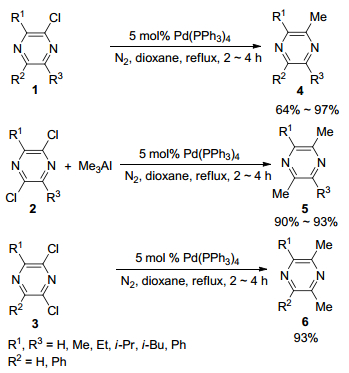
图式 1.
钯催化氯代吡嗪与AlMe3的偶联反应
Scheme 1.
Cross-coupling reaction of AlMe3 with chlorinated pyrazine catalyzed by palladium

过渡金属催化的有机金属试剂与亲电试剂的交叉偶联反应是构建C—X (X=C, N, O等)键的最强大工具之一, 其在天然产物、药物分子及新材料的合成上有着极大的应用价值[1~3].特别是在2010年, 诺贝尔化学奖颁给了从事钯催化的交叉偶联反应的先驱美国科学家Heck和日本科学家Negishi及Suzuki, 表彰他们在C—C键形成的转换研究工作中所取得的巨大成就[4].此后, 金属催化的交叉偶联反应研究及在有机合成中的应用成为了当今有机化学领域中的一个研究热点.在过去数十年的研究中, 硼、锌、镁和锡等有机金属试剂是这类交叉偶联反应中常常使用的有机金属试剂, 并且取得了十分优异的成果.然而在这类交叉偶联反应中, 很早被报道使用的有机金属铝试剂常常被忽略[5, 6].相对于有机锡试剂, 有机金属铝试剂毒性很小, 且容易制备, 因而成为一类很有吸引力的化合物.此外, 他们强烈的Lewis酸性可以使一个反应活性差的反应底物进行有效的转化反应.在某些情况下, 有机金属铝试剂不仅表现为Lewis酸催化剂, 也可作为亲核试剂参与反应.在过渡金属催化下, 有机金属铝试剂作为亲核试剂参与的交叉偶联反应为大量的有机化合物分子的合成提供了一个简便的合成方法[7~12], 同时表现出比有机锂和镁试剂更高的官能团耐受性, 可以在硝基、酯基和内酯的存在下对醛羰基进行选择性的加成反应[13].近年来许多烷基、芳基、烯基及炔基有机铝试剂已成功地应用于过渡金属催化的交叉偶联反应中, 在这些反应中, 金属钯催化的交叉偶联反应是构建C—C及C—N键的最有效的方法之一.为此, 本文重点综述金属钯催化的有机金属铝试剂作为亲核试剂参与的交叉偶联反应.
在1984年, Ohta课题组[14]报道了氯代吡嗪在金属钯的催化下与三甲基铝(Me3Al)试剂的交叉偶联反应, 合成了多种甲基取代的吡嗪化合物(Scheme 1).作者以5 mol%的Pd(PPh3)4为催化剂, 1, 4-二氧六环为反应介质, 在氮气保护下, 回流反应2~4 h即可完成2-氯代吡嗪化合物与Me3Al的交叉偶联反应, 其偶联产物收率可达64%~97%.同时, 在相同的条件下, 2, 5-及2, 6-二氯代吡嗪也可以与Me3Al进行交叉偶联反应, 并以90%~93%的收率得到双甲基化产物.但是当2, 3-二氯-5, 6-二苯基吡嗪化合物与Me3Al进行交叉偶联反应时, 回流反应4 h, 可得到单甲基化产物2-氯-5, 6-二苯基-3-甲基吡嗪, 其收率达93%, 而当回流反应时间延长为12 h, 则以93%的收率得到双甲基化产物2, 3-二甲基-5, 6-二苯基吡嗪.
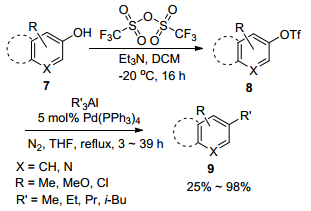
后来, Hirota[15]和Crisp课题组[16]发展了金属钯催化芳基三氟甲烷磺酸酯与三烷基铝试剂的交叉偶联反应, 这为将酚羟基转化为烷基提供了一条简便的方法.作者首先在-20 ℃的条件下, 以Et3N作碱, 二氯甲烷作溶剂, 将酚与三氟甲烷磺酸酐进行反应得到芳基三氟甲烷磺酸酯8, 然后以5 mol%的Pd(PPh3)4为催化剂, 四氢呋喃(THF)为溶剂, 氮气保护下, 加热回流反应3~39 h, 化合物8与三烷基铝试剂可以顺利地进行交叉偶联反应, 并以25%~98%的气相色谱收率得到相应的交叉偶联产物9 (Scheme 2).值得注意的是, 在该偶联反应中, 不但磺酸酯8与Me3Al及三乙基铝(Et3Al)能有效地进行交叉偶联反应, 而且与大位阻的三丙基铝(Pr3Al)及三异丁基铝(i-Bu3Al)经12~16 h的回流也可以顺利地进行交叉偶联反应.同时, 在最佳的反应条件下, 其它的杂芳环磺酸酯如吡啶、喹啉及异喹啉磺酸酯也可以与Me3Al高效的进行交叉偶联反应.虽然对于带有供电子基(CH3, CH3O)和吸电子基(Cl)的芳基三氟甲烷磺酸酯8能顺利地与Me3Al进行有效的偶联反应, 但是对于带有吸电子基(NO2)的芳基三氟甲烷磺酸酯却不能与AlMe3进行交叉偶联反应.同时, 该反应体系对于芳基磺酸酯并不适用.
具有抗病毒作用的烷基嘌呤核苷可由相应的氯代嘌呤核苷与烷基亲核试剂进行烷基化反应得到. 1992年, Hirota课题组[17]报道了由三甲基硅(TMS)保护的氯代嘌呤核苷与Me3Al亲核试剂的交叉偶联反应(Eq. 1).作者使用5 mol% PdCl2/10 mol% PPh3为催化剂, THF为溶剂, 氮气保护下回流反应2~72 h, 以13%~95%的收率得到了相应的交叉偶联产物甲基嘌呤核苷11.值得注意的是, 该偶联反应体系对具有一定空间位阻的三丙基铝(Pr3Al)及三异丁基铝(i-Bu3Al)试剂均能适用.然而, 除了Me3Al试剂外, 其它的烷基铝试剂在与溴代嘌呤核苷进行偶联反应时, 会发生脱溴的副反应, 特别是在与(i-Bu)3Al进行交叉偶联反应时, 脱溴成为主要反应.
|
|
(1) |
由于受到Hirota课题组研究成果的启发, Undheim课题组[18]报道了在温和条件下, 钯催化三烷基铝与氯代喹唑啉的偶联反应(Scheme 3).作者以7 mol%的Pd(PPh3)4为催化剂, THF或ClCH2CH2Cl为反应介质, 在氮气保护下, 回流反应24 h即可完成氯代喹唑啉化合物与三烷基铝的交叉偶联反应.当使用等量的2, 4-二氯喹唑啉12与三甲基铝(Me3Al)进行偶联反应时, 其单烷基化的偶联产物收率为76%, 而用具有一定空间位阻的三异丁基铝(i-Bu3Al), 其单烷基化的偶联产物收率也可以达到63%.作者进一步将烷基化产物13与过量的三烷基铝继续反应, 其相应的双烷基化产物14的收率为63%~86%.同时, 作者发现2, 4-二氯喹唑啉的4位选择性单取代反应受到与之偶联的三甲基铝(Me3Al)及三异丁基铝(i-Bu3Al)试剂的量的影响.在过量的铝试剂作用下, 4位选择性单取取代产物会相应减少, 二双烷基化产物会增多.然而, 在该反应条件下, 对5-溴-2, 4-二氯喹唑啉的选择性单取代却不能充分的实现, 其与铝试剂的偶联产物是4位和6位取代的混合产物.
Blum课题组[19]于1997年成功地制备了环状稳定化的单铝试剂络合物15a及双铝试剂络合物15b, 并考察了络合物15在金属钯催化下与芳基、烯基及苄基的溴及碘化合物的交叉偶联反应.结果表明, 使用2 mol% Pd(PPh3)2Cl2为催化剂, 苯为溶剂, 于30~80 ℃反应2~19 h, 交叉偶联反应产物的收率可达100% (Eq. 2).而对于带有酯基、醛基、氰基、烯基及炔基的芳基溴均能很好地与有机金属铝络合物15进行交叉偶联反应, 其偶联产物的收率为62%~96%.但是该反应体系对于2-BrC6H4CO2Et、4-BrC6H4COPh及4-BrC6H4CH2Br并不适合, 其偶联产物的收率分别只有13%, 17%和16%.同时, 作者进一步的研究表明, 铝试剂自身的配位基, 溶剂及钯催化剂的类型将影响该甲基化反应的进程.
|
|
(2) |
2006年, Woodward课题组[20]合成了新型三烷基铝试剂DABAL-AlMe3 (17), 并以钯催化剂考察了该铝试剂与芳卤的交叉偶联反应.结果表明在THF溶剂中, 1.5 mol%的Pd2(dba)3搭配3 mol%的膦配体20为催化剂, 回流反应4 h, 铝试剂17能与带有供电子基或吸电子基的溴代芳烃有效地进行交叉偶联反应, 其相应偶联产物的收率为65%~98% (Eq. 3).值得注意的是, 当在反应体系中使用富电子的膦配体20a时, 该反应的底物范围得到了有效地拓展, 铝试剂17不仅可以与芳基溴、芳基氯及芳基三氟磺酸酯进行有效的交叉偶联反应, 而且与烯基三氟磺酸酯及苄溴也可以成功地进行交叉偶联反应[21, 22].同时作者探讨了该催化体系对C—Br键和C—Cl键的选择性甲基化反应, 结果表明膦配体20b比20a更有利于碳卤键的选择性甲基化反应, 而且该催化体系对功能基团如醛基、酯基、醇羟基、烯基、甲氧基及氰基均具有很好的耐受性.在有些时候, 该体系可在0.5 mol%的催化剂或者在有氧或者未干燥的溶剂中也能有效地进行偶联反应.但是, 该催化体系对于卤代吡啶及具有烯醇结构的羰基化合物并不适用.
|
|
(3) |
后来作者又使用DABAL-Et3 (21)与芳卤进行乙基化反应, 其偶联产物的收率可达85%.同时结果表明该反应可在有氧条件及未经除水处理的THF中均可有效地进行偶联反应, 具有很好的底物适应性, 而且反应中仅有少量的β-氢消除产物生成.
|
|
在另一个相关的研究中, Woodward课题组[23]报道了离子液体参与的两相体系中的芳基卤与DABAL-Me3 (17)的交叉偶联反应.作者使用2 mol%的PdCl2-(CH3CN)2和4 mol%的膦配体24或25 (Eq. 4)为催化剂, [C4min]BF4-THF (V:V=1:4)为溶剂, 氩气保护下, 芳基卤与DABAL-Me3 (17)于65 ℃反应2~4 h, 以36%~90%的分离收率得到了相应的甲基化产物(Eq. 4).而且在该反应体系中芳基卤与DABAL-Me3 (17)的交叉偶联表现出了极高的化学选择性(甲基化与自身偶联产物比例高达100%).作者进行了离子液体催化体系的回收试验, 除了丁基二甲基咪唑盐离子液体会导致催化剂在挥发性有机化合物(volatile organic compounds, VOC)相中的损失外, 其它的离子液体催化体系的回收均非常成功.在有机溶剂和离子液体的两相混合液中能成功地进行了交叉偶联反应, 实现了交叉偶联产物的快速分离和催化剂的再循环利用.
|
|
(4) |
除了上述的烷基铝试剂能很好地应用于交叉偶联反应中, 烯基铝试剂也是交叉偶联反应中非常重要的亲核试剂之一. 1976年, Babe课题组[24]和Negishi课题组[25]报道了在室温, 以乙醚-正己烷或四氢呋喃-正己烷作溶剂, 使用5 mol%的Pd(PPh3)2Cl2催化(E)-烯基铝试剂(26)与烯基卤化物(27)的交叉偶联反应(Eq. 5).该催化体系效果优异, 能以36%~82%的收率和大于97%的立体选择性得到(E, E)-共轭二烯化合物28.在该反应过程中, 产物的构型与反应物的构型保持一致.而且, 反应中自身偶联产物的量小于2%.两年后, Negishi及其合作者[26]又报道了用锌盐作添加剂可以极大地加速烯基铝试剂与烯基卤化物的交叉偶联反应, 并能以56%~75%的收率得到相应的(E, E)-共轭二烯化合物.
|
|
(5) |
 下载:
导出CSV
下载:
导出CSV
| R | R' | R'' | X | Yield/% | E, E/E, Z |
| n-C5H11 | n-C4H9 | H | I | 74 | >99/1 |
| n-C5H11 | H | n-C4H9 | I | 55 | >99/1 |
| n-C4H9 | t-C4H9 | H | I | 82 | >99/1 |
| t-C4H9 | H | n-C4H9 | I | 36 | 97/3 |
| n-C4H9 | COOCH3 | CH3 | Br | 75 | 97/3 |
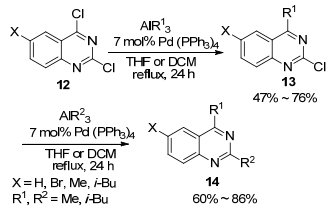
在相关的研究工作中, Negishi课题组[27]于2001年又报道了应用烯基铝试剂与烯基卤的交叉偶联反应进行β-胡萝卜素(β-carotene, 29)的全合成.在合成路线中, 作者使用了4次烯基铝试剂与烯基卤的交叉偶联反应来构建大的共轭链, 由于这些转化非常有效, 因而这成为铝试剂应用于天然产物全合成中非常具有启发性的例子.在2004年的研究中, 他们又发现在钯催化剂36催化烯基铝试剂(31及32)与1-溴-2-碘乙烯(33)进行的偶联反应中, 加入InCl3可加快反应的进行, 并能以82%~91%的收率得到交叉偶联产物.然而, 当把InCl3替换为ZnBr2时, 偶联反应的产物收率急速下降(Scheme 4)[28].
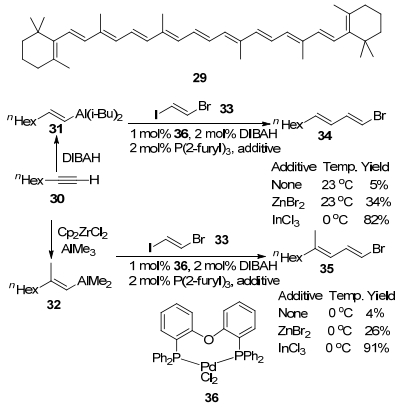
1990年, Chen及其合作者[29]报道了金属钯催化剂搭配添加剂LiCl催化芳基卤或氟代烷基磺酸酯与有机金属铝试剂的交叉偶联反应(Eq. 6).作者使用10 mol%的Pd(PPh3)4为催化剂, LiCl为添加剂, THF为溶剂, 芳基卤或氟代烷基磺酸酯与有机金属铝试剂于80~90 ℃反应8~37 h, 能以55%~96%的收率得到烷基芳基偶联产物.其中E-1-二异丁基烯基铝(38)可由端炔与DIBAL加成得到, 38与芳基卤(37)反应可以非常容易地制得E-l-烯基苯衍生物(39).然而, 在一些反应中, 也可以得到异丁基苯(40), 有时甚至是反应中唯一的产物.对于芳环上带有供电子基的亲电试剂37与二异丁基-1-己烯基或1-(苯基乙烯基)铝试剂反应时, 只得到化合物39, 然而对于芳环上带有吸电子基的亲电试剂37与二异丁基-1-己烯基或二异丁基-1-苯乙烯基铝试剂反应时, 除了得到化合物39外, 还会生成化合物40.但是, 当使用1-(3-甲氧基丙烯基)二异丁基铝与亲电试剂37进行反应时, 化合物40则是唯一的产物.这种现象在之前的二异丁基烯基铝与多种亲电试剂的反应中[30], 包括金属镍催化的烯基铝与芳基卤形成烯基-芳基偶联产物的反应中均未出现过[25].然而, 在同样条件下, 当使用芳环上带有供电子基的苯基卤作为反应底物时, 可观察到异丁基苯的形成.事实上, 烷基基团参与反应的实验现象启发了作者可以用简单的三烷基铝作为酚的去羟基化试剂.使用三异丁基铝试剂(41)与苯基氟代烷基磺酸酯反应, 可以高收率地得到预期的偶联产物异丁基苯(40).在相同的条件下, 苯基溴与三异丁基铝试剂(41)反应也能以较好的收率得到偶联产物异丁基苯(40).但是, 芳基卤及氟代磺酸酯与有机锂及Grignard试剂进行交叉偶联反应时效果并不理想, 其区域选择性很差.
|
|
(6) |
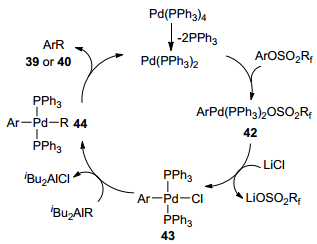
为了探究该反应的历程, 作者分离得到了苯基氟代烷基磺酸酯与金属钯的氧化加成产物, 在此基础上, 作者提出了关于产物39和40的形成机制(Scheme 5).首先是苯基氟代烷基磺酸酯对金属钯进行氧化加成得到中间体42, 接着中间体42中的氟代烷基磺酸酯被Cl-取代得到中间体43, 然后中间体43与有机铝试剂进行金属交换得到烷基芳基钯络合物44和二异丁基氯化铝(i-Bu2AlCl).最后, 络合物44经历还原消除形成目标产物39或40, 又重新产生活性Pd(0)物种进入下一次催化循环.
后来, Menicagli课题组[31]发展了钯催化2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(45)与烯基、烷基及芳基铝试剂(45~48)的交叉偶联反应(Eq. 7).作者使用10 mol%的Pd(PPh3)4或PdCl2(PPh3)2为催化剂, THF-Hexane或Et2O-THF为溶剂, 于室温或回流条件下反应5~15 h, 二异丁基烯基铝(i-Bu2AlCH=CHR) (46)能与2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(45)顺利地进行交叉偶联反应, 并以35%~62%的收率得到烯基三嗪偶联产物49.但是, 当使用具有一定空间位阻的烯基铝试剂时, 该偶联反应不能进行.而后, 作者又考察了其它有机铝试剂在该偶联反应中的反应效果.结果表明三烷基/三芳基铝(R3Al, 47)及二乙基苄基/芳基铝(REt2Al, 48)在该该反应条件下均能与2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(45)进行交叉偶联反应, 分别以43%~88%及35%~71%的收率得到偶联产物49.同时, 作者发现三烷基/三芳基铝(R3Al, 47)的反应速度要比二乙基苄基/芳基铝(REt2Al, 48)试剂的反应速度快.另一方面也表明, 在进行2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(45)的烷基化、苄基化及芳基化时, 三烷基/三芳基铝(R3Al)试剂47是一类非常有效的亲核试剂.
|
|
(7) |
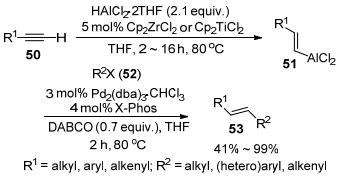
2013年, Woodward课题组[32]报道了使用2~5 mol%的Cp2ZrCl2或Cp2TiCl2为催化剂, THF为溶剂, 端位炔烃与AlHCl2(THF)2在80 ℃反应2~16 h制得了烯基铝试剂51.接着, 作者考察了烯基铝试剂51与具有sp2杂化的亲电试剂52(芳基, 杂芳基及烯基卤)的交叉偶联反应.作者使用3 mol% Pd2(dba)3•CHCl3搭配4 mol% X-Phos为催化剂, 1, 4-二氮杂二环[2.2.2]辛烷(DABCO)为添加剂, THF作溶剂, 烯基铝试剂51与卤化物52在80 ℃反应2 h, 以41%~99%的收率得到了相应的交叉偶联产物53 (Scheme 6).该反应体系可以进行克级量的合成.然而, 在该催化体系中, 一些反应底物并不适用: (ⅰ)虽然该反应体系对含有醚基和羟基等官能团的炔烃具有耐受性, 但是对于含有酯基或氰基功能团的炔烃并不适用, 会导致这些官能团的还原或降低催化剂活性. (ⅱ)在偶联组分52中可忍受酯基的存在, 但对于硝基则会受到部分进攻.而在吡啶类杂环的偶联中要得到可接受的产率需要加入过量的铝试剂和InCl3共催化才能实现. (ⅲ)在大量烯基铝的制备中如使用未纯化的炔, 由于反应中需进行氢转移则会导致有时需要较长的反应时间.
在相关的研究工作中, Zhou课题组[33]报道了钯催化的二级苄基卤亲电试剂54与烯基铝试剂55的不对称催化偶联反应(Eq. 8).作者首先使用二异丁基氢化铝与(杂)芳基乙炔反应制得烯基铝试剂55, 然后使用10 mol%的PdCl2搭配10 mol%的(R)-BINAP作为催化剂考察了二级苄基卤亲电试剂54与烯基铝试剂55的不对称催化偶联反应, 结果表明在THF溶剂中, 于-35 ℃反应48 h, 能以54%~66%的收率得到手性芳基烯目标物56, 其对映选择性可高达99%.该反应体系的优点是对于带有供电子或吸电子取代基的烯基铝试剂均能适用, 而且, α-甲基苄基溴与3-噻吩基烯基铝试剂也可以顺利地进行不对称的偶联反应, 以高达98% ee得到手性芳基烯化合物.然而, 当α-甲基苄基溴与2-吡啶基烯基铝试剂进行不对称偶联反应时, 却只能得到消旋的芳基烯化合物, 这可能是由于吡啶中杂原子N的配位作用的影响.脂肪的二异丁基-1-己烯铝试剂不适合该反应体系.
|
|
(8) |
由于受到NEt3能催化AlMe3与端炔的去质子化反应而得到二烷基炔基铝试剂的鼓舞[34], Micouin课题组[35]应用该方法制备了一系列的二甲基炔基铝试剂58, 并且评价了二甲基炔基铝试剂58与(杂)芳基卤化合物57在金属钯催化下的交叉偶联反应(Eq. 9).在以2.5 mol% Pd2(dba)3•CHCl3搭配5 mol% dppf为催化剂, DME-Heptae为溶剂的条件下得到了最佳的反应结果, 有机铝试剂58与(杂)芳基卤化合物57的交叉偶联反应能以58%~100%的收率得到相应的交叉偶联产物59.该反应体系的优点是:无论是对于脂肪的正庚炔铝试剂还是苯乙炔衍生的铝试剂均能很好地与富电子或缺电子的芳基卤进行交叉偶联反应得到相应的偶联产物.对于溴代物其反应活性比碘代物低, 反应温度需要提升至85 ℃.不过对于不同的(杂)芳基衍生物而言, 交叉偶联反应的目标物收率会受到离去基团X的极大影响, 其中只有活性较高的2-氯吡啶可以进行交叉偶联反应.
|
|
(9) |
2007年, Gau及其合作者[36]报道了金属钯催化芳基卤与三芳基铝(AlAr'3•THF, 61)的交叉偶联反应, 其最优反应条件是: 1 mol% Pd(OAc)2搭配2 mol% PCy3作催化剂, DME或甲苯作溶剂, 室温或60 ℃, 反应3~12 h (Eq. 10).反应结果表明在最优的条件下, 该催化体系不仅能非常高效地催化富电子和缺电子的芳基溴与三芳基铝(AlAr'3•THF, 61)的交叉偶联反应, 而且对于具有空间位阻的2, 6-二甲基苯基溴也能得到非常好的催化效果, 并以70%~96%的收率得到相应的交叉偶联产物62.该催化体系对于惰性的芳基氯也十分有效, 在60 ℃反应12 h能以大于90%的收率得到相应的交叉偶联产物.除了三苯基铝(AlAr'3•THF)试剂外, 其它的取代芳基铝试剂, 如大位阻的2, 4, 6-三甲基苯基铝试剂在该反应体系中也可以适用, 其具体操作与小位阻的铝试剂参与的偶联反应不同, 首先在甲苯溶剂中, 将2, 4, 6-三甲基苯基铝试剂、Pd(OAc)2及PCy3于100 ℃反应1 h, 然后加入芳基溴于40~60 ℃反应4~5 h, 能以85%~96%的收率得到相应的交叉偶联产物.而反应中不需要添加其它的碱是该催化体系的一大优点.
|
|
(10) |
由于在AlAr'3•THF参与的偶联反应中, AlAr'3•THF试剂只需提供一个芳基而导致其原子利用率较低, 为了解决这一缺点, 2009年, Gau课题组[37]使用AlAr'3•THF试剂与三乙基铝于THF中进行反应制备了二乙基芳基铝试剂AlArEt2(THF) (64), 并将其应于与芳卤的交叉偶联反应(Eq. 11).在最优反应条件下, 即1 mol% Pd(OAc)2搭配2 mol% PCy3为催化剂, 甲苯作溶剂, 芳基溴与二乙基芳基铝试剂AlArEt2(THF) (Ar = Ph, 4-MeC6H4, 4-MeOC6H4, 4-Me3SiC6H4, 2-naphthyl)可以顺利地进行交叉偶联反应, 并以14%~95%%的收率得到相应的交叉偶联产物联芳基化合物65.而其中值得注意的是在该反应体系中联芳基化合物的收率均很高, 且基本没有乙基参与偶联反应的产物生成.然而, 在使用原子利用效率较高的AlArEt2(THF)试剂时, 芳基氯则不适用, 这可能是由于它在与金属钯进行金属交换反应时的速度明显低于AlAr3•THF试剂, 同时对于吡啶卤化物需提高温度至40 ℃.该反应体系具有较广的底物适应性, 在芳基溴的芳环上无论是带有富电子基还是缺电子基或者具有一定空间位阻的功能基均能顺利的进行交叉偶联反应, 并且以较好的收率得到相应的联芳基化合物.
|
|
(11) |
同年, Knochel课题组[38]也发展了由金属钯催化芳基卤与芳基铝试剂高效合成联芳基化合物的新策略.作者使用2 mol% PEPPSI-i-Pr为催化剂, THF-Heptane为溶剂, 于室温反应1~12 h, 成功地催化了芳基溴及碘(67)与二异丁基芳基铝试剂ArAl(i-Bu)2 (66)的交叉偶联反应, 并以68%~95%的收率得到联芳基化合物68 (Eq. 12).其中芳环上带有氨基及甲硫基等功能基团的二异丁基芳基铝试剂与芳基溴及碘在室温反应1 h, 即可以68%~91%的收率得到相应的联芳基化合物, 而氨基及甲硫基等功能基不受影响.而且, 对于甲氧基、三氟甲基及酯基等功能基团在该反应体系中均能适应.
|
|
(12) |
为了拓展该研究工作, Knochel课题组[39]应用相似的反应条件考察了NHC-Pd (69)催化剂催化(杂)芳卤与铝试剂RAl2/3X (70)的交叉偶联反应.作者以1.4 mol% NHC-Pd (69)为催化剂, Zn(OAc)2作添加剂, THF-Hep-tane为溶剂, 于室温反应1~12 h, 成功地催化了多种(杂)芳基溴及碘(71)与芳基卤化铝试剂RAl2/3X (70)的交叉偶联反应, 以68%~92%的收率得到联芳基化合物72 (Eq. 13).该反应体系具有良好的原子经济性及该类铝试剂的化学选择性高的优点, 而且对于反应底物中所带的一些敏感性的功能基团如酯基、硝基、醛基及杂芳环均具有良好的适应性.
|
|
(13) |
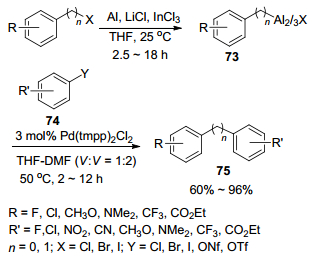
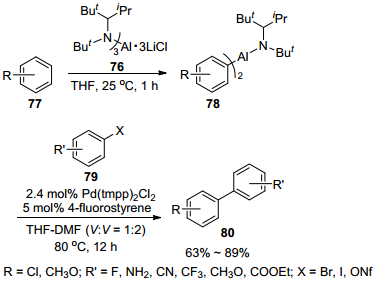
2012年, Knochel课题组[40]又发展了钯催化功能化的铝试剂与多种不饱和卤代烃的交叉偶联反应制备联芳基及二芳基甲烷的新策略.作者首先以THF为溶剂, 利用LiCl及InCl3作催化剂催化金属铝对芳基卤或苄卤进行插入反应制得有机铝试剂73.同时, 作者使用具有空间位阻的氨基铝碱76直接与取代芳基化合物进行铝化反应制备有机铝试剂78.然后作者使用3 mol% Pd(tmpp)2Cl2为催化剂, THF-DMF (V:V=1:2)为溶剂, 于50 ℃反应2~12 h, 成功地催化了有机铝试剂73与(杂)芳基碘、溴和九氟丁烷磺酸酯以及一些特殊的(杂)芳基氯和三氟甲烷磺酸酯(74)的交叉偶联反应, 并以60%~96%的收率得到多种结构的联芳基及二芳基甲烷化合物75 (Scheme 7).而有机铝试剂78与(杂)芳基碘、溴和九氟丁烷磺酸酯以及一些特殊的(杂)芳基氯和三氟甲烷磺酸酯(79)在2.4 mol% Pd(tmpp)2Cl2搭配5 mol% 4-氟苯乙烯为催化剂, THF-DMF (V:V=1:2)为溶剂的催化体系中, 于80 ℃反应12 h, 能以63%~89%的收率得到联芳基化合物(80) (Scheme 8).在这两类反应体系中, 对于芳环上带有未保护的氨基、醛基、酮基、酯基和硝基等敏感功能基均能适应.
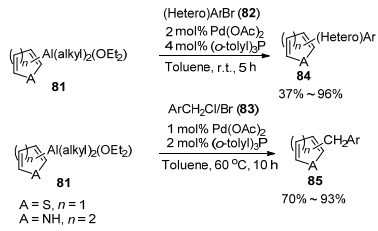
2014年, Zhou课题组[41]报道了金属钯催化(杂)芳基/苄基卤与杂芳基铝试剂的交叉偶联反应.作者应用杂芳基溴化物(2/3-溴噻吩、2/3-溴吡啶)与二烷基氯化铝((alkyl)2AlCl)在nBuLi作用下进行交换反应制得杂芳基铝试剂(81), 接着作者使用Pd(OAc)2搭配P(o-tolyl)3作催化剂考察了(杂)芳基卤与杂芳基铝试剂的交叉偶联反应.结果表明, 杂芳基铝试剂81与(杂)芳基溴(82)在2 mol% Pd(OAc)2/4 mol% P(o-tolyl)3及甲苯为溶剂的催化体系中, 于室温反应5 h, 能以37%~96%的收率得到相应的交叉偶联产物(84) (Scheme 9).该催化体系不仅能高效地催化带有供电子基或吸电子基的芳基溴(82)与杂芳基铝试剂(81)的交叉偶联反应, 而且对于具有空间位阻的2, 4, 6-三甲基溴苯也取得到非常好的催化效果, 不过对于带有吸电子基如氰基(CN)及硝基(NO2)的溴苯则需要提高催化剂的用量至4 mol% Pd(OAc)2/8 mol% P(o-tolyl)3才能得到较好的反应结果.而对于含有酯基的溴苯也能以50%~58%的收率得到相应的交叉偶联产物.但是对于2-溴苯乙酮该催化体系则不适用.同时作者又考察了杂芳基铝试剂81与取代苄基卤(83)的交叉偶联反应, 当使用1 mol% Pd(OAc)2/2 mol% P(o-tolyl)3为催化剂, 甲苯为溶剂, 于60 ℃反应10 h, 能以70%~93%的收率得到相应的交叉偶联产物二芳基甲烷化合物(85) (Scheme 9).该催化体系对于取代苄基氯及溴均能获得良好的催化效果, 而且对于芳环上带有供电子基或吸电子基的苄基卤也能得到较好的反应结果.
在过去的几十年中, 有机铝试剂已被成功地应用于许多有机合成反应中, 为大量新的有机化合物及复杂天然产物分子的合成提供了一些新的合成策略并取得了优异的成绩.虽然在过渡金属催化剂或无金属催化剂的条件下, 有机铝试剂与多种亲电试剂的交叉偶联反应已经取得了长足的发展, 但是对于反应途径的探讨, 减少过渡金属在循环过程中的浸出, 探索环保金属催化剂的使用(如Fe[42]、Cu[43]和Ni[44]等)均需要进一步地深入研究.在未来, 对于有机铝试剂参与的交叉偶联反应的研究重点将会集中在反应机理的探讨、可多次循环利用的高效催化剂的设计和开发使用绿色溶剂如离子液体、超临界流体作为反应介质来代替有机溶剂等方面.同时, 利用微波、超声波或光波来促进该类偶联反应也是一个有待发展的重要领域.
(a) Tucker. C. E. ; Vries, de J. G. Top. Catal. 2002, 19, 111.
(b) Nicolaou, K. C. ; Bulger, P. G. ; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442.
(c) Johnson, J. B. ; Rovis, T. Angew. Chem., Int. Ed. 2008, 47, 840.
(d) Torborg, C. ; Beller, M. Adv. Synth. Catal. 2009, 351, 3027.
(e) Wu, X. -F. ; Anbarasan, P. ; Neumann, H. ; Beller, M. Angew. Chem., Int. Ed. 2010, 49, 9047.
(f) Li, H. -B. ; Johansson Seechurn, C. C. C. ; Colacot, T. J. ACS Catal. 2012, 2, 1147.
(g) Sun, F. -Y. ; Lv, L. -L. ; Huang, M. ; Zhou, Z. -H. ; Fang, X. -D. Org. Lett. 2014, 16, 5024.
(h) Greco, R. ; Goessler, W. ; Cantillo, D. ; Kappe, C. O. ACS Catal. 2015, 5, 1303.
(i) Ruiz-Castillo, P. ; Buchwald, S. L. Chem. Rev. 2016, 116, 12564.
(j) Jedinák, L. ; Zátopková, R. ; Zemánková, H. ; Šustková, A. ; Cankař, P. J. Org. Chem. 2017, 82, 157.
(k) Liu, C. -W. ; Liu, Y. -M. ; Liu, R. -Z. ; Lalancette, R. ; Szostak, R. ; Szostak, M. Org. Lett. 2017, 19, 1434.
(l) Halima, T. B. ; Vandavasi, J. K. ; Shkoor, M. ; Newman, S. G. ACS Catal. 2017, 7, 2176.
(a) Yu, D. -G. ; Shi, Z. -J. Angew. Chem., Int. Ed. 2011, 50, 7079.
(b) Hirner, J. J. ; Blum, S. A. Organometallics 2011, 30, 1299.
(c) Greene, M. A. ; Yonova, I. M. ; Williams, F. J. ; Jarvo, E. R. Org. Lett. 2012, 14, 4293.
(d) Zhang, X. -Q. ; Wang, Z. -X. Synlett 2013, 24, 2081.
(e) Everson, D. A. ; Buonomo, J. A. ; Weix, D. J. Synlett 2014, 25, 233.
(f) Li, Q. -H. ; Ding, Y. ; Yang, X. J. Chin. Chem. Lett. 2014, 25, 1296.
(g) Magano, J. ; Monfette, S. ACS Catal. 2015, 5, 3120.
(h) Tarui, A. ; Shinohara, S. ; Sato, K. ; Omote, M. ; Ando, A. Org. Lett. 2016, 18, 1128.
(i) Li, Q. -H. ; Ding, Y. ; Zhang, G. ; Zhang, Z. ; Mo, S. Chin. J. Org. Chem. 2016, 36, 83(in Chinese).
(李清寒, 丁勇, 张刚, 张震, 莫松, 有机化学, 2016, 36, 83.)
(j) Matsubara, K. ; Yamamoto, H. ; Miyazaki, S. ; Inatomi, T. ; Nonaka, K. ; Koga, Y. ; Yamada, Y. ; Veiros, L. F. ; Kirchner, K. Organometallics 2017, 36, 255.
(a) Kang, S. -K. ; Yamaguchi, T. ; Kim, T. -H. ; Ho, P. -S. J. Org. Chem. 1996, 61, 9082.
(b) Mao, Z. -F. ; Wang, Z. ; Xu, Z. -Q. ; Huang, F. ; Yu, Z. -K. ; Wang, R. Org. Lett. 2012, 14, 3854.
(c) Hornillos, V. ; Pérez, M. ; Fañ anás-Mastral, M. ; Feringa, B. L. J. Am. Chem. Soc. 2013, 135, 2140.
(d) Santandrea, J. ; Bédard, A. C. ; Collins, S. K. Org. Lett. 2014, 16, 3892.
(e) Anima Bose, A. ; Mal, P. J. Org. Chem. 2015, 80, 11219.
(f) Ding, S. -Y. ; Xu, L. ; Li, P. -F. ACS Catal. 2016, 6, 1329.
(g) Singh, S. K. ; Chandna, N. ; Jain, N. Org. Lett. 2017, 19, 1322.
(h) Sahoo, H. ; Mukherjee, S. ; Grandhi, G. S. ; Selvakumar, J. ; Baidya, M. J. Org. Chem. 2017, 82, 2764.
(i) Li, Q. -H. ; Ding, Y. ; Zhang, G. ; Zhang, Z. ; Mo, S. Curr. Org. Synth. 2017, 14, 462.
(a) Baba, S. ; Negishi, E. -I. J. Am. Chem. Soc. 1976, 98, 6729.
(b) Negishi, E-i. ; Baba, S. J. Chem. Soc., Chem Commun. 1976, 596.
(a) Negishi, E-i. ; Zeng, X. ; Tan, Z. ; Qian, M. ; Hu, Q. ; Huang, Z. In Metal-catalyzed Cross-Coupling Reactions, Eds. : de Meijere, A. ; Diederich, F., Vol. 2, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004.
(b) Haas, D. ; Hammann, J. M. ; Greiner, R. ; Knochel, P. ACS Catal. 2016, 6, 1540.
(a) Andrus, M. B. ; Meredith, E. L. ; Hicken, E. J. ; Simmons, B. L. ; Glancey, R. R. ; Ma, W. J. Org. Chem. 2003, 68, 8162.
(b) Liu, B. ; Zhou, W. -S. Org. Lett. 2004, 6, 71.
(c) Elkhayat, Z. ; Safir, I. ; Dakir, M. ; Arseniyadis, S. Tetrahedron Asymmetry 2007, 18, 1589.
(d) Chanu, A. ; Safir, I. ; Basak, R. ; Chiaroni, A. ; Arseniyadis, S. Org. Lett. 2007, 9, 1351.
Woodward, S. ; Dagorne, S. Topics in Organometallic Chemistry, Vol. 41, Springer, Berlin, 2013, pp. 1~322. For the synthesis of organoaluminum compounds, see pp. 173~186, in the chapter on Preparation of organoalanes for organic synthesis, by Knochel, P. ; Blîmke, T. ; Groll, K. ; Chen, Y. -H. ; For cross-coupling reactions of organoaluminum compounds, see pp. 267~276, in chapter on Organoaluminum couplings to carbonyls, imines, and halides, by Kolb, A. ; Zezschwitz, P.
Hallwachs, W.; Schafarik, A. Justus Liebigs Ann. Chem. 1859, 109, 206. doi: 10.1002/(ISSN)1099-0690
Maruoka, K.; Yamamoto, H. Tetrahedron 1988, 44, 5001. doi: 10.1016/S0040-4020(01)86007-5
Wang, C.; Xi, Z. Chem. Soc. Rev. 2007, 36, 1395. doi: 10.1039/b608694m
Uhl, W. Coord. Chem. Rev. 2008, 252, 1540. doi: 10.1016/j.ccr.2008.01.026
(a) Mo, S. ; Shao, X. -B. ; Zhang, G. ; Li, Q. -H. RSC Adv. 2017, 7, 27248.
(b) Zhang, Z. ; Shao, X. -B. ; Zhang, G. ; Li, Q. -H. ; Li, X. -Y. Synthesis 2017, 49, 3643.
(c) Zhang, Z. ; Mo, S. ; Zhang, G. ; Shao, X. -B. ; Li, Q. -H. ; Zhong, Y. Synlett 2017, 28, 611.
Blîmke, T.; Chen, Y. H.; Peng, Z.; Knochel, P. Nat. Chem. 2010, 2, 313. doi: 10.1038/nchem.590
(a) Ohta, A. ; Inoue, A. ; Watanabe, T. Heterocycles 1984, 22, 2317.
(b) Ohta, A. ; Inoue, A. ; Ohtsuka, K. ; Watanabe, T. Heterocycles 1985, 23, 133.
Hirota, K.; Isobe, Y.; Maki, Y. J. Chem. Soc., Perkin Trans. 1 1989, 2513. https://es.scribd.com/document/224806385/Journal-of-Engineering-No-5
Crisp, G. T.; Papadopoulos, S. Aust. J. Chem. 1989, 42, 279. doi: 10.1071/CH9890279
Hirota, K.; Kitade, Y.; Kanbe, Y.; Maki, Y. J. Org. Chem. 1992, 57, 5268. doi: 10.1021/jo00045a051
Mangalagiu, I.; Benneche, T.; Undheim, K. Tetrahedron Lett. 1996, 37, 1309. doi: 10.1016/0040-4039(95)02381-X
Blum, J.; Gelman, D.; Baidossi, W.; Shakh, E.; Rosenfeld, A.; Aizenshtat, Z.; Wassermann, B. C.; Frick, M.; Heymer, B.; Schutte, S.; Wernik, S.; Herbert, S. H. J. Org. Chem. 1997, 62, 8681. doi: 10.1021/jo970822n
Biswas, K.; Chapron, A.; Cooper, T.; Fraser, P. K, Novak, A.; Prieto, O.; Woodward, S. Pure Appl. Chem. 2006, 78, 511. doi: 10.1351/pac200678020511
Vinogradov, A.; Woodward, S. Org. Synth. 2010, 87, 104. doi: 10.15227/orgsyn.087.0104
Cooper, T.; Novak, A.; Humphreys, L. D.; Walker, M. D.; Woodward, S. Adv. Synth. Catal. 2006, 348, 686. doi: 10.1002/(ISSN)1615-4169
Conte, V.; Fiorani, G.; Floris, B.; Galloni, P.; Woodward, S. Appl. Catal. A:Gen. 2010, 381, 161. doi: 10.1016/j.apcata.2010.04.005
Baba, S.; Negishi, E.-I. J. Am. Chem. Soc. 1976, 98, 6729. doi: 10.1021/ja00437a067
Negishi, E-i.; Baba, S. J. Chem. Soc., Chem. Commun. 1976, 596. https://www.sciencedirect.com/science/article/pii/B9780128000465000084
Negishi, E.-I.; Okukado, N.; King, A. O.; Van Horn, D. E.; Spiegel, B. I. J. Am. Chem. Soc. 1978, 100, 2254. doi: 10.1021/ja00475a059
Zeng, F.; Negishi, E.-I. Org. Lett. 2001, 3, 719. doi: 10.1021/ol000384y
Qian, M.; Huang, Z.; Negishi, E.-I. Org. Lett. 2004, 6, 1531. doi: 10.1021/ol049716f
Chen, Q.-Y.; He, Y.-B. Chin. J. Chem. 1990, 8, 451. doi: 10.1002/cjoc.v8.5
Zweifel, G. ; Miller, R. L. Organic Reactions, Vol. 32, John Wileys, New York, 1984, p. 375.
Samaritani, S.; Signore, G.; Malanga, C.; Menicagli, R. Tetrahedron 2005, 61, 14475. doi: 10.1016/j.tet.2008.09.056
Andrews, P.; Latham, C. M.; Magre, M.; Willcox, D.; Woodward, S. Chem. Commun. 2013, 49, 1488. doi: 10.1039/c2cc37537k
Fang, H.; Yang, Z.-Y.; Zhang, L.-J.; Wang, W.; Li, Y.-M.; Xu, X.-L.; Zhou, S.-L. Org. Lett. 2016, 18, 6022. doi: 10.1021/acs.orglett.6b02933
Feuvrie, C.; Blanchet, J.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 2333. doi: 10.1021/ol049346v
Wang, B.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 3481. doi: 10.1021/ol048741i
Ku, S.-L.; Hui, X.-P.; Chen, C.-A.; Kuo, Y.-Y.; Gau, H.-M. Chem Commun. 2007, 3847. http://www.ncbi.nlm.nih.gov/pubmed/18217667
Shu, W.-T.; Zhou, S.-L.; Gau, H.-M. Synthesis 2009, 4075. http://www.academia.edu/9431820/Concerted_Reactions_That_Produce_Diradicals_and_Zwitterions_Electronic_Steric_Conformational_and_Kinetic_Control_of_Cycloaromatization_Processes
Gao, H. J.; Knochel, P. Synlett 2009, 1321. http://growingscience.com/beta/ccl/2584-triton-b-catalyzed-efficient-and-solvent-free-approach-for-the-synthesis-of-dithiocarbamates.html
Blümke, T.; Chen, Y.-H. Peng, Z.; Knochel, P. Nat. Chem. 2010, 2, 313. doi: 10.1038/nchem.590
Groll, K.; Blümke, T. D.; Unsinn, A.; Haas, D.; Knochel, P. Angew. Chem., Int. Ed. 2012, 51, 11157. doi: 10.1002/anie.201205987
Chen, X.; Zhou, L.-M.; Li, Y.-M.; Xie, T.; Zhou, S.-L. J. Org. Chem. 2014, 79, 230. doi: 10.1021/jo4024123
(a) Nečas, D. ; Kotora, M. ; Císařová, I. Eur. J. Org. Chem. 2004, 6, 1280.
(b) Nečas, D. ; Drabina, P. ; Sedlák, M. ; Kotora, M. Tetrahedron Lett. 2007, 48, 4539.
(c) Kawamura, S. ; Ishizuka, K. ; Takaya, H. ; Nakamura, M. Chem. Commun. 2010, 46, 6054.
(d) Kawamura, S. ; Kawabata, T. ; Ishizuka, K. ; Nakamura, M. Chem. Commun. 2012, 48, 9376.
(a) Wunderlich, S. H. ; Knochel, P. Angew. Chem., Int. Ed. 2009, 48, 1501.
(b) Blümke, T. D. ; Groll, K. ; Karaghiosoff, K. ; Knochel, P. Org Lett. 2011, 13, 6440.
(c) Zhou, S. -L. ; Yang, Z. -Y. ; Chen, X. ; Li, Y. -M. ; Zhang, L. -J. ; Fang, H. ; Wang, W. ; Zhu, X. -C. ; Wang, S. W. J. Org. Chem. 2015, 80, 6323.
(d) Shrestha, B. ; Thapa, S. ; Gurung, S. K. ; Pike, R. A. S. ; Giri, R. J. Org. Chem. 2016, 81, 787.
(a) Biradar, D. B. ; Gau, H. -M. Chem. Commun. 2011, 47, 10467.
(b) Biradar, D. B. ; Gau, H. -M. Org. Biomol. Chem. 2012, 10, 4243.
(c) He, F. ; Wang, Z. -X. Tetrahedron 2017, 73, 4450.
(d) Mo, S. ; Shao, X. -B. ; Zhang, G. ; Li, Q. -H. RSC Adv. 2017, 7, 27243.

图式 1 钯催化氯代吡嗪与AlMe3的偶联反应
Scheme 1 Cross-coupling reaction of AlMe3 with chlorinated pyrazine catalyzed by palladium

图式 2 金属钯催化芳基三氟甲烷磺酸酯与三烷基铝试剂的交叉偶联反应
Scheme 2 Cross-coupling reaction of aryl trifluoromethane-sulfonates with trialkylalanes catalyzed by palladium

图式 3 钯催化三烷基铝与氯代喹唑啉的偶联反应
Scheme 3 Cross-coupling reaction of AlMe3 with chlorinated quinazoline catalyzed by palladium

图式 4 金属钯催化烯基铝试剂与烯基卤的交叉偶联反应
Scheme 4 Cross-coupling reaction of alkenylalanes with alkenyl halides catalyzed by palladium

图式 5 钯催化苯基氟代烷基磺酸酯与有机金属铝的偶联反应机理
Scheme 5 The mechanism of Pd-catalyzed cross-coupling reaction of phenyl fluoroalkane-sulfonates with organometallics

图式 6 金属钯催化烯基铝试剂与芳基(烯基)卤的交叉偶联反应
Scheme 6 Cross-coupling reactions of vinyl alanes with aryl(vinyl)halides catalyzed by palladium

图式 7 [Pd(tmpp)2Cl2]催化芳基氯化铝试剂73与各种亲电试剂74的直接交叉偶联反应
Scheme 7 Direct cross-coupling reactions of arylaluminum sesquihalides of type 73 with various electrophiles 74 catalyzed by [Pd(tmpp)2Cl2]

图式 8 [Pd(tmpp)2Cl2]催化有机铝试剂78与各种亲电试剂79的直接交叉偶联反应
Scheme 8 Direct cross-coupling of organoaluminum reagents 78 with 79 using [Pd(tmpp)2Cl2] as catalyst

图式 9 Pd(OAc)2催化2-或3-吡啶铝试剂与芳基溴的交叉偶联反应
Scheme 9 Cross-coupling reactions of 2-or 3-pyridyl aluminium reagents with aryl bromides catalyzed by Pd(OAc)2
| R | R' | R'' | X | Yield/% | E, E/E, Z |
| n-C5H11 | n-C4H9 | H | I | 74 | >99/1 |
| n-C5H11 | H | n-C4H9 | I | 55 | >99/1 |
| n-C4H9 | t-C4H9 | H | I | 82 | >99/1 |
| t-C4H9 | H | n-C4H9 | I | 36 | 97/3 |
| n-C4H9 | COOCH3 | CH3 | Br | 75 | 97/3 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们













