
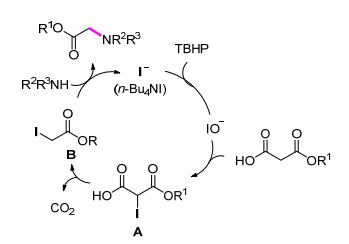
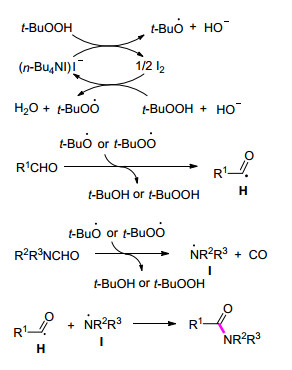
Scheme 1.
丙二酸酯类化合物合成α-氨基酸酯反应的可能机理
Scheme 1.
Proposed mechanism for the conversion of malonates into α-amino acid esters

构筑C—N键是现代有机合成的研究热点之一, 也是合成含氮化合物的主要途径[1~3].含氮化合物是药物分子、天然产物和功能材料的重要结构骨架, 它们在生物和药理活性方面扮演着极为重要的角色[4~6].因此, 不断探索简洁、高效的C—N键构筑方法, 已成为有机合成化学的研究重点.
近几十年来, 利用过渡金属催化构筑C—N键得到了快速发展, 涌现了大量的经典合成方法. Ullman、Buchwald、Hartwig等发展了通过芳卤和胺源的交叉偶联反应来构筑C—N键的方法[7~9], 为各种医药分子和功能材料地合成带来了便捷.随后, 有机合成化学家报道了氮源转变为乃春后进行的C—H键插入胺化反应[10, 11], 使得这类方法广泛的应用于天然产物和杂环合成中.最近, 通过C—H键直接活化胺化来合成含氮化合物取得了较大进展[12].相对于研究比较成熟、应用非常广泛的过渡金属催化构筑C—N键反应, 无过渡金属催化体系下构筑C—N键还远未被充分开发, 发展前景广阔.四丁基碘化铵(n-Bu4NI)/过氧化叔丁醇(t-BuOOH, TBHP)促进的C—N键形成反应具有反应条件温和、反应选择性好等优点, 因而近年来已成为构筑C—N键的一种重要策略.本文根据氮源类型的不同, 对近年来n-Bu4NI/TBHP促进的C—N键形成反应做了简单概述.
自1911年Wieland通过加热或紫外光照射四苯基肼首次得到二苯基胺自由基以来, 氮自由基在有机合成和生物转化过程中一直发挥着重要作用[13].由于氮自由基本身的高活性, 其在合成反应中的应用面临着诸多挑战.近年来, 通过有机合成工作者的不断探索, 氮自由基的应用研究取得了重大进展[14~16].咪唑并[1, 2-a]吡啶在药物化学领域应用广泛, 是构成许多医药化合物的核心结构, 其衍生物具有镇静催眠、抗肿瘤等作用[17, 18]. 2011年, Han课题组[19]报道2-氨基吡啶与β-酮酯或1, 3-二酮在n-Bu4NI促进下, 以TBHP作为氧化剂, 可以高效构建C—N键, 为咪唑并[1, 2-a]吡啶的合成提供了一条简便、经济的途径.该课题组在之前工作中发现BF3• Et2O可以催化PhI(OAc)2介导的2-氨基吡啶与1, 3-二羰基化合物之间的偶联反应[20].因此, 同样尝试将BF3• Et2O添加到该反应体系中.实验探究表明在BF3•Et2O的促进下, 目标产物收率从48%提高到81% (Eq. 1).
|
|
(1) |
2013年, Wan课题组[21]发展了一种合成α-氨基酸酯的新方法.该方法同样利用n-Bu4NI/TBHP的绿色氧化体系, 丙二酸酯类化合物经碘化/脱羧后的中间体进一步与胺反应构建C—N键, 从而得到α-氨基酸酯.值得一提的是, 该反应在最优反应条件下以良好的收率得到具有潜在药物活性的甲硝唑前体.与同类方法相比, 该反应对水不敏感, 且无需添加具有毒性的卤化物试剂.该反应TON (Turnover number)为5.0, TOF (Turnover frequency)为0.6 h-1 (Eq. 2).机理推测可能经过以下的反应过程:首先碘化物在TBHP的氧化作用下生成次碘酸盐, 随后其与丙二酸酯反应得到中间体A, 接着中间体A经脱羧形成中间体B, 最后中间体B与胺发生亲核取代反应得到最终产物α-氨基酸酯(Scheme 1).
氰是重要的有机合成中间体, 可以进一步转化为包括氨基、羧基、醛基及酯基在内的多种官能团[22].因此, 广大有机合成工作者都尝试将氰基引入到有机反应当中. 2015年, Wan课题组[23]发展了无金属催化下氰基乙酸和胺的脱羧偶联反应构建C—N键, 进而合成胺基腈的新方法.该方案底物适应性优良, 多种芳基、烷基取代的伯胺、仲胺、叔胺都能顺利地转化为相应目标产物.在该反应中, NaOAc的作用尚未明确, 推测其可能会促进脱羧反应和亲核取代反应(Eq. 3).
2011年, Nachtsheim课题组[24]在n-Bu4NI促进下, 以TBHP或H2O2作为氧化剂, 首次实现了胺和苯并噁唑在无金属催化体系下的偶联反应.在研究中发现:在无任何添加剂或添加K2CO3等碱性化合物时, 该反应收率极低或无法进行; 当添加乙酸或苯甲酸后, 收率大幅度提高; 当以H2O2作为氧化剂时, 在室温条件下就能实现该反应; 当以TBHP作为氧化剂时, 需要升温至80 ℃.该反应具有催化剂廉价易得、氧化剂易于处理等优点, 这无疑具有广泛的实际应用价值(Eq. 4).
|
|
(2) |
|
|
(3) |
|
|
(4) |
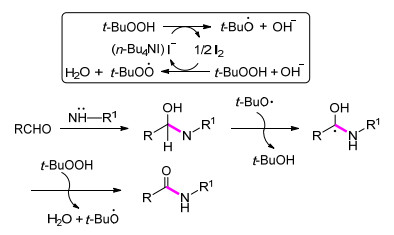
2012年, Shah课题组[25]发展了一种通过醛和胺的交叉脱氢偶联反应来合成酰胺的新方法.该策略反应条件温和, 操作过程简单.该课题组也尝试利用吡啶/ TBHP体系来构建C—N键合成酰胺, 收率中等至良好, 但在该体系下苯胺为底物时反应无法进行(Eq. 5).而在n-Bu4NI/TBHP体系中, 能够高效地实现苯胺与醛的偶联反应.可能的反应机理如下:首先, 在n-Bu4NI促进下, TBHP产生叔丁氧自由基和叔丁基过氧自由基.接着, 醛与胺发生亲核加成反应形成缩醛中间体.随后, 叔丁氧自由基进攻缩醛中间体得到烷基自由基中间体.最后, 烷基自由基中间体被氧化生成相应的目标产物酰胺(Scheme 2).
|
|
(5) |
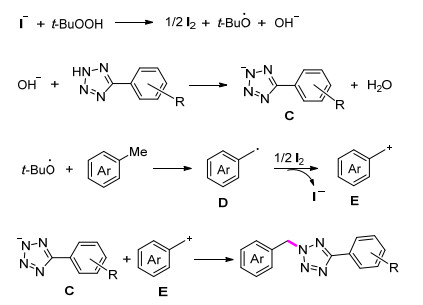
唑及其衍生物具有独特的结构特点和生理活性, 被广泛应用于生命科学、材料化学、药物化学等领域[26].由于唑类化合物对人类生活和发展的重要性, 其参与的有机反应研究备受关注. 2014年, He课题组[27]报道了以芳基甲烷和四唑为底物来构建C—N键得到苄基四唑衍生物的方法.该反应在无溶剂条件下也能得到67%的收率, 加入甲苯作为溶剂后收率提高到83%.研究发现, 芳基甲烷芳环上给电子和吸电子基团都具有良好的适应性, 并且没有显示出明显的电子效应(Eq. 6).根据实验结果, 该课题组提出了可能的机理解释: TBHP氧化n-Bu4NI得到叔丁氧自由基、碘及羟基阴离子后, 羟基阴离子将四唑去质子化, 得到四唑阴离子中间体C.同时, 芳基甲烷与叔丁氧自由基反应得到自由基中间体D, 随后其进一步被碘单质氧化产生苄基阳离子中间体E.最后, 四唑阴离子中间体C与苄基阳离子中间体E反应得到最终产物(Scheme 3).
|
|
(6) |
2015年, He课题组[28]将n-Bu4NI/TBHP体系下构建C—N键的策略拓宽到烷基醚类化合物.该反应中环醚类化合物亦具有良好的适应性.值得指出的是, 与四唑相比, 三唑的活性较低, 但在该体系下亦能实现直接胺化, 以中等收率得到目标产物.该反应TON为5.0, TOF为0.4 h-1 (Eq. 7).
|
|
(7) |
同年, Patel课题组[29]同样以四唑为氮源, 提出了在无金属催化体系下通过芳基醚和四唑的交叉脱氢偶联构建C—N键的策略.在该反应中, 芳基醚既作为反应底物又作为反应溶剂.该反应与之前He课题组[28]的反应相比, TON和TOF均有所提高, 其中TON为10.0, TOF为1.1 h-1.同时, 探究了三唑在该体系下的反应情况:通过升高温度, 增加氧化剂用量, 延长反应时间, 能以78%的收率得到苯氧甲基-1H-苯并[1, 2, 3]三唑(Eq. 8).
|
|
(8) |
2015年, Yu课题组[30]报道了一种实现(N-芳基-胺基甲酰基)-2-亚胺基乙酸酯类化合物螺环化的新策略.研究发现:该反应在氧气氛围中主要得到氮氧杂环己二烯酮, 而在氩气氛围中主要得到喹喔啉-2-酮.该反应底物适应性良好, 反应条件温和, 为螺环化合物的合成提供了一条高效的途径(Eq. 9).
自发现磺酰亚胺基是存在于药物中的有效活性基团以来[31], 广大化学工作者密切关注着磺酰亚胺基的构建研究.在已有的合成磺酰亚胺基的策略中, 通常需要预活化偶联剂或使用金属催化剂, 这不利于工业化生产. 2015年, Deng课题组[32]实现了在n-Bu4NI/TBHP氧化体系下具有重要商业价值的N-酰基磺酰亚胺类化合物的合成.该反应无需预活化偶联剂或金属催化剂, 在氧化体系下通过酰基自由基的直接偶联反应以中等到良好的收率得到目标产物(Eq. 10).
|
|
(9) |
|
|
(10) |
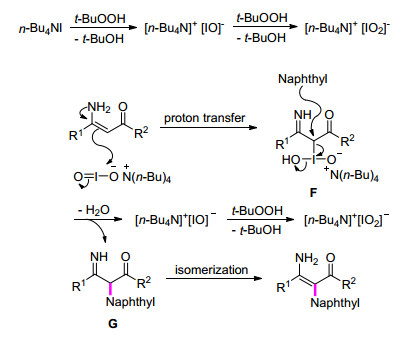
酰胺类化合物是最常见的化合物之一, 被广泛应用于药物、材料等领域.因此对其结构构建修饰一直是有机合成反应中的研究热点[33]. 2014年, Du课题组[34]实现了在n-Bu4NI/TBHP体系中, 通过烯胺与酰胺的交叉偶联反应来构建C—N的新方法.该反应能够在0.2~3.0 h内快速合成二胺基烯烃衍生物.当以苯并三唑为底物时, 得到两种互为同分异构体的产物.稍显遗憾的是, 当使用糖精或二苯胺等作为反应底物时未能得到目标产物(Eq. 11).通过在反应体系中添加自由基抑制剂四甲基哌啶证明该反应不涉及自由基反应, 于是推测可能的反应机理如下:首先, n-Bu4NI被TBHP氧化成四丁基次碘酸盐, 其进一步被氧化成四丁基碘酸盐; 随后, 四丁基碘酸盐与烯胺发生亲电加成反应得到中间体F; 最后, 酰胺进攻中间体F产生亚胺G, 其经异构化得到最终产物(Scheme 4).
|
|
(11) |
2015年, Wang课题组[35]发展了N-杂环酰胺与烯烃进行交联脱氢偶联反应合成N-烯丙基杂环的方法.与传统的由胺和羧酸衍生物缩合得到烯丙胺相比, 该策略通过C—H键和N—H键的直接氧化偶联得到烯丙胺, 大幅度提高了反应的原子经济性.此外, 该课题组尝试了将该方法应用于具有潜在药物活性的磺酰胺衍生物的合成, 如糖精与四甲基乙烯能以68%的收率得到目标产物(Eq. 12).
|
|
(12) |
大多数合成酰胺的策略都依赖于离子反应. 2012年, Wan课题组[36]以醛和N, N-二取代甲酰胺为底物, 通过自由基反应实现了酰胺的合成.该方法反应条件温和, 操作简单, 原料易合成, 底物普适性好, 这无疑是对传统合成酰胺方法的有力补充(Eq. 13).推测该自由基反应经历以下过程:在n-Bu4NI的催化作用下TBHP产生叔丁氧自由基和叔丁基过氧自由基, 这两类自由基进攻醛和N, N-二取代甲酰胺分别得到酰基自由基H和氮自由基I, 随后经自由基偶联反应得到最终产物酰胺(Scheme 5).
|
|
(13) |
2016年, Zhu课题组[37]报道了芳基异硫氰酸酯和甲酰胺合成2-胺基苯并噻唑的反应.通过控制实验发现, n-Bu4NI和TBHP在该体系中缺一不可.值得注意的是, 由于TBHP的强氧化性, 其必须分批次加入, 防止目标产物脱甲基化形成副产物N-甲基苯并噻唑-2-胺.当芳基异硫氰酸酯的芳基邻位被吸电子基团取代时, 无法得到目标产物(Eq. 14).
α-酮酰胺是构成多种重要生物分子的基本骨架[38]. 2016年, Patel课题组[39]报道了一种由芳基甲基酮和烷基磷酰胺在短时间内快速合成α-酮酰胺的方法.一系列具有不同官能团的芳基甲基酮和烷基磷酰胺均能以中等到良好的收率得到相应α-酮酰胺衍生物.在该反应中, 六甲基磷酰胺作为胺源的同时, 亦作为反应溶剂(Eq. 15).
|
|
(14) |
|
|
(15) |
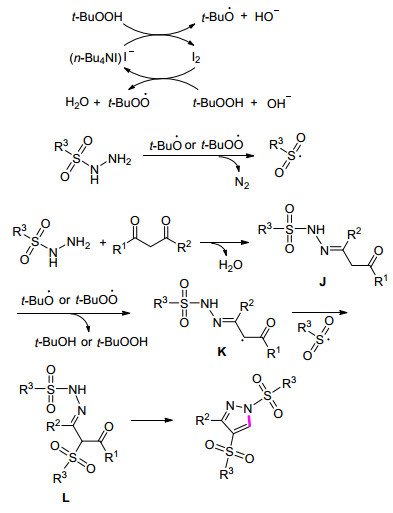
在绿色化学理念的引领下, 有机化学家期望发现合成步骤少且反应条件温和的合成路线.于是, 尝试各种不同氮源, 高效地将n-Bu4NI/TBHP催化体系应用于构筑C—N键之中. 2014年, Wan课题组[40]采用一步法策略来合成多取代的吡唑.该反应在以空气作为氧化剂时, 亦能以31%的收率得到目标产物.在n-Bu4NI/TBHP催化体系中, 磺酰肼发挥双重作用, 既用来构筑环状结构, 又提供了磺酰基前体.令人惊讶的是, 该反应在50 mmol的规模下亦能高收率的得到目标产物(Eq. 16).推测该反应经历以下过程:最初, 碘离子促进TBHP的分解产生叔丁基氧自由基和叔丁基过氧自由基; 随后, 磺酰肼被氧化生成磺酰基, 释放出N2; 接着, 磺酰肼和1, 3-二酮缩合得到中间体J; 然后叔丁基氧自由基和叔丁基过氧自由基进攻中间体J得到中间体K, 其进一步与磺酰自由基偶联得到中间体L; 最后, 中间体L经过分子内缩合得到目标产物(Scheme 6).
同年, Mal课题组[41]发展了在n-Bu4NI作为促进剂和TBHP作为氧化剂的条件下, 从醛或醇出发得到酰胺的合成策略.该反应在30 ℃下亦能以41%的收率得到目标产物.其特点在于该反应在无溶剂体系下进行, 反应条件温和, 后处理简单(Eq. 17).
|
|
(16) |
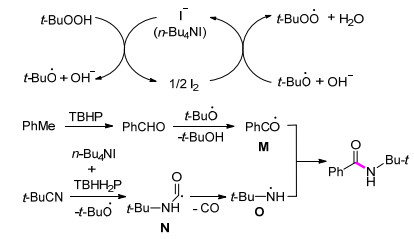
2016年, Li课题组[42]以异氰化物为氮源, 发展了一种新的构建C—N键的方法(Eq. 18).反应过程中首先碘离子促进TBHP的分解产生叔丁基氧自由基和叔丁基过氧自由基.随后, 一方面TBHP氧化芳基甲烷得到芳基甲醛, 其与叔丁基氧自由基反应得到酰基自由基M.另一方面异氰化物在n-Bu4NI和TBHP作用下得到自由基中间体N, 随后经脱羰基化得到氮自由基O.最后, 酰基自由基M和氮自由基O经自由基偶联反应得到酰胺(Scheme 7).
|
|
(17) |
|
|
(18) |
同年, Wan课题组[43]提出了使用n-Bu4NI作为促进剂和TBHP作为氧化剂, 从烯烃和重氮试剂出发直接制备吡唑的新方法.该策略经过[3+2]环加成反应/氧化脱氢反应, 高原子经济性和高选择性的得到在生物医药行业具有重要应用价值的多取代吡唑(Eq. 19).
2016年, Prabhu课题组[44]报道了1, 3-二羰基化合物在n-Bu4NI/TBHP催化体系下的叠氮化反应. 2-甲基-3-氧代-3-苯基丙酸甲酯在10 mol% n-Bu4NI作为促进剂, 2 equiv. TBHP作为氧化剂, 2 equiv. TMSN3作为氮源, 水作为溶剂的条件下, 能以98%的收率得到目标产物.实验探究表明, 1, 3-二酮类化合物在最优反应条件下无法得到目标产物, 但使用乙酸乙酯替代水作为溶剂后, 亦能以67%到82%的收率得到叠氮化产物(Eq. 20).
|
|
(19) |
|
|
(20) |
阐述了n-Bu4NI/TBHP促进的C—N键形成反应, 我们根据氮源的不同, 从胺参与、唑参与、亚胺参与、酰胺参与及其它氮源参与五个方面进行了归纳概述.这类反应的特点是利用n-Bu4NI/TBHP的催化体系构筑C—N键, 从而实现传统无过渡金属催化体系中较为困难的反应.该策略反应条件相对温和, 反应选择性好.因此, 这类反应具有潜在的发展应用空间.但是n-Bu4NI/TBHP催化体系一般需要在高温下进行, 因此实现室温下选择性构筑C—N键是亟待解决的难题.另外, 目前对于C—N键构筑的机理研究极其有限, 在一定程度上限制了C—N键构筑的更大范围应用.我们相信, 在不久的将来, 将真正意义上实现C—N键构筑的绿色工业化.
Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564. doi: 10.1021/acs.chemrev.6b00512
邱頔, 邱孟龙, 马戎, 张艳, 王剑波, 化学学报, 2016, 74, 472.Qiu, D.; Qiu, M. L.; Ma, R.; Zhang, Y.; Wang, J. B. Acta Chim. Sinica 2016, 74, 472(in Chinese).
刘薇, 郑昕宇, 曾建国, 程辟, 有机化学, 2017, 37, 1. http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I1/1Liu, W.; Zheng, X. Y.; Zeng, J. G.; Chen, P. Chin. J. Org. Chem. 2017, 37, 1(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I1/1
Collet, F.; Lescot, C.; Dauban, P. Chem. Soc. Rev. 2011, 40, 1926. doi: 10.1039/c0cs00095g
Cho, H. S.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068. doi: 10.1039/c1cs15082k
Ramirez, T. A.; Zhao, B.; Shi, Y. Chem. Soc. Rev. 2012, 41, 931. doi: 10.1039/C1CS15104E
Ullmann, F. Ber. Dtsch. Chem. Ges. 1903, 36, 2382. doi: 10.1002/(ISSN)1099-0682
Surry, D. S.; Buchwald, S. L. Angew. Chem., Int. Ed. 2008, 47, 6338. doi: 10.1002/anie.v47:34
Hartwig, J. F. Acc. Chem. Res. 2008, 41, 1534. doi: 10.1021/ar800098p
He, J.; Shigenari, T.; Yu, J.-Q. Angew. Chem., Int. Ed. 2015, 54, 6545. doi: 10.1002/anie.201502075
Gou, Q.; Liu, G.; Liu, Z.-N.; Qin, J. Chem.-Eur. J. 2015, 27, 15491. http://jbcs.sbq.org.br/imageBank/epub/jbchs_v26n12.epub
Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341. doi: 10.1021/acscatal.6b00293
Wieland, H. Liebigs Ann. Chem. 1911, 381, 200. doi: 10.1002/(ISSN)1099-0690
Zhao, Y.; Huang, B. B.; Yang, C.; Xia, W. J. Org. Lett. 2016, 18, 3326. doi: 10.1021/acs.orglett.6b01371
Zhou, L. L.; Tang, S.; Qi, X. T.; Lin, C. T.; Liu, K.; Liu, C.; Lan, Y.; Lei, A. W. Org. Lett. 2014, 16, 3404. doi: 10.1021/ol501485f
Zhao, Y. T.; Huang, B. B.; Yang, C.; Li, B.; Gou, B. Q.; Xia, W. J. ACS Catal. 2017, 7, 2446. doi: 10.1021/acscatal.7b00192
Hayakawa, M.; Kaizawa, H.; Kawaguchi, K. I.; Ishikawa, N.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S. I. Bioorg. Med. Chem. 2007, 15, 403. doi: 10.1016/j.bmc.2006.09.047
Abadi, A. H. Arch. Pharm. 2004, 337, 383. doi: 10.1002/(ISSN)1521-4184
Ma, L. J.; Wang, X. P.; Yu, W.; Han, B. Chem. Commun. 2011, 47, 11333. doi: 10.1039/c1cc13568f
Wang, X.; Ma, L.; Yu, W. Synthesis 2011, 2445. http://www.irgrid.ac.cn/handle/1471x/910802?mode=full
Zhang, J.; Jiang, J. W.; Li, Y. L.; Zhao, Y.; Wan, X. B. Org. Lett. 2013, 15, 3222. doi: 10.1021/ol401139m
Yi, H.; Zhang, X.; Qin, C.; Liao, Z. X.; Liu, J.; Lei, A. W. Adv. Synth. Catal. 2014, 356, 2873. doi: 10.1002/adsc.201400548
Wang, H. X.; Shao, Y.; Zheng, H.; Wang, H. H.; Cheng, J.; Wan, X. B. Chem.-Eur. J. 2015, 21, 18333. doi: 10.1002/chem.201502733
Froehr, T.; Sindlinger, P. C.; Kloeckner, U.; Finkbeiner P.; Nachtsheim, B. J. Org. Lett. 2011, 13, 3754. doi: 10.1021/ol201439t
Deshidi, R.; Rizvia M. A.; Shah, B. A. RSC Adv. 2015, 5, 90521. doi: 10.1039/C5RA17425B
Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. J. Med. Chem. 2016, 59, 1545. doi: 10.1021/acs.jmedchem.5b01493
Wang, L.; Zhu, K. Q.; Chen, Q.; He, M. Y. J. Org. Chem. 2014, 79, 11780. doi: 10.1021/jo502283h
Wang, L.; Zhu, K.-Q.; Wu, W.-T.; Chen, Q.; He, M.-Y. Catal. Sci. Technol. 2015, 5, 2891. doi: 10.1039/C5CY00229J
Rajamanickam, S.; Majji, G.; Santra, S. K.; Patel, B. K. Org. Lett. 2015, 17, 5586. doi: 10.1021/acs.orglett.5b02749
Li, D.; Yang, T. H.; Su, H.; Yu, W. Adv. Synth. Catal. 2015, 357, 2529. doi: 10.1002/adsc.v357.11
Zhu, Y.; Loso, M. R.; Watson, G. B.; Sparks, T. C.; Rogers, R. B.; Huang, J. X.; Gerwick, B. C.; Babcock, J. M.; Kelley, D.; Hegde, V. B.; Nugent, B. M.; Renga, J. M.; Denholm, I.; Gorman, K.; DeBoer, G. J.; Hasler, J.; Meade, T.; Thomas, J. D. J. Agric. Food. Chem. 2011, 59, 2950. doi: 10.1021/jf102765x
Qin, W.-J.; Li, Y.; Yu, X. X.; Deng, W.-P. Tetrahedron 2015, 71, 1182. doi: 10.1016/j.tet.2015.01.013
Guo, J.; Li, W. M.; Xue, W. W.; Ye, X.-S. J. Med. Chem. 2017, 60, 2135. doi: 10.1021/acs.jmedchem.6b01644
Yuan, Y. C.; Hou, W. J.; Zhang-Negrerie, D.; Zhao, K.; Du, Y. F. Org. Lett. 2014, 16, 5410. doi: 10.1021/ol5026525
Sun, J. W.; Wang, Y.; Pan, Y. J. Org. Chem. 2015, 80, 8945. doi: 10.1021/acs.joc.5b01383
Liu, Z. J.; Zhang, J.; Chen, S. L.; Shi, E. B.; Xu, Y.; Wan, X. B. Angew. Chem., Int. Ed. 2012, 51, 3231. doi: 10.1002/anie.v51.13
He, Y. M.; Li, J.; Luo, S.; Huang, J. B.; Zhu, Q. Chem. Commun. 2016, 52, 8444. doi: 10.1039/C6CC04394A
Six, D. A.; Barbayianni, E.; Loukas, V.; Constantinou-Kototou, V.; Hadjipavlou-Litina, D.; Stephens, D.; Wong, A. C.; Magrioti, V.; Moutevelis-Minakakis, P.; Baker, F. S.; Dennis, E. A.; Kokotos, G. J. Med. Chem. 2007, 50, 4222. doi: 10.1021/jm0613673
Behera, A.; Ali, W.; Tripathy, M.; Sahoo, D.; Patel, B. K. RSC Adv. 2016, 6, 91308. doi: 10.1039/C6RA16118A
Zhang, J.; Shao, Y.; Wang, H. X.; Luo, Q.; Chen, J. J.; Xu, D. M.; Wan, X. B. Org. Lett. 2014, 16, 3312. doi: 10.1021/ol501312s
Achar, T. K.; Mal, P. J. Org. Chem. 2015, 80, 666. doi: 10.1021/jo502464n
Liu, Z. Q.; Zhang, X. L.; Li, J. X.; Li, F.; Li, C. J.; Jia, X. S.; Li, J. Org. Lett. 2016, 18, 4052. doi: 10.1021/acs.orglett.6b01928
Shao, Y.; Tong, J. J.; Zhao, Y. W.; Zheng, H.; Ma, L.; Ma, M. H.; Wan, X. B. Org. Biomol. Chem. 2016, 14, 8486. doi: 10.1039/C6OB01522K
Dhineshkumar, J.; Prabhu, K. R. Eur. J. Org. Chem. 2016, 2016, 447. doi: 10.1002/ejoc.201501374

Scheme 1 丙二酸酯类化合物合成α-氨基酸酯反应的可能机理
Scheme 1 Proposed mechanism for the conversion of malonates into α-amino acid esters

Scheme 2 醛和胺合成酰胺反应的可能机理
Scheme 2 Proposed mechanism for the conversion of aldehydes and amines into amides

Scheme 3 芳基甲烷和四唑合成苄基四唑衍生物反应的可能机理
Scheme 3 Proposed mechanism for the conversion of arylmethane and tetrazoles into benzyl tetrazole derivatives

Scheme 4 烯胺和酰胺合成二胺基烯烃衍生物反应的可能机理
Scheme 4 Proposed mechanism for the conversion of enamines and amines into diaminoalkenes

Scheme 5 醛和N, N-二取代甲酰胺合成酰胺反应的可能机理
Scheme 5 Proposed mechanism for the conversion of aldehydes and N, N-disubstituted formamides into amide

Scheme 6 多取代吡唑合成反应的可能机理
Scheme 6 Proposed mechanism for the synthesis of poly-sub-stituted pyrazoles

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



















 下载:
下载:
