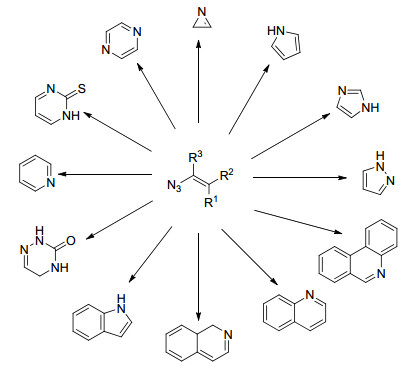
图 1.
以烯基叠氮为原料合成含氮杂环化合物
Figure 1.
Nitrogen heterocycles generated from vinyl azides

烯基叠氮化合物是指含有C=C—N3结构单元的化合物, 可以被看作为N-重氮烯胺, 结构类似烯醇, 但比烯醇稳定.其中, 碳碳双键与叠氮基团直接相连形成共轭结构, 由于叠氮基团的给电子作用, 使得碳碳双键发生极化, 从而活化了C=C双键.而与烯烃相连的叠氮基团可以充当亲电子试剂、亲核试剂或自由基接受体.因此烯基叠氮化合物的反应性质并非是叠氮基团与烯烃的简单累加, 其化学性质活泼, 能够参与不同类型的有机反应中, 是重要的合成砌块之一.
1910年, Forster等[1]首次报道了烯基叠氮化合物的合成方法.首先利用2-氯乙醇和叠氮化钠通过亲核取代合成2-叠氮基乙醇, 并进一步通过溴代或者碘代得到2-叠氮基溴/碘代乙烷, 最后在碱性条件下发生消除反应得到烯基叠氮.目前, 合成烯基叠氮的方法主要有烷基叠氮的消除反应[1]、醛/酮的缩合反应[2]、烯基卤化物的亲核取代反应[3]、联烯的亲核加成反应[4]、烯烃的加成-消除反应[5]、[3,3]单键迁移重排反应[6]以及烯丙基的异构化反应[7]等.
由于其特殊的化学结构和活泼官能团, 科学家们探索出一系列烯基叠氮参与的反应[8].近几年, 在过渡金属催化下, 烯基叠氮化合物参与的氮杂环类化合物的合成尤为突出[9], 花书贵课题组[10]也对此相关工作的研究进展进行了报道.本文综述了近20年, 烯基叠氮化物在合成含氮有机杂环化合物中的应用研究(图 1), 并对相应的反应机理进行了阐述.
2H-吖啶, 最早由Neber等[11]报道, 引起了理论以及实验化学家们的广泛关注.它是由两个碳原子和一个具有C=N双键的氮原子组成, 具有较高的环张力[12], 增强了C=N双键的反应性并促进环的裂分.在多种有机反应中充当亲核试剂, 亲电子试剂, 亲二烯体和双极性试剂.从烯基叠氮化合物出发合成2H-吖啶的方法, 根据的反应条件不同, 大致可以分为热解法、光解法、微波法、常温法等.
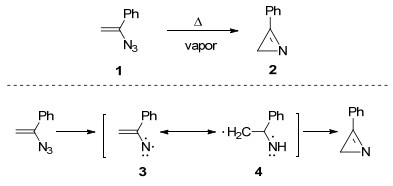
Smolinsky等[13]报道了首例乙烯基叠氮化合物的热解反应.他发现α-苯基乙烯基叠氮化合物具有光和热的不稳定性, 即在光照或者加热的条件下会脱除一分子氮气, 发生分子内环合反应, 以65%的产率得到2-苯基吖啶类化合物2(Scheme 1).
烯基叠氮的热解是目前合成2H-吖啶最常用的方法.例如, 反应在回流的甲苯溶剂中将叠氮化物5加热1.5 h后合成3-酯基吖啶6[14](Eq. 1); β-叠氮基- α, β-不饱和酯7也可在回流的甲苯中加热1 h后, 以中等至良好的收率合成3-芳酰基-2-乙氧基羰基-2H-吖啶(8)[15](Eq. 2);或在110 ℃的无水二氧六环中热解烯基叠氮化合物9合成2H-吖啶10[16](Eq. 3). 2001年, Pinhoe Melo等[17]报道了多种卤代烯基叠氮11在庚烷中加热2~3 h时, 容易转化为相应的2-卤代-2H-吖啶12.其中, 2-氯-2H-吖啶合2-溴-2H-吖啶的产率高达89%~99%, 而2-碘-2H-吖啶衍生物的产率仅为36%~85% (Eq. 4).
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
Suárez课题组[18, 19]发现, 与C(2)上携有吸电子取代基团的2H-吖啶相比较, 二取代的3-烷基-2-卤代-2H-吖啶更加稳定.此类吖啶可以通过在苯中热解或在C6D6中使用未过滤光450 W中压汞灯光解烯基叠氮进行合成. E-构型的烯基叠氮的热解反应比Z-异构体发生得更快, 所需的温度更低(Eq. 5).
|
|
(5) |
鉴于吖啶的热不稳定性, 2003年, Timen等[20]对热解法进行了优化, 他们发现在密闭环境中, 0.1 mol·L-1乙烯基叠氮化物的CH2Cl2溶液, 在150 ℃下反应20 min可得到吖啶产物.该方法的主要优点是产率高, 反应时间短, 产物易于分离(Eq. 6).
|
|
(6) |
2003年, Pastor等[21]开发了一种新颖的常温方法, 在Z-3-芳基-3-叠氮基丙-2-烯-1-醇(15)体系中加入MnO2, 能够以44%~55%产率制备2-醛基3-芳基-2H-吖啶.通过实验证明, 产品中的醛基是在环合之前生成的(Eq. 7).
|
|
(7) |
微波辐射法具有反应时间短, 产率高, 选择性高, 操作简单等优势, 近年来常被用于有机反应合成中. 2003年Gudmundsdóttir等[22]报道了无溶剂条件下微波照射烯基叠氮化物17, 实现了以高效和环境友好的方法制备2H-吖啶18, 解决了2H-吖啶合成产率低的问题(Eq. 8).
|
|
(8) |
部分性质不稳定的烯基叠氮类化合物在温度较高时会发生分解, 无法得到2H-吖啶类化合物, 为解决这个问题, 2001年, Furin等[23]报道了20 ℃下, UV照射全氟-2-甲基-3-叠氮基-2-戊烯(19), 得到产率为80%的吖啶类化合物20.他们提出吖啶20是可能通过氮烯中间体的分子内环化形成, 也可能通过不稳定的1, 2, 3-三唑啉热解形成(Eq. 9).
|
|
(9) |
2002年, Banert等[24]探索了从烯基叠氮合成氮杂螺环的方法. 6-叠氮基亚甲基环戊二烯(21)在氘代氯仿中进行低温(-40~-60 ℃)光解, 能够以80%~100%的高产率合成1-氮杂螺[2.4]庚-1, 4, 6-三烯22 (Eq. 10).在此类化合物中, 存在显著的共轭作用:即2H-吖啶氮原子的孤对轨道与环戊二烯环的π-轨道相互作用.
|
|
(10) |
2008年, Banert等[25]通过1H NMR监测发现, 紫外光照射开链的1, 2-二叠氮基烯烃在形成二氰衍生物的过程中, 历经了中间体2H-吖啶类化合物.此外, Banert等[26]还进一步实现了在-70 ℃下, 二氯甲烷中照射偕-溴碘取代的乙烯基叠氮化物26, 以90%的产率完全转化成高度不稳定的2-溴-2-碘-2H-吖啶27 (Eq. 12). 2011年, Banert等[27]又报道了双叠氮化物28在回流的氯仿溶剂中或者在相同溶剂中低温光解, 观察到几乎定量的双-2H-吖啶29的形成(Eq. 13).双叠氮化物中取代基的性质和位置显著影响反应的结果, 2和3位未被取代的1, 4-二叠氮基-1, 3-二烯30仅能合成单吖啶31 (Eq. 14), 该化合物在室温下不稳定, 但可以通过NMR检测.基于相同的光解策略, 同年, 他们继续利用光解取代的1-叠氮基环己烯32合成2, 3-桥连吖啶33[28](Eq. 15).
|
|
(11) |
|
|
(12) |
|
|
(13) |
|
|
(14) |
|
|
(15) |
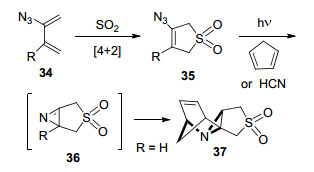
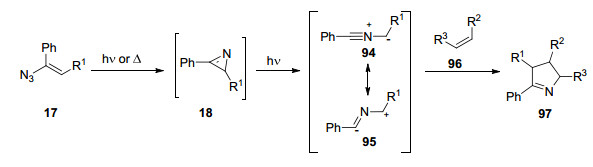
2013年, Banert等[29]发现烯基叠氮化合物的光解产物能够被另一分子捕获, 发生[4+2]环加成反应或加成开环反应.例如, 与环戊二烯反应, 生成立体选择性的产物2, 3-桥连2H-吖啶; 或与氢氰酸反应, 生成双环2-腈基氮杂环丙烷(Scheme 2).
从烯基叠氮化合物出发能够合成多种五元含氮杂环化合物, 如吡咯、咪唑、吡唑等.
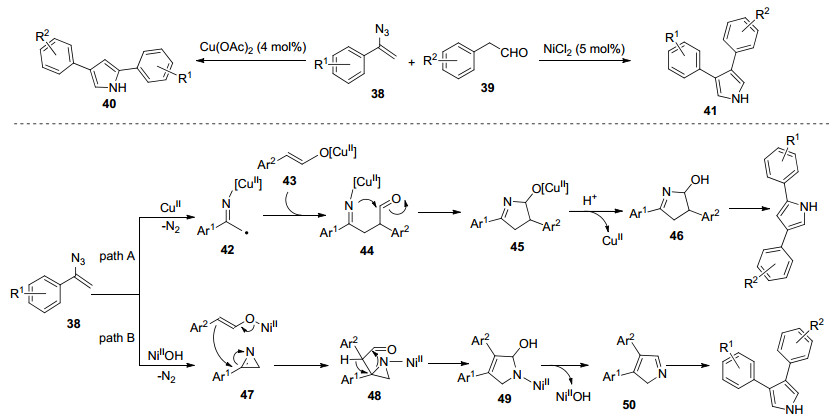
Jiao等[30]报道了一种新的、具有高度区域选择性的方法, 即从相同的底物出发, 利用不同的过渡金属催化剂构建2, 4-和3, 4-二芳基取代的吡咯.与其他报道的酸性或碱性条件相比, 该反应条件是温和中性的, 特别是这种新的方法实现了非对称的3, 4-二取代吡咯的合成, 这是传统方法很难达到的.他们提出了可能的反应机理(Scheme 3):在Cu催化的2, 4-二取代吡咯的形成中, 烯基叠氮化合物通过脱去一分子氮气, 生成自由基中间体42, 随后与醛的烯醇异构体43发生自由基偶合反应, 通过分子内氮对羰基的亲核进攻发生环合, 生成中间体45, 经过质子化和脱水, 得到目标产物40.当使用Ni(Ⅱ)作为催化剂时, 首先生成2H-吖啶, 之后接受苯乙醛Ni(Ⅱ)的烯醇化物的亲核进攻, 生成中间体48, 随后通过β-OH消除和互变异构化反应, 合成目标化合物3, 4-二取代的吡咯41.
2015年, Adimurthy等[31]利用铜催化乙烯基叠氮化物与酮类化合物的C(sp3)—H官能化来合成多取代吡咯.该反应对官能团具有良好的耐受性, 并且以高产率获得一系列2, 3, 5-三取代-1H-吡咯(Scheme 4).
Chiba等[32, 33]以烯基叠氮化合物和1, 3-二羰基化合物为起始原料, 区域选择性地制备了多官能化的吡咯. (1)烯基叠氮与1, 3-二羰基化合物在加热条件下发生1, 2-加成反应, 生成四取代吡咯59.该反应可能历经以下反应历程:烯基叠氮发生热解反应, 释放一分子氮气, 并发生分子内环合生成2H-吖啶中间体.随后, 1, 3-二羰基化合物亲电进攻2H-吖啶形成新的C—C键, 产生中间体63, 接着氮原子亲核进攻羰基, 环化形成吡咯烷中间体64, 最后脱水形成2, 3, 4, 5-四取代的吡咯59; (2)铜催化下, 烯基叠氮和乙酰乙酸乙酯发生1, 4-加成反应, 合成2, 3, 4, 5-四取代的吡咯61.与上述反应历程不同, 此反应并没有历经2H-吖啶中间体, 而是烯醇铜化合物与烯基叠氮化合物发生1, 4-加成反应, 并释放一分子氮气, 生成亚胺铜中间体65, 之后经历环化、水解、脱水等过程, 生成目标产物61 (Scheme 5).
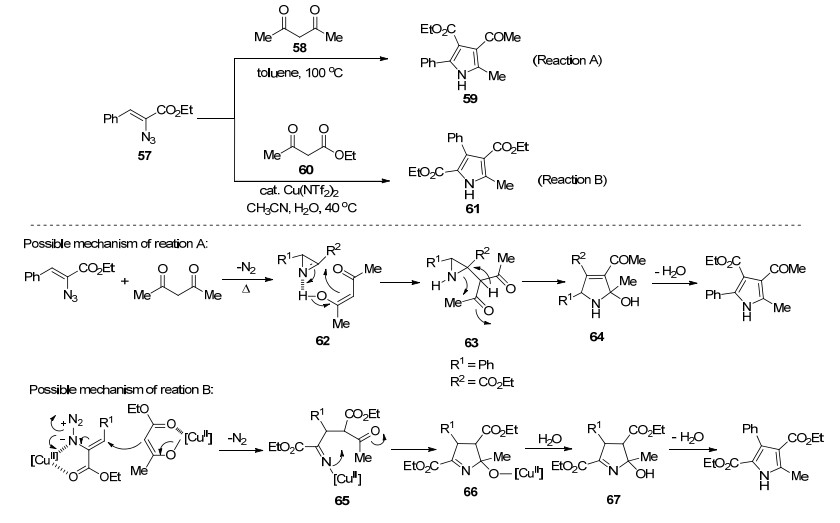
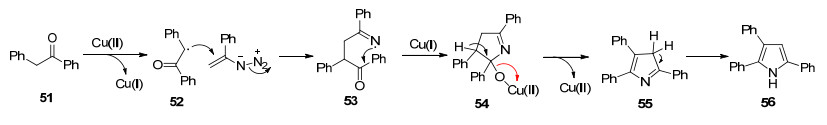
Mn(Ⅲ)盐是优异的单电子氧化剂, 已被广泛用于自由基的环化反应. 2008年, Chiba和Narasaka等[34]报道了醋酸锰催化的烯基叠氮化物和1, 3-二羰基化合物来合成多取代的N—H吡咯的反应.不同于之前报道的环加成反应, 该反应历经了自由基反应历程(Scheme 6):烯醇锰化合物69与烯基叠氮化合物发生加成反应, 伴随着Mn(Ⅱ)化合物和一分子氮气的释放, 生成亚氨基自由基70, 从而引发该自由基反应.亚氨基自由基70与羰基发生分子内加成, 生成烷氧基自由基71, 71被Mn(Ⅱ)化合物还原产生Mn(Ⅲ)醇盐72 (Path A); 或者, 亚氨基自由基70与Mn(Ⅱ)化合物反应生成中间体73, 进一步分子内的羰基亲核反应, 生成中间体72 (Path B).最后, 经历质子化、脱水过程, 得到化合物68.基于同样的反应策略, Chiba组[35]随后又实现了Mn(acac)3催化的乙烯基叠氮化物和β-酮酸来合成吡咯的反应.
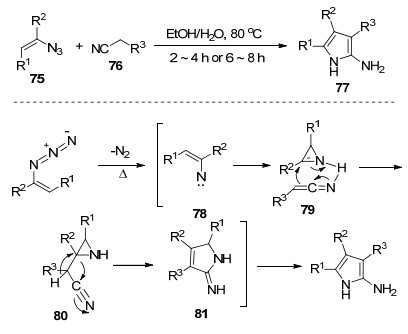
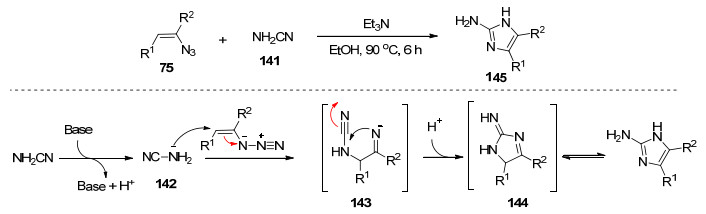
之前报道的一些合成2-氨基吡咯的方法通常具有一些不足之处, 如:需要很长的反应时间、昂贵的过渡金属催化剂、复杂的底物或较低的产率.因此, 构建2-氨基吡咯环的有效和简便的方法是有价值的. 2013年, Yu等[36]报道了以烯基叠氮化物和α-氰基衍生物76为原料合成多官能化的2-氨基吡咯77的方法.通过条件优化发现, 在80 ℃的乙醇和水(体积比1:1)共溶剂中获得最有效和环保的反应性.该反应历经分子内环合、环扩张、互变异构化反应等, 得到2-氨基吡咯产物(Scheme 7).该反应具有操作简单、环保、产率高等优点.
2012年, Yu等[37]实现了在温和的碱存在下, 甲苯磺酰甲基异氰化物和乙烯基叠氮化物反应生成2-甲苯磺酰基取代的吡咯.乙烯基叠氮化物中的叠氮基团用作离去基团, 对于中间体进行芳构化进而合成目标化合物是至关重要的.作者通过反应条件筛选优化, 找出最佳的碱为NaH (Scheme 8).
不同于Yu等合成2-氨基吡咯的方法, Huang等[38]开发了一种金催化下烯基叠氮化物和炔酰胺合成多取代2-氨基吡咯化合物的方法.与此之前的研究的反应历程不同[39, 40], Huang提出该反应历经金卡宾中间体.在实验的基础上, 他们提出了可能的反应机理:中间体89与2H-吖啶88反应得到两性离子中间体90, 接着开环得到金卡宾中间体91, 最后环合得到92 (Path A); 或者90直接关环生成92 (Path B), 通过异构化反应, 92转化为产物2-氨基吡咯, 并且再生金催化剂(Scheme 9).

2013年, Kirschning等[41]首次实现了在流动状态下, 光活化芳族乙烯基叠氮化物和活化烯烃合成二氢吡咯的方法.该方法通过光照烯基叠氮转化为2H-吖啶, 随后发生开环得到腈内鎓盐.该腈内鎓盐作为1, 3-偶极子, 可与缺电子烯烃进行[3+2]环加成产物.在研究底物使用范围时发现, 芳族乙烯基叠氮化物中芳环的电子效应对反应的影响不大(Scheme 10).
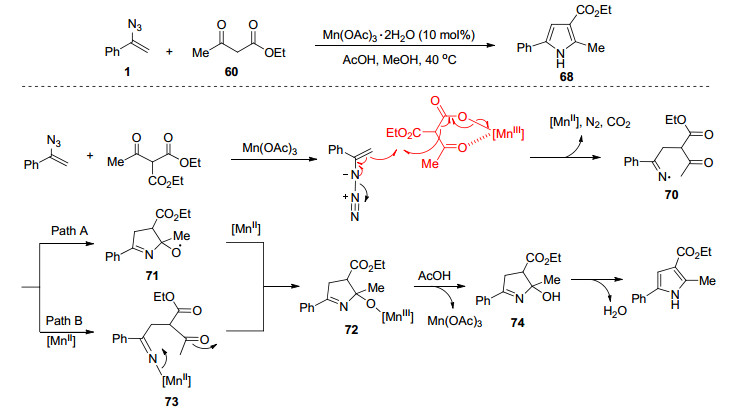
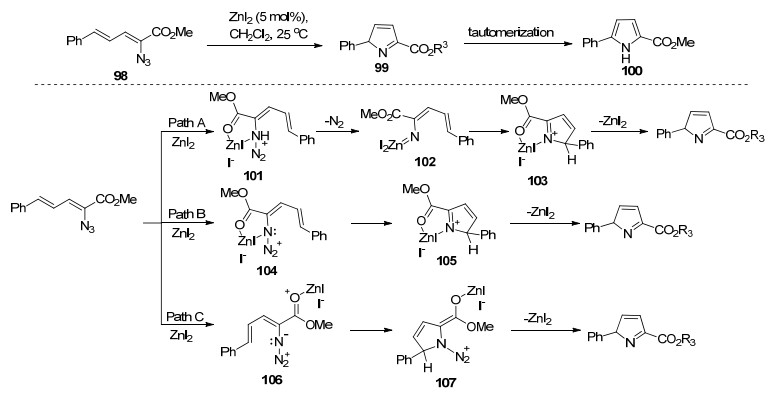
2007年, Driver课题组[42]发展了一种新的, 温和的方法, 即在室温条件下利用过渡金属催化二烯基叠氮化物发生自身分子内环合反应合成一系列2, 5-二取代和2, 4, 5-三取代吡咯.该课题组研究发现铑、铜和锌催化剂都能有效催化该吡咯合成反应, 考虑到价格因素, 最终采用ZnI2作为进一步研究的反应催化剂.作者对反应机理进行了深入研究, 提出了以下可能的三种反应机理(Scheme 11):在机理A中通过乃春化合物102进行氮原子转移.机理B与C中提出路易斯酸活化机制, 在机理B中, 锌与叠氮化物(和酯)的配位生成了化合物104, 烯烃攻击释放N2以在105中直接形成C—N键, 接着脱除碘化锌合成2H-吡咯99, 最后互变异构化合成1H-吡咯100; 在机理C中, 碘化锌与羰基的配位增加了侧链烯烃的亲电性, 叠氮化物的分子内进攻形成107, 接着互变异构化产生吡咯99.根据底物的反应性趋势, 表明反应更倾向于通过机理B进行.他们观察到, 芳基-γ-取代基越缺乏电子, 底物的反应性越低.通过在δ-或γ-位的烷基取代来提高底物的给电性质可显著提高反应速率.
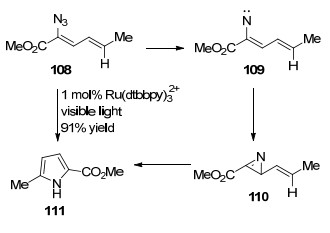
在反应过程中使用可见光活化的过渡金属催化剂目前正引起越来越多的关注. 2014年, Yoon等[43]报道了在Ru光催化下, 二烯基叠氮化物光解转化为吡咯的反应.值得一提的是, 当光催化剂为铱催化剂时, 也能得到81%的吖啶产物.作者提出可能的反应机理:烯基叠氮在可见光活化的过渡金属催化剂下, 转化为三重态的烯基叠氮, 然后释放一分子N2, 得到氮烯中间体, 随后环化得到2H-吖啶, 最后重排得到目标化合物吡咯.他们重点强调了氮烯中间体在构建C—N键以及杂环的重要性(Scheme 12).
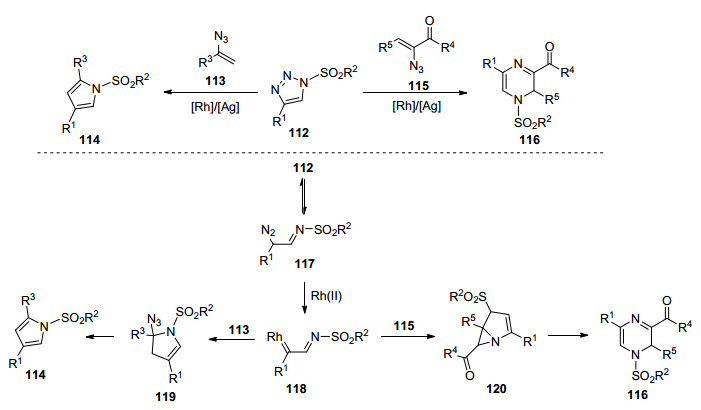
2016年, Bi课题组[44]报道了首例在Rh/Ag二元金属催化剂体系下, 乙烯基叠氮化物和N-磺酰基- 1, 2, 3-三唑的环化反应制备吡咯114和2H-吡嗪116.通过对携有不同取代基团的三氮唑进行研究, 发现不管是富电子还是缺电子的芳基、稠芳基、杂芳基取代的三氮唑均有很好的耐受性, 并且磺酰基的变化对反应结果没有显著影响.他们提出可能的反应机理如下: N-磺酰基-1, 2, 3-三唑112首先可逆地转化为重氮亚胺117, 铑催化剂催化117合成α-亚氨基铑(Ⅱ) 118.对于α-取代的乙烯基叠氮化物, 进行环丙烷化反应后, 接着分子内重排形成中间体2, 3-二氢吡咯119.最后释放一分子HN3形成吡咯114; 而对于α, β-双取代的乙烯基叠氮化物, 首先释放一分子氮气转化为2H-吖啶, 之后历经亲核加成, 开环重排过程后, 生成二氢吡嗪116 (Scheme 13).
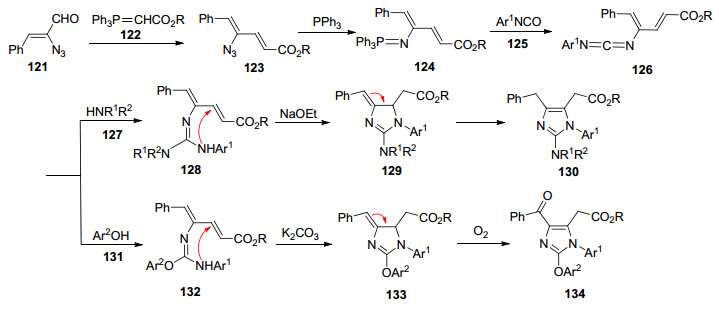
咪唑及其衍生物是在天然产物和药物中广泛存在的重要含氮杂环化合物之一, 有一些已被开发成上市药物, 如西咪替丁、氯沙坦等.在此之前, 并没有用于合成不同官能化咪唑衍生物的一般方法, 并且2-氨基或2-芳氧基取代的咪唑更不容易制备.鉴于此, Ding课题组[45]报道了烯基叠氮与芳基异氰酸酯的串联反应, 历经Aza-Wittig/迈克尔加成/异构化的过程, 合成1, 2, 4, 5-四取代的咪唑.首先由碳二亚胺中间体126与仲胺反应合成胍中间体128, 接着在乙酸钠催化下128分子内迈克尔加成生成二氢咪唑129, 最后129经历1, 3-H迁移得到2-氨基咪唑130.值得一提的是, 当酚用作亲核试剂来捕获中间体126时, 得到4-苯甲酰甲基咪唑134 (Scheme 14).
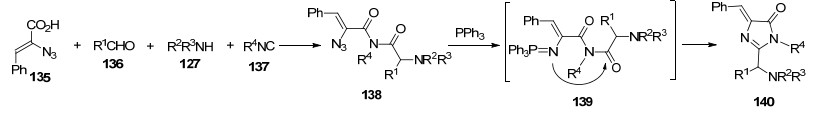
Ding课题组[46]又报道了2-叠氮基-3-芳基丙烯酸135、醛136、仲胺127和异腈137的四组分Ugi反应合成咪唑.反应首先生成烯基叠氮中间体138, 加入三苯基膦后, 乙烯基亚氨基正膦中间体139进行分子内Aza-Wittig反应, 得到咪唑类化合物140 (Scheme 15).
2014年, Yu等[47]从氨基腈和烯基叠氮化合物出发, 简单直接、高效环保地制备了2-氨基咪唑.该反应历经迈克尔加成-分子内环合-互变异构化过程(Scheme 16).
2015年, Yu等[48]进一步研究了乙烯基叠氮化物的亲电反应性, 并报道了活化亚胺与乙烯基叠氮化物的反应, 高效地合成了多官能化咪唑衍生物.与上述反应经历迈克尔加成过程不同, 该反应是通过2H-吖啶与活化亚胺串联环化的反应机制进行的(Scheme 17).
同年, Yan等[49]又报道了I2催化烯基叠氮化物和苄胺的氧化串联环化反应, 该反应新颖有效地合成多取代的咪唑化合物.研究发现单独使用I2催化时, 反应产率很低, 为了提高产率, 他们使用催化剂/氧化剂体系KI/TBHP, TBAI/TBHP和I2/TBHP作为替代, 通过筛选发现I2/TBHP体系使反应进行得更为有效.值得注意的是, 没有I2存在的反应不能进行.该反应的适用范围广且对官能团有良好的兼容性, 能以中等至良好的收率得到咪唑(Eq. 16).
|
|
(16) |
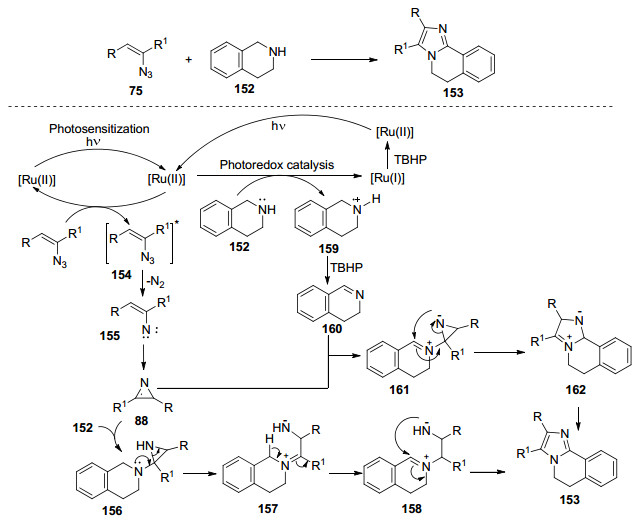
2016年, Maurya等[50]报道了在流动微反应器中, 可见光/[Ru(bpy)3][(PF6)2]促进烯基叠氮化物和仲胺的偶联反应, 合成咪唑衍生物.之前报道的多例反应涉及可见光促进的叔氮原子上的自由基阳离子的生成作为反应的关键步骤, 而可见光促进的仲氮原子上的自由基阳离子的生成和与仲氮原子相邻的C(sp3)—H键的官能化的实例是罕见的, 这也正是该反应的特色之处.在优化反应条件中发现间歇反应器中咪唑的产率不高, 而采用体表面积比大、照明均匀性高、传热能力强、安全隐患低的流动微反应器技术非常适合光化学合成转化.作者对反应过程进行探索, 并提出以下反应机理:乙烯基叠氮化物在可见光促进下生成2H-吖啶88, 胺152通过光催化氧化产生亚胺160.接下来, 2H-吖啶和亚胺160反应生成中间体161, 进一步开环和环化生成中间体162, 最后通过重排和氧化合成咪唑153.同时, 还存在另一种可能的机理, 即高反应性的2H-吖啶与胺152反应生成中间体156, 接着通过重排和光氧化还原反应合成咪唑153 (Scheme 18).同年, Maurya等[51]还进一步实现了Ag2CO3促进的烯基叠氮化物与仲胺的偶联合成稠环咪唑.
在过去几十年中, 过渡金属催化的氧化偶联为构建C—C和C—X提供了理想的合成方法.在众多过渡金属催化剂中, Pd, Rh, Ir和Ru等贵金属催化剂由于其高催化活性而经常被使用.然而, 从成本、毒性和可持续性等方面综合考虑, 铜和铁越来越受到化学家的关注. 2017年, Jiang等[52]报道了铁催化乙烯基叠氮化物和肟酸乙酯的[3+2]环化反应, 合成了2, 4-二取代-2H-咪唑.该方法涉及N—O/N—N键的裂解和两个新C—N键的形成(Scheme 19).考虑到绿色和可持续化学品的要求, 他们还使用绿色氧化剂如氧气或乙酸肟酯.该方法具有催化剂成本低、条件温和、易于处理以及高原子经济性等优点.
吡唑是具有一系列生物活性的含氮杂环化合物之一, 如抗菌、抗白血病、抗肿瘤、抗肥胖和抗炎和心脏肥大的抑制作用等.多取代吡唑还可用作一些交叉偶联反应的配体.
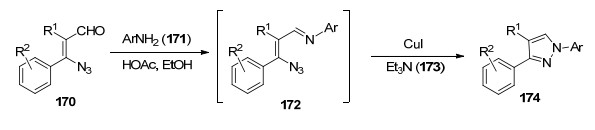
2011年, Rao等[53]以邻位乙烯基叠氮基醛170和芳胺171为起始原料, 在温和条件下, 通过简洁的两步反应实现对吡唑的直接合成(Scheme 20).
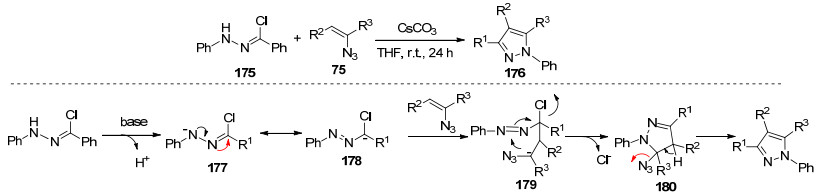
2011年, Yu等[54]报道了以腙氯化物和烯基叠氮化物为起始原料合成多取代吡唑.作者提出的可能反应机理如下:在碱的存在下, 通过去氢引发175的极化, 形成的活性腙氯178进行迈克尔加成, 随后与乙烯基叠氮化物分子内环化合成中间体179.伴随着叠氮基的离去生成目标产物176 (Scheme 21).在该反应中乙烯基叠氮化合物中的叠氮基作为离去基团和稳定化负电荷参与反应.
2013年, Yu等[55]又报道了在碱存在下, 通过烯基叠氮化物、醛以及甲苯磺酰肼三组分反应高效、区域选择性地合成3, 4, 5-三取代-1H-吡唑.在该反应中, 叠氮基仍是极好的离去基团(Scheme 22).
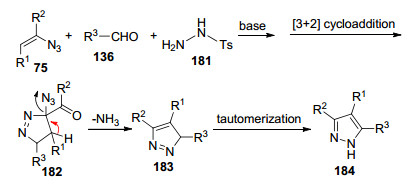
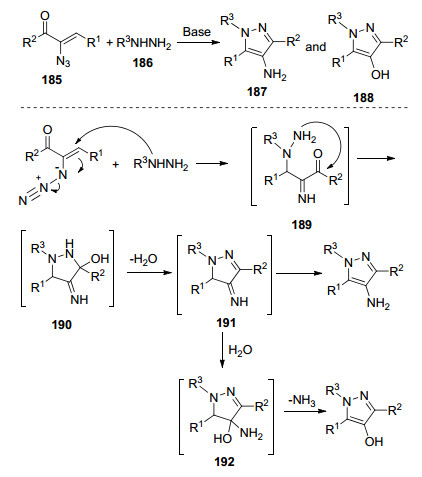
基于之前的研究成果, Yu等[56]报道了在碱存在下, 从乙烯基叠氮化物和肼出发, 简单直接地合成多取代4-氨基吡唑和4-羟基吡唑(Scheme 23).他们提出了可能的反应机理:肼186迈克尔加成到乙烯基叠氮上生成中间体189, 随后分子内缩合形成中间体190, 进一步脱水重排合成目标产物187; 当肼为水合肼时, 由于反应体系中存在过多的水, 因此通过进一步水解以及释放NH3合成目标产物188.
2011年, Chiba课题组[33]在报道了乙烯基叠氮与1, 3-二羰基化合物合成四取代吡咯的反应过程中发现, 当反应在N, N-二甲基甲酰胺(DMF)中进行, K2CO3催化乙烯基叠氮化物和1, 3-二羰基化合物进行1, 3-偶极环加成, 高效合成了1-乙烯基-1, 2, 3-三氮唑195[31](Eq. 17).
|
|
(17) |
从烯基叠氮化合物出发来合成六元含氮杂环的实例并不多, 目前主要报道的有吡啶、吡嗪、嘧啶等.
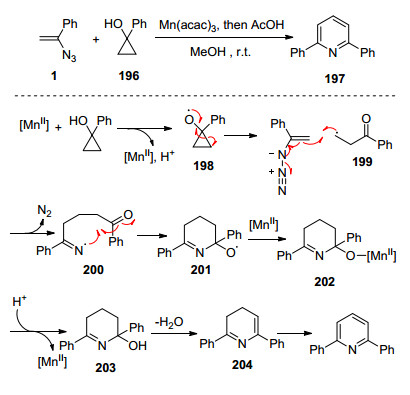
2009年, Chiba等[57a]报道了Mn(Ⅲ)催化的环丙烷醇与烯基叠氮, 合成吡啶以及2-氮杂双环[3.3.1]壬-2-烯-1-醇衍生物.在此, 他们使用环丙烷作为β-羰基的前体, 研究了它们对乙烯基叠氮化物的加成反应.他们提出反应可能经历以下历程: Mn(Ⅲ)激发1-苯基环丙醇196的单电子氧化产生的α-酮自由基199加成到乙烯基叠氮化物中, 释放N2并产生亚氨自由基200, 200分子内环化产生烷氧自由基201, 201被Mn(Ⅱ)还原, 接着被质子化合成四氢吡啶202并再生Mn(Ⅲ)物质, 最后202水合以及进一步氧化合成目标产物(Scheme 24). 2011年, Chiba课题组[57b]继续发展Mn(Ⅲ)催化的乙烯基叠氮化物与环丙醇的[3+3]环合反应形成吡啶以及其它两种含氮杂环化合物.
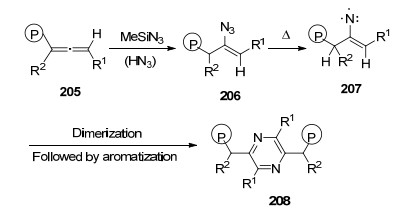
2012年, Kumara Swamy课题组[58]报道了含有磷取代基的烯基叠氮类化合物在光催化或者加热的环境中, 无溶剂、无催化剂的条件下的两分子缩合反应, 高效合成1, 4-吡嗪类化合物208.该反应可能历经氮烯中间体207的自由基机理形成吡嗪(Scheme 25).
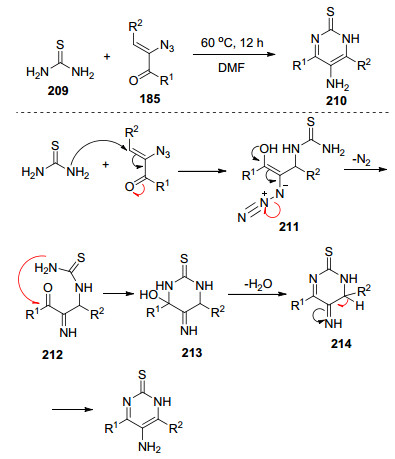
一般来说, 合成5-氨基嘧啶需要多个步骤. 2012年Zhang等[59]报道了一种简单, 直接, 有效的方法合成多取代的5-氨基嘧啶-2(1H)-硫酮, 这一新方法缩短了反应步骤.该反应历程是将硫脲迈克尔加成到烯基叠氮化物得到中间体211, 随后经过分子内环化、脱水和重排反应生成目标产物(Scheme 26).在底物扩展过程中发现芳基取代的烯基叠氮更利于反应的进行, 并且芳基上连有吸电子基时产率更高.
2015年, Nishimura等[60]报道了在PPh3存在下异氰酸酯216与乙烯基叠氮化物合成二氢嘧啶217的反应.该反应经历Staudinger反应、Aza-Wittig反应和分子内环化过程(Scheme 27).
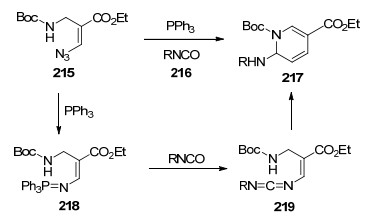
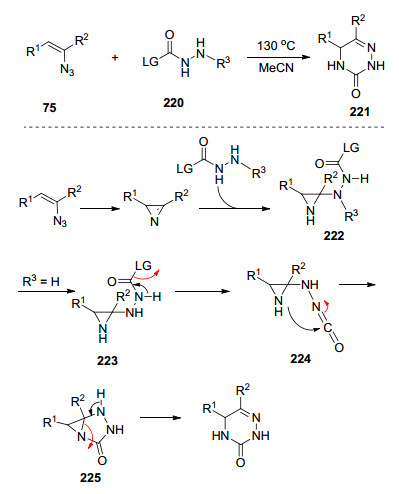
2015年, Yu等[61]实现了利用乙烯基叠氮化物作为双功能配偶体, 与肼基酸酯合成氮杂嘧啶酮.他们提出了可能的反应机理:乙烯基叠氮化物热解生成2H-吖啶, 随后, 亲核进攻肼基酸酯220生成加成产物222, 当R3为H时, 223脱去离去基团(HLG)得到异氰酸酯中间体224, 接着吖啶氮分子亲核攻击异氰酸酯生成225, 最后三元环开环合成氮杂嘧啶酮221.若R3为苄基取代, 可通过5-环闭合途径得到咪唑类化合物(Scheme 28).
从烯基叠氮化合物出发能够合成多种含氮稠杂环化合物, 如吲哚、喹啉、异喹啉、菲啶等.
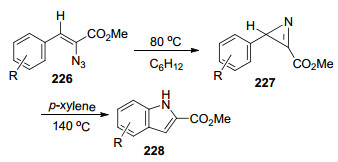
1985年, Knittel[62]报道了2-叠氮基-3-芳基丙烯酸甲酯(227)加热至80 ℃可以发生环合反应生成2H-吖啶类化合物.若继续加热至140 ℃可发生开环再环合反应合成吲哚类化合物228 (Scheme 29).
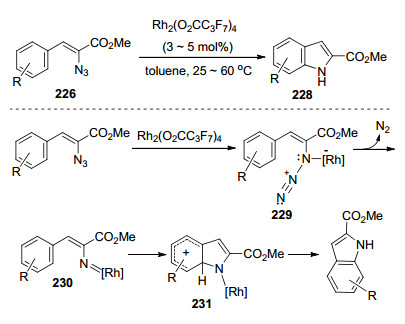
2007年, Stokes等[63]实现了铑催化下叠氮基丙烯酸酯的分子内C—H胺化反应.他们通过条件筛选优化发现只有羧酸铑是可以生成吲哚的, 并且全氟丁酸铑表现出最优反应活性.利用全氟丁酸铑与烯基叠氮配位形成金属乃春中间体229, 这一中间体的生成是构筑该类方法的重要步骤, 最后通过C—N键的构建以合成目标化合物吲哚(Scheme 30).
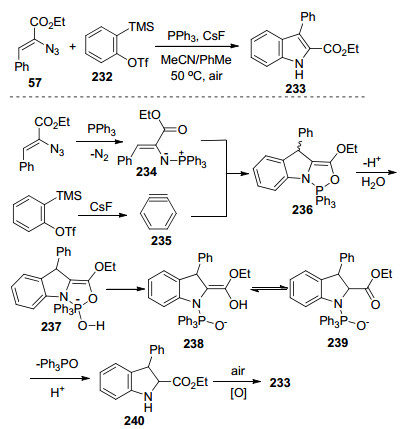
2010年Wang课题组[64]的研究表明, 2-叠氮基丙烯酸酯与邻甲硅烷基芳基三氟甲磺酸酯232在PPh3和CsF的协同促进下可以合成吲哚(Scheme 31).他们提出可能的反应机理如下:首先乙烯基叠氮化物与PPh3发生Staudinger-Meyer反应生成乙烯基亚氨基磷酰胺234, 接着与原位生成的苯炔反应, 通过亲核双环化产生中间体236, 之后通过水解、释放Ph3PO和空气氧化过程, 合成目标产物吲哚233.
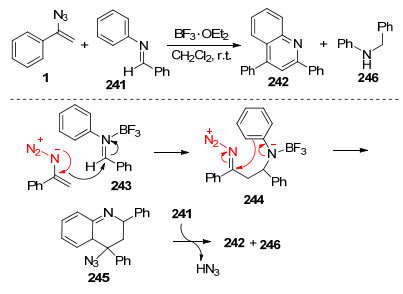
以烯基叠氮的氧自由基的反应为基础, Chiba课题组[65]进一步实现了BF3·OEt2促进的乙烯基叠氮化物和亚胺的[4+2]环合反应合成高官能化的喹啉(Scheme 32).其中乙烯基叠氮化物作为烯胺型亲核试剂, 假定的亚氨基重氮离子中间体作为亲电试剂.他们提出的反应机理如下:乙烯基叠氮化物进攻由BF3·OEt2亲电活化的亚胺构建第一个C—C键, 生成亚氨基重氮离子中间体244, 接着分子内环化生成4-叠氮基-四氢喹啉245, 通过氢迁移到醛亚胺241上, 最后释放HN3以及芳构化后合成喹啉242和N-苄基苯胺246.
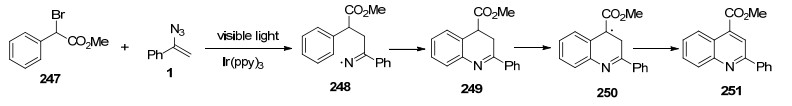
2015年, Zhou等[66]实现了可见光诱导的乙烯基叠氮化物和α-羰基苄溴的自由基反应合成喹啉化合物.该反应不仅为多取代的喹啉提供温和的反应途径, 而且还实现了可见光照射条件下使用乙烯基叠氮化物作为自由基受体来合成喹啉, 这也证明了光氧化还原催化亚氨基自由基构建多种氮杂环框架的可能性(Scheme 33).
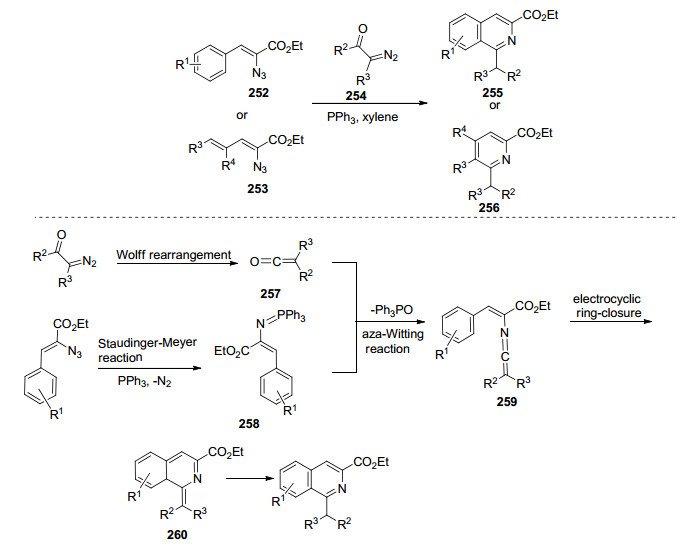
异喹啉是天然产物中常见的重要生物碱类, 也被用作多种药物的结构单元以及过渡金属催化剂的手性配体.经典的合成异喹啉的方法通常需要使用碱或者较长的反应时间.亚氨基磷烷的Aza-Wittig反应由于其在氮杂环化合物的合成中所表现出的实用性而受到越来越多的关注. Wang课题组已经大大推进了乙烯基亚氨基磷烷的使用, 并且发展了一种新的串联法来合成异喹啉. 2008和2009年, Wang课题组[67a, 67b]先后报道了通过一锅法实现2-叠氮基-3-芳基丙烯酸酯、α-重氮甲酮化合物254和三苯基膦的串联反应合成异喹啉和吡啶.此串联过程涉及Wolff重排、Staudinger-Meyer反应、Aza-Wittig反应和电环化反应, 该反应具有高效、快速且底物易于获得的优点(Scheme 34).
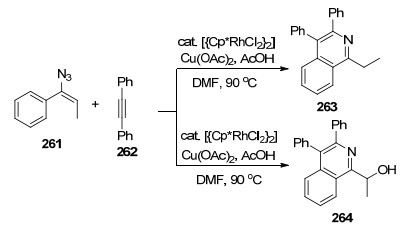
2011年, Chiba课题组[68]实现了铑/铜双金属体系催化α-芳基乙烯基叠氮化物和内炔262合成多取代的异喹啉(Scheme 35).
2014年, Chiba课题组[69]进一步发展基于乙烯基叠氮化物的自由基反应来合成含氮杂环化合物(Scheme 36).他们提出可能的反应机理:反应先由PhI(OAc)2引发, 通过Me3SiCF3的单电子氧化产生CF3自由基, 随后将CF3自由基加成到乙烯基叠氮化物的C=C键上以形成三氟甲基亚胺自由基265并释放氮气, 接着二聚得到化合物266, 最后进一步转化为5-氟吡唑268, 并且可以在Rh(Ⅱ)/Cu(Ⅱ)联合催化下, 与炔267反应生成三氟乙基取代的异喹啉269.

除Me3SiCF3外, Togni’s试剂也是一种重要的三氟甲基化试剂, 通常在Cu盐作为催化剂时, 用于三氟甲基化反应合成多种化合物. 2016年Liu等[70]实现了在Rh(Ⅲ)-Cu(Ⅱ)双金属体系催化乙烯基叠氮化物、炔烃以及Togni’s试剂的三组分串联反应.证明这种三氟甲基化/C—H活化反应能够合成三氟乙基异喹啉.作者对反应过程进行探索, 并提出了以下反应机理:在Cu(Ⅱ)与Togni’s试剂共存的反应条件下可以产生三氟甲基自由基, 随后与乙烯基叠氮化物反应, 生成中间体272, 272在被Cu(Ⅱ)捕获后与亚胺基铑中间体273反应, 生成中间体274.再与炔烃反应, 历经还原消除合成目标化合物. Rh(Ⅰ)和Cu(Ⅲ)发生氧化还原反应, 能够再生Rh(Ⅲ) (Scheme 37).
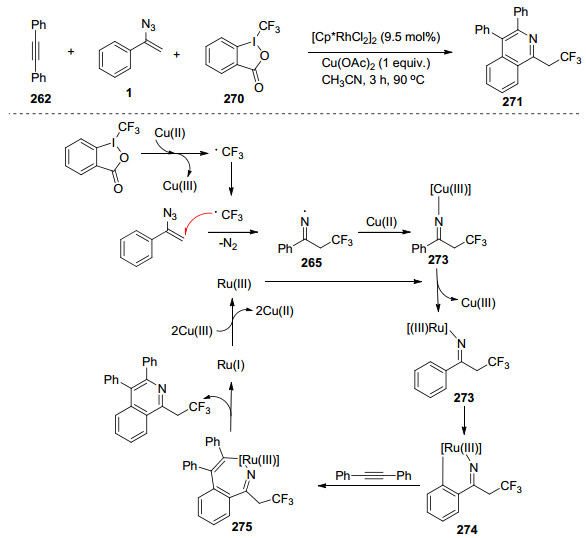
2016年, Jiang等[71]实现了钯催化的肟与乙烯基叠氮化物的环化反应来合成异喹啉(Scheme 38).肟在转化中可作为导向基团和氧化剂.该反应具有使用简单的起始材料、不需要任何的添加剂、无有毒副产物生成、操作简单和高原子经济性等优点.
在有机分子上引入全氟烷基能够改变它们的化学、物理和生物性质, 这使含氟分子在医药化学和材料科学领域中有着广泛的应用.在特定位置引入全氟烷基或多氟烷基氮杂环的合成在各个领域引起了相当大的关注.菲啶衍生物显示出广谱的生物活性和光电性质, 使得多氟烷基官能团取代的菲啶衍生物在药物和材料开发的应用中更有价值. 2014年, Chiba课题组[72]从之前的报道中获得灵感, 随后发展了α-联芳基-2-乙烯基叠氮化物的分子内自由基环化反应合成三氟乙基菲啶.其中亚胺自由基285是该反应重要的反应中间体(Scheme 39).
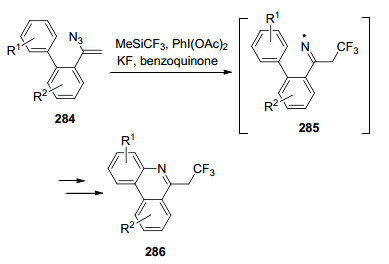
此外, Studer等[73]也报道了以乙烯基叠氮为起始原料合成氟代烷基的菲啶和喹喔啉酮的方法.以Togni’s试剂作为氟源, Bu4NI作为引发剂, 并在电子催化下发生反应.值得注意的是, 此反应不需要使用过渡金属催化剂, 也可以实现一步构建多个化学键, 从而合成含氮稠环化合物(Eq. 18).
|
|
(18) |
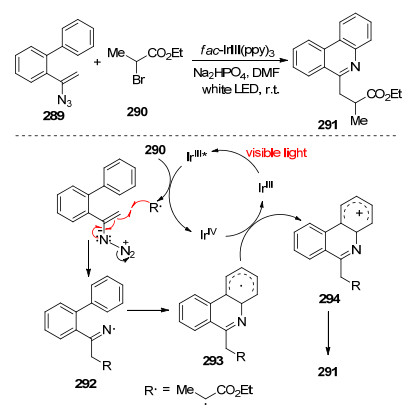
不同于Studer等的方法, 2016年Yu等[74]实现了可见光促进的乙烯基叠氮化物, 合成了6-(氟)烷基取代的菲啶衍生物.与传统使用有毒的锡试剂、紫外线照射或在微波照射下升高温度相比, 此方法可以在温和条件下进行, 并取得较好的产率.他们提出了可能反应机理:光催化剂fac-IrⅢ(ppy)3在光照下成激发态fac-IrⅢ(ppy)3*, 被溴化物290氧化猝灭后产生复合物和自由基R·, 自由基R·加成到乙烯基叠氮化物上生成亚胺自由基中间体292, 经历电环化-氧化-去质子化过程, 最终生成产物6-(氟)烷基化菲啶(Scheme 40).
2010年, Yu等[75]开发了一种新的、温和的方法来合成多官能化吡咯并[1, 2-α]吡嗪(Scheme 41).这种新的多米诺合成法首先由乙烯基叠氮化物296和2-吡咯甲醛295进行迈克尔加成, 接着在室温下分子内缩合, 生成吡咯并[1, 2-α]吡嗪297.

2014年, Adimurthy等[76]报道了首例以烯基叠氮为原料合成咪唑并[1, 2-α]吡啶的反应(Eq. 19).该方法具有仅释放N2作为良性副产物, 使用空气作为环境友好的氧化剂以及温和的反应条件(65 ℃)等优点.
|
|
(19) |
Maurya等[77]实现了无金属催化α-酮基乙烯基叠氮化物和2-氨基吡啶, 并通过简单的蒸发反应溶剂过程来合成高纯度的咪唑并[1, 2-a]吡啶(Scheme 42).该方法通过冷凝、环化和开环反应可形成三个新的C—N键, 只释放H2O和N2作为副产物, 整个过程体现了方便性以及高原子经济性.
除此之外, 化学家们还不断探索合成了一些结构复杂的含氮稠杂环化合物.例如, 用六甲基二硅烷重氮锂(LHMDS)和乙烯基叠氮化物处理异苯并呋喃酮发生[4+3]环化合成5-羟基-2-苯并氮杂酮[78]; Mn(Ⅱ)乙酸酯催化烯基叠氮化物和2-羟基萘醌合成多官能化的苯并吲哚-4, 9-二酮[79]; 锰(Ⅱ)催化乙烯基叠氮化物和4-羟基香豆素的多米诺反应合成多官能化的螺苯并呋喃酮内酰胺[80]; 2H-吖啶与二烯的分子内氮杂Diels-Alder反应[81]等.
烯基叠氮化合物作为一种具有多活性和多功能的结构单元, 在医药、农药、材料等领域都有广泛的应用, 激起了科学家们对其合成方法及应用研究的热情.综上所述, 烯基叠氮化合物可通过加热、光解、过渡金属催化、路易斯酸配位等方式形成不同类型的反应中间体, 进而构筑多种含氮杂环类化合物.
近些年, 随着烯基叠氮化合物合成方法的不断推进, 极大地促进了烯基叠氮化合物应用研究的发展, 特别是在合成不同类型的含氮杂环化合物方面.烯基叠氮化学已经取得了巨大进展, 毫无疑问, 烯基叠氮化合物的合成方法将会有更大的创新和进步, 相信在含氮杂环化合物的合成研究中必将取得更大的发展.
Forster, M. O.; Newman, S. H. J. Chem. Soc. 1910, 97, 2570. doi: 10.1039/CT9109702570
Hemetsberger, H.; Knittel, D.; Weidmann, H. Monatsh. Chem. 1969, 100, 1599. doi: 10.1007/BF00900176
Zhu, W.; Ma, D.-W. Chem. Commun. 2004, 888. https://doaj.org/article/9d9cdd3109844f508a7b753f4518a1d5
Hassner, A.; Levy, L. A. J. Am. Chem. Soc. 1965, 87, 4203. doi: 10.1021/ja01096a045
Harvey, G. R.; Ratts, K. W. J. Org. Chem. 1966, 31, 3907. doi: 10.1021/jo01350a005
Priebe, H. Angew. Chem., Int. Ed. Engl. 1984, 23, 736. doi: 10.1002/(ISSN)1521-3773
Chou, T. S.; Lee, S. J.; Peng, M. L.; Sun D. J.; Chou, S. S. P. J. Org. Chem. 1988, 53, 3027. doi: 10.1021/jo00248a023
Fu, J.-K.; Giuseppe, Z.; Edward, A. A.; Bi, X.-H. Chem. Soc. Rev. 2017, 46, 7208. doi: 10.1039/C7CS00017K
Zhang, L.; Sun, G.; Bi, X.-H. Chem.-Asian J. 2016, 11, 3018. doi: 10.1002/asia.v11.21
严珺, 姬晓悦, 花书贵, 王静, 有机化学, 2018, 38, 791. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346349.shtmlYan, J.; Ji, X.-Y.; Hua, S.-G.; Wang, J. Chin. J. Org. Chem. 2018, 38, 791(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346349.shtml
(a) Neber, P. W. ; Burgard, A. Justus Liebigs Ann. Chem. 1932, 493, 281.
(b) Neber, P. W. ; Huh, G. Justus Liebigs Ann. Chem. 1935, 515, 283.
(a) Würthwein, T. ; Hergenrö ther, T. ; Quast, H. Eur. J. Org. Chem. 2002, 1750.
(b) Banert, K. ; Meier, B. Angew. Chem., Int. Ed. 2006, 45, 4015.
(a) Smolinsky, G. J. Am. Chem. Soc. 1961, 83, 4483.
(b) Smolinsky, G. J. Org. Chem. 1962, 27, 3557.
Alves, M. J.; Bickley, J. F.; Gilchrist, T. L. J. Chem. Soc., Perkin Trans. 1 1999, 1399.
Patonay, T.; Jeko, J.; Juhasz-Toth, E. Eur. J. Org. Chem. 2008, 1441.
Kumar, R.; Nath, M.; Tyrrell, D. L. J. J. Med. Chem. 2002, 45, 2032. doi: 10.1021/jm010410d
Pinhoe Melo, T. M. V. D.; Lopes, C. S. J.; Cardoso, A. L.; Rocha Gonsalves, A. M. d'A. Tetrahedron 2001, 57, 6203. doi: 10.1016/S0040-4020(01)00598-1
Alonso-Cruz, C. R.; Kennedy, A. R.; Rodríguez, M. S.; Suárez, E. Tetrahedron Lett. 2007, 48, 7207. doi: 10.1016/j.tetlet.2007.07.190
Alonso-Cruz, C. R.; Kennedy, A. R.; Rodríguez, M. S.; Suárez, E. J. Org. Chem. 2008, 73, 4116. doi: 10.1021/jo800182y
Timen, A. S.; Risberg, E.; Somfai, P. Tetrahedron Lett. 2003, 44, 5339. doi: 10.1016/S0040-4039(03)01205-X
Alajarín, M.; Orenes, R.; Vidal, Á.; Pastor, A. Synthesis 2003, 49.
Singh, P. N. D.; Carter, C. L.; Gudmundsdóttir, A. D. Tetrahedron Lett. 2003, 44, 6763. doi: 10.1016/S0040-4039(03)01558-2
Furin, G. G.; Gatilov, Y. V.; Bagrynskay, I. Y.; Zhuzhgov, E. L. J. Fluorine Chem. 2001, 110, 21. doi: 10.1016/S0022-1139(01)00395-5
Banert, K.; Kohler, F.; Kowski, K.; Meier, B.; Muller, B.; Rademacher, P. Chem.-Eur. J. 2002, 8, 5089. doi: 10.1002/1521-3765(20021115)8:22<5089::AID-CHEM5089>3.0.CO;2-F
Banert, K.; Fotsing, J. R.; Hagedorn, M.; Reisenauer, H. P.; Maier, G. Tetrahedron 2008, 64, 5645. doi: 10.1016/j.tet.2008.04.037
Banert, K.; Hagedorn, M.; Wutke, J.; Ecorchard, P.; Schaarschmidt, D.; Lang, H. Chem. Commun. 2010, 46, 4058. doi: 10.1039/c0cc00079e
Banert, K.; Kçhler, F.; Melzer, A.; Scharf, I.; Rheinwald, G.; Ruffer, T.; Lang, H.; Herges, R.; He , K.; Ghavtadze, N.; Wurthwein, E.-U. Chem.-Eur. J. 2011, 17, 10071. doi: 10.1002/chem.v17.36
Banert, K.; Meier, B.; Penk, E.; Saha, B.; Wurthwein, E.-U.; Grimme, S.; Ruffer, T.; Schaarschmidt, D.; Lang, H. Chem.-Eur. J. 2011, 17, 1128. doi: 10.1002/chem.v17.4
Banert, K.; Ihle, A.; Kuhtz, A.; Penk, E.; Saha, B.; Würthwein, E.-U. Tetrahedron 2013, 69, 2501. doi: 10.1016/j.tet.2012.12.054
Chen, F.; Shen, T.; Cui, Y.-X.; Jiao, N. Org. Lett. 2012, 14, 4926. doi: 10.1021/ol302270z
Donthiri, R. R.; Samanta, S.; Adimurthy, S. Org. Biomol. Chem. 2015, 13, 10113. doi: 10.1039/C5OB01407G
Chiba, S.; Wang, Y.-F.; Lapointe, G.; Narasaka, K. Org. Lett. 2008, 10, 313. doi: 10.1021/ol702727j
Ng, E. P. J.; Wang, Y.-F.; Hui, B. W.-Q.; Lapointe, G.; Chiba, S. Tetrahedron 2011, 67, 7728. doi: 10.1016/j.tet.2011.08.006
Wang, Y.-F.; Toh, K. K.; Chiba, S.; Narasaka, K. Org. Lett. 2008, 10, 5019. doi: 10.1021/ol802120u
Ng, E. P. J.; Wang, Y.-F.; Chiba, S. Synlett 2011, 783. http://downloads.hindawi.com/journals/bmri/2014/248419.xml
Yu, W.-W.; Chen, W.-T.; Liu, S.; Shao, J.-A.; Shao, Z.-Y.; Lin, H.-L.; Yu, Y.-P. Tetrahedron 2013, 69, 1953. doi: 10.1016/j.tet.2012.11.041
Chen, W.-T.; Shao, J.-A.; Li, Z.; Giulianotti, M. A.; Yu, Y.-P. Can. J. Chem. 2012, 90, 214. doi: 10.1139/v11-150
Wu, Y.-F.; Zhu, L.; Yu, Y.-H.; Luo, X.-S.; Huang, X.-L. J. Org. Chem. 2015, 80, 11407. doi: 10.1021/acs.joc.5b02057
Pawar, S. K.; Sahani, R. L.; Liu, R.-S. Chem.-Eur. J. 2015, 21, 10843. doi: 10.1002/chem.v21.30
Zhu, L.; Yu, Y.-H.; Mao, Z.-F.; Huang, X.-L. Org. Lett. 2015, 17, 30. doi: 10.1021/ol503172h
Cludius-Brandt, S.; Kupracz, L.; Kirschning, A. Beilstein J. Org. Chem. 2013, 9, 1745. doi: 10.3762/bjoc.9.201
Dong, H.-J.; Shen, M.-H.; Redford, J. E.; Stokes, B. J.; Pumphrey, A. L.; Driver, T. G. Org. Lett. 2007, 9, 5191. doi: 10.1021/ol702262f
Farney, E. P.; Yoon, T. P. Angew. Chem., Int. Ed. 2014, 53, 793. doi: 10.1002/anie.201308820
Zhang, L.; Sun, G.; Bi, X.-H. Chem.-Asian J. 2016, 11, 3018. doi: 10.1002/asia.v11.21
Nie, Y.-B.; Wang, L.; Ding, M.-W. J. Org. Chem. 2012, 77, 696. doi: 10.1021/jo201846w
Wang, Y.; Xie, H.; Pan, Y.-R.; Ding, M. W. Synthesis 2014, 336.
Liu, S.; Shao, J.-A.; Guo, X.; Luo, J.; Zhao, M.-H.; Zhang, G.-L.; Yu, Y.-P. Tetrahedron 2014, 70, 1418. doi: 10.1016/j.tet.2014.01.007
Luo, J.; Chen, W.-T.; Shao, J.-A.; Liu, X.-Y.; Shu, K.; Tang, P.; Yu, Y.-P. RSC Adv. 2015, 5, 55808. doi: 10.1039/C5RA07320K
Xiang, L.-K.; Niu, Y.-N.; Pang, X.-B.; Yang, X.-D.; Yan, R.-L. Chem. Commun. 2015, 51, 6598. doi: 10.1039/C5CC01155H
Tiwari, D. K.; Maurya, R. A.; Nanubolu, J. B. Chem.-Eur. J. 2016, 22, 526. doi: 10.1002/chem.201504292
Adiyala, P. R.; Borra, S.; Kamal, A.; Maurya, R. A. Eur. J. Org. Chem. 2016, 1269.
Zhu, Z.-Z.; Tang, X.-D.; Li, J.-X.; Li, X.-W.; Wu, W.-Q.; Deng, G.-H.; Jiang, H.-F. Org. Lett. 2017, 19, 1370. doi: 10.1021/acs.orglett.7b00203
Hu, J.-T.; Cheng, Y.-F.; Yang, Y.-Q.; Rao, Y. Chem. Commun. 2011, 47, 10133. doi: 10.1039/c1cc13908h
(a) Zou, H. -B. ; Zhu, H. -J. ; Shao, J. -A. ; Wu, J. -W. ; Chen, W. -T. ; Giulianotti, M. A. ; Yu, Y. -P. Tetrahedron 2011, 67, 4887.
(b) Li, Y. ; Hong, D. ; Lu, P. ; Wang, Y. G. Tetrahedron Lett. 2011, 52, 4161.
Zhang, G.-L.; Ni, H.-C.; Chen, W.-T.; Shao, J.-A.; Liu, H.; Chen, B.-H.; Yu, Y.-P. Org. Lett. 2013, 15, 5967. doi: 10.1021/ol402810f
Huang, W.; Liu, S.; Chen, B.-H.; Guo, X.; Yu, Y.-P. RSC Adv. 2015, 5, 32740. doi: 10.1039/C5RA04371A
(a) Wang Y. -F. ; Chiba, S. J. Am. Chem. Soc. 2009, 131, 12570.
(b) Wang, Y. F. ; Toh, K. K. ; Ng, E. P. J. ; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411.
Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 8712. doi: 10.1021/jo301694n
Shao, Z.-Y.; Pan, Q.-H.; Chen, J.; Yu, Y.-P.; Zhang, G.-L. Tetrahedron 2012, 68, 6565. doi: 10.1016/j.tet.2012.05.057
Nishimura, Y.; Hidetsura, C. H. Synlett 2015, 233. https://es.scribd.com/doc/92685461/Materials-Science-and-Technology
Shao, J.-A.; Liu, X.-Y.; Shu, K.; Tang, P.; Luo, J.; Chen, W.-T.; Yu, Y.-P. Org. Lett. 2015, 17, 4502. doi: 10.1021/acs.orglett.5b02180
Knittel, D. Synthesis 1985, 186.
Stokes, B. J.; Dong, H.; Leslie, B. E.; Pum-phrey, A. L.; Driver, T. G. J. Am. Chem. Soc. 2007, 129, 7500. doi: 10.1021/ja072219k
Hong, D.; Chen, Z.-B.; Lin, X.-F.; Wang, Y.-G. Org. Lett. 2010, 12, 4608. doi: 10.1021/ol101934v
Zhu, X.; Wang, Y.-F.; Zhang, F.-L.; Chiba, S. Chem.-Asian J. 2014, 9, 2458. doi: 10.1002/asia.201402421
Wang, Q.-L.; Huang, J.; Zhou, L. Adv. Synth. Catal. 2015, 357, 2479. doi: 10.1002/adsc.v357.11
(a) Yang, Y. Y. ; Shou, W. G. ; Chen, Z. B. ; Hong, D. J. Org. Chem. 2008, 73, 3928.
(b) Chen, Z. B. ; Hong, D. ; Wang, Y. -G. J. Org. Chem. 2009, 74, 903.
Wang, Y.-F.; Toh, K.-K.; Lee, J.-Y.; Chiba, S. Angew. Chem., Int. Ed. 2011, 50, 5927. doi: 10.1002/anie.v50.26
Wang, Y.-F.; Lonca, G. H.; Chiba, S. Angew. Chem., Int. Ed. 2014, 53, 1067. doi: 10.1002/anie.201307846
Liu, K.; Chen, S.; Li, X.-G.; Liu, P.-N. J. Org. Chem. 2016, 81, 265. doi: 10.1021/acs.joc.5b01995
Zhu, Z.-Z.; Tang, X.-D.; Li, X.-W.; Wu, W.-Q.; Deng, G.-H.; Jiang, H.-F. J. Org. Chem. 2016, 81, 1401. doi: 10.1021/acs.joc.5b02376
Wang, Y. F.; Lonca, G. H.; Runigo, M. L.; Chiba, S. Org. Lett. 2014, 16, 4272. doi: 10.1021/ol501997n
Mackay, E. G.; Studer, A. Chem.-Eur. J. 2016, 22, 13455. doi: 10.1002/chem.201602855
Sun, X.-Y.; Yu, S.-Y. Chem. Commun. 2016, 52, 10898. doi: 10.1039/C6CC05756J
Chen, W.-T.; Hu, M.; Wu, J.-W.; Zou, H.-B.; Yu, Y.-P. Org. Lett. 2010, 12, 3863. doi: 10.1021/ol101538x
Donthiri, R. R.; Pappula, V.; Reddy, N. N. K.; Bairagi, D.; Adimurthy, S. J. Org. Chem. 2014, 79, 11277. doi: 10.1021/jo5021618
Adiyala, P. R.; Mani, G. S.; Nanubolu, J. B.; Shekar, K. C.; Maurya, R. A. Org. Lett. 2015, 17, 4308. doi: 10.1021/acs.orglett.5b02124
Dinda, B. K.; Jana, A. K.; Mal, D. Chem. Commun. 2012, 48, 3999. doi: 10.1039/c2cc30279a
Guo, S.-S.; Chen, B.-H.; Guo, X.; Zhang, G.-L.; Yu, Y.-P. Tetrahedron 2015, 71, 9371. doi: 10.1016/j.tet.2015.08.067
Guo, S.-S.; Chen, B.-H.; Zhao, D.-H.; Chen, W.-T.; Zhang, G.-L. Adv. Synth. Catal. 2016, 358, 3010. doi: 10.1002/adsc.v358.19
Xu, H.-D.; Zhou, H.; Pan, Y.-P.; Ren, X.-T.; Wu, H.; Han, M.; Han, R.-Z.; Shen, M.-H. Angew. Chem., Int. Ed. 2016, 55, 2540. doi: 10.1002/anie.201510096

Scheme 2 2-叠氮基-1, 3-丁二烯和二氧化硫的螯变反应
Scheme 2 Chelating reaction of 2-azidobuta-1, 3-dienes and sulfur dioxide

Scheme 3 铜和镍催化合成2, 4-和3, 4-二取代吡咯的反应
Scheme 3 Reaction of 2, 4- and 3, 4-disubstituted pyrroles catalyzed by Cu and Ni

Scheme 4 铜催化的酮与乙烯基叠氮化物C(sp3)—H官能化反应
Scheme 4 Copper-catalyzed C(sp3)—H functionalization of ketones with vinyl azides

Scheme 5 乙烯基叠氮化物和1, 3-二羰基化合物的反应
Scheme 5 Reactions of vinyl azides and 1, 3-dicarbonyl compounds

Scheme 6 锰催化的烯基叠氮和1, 3-二羰基化合物的反应
Scheme 6 Mn(Ⅲ)-catalyzed reactions of vinyl azides with 1, 3-dicarbonyl compounds

Scheme 9 金催化的烯基叠氮和炔酰胺的反应
Scheme 9 Gold-catalyzed reactions of vinyl azides with alkynes

Scheme 11 Zn催化下合成2, 5-二取代和2, 4, 5-三取代吡咯的反应
Scheme 11 Reaction of 2, 5-disubstituted and 2, 4, 5-trisubstituted pyrroles catalyzed by Zn

Scheme 13 铑/银共催化N-磺酰基-1, 2, 3-三氮唑与烯基叠氮的反应
Scheme 13 Rhodium/silver-cocatalyzed transannulation process of N-sulfonyl-1, 2, 3-triazoles with vinyl azides

Scheme 15 烯基叠氮与醛、仲胺、异腈的反应
Scheme 15 Reaction of vinyl azides, aldehydes, secondary amines and isocyanates

Scheme 18 烯基叠氮与仲胺偶联的反应
Scheme 18 Coupling reaction of secondary amines with vinyl azides

Scheme 22 烯基叠氮与苯甲醛、甲苯磺酰肼的反应
Scheme 22 Reaction of vinyl azides, aldehyde and tosylhydrazine

Scheme 31 乙烯基叠氮化物和邻甲硅烷基芳基三氟甲磺酸盐的反应
Scheme 31 Reaction of vinyl azides and ortho-silyl aryltriflates

Scheme 32 烯基叠氮化物与亚胺的[4+2]环合反应
Scheme 32 Formal [4+2] annulation of vinyl azides with aldimines

Scheme 33 烯基叠氮化物和α-羰基苄溴的反应
Scheme 33 Reaction of vinyl azides and α-carbonyl benzyl bromides

Scheme 34 烯基叠氮与α-二氮羰基化合物反应
Scheme 34 Reaction of vinyl azide with α-dicarbonyl compounds

Scheme 36 乙烯基叠氮化物与Me3SiCF3的自由基三氟甲基化反应
Scheme 36 Radical trifluoromethylation of vinyl azides with Me3SiCF3

Scheme 37 烯基叠氮化物、炔烃和Togni's试剂的反应
Scheme 37 Reaction of vinyl azides, alkynes and Togni's reagent

Scheme 39 PhI(OAc)2促进的乙烯基叠氮化物的自由基环化反应
Scheme 39 PhI(OAc)2-mediated free radical cyclization of vinyl azides

Scheme 40 乙烯基叠氮化物在光氧化还原催化条件下合成菲啶的反应
Scheme 40 Synthesis of phenanthroline from vinyl azide under photooxidative reduction

Scheme 41 乙烯基叠氮化物和2-吡咯甲醛的[3+3]环化反应
Scheme 41 [3+3] cyclization of vinyl azides and pyrrole-2- carboxaldehyde

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
























 下载:
下载:





















