Citation:
Wang Wei, Lü Mengjiao, Zhao Lixia, Zhang Yaling, Li Benhao, Li Baolin. Synthesis of New Matrine Derivatives in Aqueous Phase and Their Crystal Structures[J]. Chinese Journal of Organic Chemistry,
2018, 38(4): 883-889.
doi:
10.6023/cjoc201709021
Key Laboratory of Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi'an 710062
Received Date:
13 September 2017 Revised Date:
11 October 2017 Available Online:
01 April 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21272144) and the Fundamental Research Funds for the Central Universities (Nos. 1301030054, X2015YB06)
Abstract:
Thirteen matrine derivatives were synthesized with high yield with one step in aqueous phase. The chemical structures of the synthesized compounds were characterized by 1H NMR, 13C NMR and HRMS. Three crystals of compound 13-(piperidin-1-yl)matrine (a), 13-(piperidin-1-yl)carbodithioate matrine (k) and 13-(morpholin-4-yl) carbodithioate matrine (m) were obtained and X-ray diffraction data showed that their structures included five chiral carbon atoms with the absolute configuration of 5(S), 6(S), 7(R), 11(R) and 13(S). Meanwhile, there were three classical hydrogen bonds:O(2)-H(21B)…N(2), O(2)-H(21A)…O(1) and C(9)-H(9A)…O(1) by analyzing the data of compound 13-(piperidin-1-yl) matrine (a). These strong hydrogen bonds can play a key role in the accumulation of crystals. Biological studies indicated that the synthetic derivatives had some inhibitory effect against SW480, A549 and A431 cells. The introduction of N or S atom at C-13 position of matrine could improve the antitumor activity.
Matrine (1) is one of the major active alkaloids isolated from Sophora alopecuroides L, which is a useful Chinese herbal drug. Matrine possesses good biological activities. It not only has been used for the treatment of lipopolysaccharide-induced liver injury, [1] but also could regulate immunity, treat cardiac arrhythmia, anti-inflammatory, [2] antitumor, [3~6] antipyretic[7] and hepato-protective. The study on biological activity showed that matrine derivatives and sophora acid derivatives have been used in the prevention and treatment of viral diseases of hepatitis B, hepatitis C, human immunodeficiency virus (HIV) and anti-hepatitis B virus (HBV) with greater inhibitory activity than both matrine and sophocarpine.[8, 9] He et al.[10] has synthesized a series of furoxan-based NO-donating matrine derivatives. These have been evaluated with stronger cytotoxic activities than 5-fluorouracil against human hepatoma cells (HepG2). Some other researchers have synthesized matrinic acid by breaking amide bond.[3, 11]
Obviously, modification of matrine is becoming one of the most interesting fields for researchers due to the potential pharmacological activities. Matrine derivatives at C-13 position are mainly used as a disinfectant and have effect on Escherichia coli, Staphylococcus aureus, Bacillus subtilis variety with the average sterilization rate of 99.9%. Therefore, introducing a substituent containing N or S atom at C-13 position of matrine is regarded to be important for improving the biological activities. Duan and Zhang et al.[12, 13] had synthesized some matrine derivatives containing S, O or N atom at C-13 position of matrine. However, as we know, the solubility of matrine is not ideal and many matrine derivatives are restricted in experiments on activity measurement and further clinical applications.[14] Li et al.[15] successfully synthesized sodium matrine sulfonate which is obviously more hydropholic than matrine. Matrine derivatives with better water solubility are suitable for industrial application and could be promising antitumor drugs in medical fields. And for all we know, there is no research about synthesis of matrine derivatives at C-13 position in aqueous phase by one step. Hence, we describe a simple method for synthesis of matrine derivatives at C-13 position with sophocarpine (2) as the starting material in aqueous phase by one step (Scheme 1). Synthetic method is simple with high yield. Especially, the target compounds a~j have better water solubility.
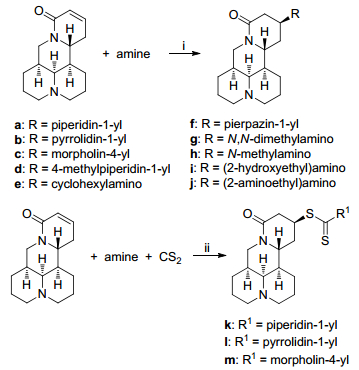
Scheme 1
Scheme 1.
Synthesis of compounds a~m
Reagents and conditions: (ⅰ) sophocarpine (1.25 mmol), amine (2.5 mmol), 2 mL of H2O, 45~80 ℃, 3~24 h; (ⅱ) sophocarpine (1.25 mmol), amine (5 mmol), CS2 (10 mmol), MeOH (2 mL)/H2O (4 mL), 60 ℃, 24 h.
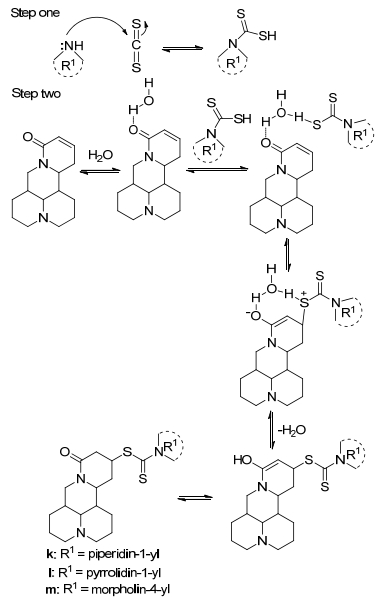
We designed and synthesized thirteen matrine derivatives with matrine as the parent compound. Sophocarpine (2) was used as the starting material and N-containing substituents were introduced via one step classical Michael addition in aqueous phase. Moreover, carbon disulfide participated in the chemical reactions and two S atoms were introduced into compounds k, l and m. The reaction mechanism might be that CS2 reacted firstly with amine to give thiol compound, which attacked the C-13 position of sophocarpine via Michael addition as shown in Scheme 2. In step two, hydrogen bonds were formed between H2O and carbonyl group of sophocarpine, and the electrophilicity of β-C in α, β-unsaturated double bond was increased, while hydrogen bonds could also be formed between H atom in thiol group and O atom in H2O, which increased the nucleophilicity of S atom in thiol. Above all, it could be seen that the reaction yield was increased with H2O as catalyst.
Scheme 2
Scheme 2.
Synthetic reaction mechanism of compounds k, l and m
The optimization for the synthesis of compound l is listed in Table 1. Firstly, the mixture of pyrrolidine (5.00 mmol), CS2 (5.00 mmol) and methanol (2 mL) was stirred for 0.5 h at 45 ℃, and then sophocarpine (1.25 mmol) and distilled water (2 mL) were added, and reacted for another 24 h, to generate compound l with a yield of 39%. While the amount of reactants were increased from 5.00 to 10.00 mmol for CS2 and from 2 to 4 mL for distilled water, the yield was improved to 63%. By changing the temperature, 60 ℃ was found to be the best reaction temperature. The optimized synthetic conditions of compound k were listed in Table 2. Similarly, compound k was synthesized in the same method.
Table 1
Table 1.
Optimization for the synthesis of compound la
aReaction condition: 1.25 mmol of sophocarpine, time was 24 h. b Isolated yield.
Experimental data of the target compounds were described in detail in Table 3. Their chemical structures were further confirmed by 1H NMR, 13C NMR and HRMS. Moreover, the optical rotations of compounds a, k and m as single crystals were measured, and the values of specific rotation [α]D20 were +38.1 (c 1.00, CH3OH), +47.6 (c 1.00, CHCl3) and +42.6 (c 1.00, CHCl3), respectively.
Table 3
Table 3.
Experimental data of the target compounds
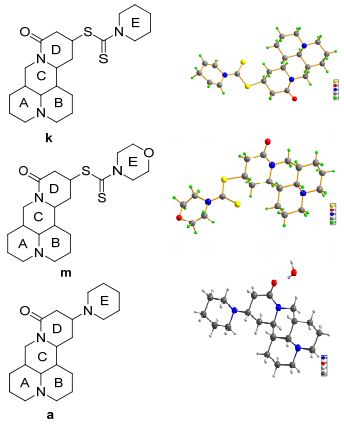
In compound k, the structure includes five chiral carbon atoms and has the absolute configuration of 5(S), 6(S), 7(R), 11(R), 13(S) (Figure 1). Single crystal data analysis also reveals that A and B rings present chair conformation and they are perpendicular to the C ring by paralleling with the D ring. There is no stacking interactions exist between the molecules. The three special interactions, C(151)—H(15A)…O(11)[2.716(7) Å , 101°], C(41)—H(41)…O(11) [3.318 (13) Å , 142°] and C(71)—H(71)…S(21) [3.108(6) Å , 113°], were existing in the intermolecule. These bonds are not in agreement with the typical hydrogen bond distances.[16] But, the bonds have a positive effect on the stability of the crystal. The C=O and C=S bond lengths are 1.219(6) and 1.652(5) Å , respectively. The bond angles of N(11)—C(61)—S(11) and S(21)—C(61)—S(11) are 113.0(4)° and 122.3(3)° (Table 4). The crystal system and space group is monoclinic.
Table 4
Table 4.
Selected bond distances (Å ) and angles (°) for compounds k, m and aa
a In compounds k and m: symmetry code: i=-x, y+1/2, -z. In compound a: symmetry code: i=-x+1/2,
-y, z+1/2, ii=x+1/2, -y+1/2, -z, iii=-x, y+1/2, -z+1/2.
In compound m, the structure includes five chiral carbon atoms and has the absolute configuration of 5(S), 6(S), 7(R), 11(R), 13(S) (Figure 1). Single crystal data analysis proves that A and B rings present chair conformation and they are perpendicular to the C ring by paralleling with the D ring. Ring D have the chair form and ring C has the half-chair form. Crystal structural investigation reveals the absence of intermolecular hydrogen bonds. A and B rings are at the side of the C ring, but the D ring is at the other side of the C ring. Such structural features can be confirmed by H(1), H(3) and H(4) lying at different sides of C ring. The C=O and C=S bond lengths are 1.226(6) and 1.659(5) Å . The bond angles of S(1)—C(16)—S(2) and N(3)—C(16)—S(1) are 122.2(3)° and 113.2(4)° (Table 4). The crystal system and space group are monoclinic.
In compound a, the structure includes five chiral carbon atoms and has the absolute configuration of 5(S), 6(S), 7(R), 11(R), 13(S) (Figures 1 and 2). A single crystal with approximate dimensions of 0.47 mm×0.27 mm×0.13 mm was chosen for X-ray diffraction (XRD) studies. The molecules are interlinked by two kinds of hydrogen bonds, O(2)—H(21B)…N(2) and O(2)—H(21A)…O(1), with distances of 2.830(3) and 3.038(3) Å which are in the range of classical hydrogen bonds. In addition, the intramolecular hydrogen bond was expressed as C(9)—H(9A)…O(1), the length is 2.749(3) Å , which is in the range of classical hydrogen bonds, too. Their bond angles are 164° [O(2)—H(21A)…O(1)] and 171° [O(2)—H(21B)…N(2)], which is in the range of classic bond angle of 150°~180°. The intramolecular hydrogen bond angle [C(9)—H(9A)…O(1)] is 102°. These strong hydrogen bonds can play a key role in the accumulation of crystals.[17, 18] The four saturated rings have chair form configurations, but the caprolactam ring has a half-chair conformation. Through the data (Tables 4, 5), it is not difficult to find that C(1)—C(2) bond length is longer than O(1)—C(1) bond length. The crystal system space group of compound a is orthorhombic.
a Hydrogen bonds between compound a and water molecules. bHydrogen bonds between molecules of compound a.
1.3
Antiproliferation activity
Antiproliferation activities of synthesized matrine derivatives were evaluated by methyl thiazolyl tetrazolium (MTT) assay. Sophocarpine and matrine were used as positive control. The matrine derivatives (compounds k•HCl, l•HCl, m•HCl) have been prepared as a salt to achieve higher water solubility and improve antiproliferation activity. Among the tested compounds, k•HCl displayed the most effective antiproliferation activity against A431 cell lines with inhibition rates above 94% at the tested concentration of 120 μmol•L-1. Meanwhile, compound k•HCl displayed inhibition rates above 65% against SW480 cells and above 95% against A549 at 330 μmol•L-1. Furthermore, the antiproliferation activities of k•HCl, l•HCl, m•HCl against the three cancer cell lines were much stronger than that of matrine.
Table 6 shows that the resulting compounds a, c, e, f, k and lexhibited stronger antitumor activities against the three cancer cell lines with IC50 values of 1.3 fold to 16 fold higher than that of matrine. Compounds k and l showed stronger inhibitory effect against A549 cells with an IC50 value of 64.2 and 91.2 μmol•L-1, respectively. It was clear that introduction of substituents containing N or S atom at C-13 position of matrine derivatives could improve the biochemical profile. Moreover, the S atom had better contribution than N atom to the inhibitory effect. Compared to compounds k, l and m, the hydrochloride salts have different inhibition activities, possibly because the different lipophilicity of these derivatives influenced their interaction with tumor cells.[19] Among all these compounds, k•HCl showed the highest effect against the three cancer cells, with IC50 values of 43.2 μmol•L-1 against A431 cells.
Table 6
Table 6.
IC50 of the target compounds against SW480, A549 and A431 cells
In summary, thirteen matrine derivatives have been successfully synthesized and characterized by 1H NMR, 13C NMR and HRMS. And the preparation method with one step in aqueous phase is simple with mild reaction conditions, low cost and high yields. Organic solvents and expensive catalysts are not needed for the synthesis of these derivatives. According to analysis of single crystal X-ray diffraction, the absolute configurations of the chiral carbon atoms for target compounds a, k and m are 5(S), 6(S), 7(R), 11(R), 13(S). There are no stacking interactions in the three new crystal structures. In compound m, rings A, B and D have chair forms while ring C has the half-chair form. The compound k reveals the existence of interaction between molecules as follows: C(151)—H(15A)…O(11), C(41)—H(41)…O(11), C(71)—H(71)…S(21). These special interaction between molecules can play an important role in the growth of crystals. In compound a, the molecules are interlinked by three kinds of hydrogen bonds: O(2)—H(21B)…N(2), O(2)—H(21A)…O(1) and C(9)—H(9A)…O(1). These interactions link the molecules into a two-dimensional network parallelly. Moreover, the values of specific rotation of compounds a, k and m indicated that the rotatory direction is right handed. Biological studies of the synthetic compounds suggest that introducing substituents containing N or S atom at the C-13 position of matrine has a strong effect on its antiproliferation activity. Matrine derivatives such as compound k•HCl exhibited some activities against three cancer cells including SW480, A549 and A431 cells. The results of our study may be helpful to explore structural modifications of matrine at C-13 position and the synthesis of potential new antitumor agents.
3.
Experimental
3.1
General procedure
Sophocarpine was purchased from Ningxia Doushun Biological Technology Co., Ltd (Yanchi, China). Human colorectal carcinoma cells SW480, human lung cancer cells A549 and human epidermoid carcinoma cells A431 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Dulbecco's Modified Eagle's Medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco. Trypsin, penicillin-streptomycin (P/S) and L-glutamate were purchased from Sigma-Aldrich. All other reagents and solvents were analytical grade, which were supplied by local commercial suppliers and used without further purification unless otherwise noted. lH NMR and 13C NMR spectra were recorded on 400, 600 MHz (Bruker Avance) instruments. The high-resolution mass spectra were measured using a Bruker Esquire 3000 plus mass spectrometer. Melting points were determined using a digital melting point apparatus (Beijing Tektronix Instrument Co. Ltd., Zhengzhou, China). Optical rotation were measured using America Rudolph (Autopol Ⅳ-T) instruments.
The crystallographic measurement of the target compounds were collected at 273 K on a Bruker Smart-1000CCD diffractometer with graphite-monochromated Mo Kα radiation (λ=0.071073 Å ) using ω-2θ scan technique. The structures were solved by direct methods and refined on F2 by full matrix least squares with the Bruker's SHELXL-97 program.
3.2
Synthesis of the target compounds a~j
Sophocarpine (1.25 mmol) and amines (2.5 mmol) were dissolved in water (2.0 mL) with stirring. The reaction mixture was stirred at different temperatures. After completion of the reaction, water was removed under reduced pressure. The residue was purified with column chromatography on silica gel using ethyl acetate/methanol (V: V=4:1) as eluent to give a white solid or colorless oil. The target compound was obtained by recrystallization from petroleum ether.
Carbon disulfide (10.00 mmol) was added to a mixture of methanol (2.0 mL) and amines (5.0 mmol). The mixture was stirred vigorously at 45 ℃ for 0.5 h and then sophocarpine (1.25 mmol) and water (4.0 mL) were added. The reaction mixture was heated for 24 h at 60 ℃. In the reaction process, a small balloon is sealed at the upper end of the spherical condenser tube in order to avoid the volatile of carbon disulfide. After the end of reaction, MeOH and water were removed under reduced pressure. The residue was purified with column chromatography on silica gel using ethyl acetate/methanol (V:V=15:1) as eluent to give a white solid. The pure compounds k~m were obtained by recrystallization from ethanol.
The antiproliferation activities of a~m against SW480, A549 and A431 cells were evaluated in vitro by the MTT assay, with slight modifications. SW480, A549 and A431 cells were cultured using DMEM supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin and 2 mmol•L-1L-glutamate at 37 ℃ and 5% CO2 with 95% humidity. The cells were collected, and then seeded into 96-well microtiter plates with 100 μL of fresh medium overnight. Solutions of each compound at different concentrations were added to the cells, and cultured for another 72 h. Afterwards, the cells were incubated at 37 ℃ for another 4 h with 20 μL of 5 mg/mL MTT. Finally, the media was washed out, and reduced MTT product (blue formazan product) was solubilized by adding 150 μL of dimethylsulfoxide (DMSO) to each well. Plates were agitated for 10 min at room temperature. The optical density (OD) of the solubilized products was measured using a microplate reader (model Multiskan MK3, Thermo, MA, USA) at 570 nm with background subtraction at 630 nm. The inhibition rate of the compounds was calculated according to the following formula:
[1-(ODS-ODNC)/(ODPC-ODNC)]×100%
where ODPC and ODS are the optical densities of the control and test groups at 570 nm, respectively. ODNC is the optical densities at 630 nm. All assays were done in triplicate. Experimental results are presented as the mean±standard deviation (SD) of three parallel measurements. IC50 values of the compounds against the tumor cell lines were generated using GraphPad Prism 5.
Supporting Information PDF files of NMR, HRMS spectra and crystallographic data of compounds a~m. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
Hu, H. G.; Wang, S. Z.; Zhang, C. M.; Wang, L.; Ding, L.; Zhang, J. P.; Wu, Q. Y. Bioorg. Med. Chem. Lett. 2010, 20, 7537. doi: 10.1016/j.bmcl.2010.09.075
[3]
Wang, L. S.; You, Y. J.; Wang, S. Q.; Liu, X.; Liu, B. M.; Wang, J. N.; Lin, X.; Chen, M. S.; Liang, G.; Yang, H. Bioorg. Med. Chem. Lett. 2012, 22, 4100. doi: 10.1016/j.bmcl.2012.04.069
[4]
Huang, Z. S.; Zhou, X. H. Med. Recapitulate2009, 15, 1701.
[5]
Zhang, L. J.; Wang, T. T.; Wen, X. M.; Wei, Y.; Peng, X. C.; Li, H.; Wei, L. Eur. J. Pharmacol. 2007, 563, 69. doi: 10.1016/j.ejphar.2007.01.073
Du, N. N.; Li, X.; Wang, Y. P.; Liu, F. Y.; Liu, X. C.; Li, X.; Peng, Z. G.; Gao, L. M.; Jiang, J. D.; Song, D. Q. Bioorg. Med. Chem. Lett. 2011, 21, 4732. doi: 10.1016/j.bmcl.2011.06.071
[9]
Gao, L. M.; Han, Y. X.; Wang, Y. P.; Li, Y. H.; Shan, Y. Q.; Li, X.; Peng, Z. G.; Bi, C. W.; Zhang, T.; Du, N. N.; Jiang, J. D.; Song, D. Q. J. Med. Chem. 2011, 54, 869. doi: 10.1021/jm101325h
[10]
He, L. Q.; Liu, J.; Yin, D. K.; Zhang, Y. H.; Wang, X. S. Chin. Chem. Lett. 2010, 21, 381. doi: 10.1016/j.cclet.2009.11.033
[11]
Chao, F.; Wang, D. E.; Liu, R.; Tu, Q.; Liu, J. J.; Wang, J. Y. Molecules2013, 18, 5420. doi: 10.3390/molecules18055420
Ci, Y. ; Han, X. L. ; Ma, A. M. ; Wang, L. ; Cheng, C. T. ; Li, J. Y. ; Wen, Y. J. CN 102603744, 2012[Chem. Abstr. 2012, 157, 295336].
[15]
Li, Z. B. ; Liao, D. D. ; Zhao, C. G. ; Zuo, H. ; He, X. Y. ; Deng, L. ; Chen, M. ; Liu, Q. W. ; Tian, X. ; Yang, J. F. CN 101585838, 2009[Chem. Abstr. 2009, 152, 57453].
Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525. doi: 10.1021/ja00170a016
[18]
Kaafarani, B. R.; Pinkerton, A. A.; Neckers, D. C. Tetrahedron Lett. 2001, 42, 8137. doi: 10.1016/S0040-4039(01)01757-9
[19]
Lo, C. Y.; Hsu, L. C.; Chen, M. S.; Lin, Y. J.; Chen, L. G.; Kuo, C. D.; Wu, J. Y. Bioorg. Med. Chem. Lett. 2013, 23, 305. doi: 10.1016/j.bmcl.2012.10.098
Scheme 1
Synthesis of compounds a~m
Reagents and conditions: (ⅰ) sophocarpine (1.25 mmol), amine (2.5 mmol), 2 mL of H2O, 45~80 ℃, 3~24 h; (ⅱ) sophocarpine (1.25 mmol), amine (5 mmol), CS2 (10 mmol), MeOH (2 mL)/H2O (4 mL), 60 ℃, 24 h.
Table 4.
Selected bond distances (Å ) and angles (°) for compounds k, m and aa
Compound k C21H33N3OS2
S(11)—C(61)
1.769(5)
C(61)—S(11)—C(71)
103.7(2)
S(11)—C(71)
1.804(5)
N(11)—C(61)—S(11)
113.0(4)
S(21)—C(61)
1.652(5)
S(21)—C(61)—S(11)
122.3(3)
N(11)—C(61)
1.316(7)
N(11)—C(61)—S(21)
124.7(4)
Compound m C20H31N3O2S2
S(1)—C(21)
1.806(4)
C(12)—S(1)—C(16)
104.2(2)
S(1)—C(16)
1.765(5)
N(3)—C(16)—S(1)
113.2(4)
S(2)—C(16)
1.659(5)
S(1)—C(16)—S(2)
122.2(3)
N(3)—C(16)
1.328(7)
N(3)—C(16)—S(2)
124.6(4)
Compound a C20H33N3•H2O
C(3)—C(2)
1.528(4)
C(3)—C(4)
1.506(4)
N(2)—C(3)
1.483(3)
N(1)—C(1)
1.346(4)
N(2)—C(16)
1.452(4)
N(2)—C(20)
1.461(4)
O(1)—C(1)
1.229(3)
C(1)—C(2)
1.505(4)
C(16)—N(2)—C(20)
109.0(3)
C(3)—N(2)—C(20)
111.1(2)
C(2)—C(3)—N(2)
111.0(2)
N(2)—C(3)—C(4)
111.3(2)
C(3)—N(2)—C(16)
112.8(2)
C(4)—C(3)—C(2)
107.3(2)
a In compounds k and m: symmetry code: i=-x, y+1/2, -z. In compound a: symmetry code: i=-x+1/2,
-y, z+1/2, ii=x+1/2, -y+1/2, -z, iii=-x, y+1/2, -z+1/2.


 下载:
下载:

 下载:
下载:





