
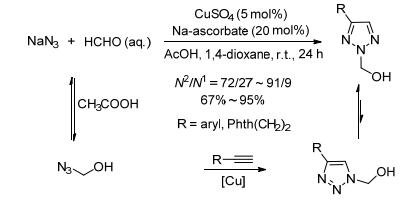
图式 1.
N2-烷基-1, 2, 3-三唑的合成
Scheme 1.
Synthesis of N2-alkyl-1, 2, 3-triazoles

1, 2, 3-三唑是一类具有重要生物活性的含氮五元杂环化合物, 虽然该类化合物已问世一百多年, 但科学家对合成此类关键结构的兴致有增无减[1].早期用于合成1, 2, 3-三唑化合物的方法是基于Huisgen类型的叠氮/炔的偶极环加成反应, 但该合成方法区域选择性较差[2].直到2002年, Sharpless等[3]对上述方法进行了改进, 通过Cu/Ru催化选择性地合成了1, 4-及1, 5-取代1, 2, 3-三唑化合物, 该方法的发展促进了结构多样性的新型N1-取代1, 2, 3-三唑的合成, 为此类化合物在药物分子、聚合物及生物材料中的广泛应用奠定了基础[4].
虽然N2-取代1, 2, 3-三唑和N1-取代1, 2, 3-三唑是区域选择性异构体, 但前者却展现出了诸多特殊性质[5].例如, N2-取代1, 2, 3-三唑作为配体, 能够形成具有显著荧光性质的一维链状配位聚合物; 另外, 二者的碱性不同, 使其在生物体系中的表达形式也不同, 这在药物研究领域开启了一个新的方向[6].因此, 发展简便、高效、高选择性合成N2-取代1, 2, 3-三唑的方法备受关注.本文根据不同合成策略, 分别从选择性N2-功能化及氧化环化两个方面, 对合成N2-取代1, 2, 3-三唑的研究成果进行了较为详细的综述, 基于反应类型的相似性, 某些N1-取代1, 2, 3-三唑的研究进展也做了一些探讨.
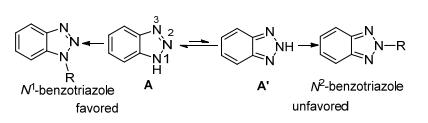
1, 2, 3-三唑N(1) [N(3)]与N(2)异构体之间存在平衡, 且N(1) [N(3)]位电子云更加富集, 容易发生反应, 因此, 传统方法一般得到N1-和N2-取代1, 2, 3-三唑的混合物, 其中以N1-取代产物为主.现有研究报道中, 主要通过以下两种方法控制反应的N2-选择性:调整底物或配体结构和分子间氢键诱导.史晓东课题组、陈保华课题组及王晓军课题组做了许多开创性工作.
2008年, 史晓东课题组[7]报道了在K2CO3作用下, 促使4, 5-双取代1, 2, 3-三唑和卤代烃反应, 实现了1, 2, 3-三唑的高选择性N2-烷基化.首次通过单晶衍射对各种N-异构体的结构进行了表征.作者从产物单晶结构分析中发现C(4)位芳基和三唑环接近于共平面(二面角大于140°), 导致N(3)位在亲电加成时受阻.当增加C(5)位取代基的体积时, 得到了更高的N2-选择性.其中, C(5)是芳基酮或烷基酮时, 能得到单一选择性的N2-烷基化产物, 通过X单晶衍射结果发现, 为了避免羰基O和N(1)之间的电子排斥力, C(5)的苯甲酰基和三唑环几乎处于共平面的结构(二面角为169°), 导致N(1)位也受阻, 使得N(2)位成为最有利的进攻位点, 从而保证了反应的高N2-选择性.在后期有关选择性N2-官能化的报道中, 此类底物成为了非常经典的研究体系.另外, C(5)位的羰基还可转化为其他官能团, 进一步扩展了这种选择性N2-烷基化策略的潜在应用范围.该反应条件简单、温和.然而, 对于烷基取代的底物, 反应体系比较复杂(Eq. 1).
|
|
(1) |
2008年, Fokin小组[8]报道了Cu(Ⅰ)催化炔和叠氮化钠、甲醛的三组分环加成反应, 选择性得到了N2-羟甲基-1, 2, 3-三唑.该反应中, 弱酸条件下, 质子化的甲醛和叠氮化钠形成中间体HO-CH2-N3, 随后在Cu(Ⅰ)的作用下和炔反应生成N1-羟甲基-1, 2, 3-三唑, 由于该产物不稳定且和1, 2, 3-三唑及甲醛之间存在动力学平衡, N1-取代产物最终会重排得到更加稳定的N2-取代产物.另外, 羟甲基也易被还原脱去, 这也为NH-1, 2, 3-三唑的合成提供了一种便捷的方法.该反应条件温和, 分离产率高(当底物量为1 mol时, 仍能以高达92%的收率得到目标产物), 但反应时间较长(Scheme 1).
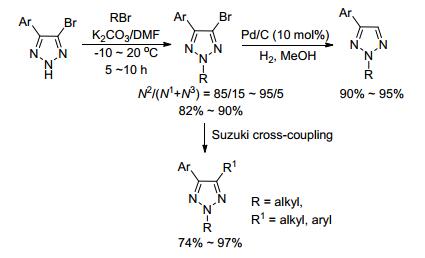
2009年, 王晓军课题组[9]报道了在K2CO3作用下, 四氢呋喃(THF)作溶剂, 实现了溴原子导向的1, 2, 3-三唑选择性N2-烷基化.产物中溴原子不仅可以通过Suzuki偶联反应得到2, 4, 5-三取代1, 2, 3-三唑, 也可以被还原生成2, 4-二取代1, 2, 3-三唑类化合物.作者认为, 溴原子的引入能够抑制N3-烷基化产物的生成, 从而使得生成N2-产物更占优势, 这与史晓东课题组[7, 27]的观点类似(Scheme 2).
2010年, 王晓军课题组[10]进一步系统研究了1, 2, 3-三唑的选择性N2-烷基化.其中4, 5-双溴代、4-溴-5-三甲基硅基及4, 5-双三甲基硅基都能很好地控制反应的N2-选择性, 这些结构片段也易转化为其它官能团, 用于2-单取代、2, 4-双取代及2, 4, 5-多取代1, 2, 3-三唑的合成.结合前线分子轨道理论(FMO)、位阻效应及静电导向对三唑环的影响, 解释了N-选择性产物的形成原因, 该反应条件简单、温和, 反应时间短, 且对各种烷基卤代物都具有广泛的适应性(Scheme 3).
2011年, 史晓东课题组[11]选用醇作烷基化试剂, THF作溶剂, 在偶氮二甲酸二异丙酯(DⅠAD)和三苯基磷(PPh3)作用下, 通过Mitsunobu反应实现了1, 2, 3-三唑的选择性N2-官能化.不同于前期通过底物结构控制反应N2-选择性, 他们认为Mitsunobu反应更加倾向于形成动力学稳定的产物(即N2-异构体), 即使是极易形成N1-取代产物的苯并三唑也不例外, 这为N2-烷基-1, 2, 3-三唑的合成提供了一种新的思路.另外, 通过这种方法也实现了手性三唑化合物的合成.该反应对各种烷基、烯丙基、炔丙基醇具有广泛的适应性, 且反应条件温和、反应时间短(Eq. 2).
|
|
(2) |
2014年, Rangappa和Shashikanth小组[12]以40 mol% ZrO2纳米粒子负载的Cu(Ⅱ)-β-环糊精配合物作催化剂, 芳酸烷基酯作烷基化试剂, 在无任何添加剂的条件下, 实现了系列N2-烷基-1, 2, 3-三唑的合成.该反应的催化剂通过空气氧化即可再生, 且重复使用四次后活性仍无明显变化.但反应时间较长, 且增加烷基链的长度时反应活性有所降低(Eq. 3).
|
|
(3) |
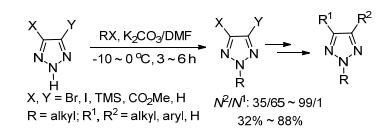
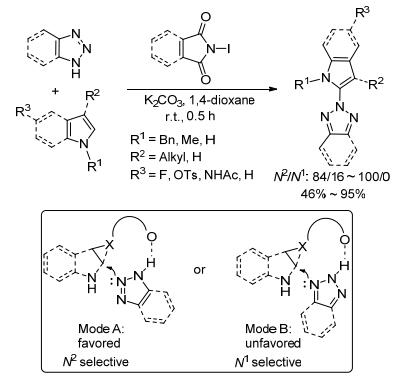
2016年, 我们课题组[13]报道了在对甲基苯磺酸(TsOH)的作用下, N1-Ts-1, 2, 3-三唑和烯烃反应, 选择性合成了系列N2-烷基-1, 2, 3-三唑.反应中Ts-既是离去基团又是阻力基团, 通过N(1)位Ts-的位阻效应实现了反应的高区域选择性.苯并三唑作为底物时, 仍然能得以4/1 (N2/N1)的选择性得到目标产物, 与已有研究报道(N2/N1接近1/1)相比[36], N2-选择性有了很大提高.另外, 底物N1-Ts-1, 2, 3-三唑通过Click反应即可获得, 增加了反应的实用性.由于C(4)位烷基取代的N1-Ts-三唑不稳定, 未得到目标产物.简单的烷基烯烃参与反应时, 体系比较复杂(Eq. 4).
|
|
(4) |
2016年, 施敏课题组[14]报道了碱作催化剂, 双(二烷胺基)甲烷作亲电试剂, N1-磺酰基-1, 2, 3-三唑作底物, 选择性得到了系列N2-烷基-1, 2, 3-三唑.研究结果表明, 高温条件下, N1-磺酰基-1, 2, 3-三唑在碱的作用下可消除得到4-苯基-1H-1, 2, 3-三唑, 并进一步转化成更加稳定的4-苯基-2H-1, 2, 3-三唑, 随后通过2位N原子捕获亲电试剂, 实现反应的高N2-选择性.该反应对各种芳基、烷基取代的N1-磺酰基-1, 2, 3-三唑都具有广泛的适应性, 其中缺电子芳基取代的收率更高.且反应时间短, 操作简便(Scheme 4).
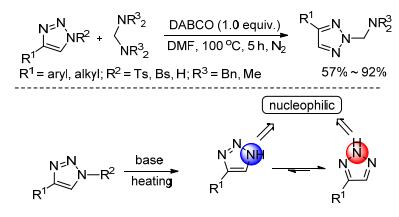
分子间氢键是选择性有机合成反应中的常用策略, 在烯烃的不对称卤化亲核加成反应中被经常运用[15]. 1, 2, 3-三唑可作为一种Brönsted酸, 与产生的鎓离子中间体形成氢键; 同时, N(1) [N(3)]位氮原子上电子云密度较高, 更加利于氢键形成, 从而生成N2-选择性产物. 2016年, 我们课题组[16]利用分子间氢键活化的策略, 实现了1, 2, 3-三唑和烯烃的选择性N2-烷基化.值得一提的是, 以水作溶剂时, 仍能以46%的收率和4/1的选择性得到目标产物, 这使得反应更加绿色.同时, 该反应也适用于氨基酸衍生物合成中, 这将为多肽的环化提供一种简单、绿色、便捷的途径.该反应对各类芳基取代、烷基取代的三唑和单取代、多取代、开链或环状及氨基酸衍生的烯烃都具有广泛的适应性.反应具有操作简便、对空气和水耐受性强、时间极短等特点, 且具有高的N2-选择性和高的分离收率(Scheme 5).
2017年, 王飞军课题组[17]报道了TsOH催化烯烃的氢胺化反应, 合成了系列N2-烷基-1, 2, 3-三唑.作者认为, 电子云密度较高的N(1)原子, 在酸性条件下优先与氢离子结合, 从而阻碍了亲电试剂的进攻, 使得N(2)位成为优势的进攻位点, 从而得到了高N2-选择性产物.另外, 2H-1, 2, 3-三唑和1H-1, 2, 3-三唑具有相似的N2-选择性.该反应条件温和, 无金属参与, 对官能团的容忍度高, 与相似类型的反应相比, 催化剂用量少, 原料简单易得, 但反应时间较长(Eq. 5).
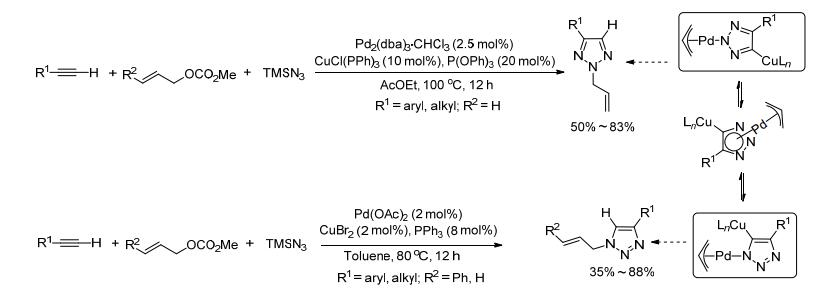
2003年, Yamamoto小组[18]报道了Pd(0)-Cu(Ⅰ)双金属催化未活化的端炔、烯丙基碳酸酯和三甲基硅基叠氮(TMSN3)的三组分偶联反应, 以50%~83%的收率选择性得到了N2-烯丙基-1, 2, 3-三唑化合物, 其经过进一步的去烯丙基化可得到简单的三唑化合物, 避免了使用吸电子基团对炔基或叠氮的活化. 2004年, 该小组[19]进一步完善了Pd-Cu双金属催化的合成方法, 分别在Pd2(dba)3•CHCl3-CuCl(PPh3)3-P(OPh)3和Pd(OAc)2-CuBr2-PPh3催化下实现了N2-烯丙基-1, 2, 3-三唑和N1-烯丙基-1, 2, 3-三唑的选择性合成.其中, Pd-Cu的共同催化对反应起到非常重要的作用, Pd-催化剂主要用于催化形成π-烯丙基-Pd-N3配合物, Cu-催化剂通过形成炔- Cu(Ⅰ)叶立德实现了对端炔的活化, 并进一步促进其和叠氮配合物中间体的[3+2]环加成反应, 另外, 反应的区域选择性取决于配体的性质(Scheme 6).
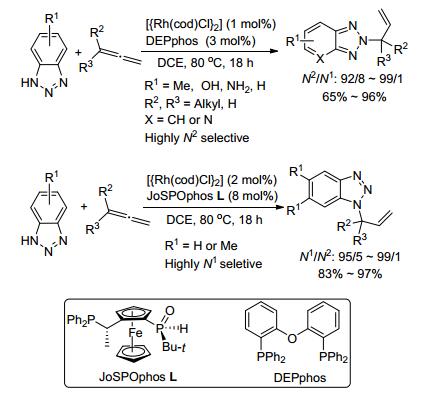
2014年, Breit小组[20]首次报道了利用Rh(Ⅰ)/DPE phos催化实现苯并三唑和联烯的N2-选择性偶联.同时, 在Rh(Ⅰ)/JoSPOphos的催化条件下也以高收率和高N1-选择性合成了N1-烯丙基化产物.研究表明, 苯并三唑的N—H首先和Rh(Ⅰ)发生氧化加成生成Rh(Ⅲ)配合物, 随后与位阻较小的双键部分发生氢金属化形成σ-allyl-Rh中间体, 其经过还原消除得到N-烯丙基化产物.另外, 反应的N-选择性由两个催化循环中的能量差决定.这与Buchwald课题组的观点类似[36], 即通过调控N2-配合物中间体和N1-配合物中间体的能量差, 可以实现高N2-选择性.这不仅是一种原子经济性高的合成方法, 而且也为这类化合物的活性研究奠定了基础, 同时将进一步扩展烷基取代苯并三唑的应用范围(Scheme 7).
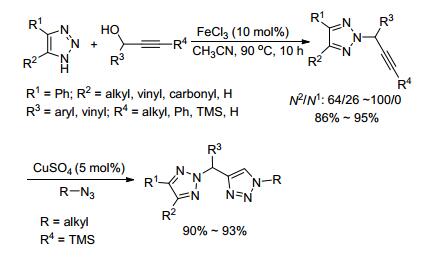
2010年, 史晓东课题组[21]报道了FeCl3催化活化C—O键实现1, 2, 3-三唑的选择性炔丙基化反应.该反应能以较高的收率获得N2-炔丙基-1, 2, 3-三唑.实验结果表明, 反应的N-选择性和三唑的性质有关, 非苯并三唑类底物主要得到N2-选择性产物; 而苯并三唑类, 主要得到N1-选择性产物(在后期的研究报道[22, 23]中也进一步得到了证实), 收率为85%~96%. N-炔丙基-1, 2, 3-三唑化合物可以进一步衍生合成系列不对称1, 1-双三唑, 这类化合物可作为一种新型的双齿配体, 用于过渡金属的配位或催化反应.该反应适用于各种炔醇, 反应条件温和, 操作简便(Scheme 8).
2009年, 史晓东课题组[22]报道了在Au(Ⅰ)作用下1, 2, 3-三唑和炔烃反应合成N2-乙烯基-1, 2, 3-三唑的方法.该反应得到了单一的反式加成产物, C(4), C(5)位的取代基仍然是控制反应N2-选择性的关键因素.当C(4)位为苯基取代, C(5)位为羰基取代时, 得到了单一选择性N2-烯基化产物, 这与他们前期报道的N2-烷基化和芳基化的结果相似[7, 27].另外, 在Lewis酸的作用下, 苯并三唑和炔烃的反应, 也能以70%~96%的收率得到单一选择性的N1-烯基化产物.该反应催化剂用量少, 原料简单易得(Eq. 6).
|
|
(6) |
基于对Fe(Ⅲ)催化活化C—O键的研究, 2012年, 史晓东课题组[23]通过增加炔丙位的位阻, 使得三唑选择性进攻炔基从而得到了联烯-三唑.值得一提的是, 与苯并三唑反应时, 能以74%~95%的收率得到了单一选择性N1-联烯化产物; 而与非苯并的三唑反应时, 能以高达96%的收率得到N2-联烯化产物.与其它简单联烯相比, 三唑修饰的联烯稳定性更强, 这类片段可作为骨架结构用于设计新型催化剂(Eq. 7).
|
|
(7) |
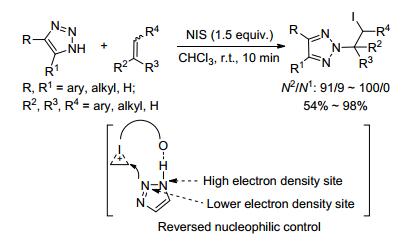
2013年, 王少仲课题组[24]在研究富电子芳炔的双功能化反应中, 通过N-碘代丁二酰亚胺(NⅠS)诱导实现了苯并三唑的选择性N2-烯基化.其中炔烃上取代基的位阻效应同时控制了反应的N2-选择性及产物的Z/E构型, 而炔烃的电子效应决定了反应的活性.另外, 反应中使用蠕动泵滴加苯并三唑, 降低了体系中苯并三唑的浓度, 有助于进一步提高反应的N2-选择性.该反应条件温和、无金属参与、室温下反应就能得到单一的N2-选择性产物.然而, 对于苯乙炔、4-甲基苯乙炔、4-硝基苯乙炔这些电子云密度稍低的炔烃, 无法得到目标产物(Eq. 8).
|
|
(8) |
与非苯并的1, 2, 3-三唑相比, 合成N2-取代苯并三唑是一项更具挑战性的工作, 除了N(1) [N(3)]与N(2)异构体之间存在平衡, 苯并三唑的N2-异构体类似醌型, 其芳香性比非苯并三唑的要低[25].因此, 传统方法主要得到N1-取代产物(Scheme 9).尽管这类化合物不易合成, 但是经过努力, 科学家们也取得了突破性的研究成果.
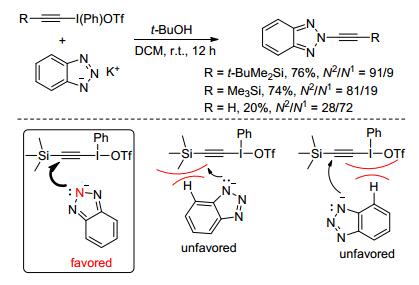
2011年, Kitamura小组[26]选用三氟甲磺酸硅基乙炔碘盐作为炔基化试剂, 叔丁醇和二氯甲烷作溶剂, 室温下与苯并三唑的钾盐反应, 首次实现了苯并三唑的选择性N2-官能化.作者认为, 反应的N2-选择性很可能是由底物的位阻效应所控制(Sheme 10).
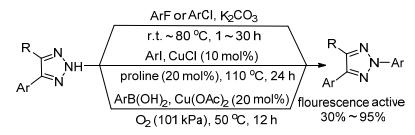
基于调节底物结构能实现1, 2, 3-三唑的选择性N2-烷基化反应研究. 2008年, 史晓东课题组[27]继续发展了C(4), C(5)位双取代1, 2, 3-三唑的N2-芳基化反应.分别通过SNAr、Cu(Ⅰ)催化的芳胺化及Cu(Ⅱ)诱导的硼酸偶联反应实现了高效、高选择性N2-芳基1, 2, 3-三唑的合成(Scheme 11).作者发现, N2-芳基-1, 2, 3-三唑是一类能发出蓝紫光的有机小分子化合物, 并在2011年对其光学性质进行了初步研究[28], 理论计算结果表明该类化合物中三唑环和芳环的共平面程度对其发光性能起到了重要影响, 作者认为N2-芳基-1, 2, 3-三唑(NAT)很可能是通过平面分子内电荷转移(PICT)机制发光. 2015年, 史晓东课题组和陈敏勇课题组[29]合作进一步报道了萘环桥连的双N2-芳基-1, 2, 3-三唑(NBT)荧光团的合成及荧光性质的研究工作, 与NAT通过PICT机制发光不同, NBT中三唑环和萘环虽然不共平面(二面角为61.5°), 但该分子仍然能通过扭曲的分子内电荷转移(TICT)实现发光, 同时, 三唑环上取代基的电子效应对NBT的发射波长无明显影响.二者都证实了N2-芳基取代是影响荧光发射的关键.
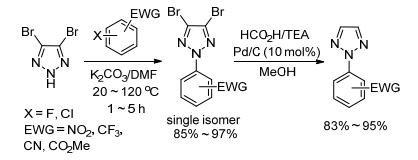
2009年, 王晓军课题组[30]继续报道了4, 5-未取代N2-芳基-1, 2, 3-三唑的合成方法.在K2CO3作用下, 以C(4), C(5)位双溴代1, 2, 3-三唑作原料, 与缺电子芳基卤代物发生SNAr反应, 最后将溴原子还原氢化得到4, 5-未取代N2-芳基-1, 2, 3-三唑.作者认为C(4), C(5)位溴原子的位阻效应和电子效应共同诱导了反应的高N2-选择性(Scheme 12).
2010年, 陈保华课题组[31]以炔酮、叠氮化钠及卤代物为原料, 一锅合成2, 4, 5-三取代1, 2, 3-三唑化合物.其中N2-芳基化的选择性较N2-烷基化的高, 作者认为这可能是因烷基卤代物的位阻过大.该反应体系简单、无金属催化剂和碱参与、环境友好、原子经济性高, 且对各种芳基及杂芳基取代的炔酮具有广泛的适应性, 同时能以非常高的收率分别实现选择性N2-烷基化、芳基化及杂芳基化产物(Eq. 9).
|
|
(9) |
2013年, 陈保华课题组[32]继续改进了上述方法, 使用酰氯和端炔现制炔酮, 产物无需分离, 随后与叠氮化钠、卤代物反应, 通过Sonagashira偶联/1, 3-偶极环加成/C—N键偶联的三步串联反应, 一锅合成了2, 4, 5-三取代1, 2, 3-三唑化合物.这种多步串联的一锅合成方法反应时间短、纯化过程少、环境友好、原子经济性高, 且能以高选择性和高收率实现了目标化合物的合成, 是现代有机化学发展的趋势(Eq. 10).
|
|
(10) |
基于叠氮-查尔酮的氧化环化及原位三唑的官能团化合成N2-取代1, 2, 3-三唑是一种高效、便捷、原子经济性高的方法. 2012年, 陈保华课题组[33]以CuO作催化剂, 查尔酮作原料, N, N-二甲基甲酰胺(DMF)作溶剂, 以高达98%的收率和单一的N2-选择性合成了系列2, 4, 5-三取代1, 2, 3-三唑化合物.对照实验结果表明, 反应中CuO不仅是氧化剂, 也是催化剂, 通过空气氧化再生.值得一提的是, 该反应操作简便、收率高、选择性单一, 对各种取代的芳基具有广泛的适应性, 且原料和催化剂简单易得.这种合成方法对于构建具有潜在生物活性的三唑骨架结构具有重要的借鉴价值(Eq. 11).
|
|
(11) |
选用相似的体系, 2013年, Kamal小组[34]选用5 mol%纳米Fe2O3为催化剂, 以59%~92%的收率得到了N2-芳基-1, 2, 3-三唑化合物, 实现了此类化合物的催化合成, 且催化剂环境友好、易于分离、重复使用三次以上活性仍无明显变化.然而, Ar1为吡咯和噻吩基时, 不能得到目标产物(Eq. 12).
|
|
(12) |
2016年, Singh小组[35]合成了一种基于吲哚衍生物的双核Cu(Ⅰ)配合物, 在催化剂量低至1 mol%的条件下, 仍然以较好的收率实现N2-芳基-1, 2, 3-三唑化合物的合成.与相同类型反应相比, 该反应催化剂用量少, 但具有催化剂昂贵、不易获得, 反应温度高、时间长的不足之处(Eq. 13).
|
|
(13) |
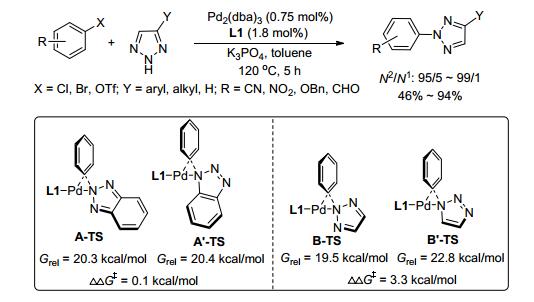
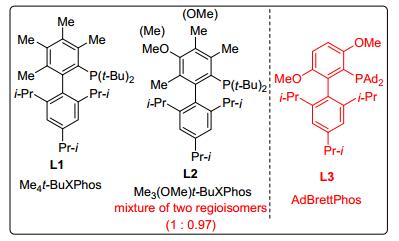
在过渡金属催化的有机化学反应中, 配体扮演了一个非常重要的角色. 2011年, Buchwald小组[36]选用L1 (Me4t-BuXPhos)作为配体(图 1), Pd2(dba)3作催化剂, 实现了4, 5-未取代及4-取代1, 2, 3-三唑和卤代苯的选择性偶联, 合成了系列N2-芳基-1, 2, 3-三唑, N2/N1高达99/1, 但对于苯并三唑, N2/N1接近1/1.理论计算结果表明, 反应具有高选择性很可能是因为中间体N2-1, 2, 3-三唑/Pd和N1-1, 2, 3-三唑/Pd配合物的能量差较大, 使得前者能够快速还原消除得到N2-芳基化产物; 而对于苯并三唑, 由于其能量接近导致选择性大大降低.该反应对各种芳基卤代物、芳基三氟甲磺酸酯及芳基、烷基、杂环取代的1, 2, 3-三唑具有广泛的适应性, 且催化剂用量低、反应时间短, 但配体不易获得(Scheme 13).
2012年, Buchwald小组[37]以廉价易得的2, 3, 6-三甲基苯酚作为原料, 合成了一种新型配体L2(Me3(OMe)- XPhos), 虽然该混合物比例接近1/1, 但无需分离就可用于催化反应.该合成过程中只需要通过简单结晶即可实现产物纯化, 且三步总产率高达54%.对照实验结果表明, 在基于L1作为配体的Pd-催化C—N和C—O键偶联反应中, 配体L2展现出了相似的催化活性, 其可以作为配体L1的替代物.因此, Pd-催化的N2-选择性芳基化反应可以更加简便、经济, 符合现代有机化学发展的方向(图 1).
2016年, Miranda小组[38]也报道了相关研究成果, 他们选用类似的配体L3(图 1)实现了C-核苷取代的1, 2, 3-三唑选择性N2-芳基化. C-核苷是一类非常重要的具有抗菌和抗癌活性的核苷化合物之一, 同时, 三唑杂环常用于修饰核苷类似物中的碱基, 通过三唑核苷的选择性官能团化首次实现了这类新型骨架结构的合成.反应对富电子、缺电子、杂环的芳基卤代物具有广泛的适应性, 且产率高、选择性单一, 但配体昂贵(Eq. 14).
2014年, 我们课题组[39]利用分子间氢键活化策略, 分别实现了1, 2, 3-三唑和吲哚及吡咯的选择性N2-芳基化.在该反应中, 单取代、双取代和未取代的1, 2, 3-三唑都能以较好的收率和N2-选择性得到目标产物(Scheme 14). 2017年, 课题组[40]利用该方法进一步实现了含有色氨酸片段多肽的原位修饰, 合成了系列新型的具有荧光的三唑-多肽化合物.另外, 反应中添加(CF3)2CHOH能有效提高反应收率.同时, 我们课题组[41]也对N2-吲哚基-1, 2, 3-三唑衍生物的荧光性质进行了初步研究.
2012年, Punniyamurthy小组[42]报道了Cu(Ⅱ)催化双芳腙的氧化C—H键官能化, 选择性合成2, 4, 5-三芳基-1, 2, 3-三唑.研究表明, 该反应可能是一个自由基历程, 并且他们顺利分离到了反应中间体(芳腙二聚物), 其进一步氧化环化即可得到目标产物.反应中以空气为氧化剂, 条件温和, 且原料放大到10 mmol时仍然能以75%的收率得到2, 4, 5-三苯基-1, 2, 3-三唑, 但是烷基芳腙在该条件下不稳定, 易分解成相应的醛(Scheme 15).
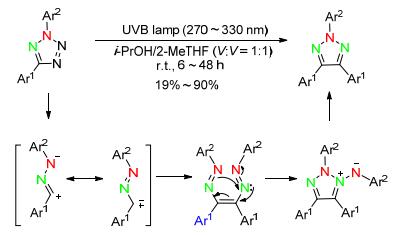
2014年, Harris和Jamieson小组[43]报道了光催化条件下, 2, 5-二取代四唑为原料, 异丙醇/2-甲基四氢呋喃(1:1)作溶剂, 室温下反应得到N2-芳基-1, 2, 3-三唑.该反应通过2, 5-二取代四唑在光催化下裂解形成氰亚胺中间体, 其共振式即卡宾中间体二聚后环化, 随后N-N键断裂得到目标产物.反应条件温和, 但对官能团的容忍性较差, 缺电子取代的收率很低(Scheme 16).
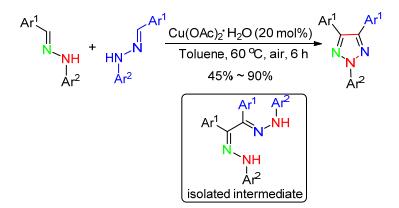
2015年, 林强课题组[44]报道了Cu(Ⅱ)催化2-芳基腙酮氧化环化得到系列N2-芳基-1, 2, 3-三唑.首先2-芳基腙酮和NH4Ac缩合形成C—N键, 随后Cu(Ⅱ)催化单电子氧化形成氮正离子自由基中间体, 进一步通过分子内环化构建N—N键, 经过Cu(Ⅱ)催化的另一个单电子氧化从而得到目标产物.该反应对各种芳基、烷基、酯基、氰基、磺酸基取代的芳腙具有广泛的适应性.反应中选用廉价易得的NH4Ac作为氮源, 氧气作为氧化剂, 这是一种高效、低廉、绿色的合成方法(Scheme 17).
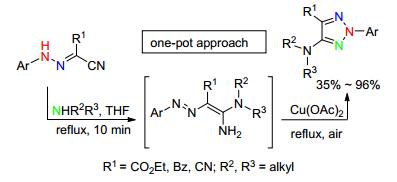
2016年, Belskaya和Benassi小组[45]报道了一锅法合成2-芳基-5-氨基-1, 2, 3-三唑.该反应先经过烷基胺对氰基的亲核加成, 然后在Cu(Ⅱ)和空气作氧化剂的条件下原位氧化环化得到N2-芳基-1, 2, 3-三唑化合物.该反应具有产率高、区域选择性好、方法简便、反应条件温和、原料易得等特点, 且克级反应的收率高达71%, 为N2-芳基-1, 2, 3-三唑类化合物的合成提供了一种新的方法(Scheme 18).
苯并三唑类化合物是一种高效的紫外吸收剂, 在塑料、聚合物、涂料及润滑油、染色/印花纺织品的后处理中, 常作为典型的光稳定添加剂, 目前市场上常用的有UV-326, UV-327, UV-328, UV-329等, 且需求量极大.苯并三唑类紫外吸收剂的核心结构是N2-(邻羟基苯基)-苯并三唑(TIN-P)[46], 其作为紫外吸收剂是由于形成了分子内氢键螯合环, 当分子吸收紫外线的能量后, 电子由基态越迁至激发态, 然后通过分子内氢键实现质子转移, 经过系列无辐射跃迁回到基态, 有效地将激发能转变成无害的热能.
2007年, 刘国斌课题组[47]报道了碱性水溶液中锌粉催化邻硝基苯基偶氮苯酚还原环化, 以较高收率得到N2-芳基-苯并三唑, 一步构建了苯并三唑类紫外吸收剂的核心结构.该反应时间短、操作简便、条件温和, 以水作溶剂, 不仅绿色环保, 而且避免产生传统有机溶剂条件下易生成的过度还原产物(如去氯化产物)或苯并三唑氮-氧化物(Eq. 15).
|
|
(15) |
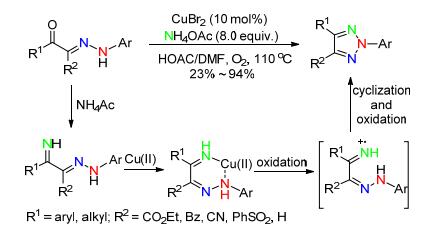
2014年, 孙培培课题组[48]首次以NO2作氮源, 用于过渡金属催化的C—H键官能化反应, 且产物经过简单还原可转化成邻氨基偶氮苯或2-芳基苯丙三唑衍生物.反应条件温和, 氮源清洁, 对各种取代的芳基偶氮苯具有广泛的适应性, 值得一提的是, 不对称的偶氮苯也能以较好的收率得到目标产物, 且硝化发生在相对富电子的芳环上.这对合成不对称的N2-芳基苯并三唑类衍生物具有重要的指导意义(Scheme 19).
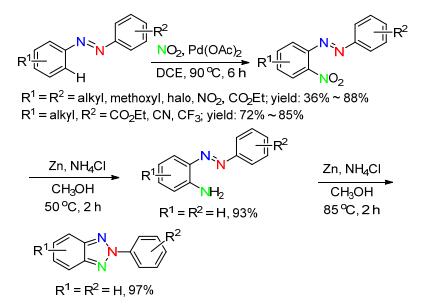
芳基三氮烯常作为合成杂环化合物的前体[49~53]. 2014年, 陈万芝课题组和邱化玉课题组共同[51]报道了Cu(Ⅰ)作催化剂, 2-卤代芳基三氮烯作原料, 叠氮化钠作氮源, 实现了在水溶液中选择性合成系列N2-胺基-苯并三唑和N2-芳基-苯并三唑类化合物.反应首先经过Cu(Ⅰ)催化C—N键偶联得到邻叠氮基偶氮苯, 其热解释放一分子氮气形成氮宾活性中间体, 然后通过分子内氮宾对N=N键加成构建N—N键.该反应操作简便, 原料和催化剂简单易得, 条件温和, 以水作溶剂, 绿色环保, 但反应温度高(Scheme 20).
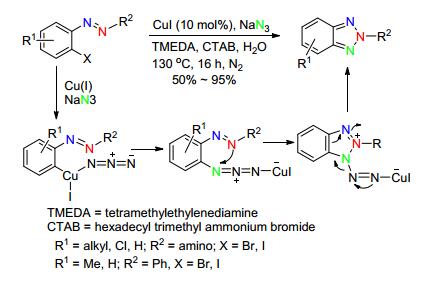
很有趣的是, 同样采用Cu(Ⅰ)作催化剂, 2-卤代芳基三氮烯作原料, 2010年, 朱永明课题组[52]选用二甲亚砜(DMSO)作溶剂、叔丁醇钠作碱, 1, 10-邻菲啰啉作配体, 不添加氮源, 得到了区域选择性不同的产物, 即N1-芳基-苯并三唑.该反应选择性单一, 对各种卤素、硝基、烷基的芳香三氮烯都具有广泛的适应性(Scheme 21).
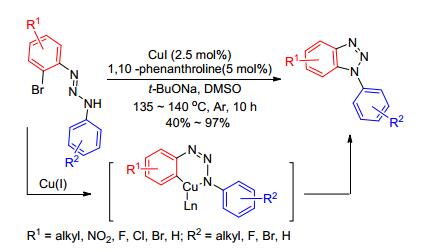
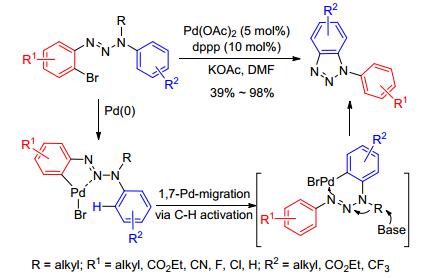
同样, 2011年, 任红军课题组[53]报道了2-溴芳基三氮烯作原料, Pd(OAc)2作催化剂, 醋酸钾作碱, dppp作配体, DMF作溶剂, 选择性得到了N1-芳基-苯并三唑.反应受电子效应和位阻效应的影响, 但通过提高反应温度, 可以增加反应收率.值得一提的是, 与朱永明课题组[51]的报道不同, 该反应通过Pd催化剂与2位氮原子配位, 使其靠近另一芳环的C—H键, 进而通过C—H键活化实现Pd的1, 7-迁移, 然后环化去烷基化, 还原消除得到目标产物.该反应途径新颖、选择性单一, 对各种卤素、氰基、酯基、烷基的芳香三氮烯都具有广泛的适应性, 且所得芳基化产物都是不对称的, 这为今后探索不对称及缺电子取代的N2-芳基-苯并三唑的合成提供了一个新的思路(Scheme 22).
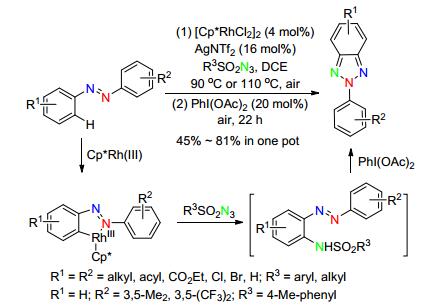
2014年, Lee小组[54]又继续报道了Rh(Ⅰ)作催化剂, 未官能化偶氮苯作原料, N-磺酰基叠氮作氮源, AgNTf2和PhI(OAc)2作氧化剂, 一锅实现了选择性N2-芳基-苯并三唑类化合物的合成.反应首先经过AgNTf2氧化[Cp*RhCl2]2得到Rh(Ⅲ)配合物, 其通过偶氮导向的C—H活化得到五元C—Rh环中间体, N-磺酰胺插入C—Rh键得到Rh(Ⅲ)氨基配合物, 随后质子化生成胺化中间体, 进一步通过PhI(OAc)2氧化环化得到目标产物.同时, 作者监测到了非常明显的同位素效应(kH/kD=2.1), 这说明偶氮苯邻位C—H键的断裂很可能是反应决速步骤.另外, 作者也对此类产物的荧光性质进行了研究, 发现该N2-芳基-苯并三唑的Stock位移较大(≈60 nm), 摩尔消光系数高达3×104 L•mol-1•cm-1, 是一类潜在的生物荧光探针.该反应对各种烷基、酯基、卤素取代的偶氮化合物具有广泛的适应性, 且能以较高的收率得到不对称及缺电子的芳基化产物, 但反应条件复杂、时间长(Scheme 23).
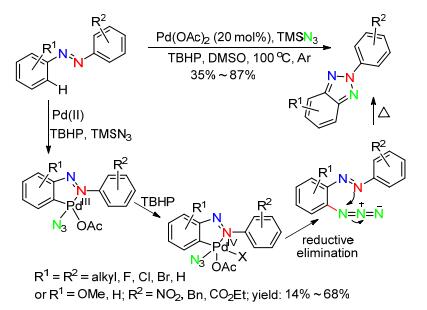
2015年, Patel小组[55]也报道了类似反应, 选用TMSN3作为氮源, 过氧化叔丁醇(TBHP)为氧化剂, 通过Pd(Ⅱ)催化活化实现偶氮邻位C(sp2)—H键胺化氧化, 选择性合成N2-芳基-苯并三唑.在该反应中, 通过底物导向的分子间叠氮化得到偶氮苯邻位C(sp2)—H键官能化中间体(构建C—N键), 然后通过分子内环化(构建C—N键)得到N2-芳基-苯并三唑化合物.另外, 对于不对称偶氮化合物, 相比中性或缺电子的芳环, 环化优先发生在富电子的芳环上(选择性为57/43~78/22).该方法适应性范围广、官能团容忍性高, 可以作为合成N2-芳基-苯并三唑的一种合适的替代方法(Scheme. 24).
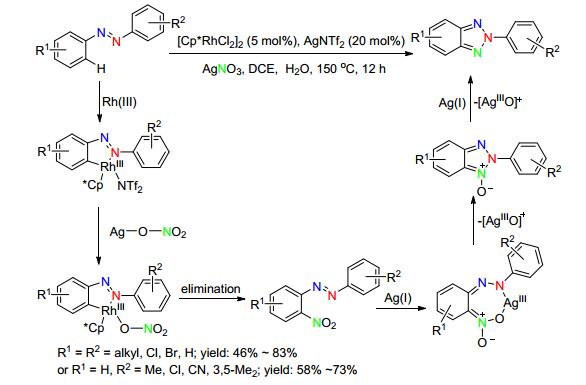
2016年, 蒋高喜课题组[56]进一步改进了该合成方法, 首次使用AgNO3代替传统叠氮试剂作为氮源, Rh(Ⅲ)催化未官能化偶氮化合物反应合成N2-芳基-苯并三唑化合物.初步的机理研究表明, Rh(Ⅲ)催化首先实现了偶氮导向的邻位C—H键硝化, 随后AgNO3在硝基N—O键的断裂和促进偶氮氧化环化过程中都起到了重要作用.该反应对各种烷基、酯基、卤素取代的偶氮具有广泛的适应性, 富电子取代的反应活性更高; 对于不对称的偶氮化合物, 电子效应和空间效应同时控制了反应的选择性, 环化优先发生在富电子取代或位阻小的芳环上(选择性为50/50~100/0), 这与Patel小组的观点类似.与同类型的反应相比, 所用氮源简单易得、安全, 且反应无需添加额外氧化剂, 反应时间短, 但反应温度高(Scheme 25).
继铜催化的“Click”反应问世以来, N1-取代1, 2, 3-三唑化合物的合成方法得到快速发展, 且相应的化合物也已得到广泛应用.而其异构体N2-取代1, 2, 3-三唑展现出的独特物理化学性质引起了科学家的广泛关注, 然而传统方法一般得到N1-和N2-取代1, 2, 3-三唑混合物, 且以N1-取代产物为主.但自2000年以来, 涌现出了系列研究报道, 特别是选择性合成N2-取代1, 2, 3-三唑的研究方法, 在控制反应N2-选择性方面取得较大突破, 呈现出选择性单一、条件温和、绿色环保、原料易得等特点.这些研究成果必将为三唑类化合物在药物化学、生物科学、材料科学等领域的应用起到了更重要的推动作用.
目前已有合成方法仍然存在一些局限性.如: N2-选择性官能化反应中部分底物的结构局限于C(4)、C(5)位双取代1, 2, 3-三唑; 氧化环化反应构建苯并三唑环时倾向于得到对称的或富电子基取代的产物.对于选择性合成N2-取代苯并三唑的方法仍需进一步探索.随着三唑化学和计算化学的不断发展, 简洁、高效、高选择性合成N2-取代1, 2, 3-三唑的方法将会进一步完善, 为其在相关领域的研究奠定基础.
(a) Jewetta, J. C. ; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272.
(b) Thirumurugan, P. ; Matosiuk, D. ; Jozwiak, K. Chem. Rev. 2013, 113, 4905.
(c) Katritzky, A. R. ; Rachwal, S. Chem. Rev. 2010, 110, 1564.
(d) Wang, D. ; Gautam, L. N. S. ; Bollinger, C. ; Harris, A. ; Li, M. ; Shi, X. Org. Lett. 2011, 13, 2618.
(e) Huang, J. ; Zhou, H. ; Chen, Z. Chin. J. Org. Chem. 2016, 36, 1555(in Chinese).
(黄家翩, 周豪, 陈知远, 有机化学, 2016, 36, 1555. )
(f) Chen, Y. ; Zheng, C. ; Peng, X. ; Fu, Q. ; Wu, L. ; Lin, Q. Chin. J. Org. Chem. 2016, 36, 1779(in Chinese).
(陈昱学, 郑超, 彭潇楚, 符清坛, 吴禄勇, 林强, 有机化学, 2016, 36, 1779. )
(g) Gavlik, K. D. ; Lesogorova, S. G. ; Sukhorukova, E. S. ; Subbotina, J. O. ; Slepukhin, P. A. ; Benassi, E. ; Belskaya, N. P. Eur. J. Org. Chem. 2016, 2700.
(a) Huisgen, R. Angew. Chem., Int. Ed. 1963, 2, 565.
(b) Huisgen, R. 1, 3-Dipolar Cycloaddition Chemistry, Ed. : Padwa, A., Wiley, New York, 1984, pp. 1~176.
(a) Kolb, H. C. ; Finn, M. G. ; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004.
(b) Rostovtsev, V. V. ; Green, L. G. ; Fokin, V. V. ; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596.
(c) Zhang, L. ; Chen, X. ; Xue, P. ; Sun, H. H. Y. ; Williams, I. D. ; Sharpless, K. B. ; Fokin, V. V. ; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998.
(d) Rasmussen, L. K. ; Boren, B. C. ; Fokin, V. V. Org. Lett. 2007, 9, 5337.
(a) Ma, Z. ; Lin, Y. ; Cheng, Y. ; Wu, W. ; Cai, R. ; Chen, S. ; Shi, B. ; Han, B. ; Shi, X. ; Zhou, Y. ; Du, L. ; Li, M. J. Med. Chem. 2016, 59, 2151.
(b) Chow, H. -F. ; Lo, C. -M. ; Chen, Y. Top. Heterocycl. Chem. 2012, 28, 137.
(c) El-Sagheerab, H. A. ; Brown, T. Chem. Soc. Rev. 2010, 39, 1388.
(d) Xu, L. ; Li, Y. ; Li, Y. Asian J. Org. Chem. 2014, 3, 582.
(e) Xiao, L. ; Ren, P. ; Jing, X. ; Ren, L. ; Li, Z. Chin. J. Org. Chem. 2007, 37, 3085(in Chinese).
(肖立伟, 任萍, 景学敏, 任丽磊, 李政戴, 有机化学, 2007, 37, 3085. )
(f) Teders, M. Pitzer, L. ; Buss, S. ; Glorius, F. ACS Catal. 2017, 7, 4053.
(a) Schulze, B. ; Schubert, U. S. Chem. Soc. Rev. 2014, 43, 2522.
(b) Wu, J. M. S. Thesis, Wuhan Institute of Technology, Wuhan, 2014 (in Chinese).
(吴俊, 硕士论文, 武汉工程大学, 武汉, 2014. )
(c) Cai, R. ; Yan, W. ; Bologna, M. G. ; Silva, K. ; Ma, Z. ; Finklea, H. O. ; Petersen, J. L. ; Li, M. ; Shi, X. Org. Chem. Front. 2015, 2, 141.
(d) Chen, Y. ; Wu, J. ; Ma, S. ; Zhou, S. ; Meng, X. ; Jia, L. ; Pan, Z. J. Mol. Struct. 2015, 1089, 1.
(e) Shi, S. ; Kuang, C. J. Org. Chem. 2014, 79, 6105.
(f) Lv, Y. ; Zhu, L. -L. ; Liu, H. ; Wu, Y. ; Chen, Z. ; Fu, H. ; Tian, Z. Anal. Chim. Acta 2014, 839, 74.
(g) Liu, H. ; Ding, H. ; Zhu, L. ; Wang, Y. ; Chen, Z. ; Tian, Z. J. Fluoresc. 2015, 25, 1259.
(h) Jo, J. ; Lee, H. Y. ; Liu, W. ; Olasz, A. ; Chen, C. -H. ; Lee, D. J. Am. Chem. Soc. 2012, 134, 16000.
(i) Gavlik, K. D. ; Sukhorukova, E. S. ; Shafran, Y. M. ; Slepukhin, P. A. ; Benassi, E. ; Belskaya, N. P. Dyes Pigm. 2017, 136, 229.
(a) Mangion, I. K. ; Sherry, B. D. ; Yin, J. ; Fleitz, F. J. Org. Lett. 2012, 14, 3458.
(b) Adibekian, A. ; Martin, B. R. ; Wang, C. ; Hsu, K. -L. ; Bachovchin, D. A. ; Niessen, S. ; Hoover, H. ; Cravatt, B. F. Nat. Chem. Biol. 2011, 7, 469.
(c) Cox, C. D. ; Breslin, M. J. ; Whitman, D. B. ; Schreier, J. D. ; McGaughey, G. B. ; Bogusky, M. J. ; Roecker, A. J. ; Mercer, S. P. ; Bednar, R. A. ; Lemaire, W. ; Bruno, J. G. ; Reiss, D. R. ; Harrell, C. M. ; Murphy, K. L. ; Garson, S. L. ; Doran, S. M. ; Prueksaritanont, T. ; Anderson, W. B. ; Tang, C. ; Roller, S. ; Cabalu, T. D. ; Cui, D. ; Hartman, G. D. ; Young, S. D. ; Koblan, K. S. ; Winrow, C. J. ; Renger, J. J. ; Coleman, P. J. J. Med. Chem. 2010, 53, 5320.
(d) Baxter, C. A. ; Cleator, E. ; Brands, K. M. J. ; Edwards, J. S. ; Reamer, R. A. ; Sheen, F. J. ; Stewart, G. W. ; Strotman, N. A. ; Wallace, D. Org. Process Res. Dev. 2011, 15, 367.
Chen, Y.; Liu, Y.; Petersena, J. L.; Shi, X. Chem. Commun. 2008, 3254.
Kalisiak, J.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2008, 10, 3171. doi: 10.1021/ol8006748
Wang, X.-J.; Sidhu, K.; Zhang, L.; Campbell, S.; Haddad, N.; Reeves, D. C.; Krishnamurthy, D.; Senanayake, C. H. Org. Lett. 2009, 11, 5490. doi: 10.1021/ol902334x
Wang, X.-J.; Zhang, L.; Krishnamurthy, D.; Senanayake, C. H.; Wipf, P. Org. Lett. 2010, 12, 4632. doi: 10.1021/ol101965a
Yan, W.; Liao, T.; Tuguldur, O.; Zhong, C.; Petersen, J. L.; Shi, X. Chem.-Asian J. 2011, 6, 2720. doi: 10.1002/asia.v6.10
Girish, Y. R.; Kumar, K. S. S.; Muddegowda, U.; Lokanath, N. K.; Rangappa, K. S.; Shashikanth, S. RSC Adv. 2014, 4, 55800. doi: 10.1039/C4RA09970B
Shi, J.; Zhu, L.; Wen, J.; Chen, Z. Chin. J. Catal. 2016, 37, 1222. doi: 10.1016/S1872-2067(15)61107-X
Jiang, Y.; Wang, Q.; Sun, R.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2016, 3, 744. doi: 10.1039/C6QO00102E
Denmark, S. E.; Kuester, W. E.; Burk, M. T. Angew. Chem., Int. Ed. 2012, 51, 10938. doi: 10.1002/anie.201204347
Zhu, L.-L.; Xu, X.-Q.; Shi, J.-W.; Chen, B.-L.; Chen, Z. J. Org. Chem. 2016, 81, 3568. doi: 10.1021/acs.joc.6b00185
Wei, H.; Hu, Q.; Ma, Y.; Wei, L.; Liu, J.; Shi, M.; Wang, F. Asian J. Org. Chem. 2017, 6, 662. doi: 10.1002/ajoc.v6.6
Kamijo, S.; Jin, T.; Huo, Z.; Yamamoto, Y. J. Am. Chem. Soc. 2003, 125, 7786. doi: 10.1021/ja034191s
Kamijo, S.; Jin, T.; Huo, Z.; Yamamoto, Y. J. Org. Chem. 2004, 69, 2386. doi: 10.1021/jo035292b
Xu, K.; Thieme, N.; Breit, B. Angew. Chem., Int. Ed. 2014, 53, 7268. doi: 10.1002/anie.201403682
Yan, Wu.; Wang, Q.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2010, 12, 3308. doi: 10.1021/ol101082v
Duan, H.; Yan, W.; Sengupta, S.; Shi, X. Bioorg. Med. Chem. Lett. 2009, 19, 3899. doi: 10.1016/j.bmcl.2009.03.096
Yan, W.; Ye, X.; Weise, K.; Petersen, J. L.; and Shi, X. Chem. Commun. 2012, 48, 3521. doi: 10.1039/c2cc17522c
Zhang, Z.; Chang, L.; Wang, S.; Wang, H.; Yao, Z.-J. RSC Adv. 2013, 3, 18446. doi: 10.1039/c3ra43075h
Poznański, J.; Najda, A.; Bretner, M.; Shugar, D. J. Phys. Chem. A 2007, 111, 6501.
Kitamura, T.; Morshed, M. H.; Tsukada, S.; Miyazaki, Y.; Iguchi, N.; Inoue, D. J. Org. Chem. 2011, 76, 8117. doi: 10.1021/jo2015467
Liu, Y.; Yan, W.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 5389. doi: 10.1021/ol802246q
Yan, W.; Wang, Q.; Lin, Q.; Li, M.; Petersen, J. L.; Shi, X. Chem.-Eur. J. 2011, 17, 5011. doi: 10.1002/chem.v17.18
Zhang, Y.; Ye, X.; Petersen, J. L.; Li, M.; Shi, X. J. Org. Chem. 2015, 80, 3664. doi: 10.1021/acs.joc.5b00006
Wang, X.-J.; Zhang, L.; Lee, H.; Haddad, N.; Krishnamurthy, D.; Senanayake, C. H. Org. Lett. 2009, 11, 5026. doi: 10.1021/ol9019875
Li, J.; Zhang, Y.; Wang, D.; Wang, W.; Gao, T.; Wang, L.; Li, J.; Huang, G.; Chen, B. Synlett 2010, 1617. http://growingscience.com/beta/ccl/2584-triton-b-catalyzed-efficient-and-solvent-free-approach-for-the-synthesis-of-dithiocarbamates.html
Liu, X.; Li, J.; Chen, B. New J. Chem. 2013, 37, 965. doi: 10.1039/c3nj40912k
Zhang, Y.; Li, X.; Li, J.; Chen, J.; Meng, X.; Zhao, M.; Chen, B. Org. Lett. 2012, 14, 26. doi: 10.1021/ol202718d
Kamal, A.; Swapna, P. RSC Adv. 2013, 3, 7419. doi: 10.1039/c3ra22485f
Singh, D. P.; Allam, B. K.; Singh, R.; Singh, K. N.; Singh, V. P. RSC Adv. 2016, 6, 15518. doi: 10.1039/C5RA27907K
Ueda, S.; Su, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2011, 50, 8944. doi: 10.1002/anie.v50.38
Ueda, S.; Ali, S.; Fors, B. P.; Buchwald, S. L. J. Org. Chem. 2012, 77, 2543. doi: 10.1021/jo202537e
Lopes, A. B.; Wagner, P.; Souza, R. O. M. A.; Germain, N. L.; Uziel, J.; Bourguignon, J.-J.; Schmitt, M.; Miranda, L. S. M. J. Org. Chem. 2016, 81, 4540. doi: 10.1021/acs.joc.6b00323
Wen, J.; Zhu, L.-L.; Bi, Q.-W.; Shen, Z.-Q.; Li, X.-X.; Li, X.; Wang, Z.; Chen, Z. Chem.-Eur. J. 2014, 20, 974. doi: 10.1002/chem.201302761
Gu, C.-X.; Bi, Q.-W.; Gao, C.-K.; Wen, J.; Zhao, Z.-G.; Chen, Z. Org. Biomol. Chem. 2017, 15, 3396. doi: 10.1039/C7OB00329C
Zhang, Y.-C.; Jin, R.; Li, L.-Y.; Chen, Z.; Fu, L.-M. Molecules 2017, 22, 1380. doi: 10.3390/molecules22091380
Guru, M. M.; Punniyamurthy, T. J. Org. Chem. 2012, 77, 5063. doi: 10.1021/jo300592t
Stewart, S.; Harris, R.; Jamieson, C. Synlett 2014, 2480. http://sk.sagepub.com/cqpress/washington-information-directory-2013-2014
Wu, L.; Guo, S.; Wang, X.; Guo, Z.; Yao, G.; Lin, Q.; Wu, M. Tetrahedron Lett. 2015, 56, 2145. doi: 10.1016/j.tetlet.2015.03.019
Gavlik, K. D.; Lesogorova, S. G.; Sukhorukova, E. S.; Subbotina, J. O.; Slepukhin, P. A.; Benassi, E.; Belskaya, N. P. Eur. J. Org. Chem. 2016, 2700.
艾文, 硕士论文, 大连理工大学, 大连, 2008.Ai, W. M. S. Thesis, Dalian University of Technology, Dalian, 2008 (in Chinese).
Liu, G.-B.; Zhao, H.-Y.; Yang, H.-J.; Gao, X.; Li, M.-K.; Thiemannb, T. Adv. Synth. Catal. 2007, 349, 1637. doi: 10.1002/(ISSN)1615-4169
Dong, J.; Jin, B.; Sun, P. Org. Lett. 2014, 16, 4540. doi: 10.1021/ol502090n
Zhou, Z.; Liu, Q.-L.; Li, W.; Zhu, Y.-M. Heterocycles 2011, 83, 2057. doi: 10.3987/COM-11-12264
Kale, R. R.; Prasad, V.; Hussain, H. A.; Tiwari, V. K. Tetrahedron Lett. 2010, 51, 5740. doi: 10.1016/j.tetlet.2010.08.083
Shang, X.; Zhao, S.; Chen, W.; Chen, C.; Qiu, H. Chem.-Eur. J. 2014, 20, 1825. doi: 10.1002/chem.v20.7
Liu, Q.-L.; Wen, D.-D.; Hang, C.-C.; Li, Q.-L.; Zhu, Y.-M. Helv. Chim. Acta 2010, 93, 1350. doi: 10.1002/hlca.v93:7
Zhou, J.; He, J.; Wang, B.; Yang, W.; Ren, H. J. Am. Chem. Soc. 2011, 133, 6868. doi: 10.1021/ja2007438
Ryu, T.; Min, J.; Choi, W.; Jeon, W. H.; Lee, P. H. Org. Lett. 2014, 16, 2810. doi: 10.1021/ol501250t
Khatun, N.; Modi, A.; Ali, W.; Patel, B. K. J. Org. Chem. 2015, 80, 9662. doi: 10.1021/acs.joc.5b01706
Li, J.; Zhou, H.; Zhang, J.; Yang, H.; Jiang, G. Chem. Commun. 2016, 52, 9589. doi: 10.1039/C6CC04341K

图式 2 溴原子导向1, 2, 3-三唑的N2-烷基化
Scheme 2 Bromo-directed N2-alkylation of 1, 2, 3-triazoles

图式 5 烯烃和1, 2, 3-三唑的N2-选择性碘官能化
Scheme 5 N2-Selective iodofunctionalization of olefins with 1, 2, 3-triazoles

图式 6 Pd-Cu双金属催化选择性合成N-烯丙基-1, 2, 3-三唑
Scheme 6 Selective synthesis of N-allyl-1, 2, 3-triazole via Pd-Cu bimetallic catalysis

图式 11 N2-芳基-1, 2, 3-三唑荧光团的合成
Scheme 11 Synthesis of N2-aryl-1, 2, 3-triazole fluorophores

图式 12 4, 5-双溴-1, 2, 3-三唑的高区域选择性N2-芳基化
Scheme 12 Highly regioselective N2-arylation of 4, 5-dibromo- 1, 2, 3-triazole

图式 13 Pd(0)催化1, 2, 3-三唑的N2-芳基化
Scheme 13 Palladium(0)-catalyzed N2-arylation of 1, 2, 3-triazole

图式 15 Cu(Ⅱ)催化芳腙氧化合成N2-芳基-1, 2, 3-三唑
Scheme 15 Synthesis of N2-aryl-1, 2, 3-triazoles from arylhydrazones via copper(Ⅱ) catalysis

图式 16 光催化2, 5-二取代四唑合成N2-芳基-1, 2, 3-三唑
Scheme 16 Synthesis of N2-aryl 1, 2, 3-triazoles from 2, 5-di-substitutedtetrazoles via photochemistry

图式 17 Cu(Ⅱ)催化芳腙氧化合成N2-芳基-1, 2, 3-三唑
Scheme 17 Synthesis of N2-aryl-1, 2, 3-triazoles from arylhydrazones via Cu(Ⅱ) catalysis

图式 18 芳腙氧化合成N2-芳基-1, 2, 3-三唑
Scheme 18 Synthesis of N2-aryl-1, 2, 3-triazoles from arylhydrazones

图式 19 NO2作氮源实现偶氮苯的邻位硝化
Scheme 19 Ortho-nitration of azoarenes using NO2 as nitro source

图式 20 Cu(Ⅰ)催化2-卤代芳基三氮烯合成N2-氨基-苯并三唑
Scheme 20 Tandem synthesis of N2-amino-benzotriazoles from 2-haloaryltriazenes via copper(Ⅰ) catalysis

图式 21 Cu (Ⅰ)催化2-溴芳基三氮烯合成N1-芳基-苯并三唑
Scheme 21 Synthesis of N1-aryl-benzotriazoles from 2-bromo- aryltriazenes via copper(Ⅰ) catalysis

图式 22 Pd(Ⅱ)催化2-溴芳基三氮烯合成N1-芳基-苯并三唑
Scheme 22 Tandem synthesis of N1-aryl-benzotriazoles from 2-bromoaryltriazenes via palladium(Ⅱ) catalysis

图式 23 Rh(Ⅲ)催化偶氮苯合成N2-芳基-苯并三唑
Scheme 23 Synthesis of N2-aryl-benzotriazoles from azobenzenes via rhodium(Ⅲ) catalysis

图式 24 Pd(Ⅱ)催化偶氮苯合成N2-芳基-苯并三唑
Scheme 24 Synthesis of N2-aryl-benzotriazoles from azobenzenes via Pd(Ⅱ) catalysis

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:













 下载:
下载:








