Dao, P.; Smith, N.; Tomkiewicz, R. C.; Yen, P. E.; Camacho, A. M.; Lietha, D.; Herbeuval, J. P.; Coumoul, X.; Garbay, C.; Chen, H. X. J. Med. Chem. 2015, 58, 237. doi: 10.1021/jm500784e
Nair, V.; Lyons, A. G.; Purdy, D. F. Tetrahedron 1991, 47, 8949. doi: 10.1016/S0040-4020(01)86501-7
Dao, P.; Garbay, C.; Chen, H. X. Tetrahedron 2013, 69, 3867. doi: 10.1016/j.tet.2013.03.039
Voegel, J. J.; Altorfer, M. M.; Benner, S. A. Helv. Chim. Acta 1993, 76, 2061. doi: 10.1002/(ISSN)1522-2675
Rao, P.; Benner, S. A. J. Org. Chem. 2001, 66, 5012. doi: 10.1021/jo005743h
Li, J. J.; Song, C.; Cui, D. Mei.; Zhang, C. Org. Biomol. Chem. 2017, 15, 5564. doi: 10.1039/C7OB01018D
Holtwick, J. B.; Golankiewicz, B.; Holmes, B. N.; Leonard, N. J. J. Org. Chem. 1979, 44, 3858. doi: 10.1021/jo01336a023
Hosmane, R. S.; Bakthavachalam, V.; Leonard, N. J. J. Am. Chem. Soc. 1982, 104, 235. doi: 10.1021/ja00365a043
Balicki, R.; Hosmane, R. S.; Leonard, N. J. J. Org. Chem. 1983, 48, 3. doi: 10.1021/jo00149a002
Wang, Z. J.; Huynh, H. K.; Han, B.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2003, 5, 2067. doi: 10.1021/ol030044n
Han, B.; Jaun, B.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2004, 6, 3691. doi: 10.1021/ol048649m
Wagner, T.; Han, B.; Koch, G.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta 2005, 88, 1960. doi: 10.1002/(ISSN)1522-2675
Novinson, T.; Senga, K.; Kobe, J.; Robins, R. K.; O'Brien, D. E.; Albert, A. A. J. Heterocycl. Chem. 1974, 11, 691. doi: 10.1002/jhet.v11:5
Brandt, T. A.; Caron, S.; Damon, D. B.; DiBrino, J.; Ghosh, A.; Griffith, D. A.; Kedia, S.; Ragan, J. A.; Rose, P. R.; Vanderplas, B. C.; Wei, L. L. Tetrahedron 2009, 65, 3292. doi: 10.1016/j.tet.2008.10.067
Strohmeyer, T. W.; Sliskovic, D. R.; Lang, S. A.; Jr.; Lin, Y. J. Heterocycl. Chem. 1985, 22, 7. doi: 10.1002/jhet.v22:1
Insuasty, H.; Estrada, M.; Corte's, E.; Quiroga, J.; Insuasty, B.; Abonı'a, R.; Nogueras, M.; Cobo, J. Tetrahedron Lett. 2006, 47, 5441. doi: 10.1016/j.tetlet.2006.05.168
Lim, F. P. L.; Luna, G.; Dolzhenko, A. V. Tetrahedron Lett. 2014, 55, 5159. doi: 10.1016/j.tetlet.2014.07.105
Norman, R. E.; Perkins, M. V.; Liepa, A. J.; Francis, C. L. Aust. J. Chem. 2016, 69, 61. doi: 10.1071/CH14547

Scheme 2 四氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物的合成
Scheme 2 Synthesis of tetrazolo[1, 5-a]-1, 3, 5-triazine derivatives

Scheme 3 以6-氯-1, 3, 5-三嗪为原料合成四氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物
Scheme 3 Synthesis of tetrazolo[1, 5-a]-1, 3, 5-triazine derivatives from 6-chloro-1, 3, 5-triazine

Scheme 4 1-脒基-5-氨基-1, 2, 4-三氮唑参与合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物
Scheme 4 Synthesis of 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives from 1-amidine-5-amine-1, 2, 4-triazole

Scheme 5 2-芳基-5-三氯甲基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 5 Synthesis of 2-aryl-5-trichloromethyl-1, 2, 4-triazolo- [1, 5-a]-1, 3, 5-triazine

Scheme 6 2-取代-7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物的合成
Scheme 6 Synthesis of 2-substituted-7-amino-1, 2, 4-triazolo- [1, 5-a]-1, 3, 5-triazine derivatives

Scheme 7 7-氧(硫)代-5-硫(氧)代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 7 Synthesis of 7-oxo(thio)-5-thio(oxo)-1, 2, 4-triazolo- [1, 5-a]-1, 3, 5-triazine

Scheme 8 5, 7-二(二甲氨基)-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 8 Synehesis of 5, 7-doubledimethylamino-1, 2, 4-tria- zolo[1, 5-a]-1, 3, 5-triazine

Scheme 9 以2-氮杂-1, 3-丁二烯和5-氨基-1, 2, 4-三氮唑为原料合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物
Scheme 9 Synthesis of 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives from 2-azabuta-1, 3-dienes and 5-amino-1, 2, 4-triazine

Scheme 10 5-取代-2-氨基-7-氧代-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物的合成
Scheme 10 Synthesis of 5-substituted-2-amino-7-oxo-1, 2, 4- triazolo[1, 5-a]-1, 3, 5-triazine derivatives

Scheme 11 形成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物可能的反应机理
Scheme 11 Mechanism proposed for the formation of tris- 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives

Scheme 12 以6-肼基-1, 3, 5-三嗪与原甲酸三乙酯为原料合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物
Scheme 12 Synthesis of 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives by 6-hydrazinyl-1, 3, 5-triazine and triethyl orthoformate

Scheme 13 以6-肼基-1, 3, 5-三嗪与苯甲醛为原料合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物
Scheme 13 Synthesis of 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives from 6-hydrazinyl-1, 3, 5-triazine and benzaldehyde

Scheme 14 6-肼基-1, 3, 5-三嗪和苯甲醛类化合物参与合成1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪衍生物可能的反应机理
Scheme 14 Mechanism proposed for the formation of 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine derivatives from 6-hydrazinyl- 1, 3, 5-triazine and benzaldehyde

Scheme 15 2, 2'-二取代-7-氨基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 15 Synthesis of 2, 2'-disubstituted-1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine

Scheme 16 7-取代-2-甲基-5-甲硫基-1, 2, 4-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 16 Synthesis of 7-substituted-2-methyl-5-methylthio- 1, 2, 4-triazolo[1, 5-a]-1, 3, 5-triazine

Scheme 17 2-肼基-1, 3, 5-三嗪参与合成1, 2, 4-三氮唑并[4, 3-a]- 1, 3, 5-三嗪衍生物
Scheme 17 Synthesis of 1, 2, 4-triazolo[4, 3-a]-1, 3, 5-triazine derivatives from 2-hydrazinyl-1, 3, 5-triazine

Scheme 18 5-二硝基甲基-7-烷氨基-1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪钾盐的合成
Scheme 18 Synthesis of the potassium salts of 7-alkylamino- 5-dinitromethyl-1, 2, 4-triazolo[4, 3-a]-1, 3, 5-triazine

Scheme 19 微波辐射下一锅法合成1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物
Scheme 19 One pot synthesis of 1, 2, 4-triazolo[4, 3-a]-1, 3, 5- triazine derivatives under microwave irradiation

Scheme 20 1, 2, 4-三氮唑并[4, 3-a]-1, 3, 5-三嗪类化合物的合成
Scheme 20 Synthesis of 1, 2, 4-triazolo[4, 3-a]-1, 3, 5-triazine derivatives

Scheme 21 4-甲基-8-氧代-6-硫代-1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 21 Synthesis of 4-methyl-8-oxo-6-thio-1, 2, 3-triazolo- [1, 5-a]-1, 3, 5-triazine

Scheme 22 通过迁移重排合成4-甲基-8-氨基-1, 2, 3-三氮唑并[1, 5-a]-1, 3, 5-三嗪
Scheme 22 Synthesis of 8-amino-4-methyl-1, 2, 3-triazolo- [1, 5-a]-1, 3, 5-triazine via translocative rearrangement

Scheme 23 2-三氯甲基苯并咪唑[1, 2-a]-1, 3, 5-三嗪的合成
Scheme 23 Synthesis of 2-trichloromethylbenzimidazo- [1, 2-a]-1, 3, 5-triazine

Scheme 24 4-氨基苯并咪唑[1, 2-a]-1, 3, 5-三嗪的合成
Scheme 24 Synthesis of 4-aminobenzimidazo[1, 2-a]-1, 3, 5- triazine

Scheme 25 一锅法合成苯并咪唑[1, 2-a]-1, 3, 5-三嗪衍生物
Scheme 25 One pot synthesis of benzimidazo[1, 2-a]-1, 3, 5- triazine derivatives

Scheme 26 合成咪唑并[1, 2-a]-1, 3, 5-三嗪衍生物可能的反应机理
Scheme 26 Mechanism proposed for the formation of imidazo[1, 2-a]-1, 3, 5-triazine derivatives

Scheme 27 咪唑并[1, 5-a]-1, 3, 5-三嗪类化合物的合成
Scheme 27 Synthesis of imidazo[1, 5-a]-1, 3, 5-triazine derivatives

Scheme 28 通过迁移重排合成8-甲基-4-氨基咪唑并[1, 5-a]- 1, 3, 5-三嗪
Scheme 28 Synthesis of 4-amino-8-methylimidazo[1, 5-a]- 1, 3, 5-triazine via translocative rearrangement

Scheme 29 两种方法合成2, 4-二氨基咪唑并[1, 5-a]-1, 3, 5-三嗪
Scheme 29 Synthesis of 2, 4-diaminoimidazo[1, 5-a]-1, 3, 5-tria- zine with two methods

Scheme 30 几种咪唑并[1, 5-a]-1, 3, 5-三嗪类化合物的合成
Scheme 30 Synthesis of several imidazo[1, 5-a]-1, 3, 5-triazine derivatives

Scheme 31 2, 4, 7-三取代吡唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 31 Synthesis of 2, 4, 7-trisubstituted-pyrazolo[1, 5-a]- 1, 3, 5-triazine

Scheme 32 7-苯基-2-三氯甲基吡唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 32 Synthesis of 7-phenyl-2-trichloromethylpyrazolo- [1, 5-a]-1, 3, 5-triazine

Scheme 33 7, 8-二取代-4-氨基吡唑并[1, 5-a]-1, 3, 5-三嗪的合成
Scheme 33 Synthesis of 7, 8-disubstituted-4-aminopyrazolo- [1, 5-a]-1, 3, 5-triazine

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
































 下载:
下载:


