引用本文:
华煜晖, 张弘, 夏海平. 芳香性:历史与发展[J]. 有机化学,
2018, 38(1): 11-28.
doi:
10.6023/cjoc201709009

Citation: Hua Yuhui, Zhang Hong, Xia Haiping. History and Development[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 11-28. doi: 10.6023/cjoc201709009
Citation: Hua Yuhui, Zhang Hong, Xia Haiping. History and Development[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 11-28. doi: 10.6023/cjoc201709009
芳香性:历史与发展
English
History and Development
Abstract:
Aromaticity is one of the most fundamental concept in organic chemistry. Aromatic compounds generally present special thermodynamic stability. Research on aromaticity can help us to understand the stability essence of aromatic compounds, and thus enables the further prediction and construction of species with stabilization or destablization. The endless richness of aromaticity researches usually originates the nature and criterion of aromaticity. The main emphasis of this review is on a discussion of historical discoveries, definitions and classification of aromaticity-related structural types, as well as various theoretically and experimentally criterions. Furthermore, this review contains the recent development of aromaticity illustrated by recent representative examples.
-
Key words:
- aromaticity
- / anti-aromaticity
- / definition
- / criterion
- / classification
-
1 绪论
“芳香性”是用于描述一些环状化合物特殊稳定性的概念.在有机化学中, 尤其是针对不饱和环体系、多环体系研究中居重要地位.自1825年Michael Faraday从煤焦油中分离出苯开始, 芳香化学至今已有近200年的历史.为了深入了解“芳香性”及其在自然学科中产生的深远影响, 本文简述了其发展历史与本质, 并对芳香性的判断方法进行归类总结.除了列举实例, 进一步对芳香化合物进行分类以外, 还基于本课题组近年的研究成果, 提出一些新观点.
1.1 芳香性简史
1825年, Faraday[1]首次分离得到了一种具有“芳香气味”的纯净物——苯.因这类化合物通常具有芳香的气味, 因而得名“芳香的(Aromatic)”化合物.历经近200年的发展, “芳香性”的定义日趋丰富, 并且实验上也不断衍生出全新的结构与性质.关于芳香性的发展历史, 已有很多综述性文章展示[2~4], 表 1列出了芳香性发展的简单历史进程.
 表 1
芳香性简史
Table 1.
A brief history of aromaticity
表 1
芳香性简史
Table 1.
A brief history of aromaticity
年份/年 事件 关键词 1825前 人们将一类具有芳香气味的化合物称为“芳香化合物” 芳香化合物 1825 Faraday分离出一种具有芳香气味的化合物——苯[1] 苯 1865前 不饱和的、具有高C/H比的稳定化合物称为“芳香性化合物” 稳定性 1865 Kekulé提出著名的苯环的“Kekulé式”, 认为苯环是平面、环状分子, 并且具有3个C=C双键和3个C—C单键交替构成[5] Kekulé式 1866 Erlenmeyer提出了芳香性的化学判据:在实验上, 芳香化合物的取代反应比加成反应更易发生[6] 取代反应 1910 Pascal提出芳香性化合物的抗磁磁化率升高, 即该类化合物具有极强的抗磁性[7] 抗磁磁化率 1925 Armit和Robinson提出苯环为“六电子体(Electron Sextet)”, 认为苯环的性质与其电子有关, 并提出了杂原子参与的芳香性[8] 六电子体杂环芳香性 1931 Hückel提出著名的[4n+2]规则:在sp2杂化碳原子平面单环体系中, 含有(4n+2)个π电子的化合物具有芳香性[9] [4n+2]规则 1936 Pauling提出环电流理论:在一定条件下离域π电子可在苯环及同系稠环上形成环电流[10] 环电流 1937 Wheland根据大量实验数据总结提出了各种芳香烃共振能(ERE)的估算值[11] 芳香烃共振能(ERE) London提出了London抗磁性, 通过LCAO分子轨道法计算了π电子环电流对磁化率的贡献[12] London抗磁性 1956 Pople提出环流对核磁共振(NMR)化学位移的影响[13] NMR 1958 Craig首次提出了具有Craig模型的芳香性, 即如今的平面Möbius芳香性[14] Craig-Möbius芳香性 1964 Heilbronner首次提出:环状4n电子轮烯可能具有Möbius带扭曲结构的Möbius芳香性[15] 扭曲结构的Möbius芳香性 1965 Dewar提出基于原子化热的共振能(EDRE)作为芳香性的能量判据[16] Dewar芳香烃共振能 1969 Dauben进行抗磁磁化率的近代研究, 认为芳香化合物有较大的抗磁磁化率升高[17] 抗磁磁化率 1970 Flygare研究了芳香化合物磁化率的各向异性[18] 磁化率各向异性 1979 Hoffmann等预测了金属苯的芳香性[19] 金属苯 1980 Kutzelnigg通过IGLO量子化学计算了芳香化合物的磁性质:化学位移、磁化率、磁化率的各向异性等[20] 芳香化合物的磁性质 1982 Roper组报道首例金属苯的合成与表征, 并研究其芳香性[21] 金属苯 1996 Schleyer提出使用核独立化学位移(NICS)作为芳香性的判据[22] NICS Wallenborn首次将诱导电流密度的各向异性(AICD)用于芳香性领域, 并研究了富勒烯衍生物过渡态的芳香性[23] AICD 2001 贾国成组报道首例金属苯炔, 并研究了其芳香性[24] 金属苯炔 2003 Herges组首次合成了具有Möbius拓扑结构的Möbius芳香性化合物[25] 扭曲结构的Möbius芳香性 2013 夏海平组首次制备了金属杂戊搭炔, 并与朱军组合作研究了其芳香性, 发现可以通过掺杂金属实现分子骨架由反芳香到芳香的反转[26] 芳香性反转 2017 席振峰组报道首例螺芳香性化合物, 并研究其芳香性[27] 螺芳香性 1.2 芳香性的含义
“芳香性”一词自使用以来, 科学家们不断发现具备该性质的化合物的各种特征, 并进行了总结与归纳.这些特征, 一部分是规律的总结, 例如[4n+2]规则; 另一部分则是对化合物本身特征的描述, 例如平面性、键长平均化等.随着科学研究的不断推进, 其中一些观点被修正甚至推翻, 但仍有一些经典的描述被保留并沿用至今.仔细考察芳香性的历史, 我们发现“芳香性”事实上是用于描述化合物的特殊稳定性的概念.换句话说, “芳香性”是一个描述行为特征, 而非表达理论规律的术语.
因此, “芳香性”应该由此类化合物的本质属性决定, 而“芳香性”规律则是对芳香化合物普遍性质的总结.本文就“传统芳香化合物”的行为特征进行了总结(指Hückel平面芳香性), 并列于下表中(表 2).另外, 对近年发展的一些芳香性的独特类型, 本文也将结合实例逐一介绍.
表 2
传统芳香性化合物的特征
Table 2.
Features of traditional aromatics
特征 芳香化合物 反芳香化合物 非芳香化合物 平面性 环共平面趋势 环平面扭曲趋势 一般为非平面 键长交替 平均化 单双键交替明显 与一般化合物类似 稳定性 稳定化 去稳定化 中等 环外1H NMR 去屏蔽 屏蔽 中等 反应性 相对更易取代、加成 相对更易加成 都可能发生 外加磁场 产生抗磁环电流 产生顺磁环电流 产生多向电流 2 芳香性的判据
“芳香性”自从创立至今, 仍未发展出一种公认的精准定义.实验科学家们不断地通过发展新的实验手段、表征技术来探索各种实验现象并分析其原因.理论化学家们通过密度泛函理论(DFT)等理论计算方式建立并发展研究芳香性的理论手段.因此, 对于“芳香性”的判据成为了研究芳香化合物的重要内容, 而研究芳香性判据的过程则是深入理解化合物本质的过程.本文将“芳香性”的判据分为实验判据和理论判据两个部分进行归纳总结.
2.1 芳香性的实验判据
“芳香性”的实验判据目前主要有:结构判据、反应性判据、电磁性质判据这三类.这三类研究均是通过对芳香化合物本质特征来开展, 其研究的主体框架与表 2总结的内容一致, 本节就此内容做一个详细的解释与归纳.
2.1.1 结构判据
结构特征是芳香性最直观的判据, 也是最早诞生的判据之一, 被广泛用于对芳香化合物的性质描述.已有综述对芳香化合物的结构特征做了总结[28], 下面介绍几种常用的结构特征判据:
2.1.1.1 共平面性
传统芳香性化合物与非芳香体系相比具有更好的平面性, 而与反芳香体系相比则更趋向于保持环共平面结构. “反芳香性”最早被Breslow定义为结构上共平面的、能量上去稳定化的结构特征[29], 并撰写了一篇综述描述总结了反芳香性质[30].
通常情况下, 我们认为“反芳香性”会使分子的能量升高, 导致去稳定化.因此, 反芳香性的分子会通过自身拓扑结构的扭曲, 以减少共平面性, 从而降低反芳香带来的能量劣势.例如, 1, 3, 5, 7-环辛四烯分子的8个碳原子是一个非平面环的结构(Scheme 1), 实验结果证明其翻转需要越过一个13.7 kcal/mol的能垒[31].在翻转过程中, 其环上的8个碳原子会经历一个同平面过程, 形成8个π电子的反芳香体系, 热力学上相对更不稳定.因此, 芳香性化合物比起反芳香性化合物, 具有更好的共平面性趋势(表 2).其原因可以解释为:当一个环状分子的原子共平面时, 其原子轨道中用于形成π键的部分(如果是全碳体系, 是一组平行的p轨道)能更好地以“肩并肩”的形式重叠交盖, 从而使π电子离域到整个体系, 芳香化合物的离域可以稳定分子结构; 而反芳香化合物则通过环平面的扭曲以减少这种形式的交盖, 从而降低“反芳香性”带来的热力学上的不稳定性.
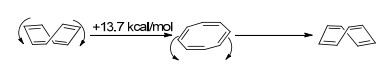 图式 1
1, 3, 5, 7-环辛四烯的构象转换
Scheme1.
Ttransformation of conformations of 1, 3, 5, 7-cyclooctatetraene
图式 1
1, 3, 5, 7-环辛四烯的构象转换
Scheme1.
Ttransformation of conformations of 1, 3, 5, 7-cyclooctatetraene
但是, 这类判据也有例外.如我们熟知的足球烯C60, 其是一个球型芳香性分子, 而它却是非平面结构.有关球芳香性的研究已经有综述报道[32].此外, 具有“Möbius芳香性”的[16]轮烯结构也是如此[25].
2.1.1.2 键长平均化趋势
芳香性化合物环内的碳碳键长因为π电子的高度离域会趋于平均化.
对于研究者, 这一判据的难点在于:键长数据通常只能通过测定化合物的晶体结构而获知, 不同于电子规则一般可以进行简便地预测.然而这一判据具有良好的置信度和普遍性, 是目前被广泛采用的实验判据.不同空间构型、不同芳香性类型的的分子, 通常都满足这一判据.值得注意的是, 平均化程度通常只能与同体系分子进行比较, 在不同体系中使用可能会出现误导.例如, 环硼氮烷[33](图 1) (borazine, 化学式为B3N3H6, 又被称为“无机苯”)为等键长环系(所有B—N键长均为1.43 Å), 而萘分子并不是等键长体系.从我们通常的理解和其他判据的角度来看, 后者应当是比前者是更芳香的.因此, 在此处使用键长交替这种判据来判断, 会导致误解[34].同体系中, 苯的六个碳碳键长均为1.39 Å, 键长完全平均化; 而反芳香化合物环丁二烯根据Dixon的计算结果来看, 其两组碳碳键长则分别为1.48和1.35 Å[35](由于首例环丁二烯分子为硝酸银配合物[36], 其单独存在时极其不稳定且容易聚合, 因此此处键长数据使用计算结果), 近乎完全的单双键键长交替状态[传统C(sp3)—C(sp3)单键为1.54 Å, C(sp2)=C(sp2)双键为1.35 Å].
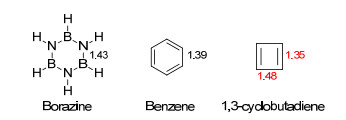 图1
一些化合物的键长数据
Figure1.
Bond length data of some compounds
图1
一些化合物的键长数据
Figure1.
Bond length data of some compounds
键长平均化趋势在芳香性实验判据中被广泛采用, 是因为不论分子结构是否共平面, π电子的离域均能进一步地稳定整个环系.而π电子离域性加强, 则分子中单双键交替的现象自然会被削弱, 从而导致环内键长趋于平均化.
2.1.2 反应性判据
通常, 芳香环由于其特殊的稳定性, 化学反应性相对其他非芳香环系要弱得多.早在1866年, Erlenmeyer[6]就已经提出了芳香性的反应性判据.在一般不饱和体系极易发生的反应, 如加成、开环反应中, 在芳香体系均较难实现.通常芳香环更容易发生取代反应(Scheme 2).
 图式 2
一般双键和芳香环双键的反应特性
Scheme2.
Reaction performances of a common C—C double and one on aromatic ring
图式 2
一般双键和芳香环双键的反应特性
Scheme2.
Reaction performances of a common C—C double and one on aromatic ring
这是由于开环、加成反应均会破坏原有体系的芳香性所提供的稳定化作用, 而取代反应只改变环上的取代基, 并没有完全影响环系原有的芳香性.针对取代反应而言, 芳香环上具有较大的不饱和度, 因此通常发生亲电加成反应.值得注意的是, 在发生取代反应时, 反应涉及的中间体也有可能导致环系原有的芳香性被暂时破坏, 因此, 芳香化合物的取代反应虽然相对于其加成、开环反应更容易, 但由于其固有的芳香性质, 往往也需要特定的反应条件.
这一判据也存在一些例外, 例如C60等富勒烯分子(fullerenes)很容易发生各种加成反应(Scheme 3), 而其取代反应却相对困难.这可能是因为其存在一定的球面张力, 导致其反应性质与常规的不饱和体系类似[37~39].
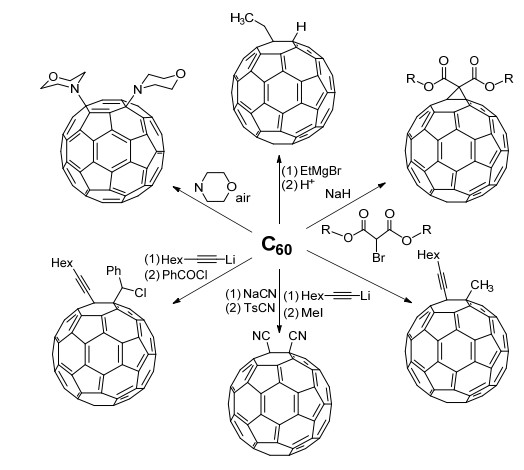 图式 3
富勒烯C60的一些加成反应
Scheme3.
Some addition reactions of C60 fullerene
图式 3
富勒烯C60的一些加成反应
Scheme3.
Some addition reactions of C60 fullerene
2.1.3 电磁性质判据
电磁性质判据来源于芳香化合物环结构与电子离域性质的结合.
以苯分子为例(图 2), 由于其良好的环内电子离域性, 在外加磁场B0下相当于一根环形的导线, 会产生分子环电流i, 从而产生感应磁场B'.根据物理学楞次定律, B'在环内具有抵抗外界磁场的方向, 在环外有和外界磁场一致的方向.因此, 这样的分子环电流i也被称为“抗磁环电流”.不难理解, 由于这样的抗磁性质, 苯环的外侧会产生去屏蔽区域, 表观磁场将增大为(B0+B'), 而内侧则是去屏蔽区域, 表观磁场将减小至(B0-B')(此处B'仅仅指代一个正值, 并非内外增减的数值完全相等).
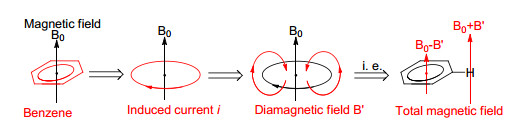 图2
苯环产生屏蔽区域的过程
Figure2.
Generation process of shielding field in benzene
图2
苯环产生屏蔽区域的过程
Figure2.
Generation process of shielding field in benzene
2.1.3.1 核磁共振化学位移(NMR Chemical Shift)
芳香化合物产生的独特抗磁环电流, 会导致周围的磁场环境发生变化.例如苯分子的6个H原子均处于抗磁环电流的去屏蔽区域(图 2), 因此其1H NMR的化学位移值会向低场移动(δ 7.3, 环己烯的sp2碳原子所连的H原子δ 5.6). Pople也通过一些理论计算, 研究了苯环不同的化学位移[13].而更显著的例子是[18]轮烯(图 3), 其含有6个向环内的H原子处在上述的屏蔽区域, 另外12个H原子则处于外侧的去屏蔽区.其环内H原子的1H NMR化学位移值明显向高场偏移(δ-3.0), 而环外H原子则明显向低场移动(δ+9.2)[40, 41].
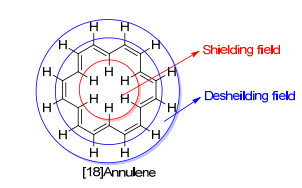 图3
[18]轮烯的屏蔽和去屏蔽区
Figure3.
Shielding and deshielding areas of [18]annulene
图3
[18]轮烯的屏蔽和去屏蔽区
Figure3.
Shielding and deshielding areas of [18]annulene
值得注意的是, 该判据需要具有环上H原子才能较好地运用, 而许多芳香分子, 如C60等就无法采用这一判据.此外, 在使用化学位移值判断化合物芳香性时, 还需要进一步排除其他结构因素或邻近基团对化学位移的影响.
2.1.3.2 偶合常数(J)
邻近H原子核之间的相互作用称为自旋耦合, Karplus理论研究表明自旋耦合主要是沿着共轭链传递的, 且偶合常数随着C—C键长的增加而减小[42].因此, 通过偶合常数的大小可以估算键长的交替程度, 可作为键长交替趋势的一种间接方法.
2.1.3.3 磁各向异性
如上所述, 由于芳香性分子的环状离域电子特征, 会在外加磁场下产生抗磁环电流, 导致其垂直于环平面方向的磁化率χz(假设分子处于xy平面)会比平面内其他方向的磁化率(如χx和χy)高[43].这可以作为平面芳香分子的磁性判据之一.类似地, 在非平面体系如C60等结构中, 这一判据的使用是很困难的.
2.1.3.4 反磁磁化率升高(Λ)
反磁磁化率升高是一种实验与理论结合的判据.反磁磁化率升高Λ定义为:实测分子磁化率χ和由参比体系计算得到的χ0之差. Pascal[7]提出, 对于不存在π电子离域的分子, 其磁化率具有加和性, 因此简单加和得到的χ0与实际测得的χ基本一致; 而对于芳香性分子, 因其具有环电流, 因此实测的绝对值χ通常会比χ0更大. Dauben等[17]分析了一些列化合物后认为, 芳香性化合物确实具有较大的Λ.而相反的, 反芳香化合物的顺磁磁化率会升高.基于这一判据, 很多文献也将Λ作为芳香性判据, 认为具有较大的负值Λ的为芳香性分子, 而具有较大的正值Λ的为反芳香性分子.
Schleyer等[22]认为, 反磁磁化率升高是唯一和芳香性相关联的判据, 并基于这个概念提出了“核独立化学位移”判据, 将在本文后续小节中介绍.
2.2 芳香性的理论判据
随着计算机科学的发展, 计算机和化学的学科交叉给化学研究带来了新的便利和促进.当一个新化合物结构通过核磁共振(NMR)、单晶X射线衍射(SXRD)、质谱(MS)等实验手段进行系统表征后, 可以通过建立结构模型, 使用计算机模拟并研究其相关性质.经过近几十年的飞速发展, 理论化学的可信度不断提高, 已能较为准确地用于体系不太大的分子结构及电子结构的研究.同样, 芳香性化合物的理论研究也可以借助计算化学来推进.本节对传统“芳香性”的判断方式进行小结, 并对近年发展的“芳香性”的计算化学判据也进行归纳与总结.
2.2.1 π体系电子数: Hückel规则与分子轨道图形
Hückel规则([4n+2]规则)认为:在sp2杂化碳原子平面单环体系中, 含有(4n+2)个π电子的化合物具有芳香性, 含有4n个π电子的化合物具有反芳香性, 其他电子数为非芳香性[9].受限于当时化学理论和计算工具的水平, 无法通过复杂的计算以获得接近物理事实的规律.因此在当时, 其提出的规则作为一种可以仅通过实验获得的结构进行简单π电子数计算, 而不进行理论模拟就可以使用的方法, 获得了广泛的认可.下面就这一经典规则, 举例简单说明:苯分子、环戊二烯负离子和环庚三烯正离子均为6π电子体系, 属于芳香结构; 而环丁二烯分子为4π电子体系, 属于反芳香结构; 环戊二烯等分子则属于非芳香结构(非共平面)[9, 29].该规则起初只针对有机全碳单环体系如苯, 简单的杂原子体系如呋喃、吡啶、噻吩.后来拓展到了周期数更大的杂原子体系(如磷、硅)、多稠环体系(如芘)、金属杂环体系(如锗、锡)等, 并且计算结果与实验结果吻合得很好[44~47].
然而, 这一规则仍有特例.例如当分子的拓扑结构发生扭曲或含有d轨道的原子的引入时, 会产生相反的情况.在此基础上产生了被称为“Möbius芳香性”的4n电子规则.与Hückel规则相反, 在“Möbius芳香性”规则中, 含有4n个π电子的化合物才具有芳香性. Rzepa[48]对这一“Möbius芳香性”进行了综述总结.
对于简单的单环体系(如苯、环丁二烯等), 我们可以利用这一经典规则, 迅速地算出π电子数, 判断该分子为芳香的或者反芳香的.然而对于双环或多环的复杂体系、或分子中含有非碳原子时, 判断其具体电子结构则相对困难得多.因此, 近年来, 借助分子轨道理论, 通过计算研究其分子轨道成为判断芳香性的一种新方式.这种判断方法也属于结构判据的范畴.分子轨道(MO)由原子轨道组合而成, 表现分子整体性质.和原子轨道一样, 每个分子轨道上可填充1对电子, 能量由低到高排列[49].
当体系中全充满的π分子轨道为(2n+1)个时, π电子数即为(4n+2)个, 则判断该化合物为芳香性化合物.使用现代可视化的分子轨道方法, 除了可以计算π电子数, 还可以分析体系的离域性质.以萘环为例(图 4), 通过分子轨道理论的理解以及直观图像的观察, 可知萘分子的π体系为一个整体, 应当按照整体计算, 具有5个π-MOs, 佐证其为10电子体系.该结构中所有分子轨道的节点数目均为偶数, 具有典型的Hückel芳香性.
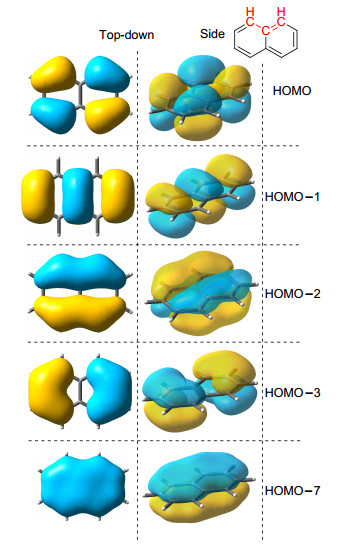 图4
萘的π分子轨道图像(占据轨道)
Figure4.
π-Orbitals of a naphthalene (occupied)
图4
萘的π分子轨道图像(占据轨道)
Figure4.
π-Orbitals of a naphthalene (occupied)
这一方法还可以用于研究复杂的结构, 甚至是非平面的C6010+(图 5).研究其10+价态的π-MOs可以发现, 其为一个球共轭结构, 分子轨道显示是C60分子整体体现的共轭(25个占据轨道, 50个共轭电子), 并非各个苯环碎片的单独作用[54].但是, 由于每个分子轨道在一些体系的贡献度无法加以确定, 例如金属杂单环平面体系, 其芳香性虽然已被广泛认可, 但是对于具体结构的π电子数仍然存在大量的争议[19, 55, 56]. 2001年, 贾国成课题组[24]报道了首例金属苯炔化合物, 其与金属苯具有类似的芳香性.
 图5
计算得到的C6010+离子的π分子轨道
Figure5.
The calculated π-molecular orbitals of C6010+
图5
计算得到的C6010+离子的π分子轨道
Figure5.
The calculated π-molecular orbitals of C6010+
2.2.2 芳香稳定化能
芳香稳定化能(ASE)是一种基于能量的理论判据, 属于异构稳定化能(Isomerization stabilization energy, ISE)的计算方法.最早的能量判据是进行共振能和各种类型稳定化能的计算[57, 58]. ASE从构筑反应的原理上来说, 是与之一致的.该方法可以概括表述为:设计一个反应, 使得反应式左右两侧的化合物尽量满足唯一差别仅为“芳香性因素的存在与否”, 而其分子组成的原子及化学键的数量和种类保持一致.这一方法实际上为等键反应(Isodesmic reaction)[59~61], 因为考虑的能量差别因素仅为芳香性, 所以被称为“芳香稳定化能”.由于ASE方法的核心仅是构筑一个释放芳香性带来的特殊稳定化能的反应, 因此在这一原则不变的情况下, 结构的改变具有多样性.这一判据近年来发展出多种较为常用的模型, 下面介绍其中主要的两种.
2.2.2.1 甲基-亚甲基法
这种方法是Schleyer等[62]在2002年建议使用的, 其相当于单分子的等键反应.与此前的等键反应的唯一区别是, 不需要引入一个新的分子, 而是直接在考察的体系上引入一个甲基取代基或借助体系中已有的甲基取代基(Scheme 4), 然后进行“H-迁移”的方式将这个甲基转变成亚甲基, 从而使得原有芳环内的一个sp2碳原子转变为一个sp3碳原子, 从而破坏了共轭, 变为非芳香结构.这一计算方法获得的能量差值就是仅考量芳香性变化的异构稳定化能(ISE)——芳香稳定化能(ASE). ASE可以采用芳香性破坏前与破坏后的能量差表示, 也可采用芳香性破坏后与破坏前的能量差表示.本文中所提及的ASE均使用考量体系的结构向非芳香参考系转变的能量, 故本文中涉及的芳香结构的ASE为正值, 反芳香结构的ASE为负值.
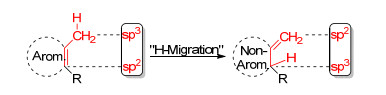 图式 4
使用甲基-亚甲基法研究ASE
Scheme4.
Studying the ASE by methyl-methylene method
图式 4
使用甲基-亚甲基法研究ASE
Scheme4.
Studying the ASE by methyl-methylene method
这种方法在加入甲基时有一定任意性, 并且H原子的“迁移”也因为芳香性化合物的共轭可以使sp3碳原子传导到环内多个位置.例如, Schleyer等[62]计算了吡啶的ASE (Scheme 5), 其建立甲基-亚甲基法模型时有多种情况, 一一列举并进行校正计算后, 发现去芳构化的结构比原本芳香结构的能量升高, 说明了芳香性提供了稳定化能.
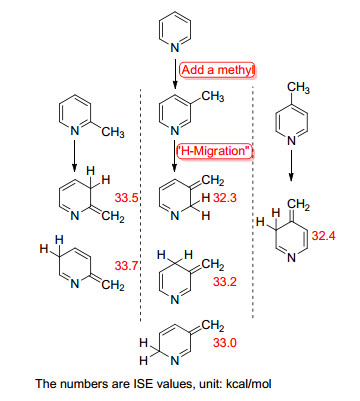 图式 5
使用甲基-亚甲基法研究吡啶的ASE
Scheme5.
Studying the ASE of pyridine by methyl-methylene method
图式 5
使用甲基-亚甲基法研究吡啶的ASE
Scheme5.
Studying the ASE of pyridine by methyl-methylene method
2.2.2.2 茚-异茚法
由于甲基-亚甲基法研究ASE反应时并未充分考虑环的张力和扭曲, 并且等键反应设置的任意性较大, 在2003年Schleyer在研究[4n]轮烯时又提出使用了茚-异茚法(Indene-isoindene isomerization method)构筑ASE反应[63].这种方法可以看做是甲基-亚甲基法的延伸, 其原理(Scheme 6)是考虑到茚环有一个饱和碳原子, 可以被当做上述“H-迁移”的对象.
 图式 6
使用茚-异茚法构筑ASE
Scheme6.
Building the ASE by indene-isoindene method
图式 6
使用茚-异茚法构筑ASE
Scheme6.
Building the ASE by indene-isoindene method
该方法的优点是sp3碳原子碳原子从五元环的一个碳上转移到了另一碳上, 减小环张力变化的因素, 并且在不改变主环碳原子种类的同时破坏了主环的共轭性. Schleyer的计算结果表明, 茚分子和异茚分子的能量差为29.0 kcal/mol, 说明这一改变确实破坏了芳香分子特殊的稳定性.
文中以[4n]轮烯为研究对象(Scheme 7), 也研究了该体系的芳香稳定化能.可以看出, 具有4n个π电子的轮烯结构, 除了环丁二烯是反芳香的, 其他分子都是接近非芳香性的.由于环系足够大, 这些分子可以通过结构的扭曲来破坏共平面性, 从而减少反芳香性导致的去稳定化作用[63].
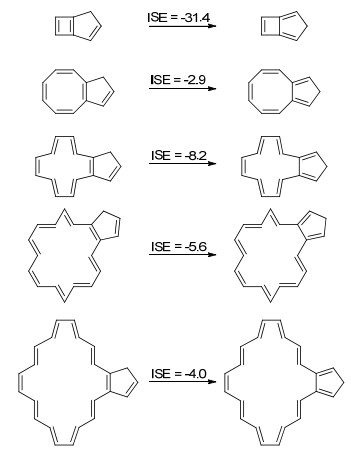 图式 7
使用茚-异茚法研究[4n]轮烯的ASE
Scheme7.
Studying the ASE of [4n]annulenes by indene-iso-indene method
图式 7
使用茚-异茚法研究[4n]轮烯的ASE
Scheme7.
Studying the ASE of [4n]annulenes by indene-iso-indene method
能量的变化是最接近芳香性本质的判据.除了上述两种计算方法外, 还有使用多组自由基的更加复杂的模型[64].然而值得注意的是, 单纯由于芳香性提供的稳定化能, 是很难独立计算的.在建立“ASE”计算模型时, 总是不可避免地引入角张力、极性、位阻等因素, 而在进行大体系计算时则会产生更多的问题.因此, 构筑ASE反应时, 最好使用合理的参照结构, 并尽量使用多个ASE计算体系进行对照, 从而提高计算数据的可信度.而且, 结合其他判据共同使用, 进行多种判据互相印证, 可以提高判断的准确性.
2.2.3 核独立化学位移
前述的可信度较高的电磁判据磁化率的变化Λ, 被广泛认为是比较接近“芳香性”本质的判断方法.但是, 由于该方法需要一个非芳香体系(即理论线性加和值)作为参比, 在实际操作上较为困难.并且, 有研究表明Λ还与环的尺寸大小有关系[17c, 22], 这导致了磁化率的变化Λ无法有一个相对稳定的衡量标准, 更适用于定性判断.而测定1H NMR的化学位移也被认为是一种较好的办法, 但只能针对含环外H的环状体系.而且由于影响化学位移的因素较多, 导致不同的芳香与非芳香体系化学位移有时差距不大, 因此也存在一定的局限性.
Schleyer等[22]在1996年提出了一种全新的理论判据“核独立化学位移”(Nucleus independent chemical shifts, 简称NICS). Schleyer等建议采用量子化学的方法计算出一个分子的绝对屏蔽值.该屏蔽值即化学位移的体现, 不同于实验测得的1H NMR、13C NMR等的化学位移, 它不依赖于核, 而是表征一个位置的屏蔽效果, 故命名为“核独立”.但就本质而言, 它仍然是对环电流所产生效果的表征, 属于电磁判据.
NICS通常是针对一个体系的几何中心位置(通常是环系, 即是环中心)进行计算.以单环体系为例, 判断过程通常如下:找到环的几何中心位置, 计算该位置的NICS(0)值, 如为负值, 则认为该环具有芳香性, 且通常认为负值越大, 芳香性越大; 反之则为反芳香.如果NICS值接近于0, 则认为该环为非芳香环. NICS(0)中的“0”代表距离环几何中心0埃的位置(也就是几何中心本身).
Schleyer等[22]还对NICS(0)和ASE的大小关系进行比较, 发现具有较好的线性关系.如图 6所示, 环戊二烯中性分子的NICS(0)接近于零, 而环戊二烯负离子则具有较负的NICS(0)(其值为-14.3).按NICS(0)值判断, 吡咯、噻吩、呋喃等都是芳香性化合物, 而硼杂环戊二烯、铝杂环戊二烯、硅杂环戊二烯正离子等都是反芳香化合物, 这与经典的归类方式相符.
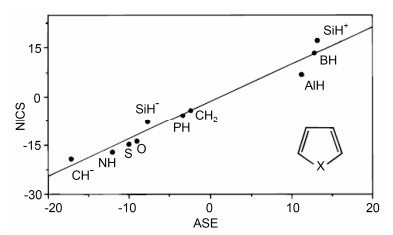 图6
一系列五元环化合物(C4H4X)的NICS(0)和ASE的对照
Figure6.
NICS(0) and ASE values of a series of five-membered ring heterocycles C4H4X
图6
一系列五元环化合物(C4H4X)的NICS(0)和ASE的对照
Figure6.
NICS(0) and ASE values of a series of five-membered ring heterocycles C4H4X
Schleyer等[22]在此方法中将这个绝对屏蔽值取反, 将NICS值为负的判定为芳香化合物, 是为了和上面提及的磁化率变化Λ取同号(磁化率变化Λ是负值体现芳香性, 正值体现反芳香性). Schleyer等[22]也在文中提到, 与Λ相比, NICS的优势在于其对环大小的依赖性较小.
Schleyer等[65]后续对NICS规则进行了仔细研究并指出部分体系的适用性较差, 认为其主要原因是该值为各项同性的值而非是垂直于环平面的分量.他们进一步研究认为, 由于π电子主要分布在环的上下平面而非环自身的平面, 且理论计算表明π电子分布较好的位置大约在距离环平面上下1 Å的位置.因此, 他们认为在环中心平面上下1 Å位置的NICS值相对更能反映芳香性大小, 定义为NICS(1).并且, 由于NICS作为一个张量且具有各向异性, 其zz方向(垂直于环平面的方向)的值具有较好的代表性, 即NICS(1)zz.考虑到多数类似的有机小分子的芳香性都来源于π体系的贡献, 将其π轨道分量提取出来可以算得NICS(1)πzz, 可更加准确地反映体系芳香性的强弱.由于拆分出π轨道进行计算的过程过于冗杂, 且NICS(1)zz具有与ASE较为对应的关系, 因此他们推荐使用NICS(1)zz判据作为一种简便的工具[65].
目前使用NICS值作为芳香性的理论判据是广泛认可的方法之一.但是仅采用NICS值并不一定能够准确地反映所有体系的芳香性程度[66], 还需要结合多个判据进行综合考量.
2.2.4 各向磁感应电流密度
前述“磁化率各向异性”的判据, 是通过电磁作用对芳香性反映程度的描述.然而迄今仍无通用的方法描述电子离域程度的大小, 也无可靠方法用于区分定域电子和离域电子.磁感应电流密度(ICD)与磁化率类似, 由于感应电流对分子平面及分子的垂直方向的作用不同, 同样具有各向异性(Anisotropy).因此, 各向磁感应电流密度(Anisotropy of the induced current density, AICD)可用于量化描述一个体系的离域程度, 并可转化为可视化图像, 方便比较. Wallenborn等[23]首次使用它并研究了富勒烯衍生物的芳香性. AICD的相关综述已经介绍的很详细[67, 68], 本文在此仅做简要介绍.
如图 7所示, AICD的使用原理为:将考察体系的平面置于纸面, 施加一个垂直纸面向外的磁场, 环系会受诱导产生环电流, 如右图中的氢原子或苯分子.当该体系具有芳香性时, 将会产生抗磁环电流, 根据左手定则, 产生的是顺时针环电流, 右图的苯分子就是这种情况; 反之, 如体系具有反芳香性时, 则会产生逆时针环电流; 如果体系为非芳香性, 则产生表观上各项同性的电流, 即微观上各个方向均存在电流[68].
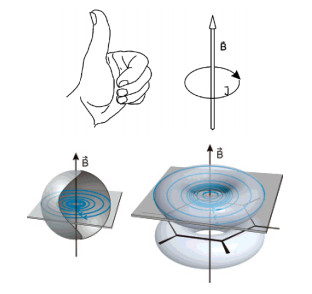 图7
“左手定则”(上)及氢原子的自身感应电流密度和苯环的离域感应电流密度(下)
Figure7.
"Left-hand rule" (up); a local current density of hydrogen atom and a delocalized one of benzene (down)
图7
“左手定则”(上)及氢原子的自身感应电流密度和苯环的离域感应电流密度(下)
Figure7.
"Left-hand rule" (up); a local current density of hydrogen atom and a delocalized one of benzene (down)
以芳香模型分子苯为例[68, 69], 总的AICD图(图 8)显示环电流箭头主要为顺时针方向, 这说明苯是具有抗磁环电流的.根据上面的推断, 具有抗磁环电流的为芳香性分子, 与事实情况相符.
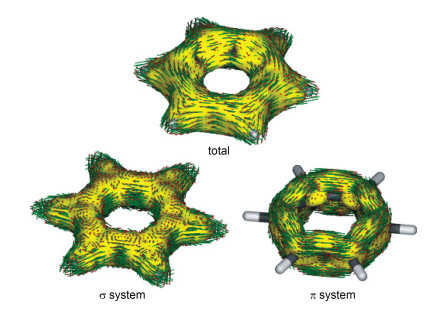 图8
苯环的AICD图像:总贡献、σ贡献及π贡献
Figure8.
AICD isosurfaces of benzene: total, σ, and π contributions.
图8
苯环的AICD图像:总贡献、σ贡献及π贡献
Figure8.
AICD isosurfaces of benzene: total, σ, and π contributions.
如果将AICD结合分子轨道情况进一步深入研究其芳香性, 则可以判别其芳香性的来源.可以将苯环的π轨道和σ轨道分开, 并分别考量其AICD图像, 可以发现: σ轨道的环上感应电流指向各个方向, 杂乱无章; 而π轨道却非常有序地以顺时针方向排列.这表明苯环的芳香性, 主要是由π轨道贡献的.因此, AICD作为一种全分子的可视化判据, 可以获知更多有关于芳香性的量化信息, 因而相对于无法可视化的、无法确定合适位点的NICS值, 更为清晰.
2.2.5 其他理论判据
除了上述常用的几种理论判据以外, 理论化学家还创造了很多可以佐证芳香性的方法和模型, 在综合考量芳香性时也曾被多次采用.下面简单介绍一些其他较为常用的、适用性较好的理论判据.
2.2.5.1 芳香性谐振模型
芳香性谐振模型(HOMA)是一种基于结构的芳香性判据, 沿用历史悠久[70].其表达式为
表达式中各符号含义为: N为成环原子数, i表示环上的第i个原子, j是它的下一个原子, R表示键长. α和RRef为和键的类型有关的常数, RRef为理想芳香性体系的平衡键长.换句话说当RRef和Ri, j相等, 那么HOMA为1, 也就是所有键长都等于理想芳香体系的键长.很容易想到, 苯环就是所有键长完全均一化的体系, 其HOMA即为1.另外, 环戊二烯负离子的HOMA值为0.764, 环庚三烯正离子为0.966等[71].
2.2.5.2 电子定域化函数
电子定域化函数(ELF)是Becke等[72]在1990年提出的一种衡量电子定域性的实空间函数, 其后也有文献总结了这一定域函数的用法[73], 用途比较广泛.这种方法的原理比较复杂, 在此仅简要其在芳香化学领域的应用[74].
在MO理论中, 分子的电子云是分散在体系, 其电子离域性越好, 电子分散程度就越大.类似于山川的等高线, 如在三维环境中对电子云密度选取等值面, 选取的值不同, 则面的囊括范围也不同. ELF就是基于此原理发展的判据方法.以苯为例, 当ELF等值面的值(isovalue)越大, 其等值面内的空间就越小(图 9)[74], 随着值的增大, 各个碳原子之间的离域空间就会被断开, 变为每个碳相对独立的电子云区域.处于刚好断开的临界值被称为ELF值, 如苯的ELF为0.91, 萘为0.78(图 9).对于类似体系, ELF值越大, 反映电子在改ELF值区域内离域程度越好.
 图9
苯(上)和萘(下)的等值面(0.3, 0.7和ELF点)
Figure9.
Isovalues of benzene (top) and naphthalene (bottom) (0.3, 0.7 and ELF points)
图9
苯(上)和萘(下)的等值面(0.3, 0.7和ELF点)
Figure9.
Isovalues of benzene (top) and naphthalene (bottom) (0.3, 0.7 and ELF points)
除了上述这些判据以外, 文献中还有许多其他的判据, 如适应性自然密度划分(AdNDP, Adaptive natural density partitioning)[75], 芳环化学屏蔽(ARCS, Aromatic ring chemical shieldings)[76], Bird芳香性指数[77, 78], 谐振稳定化能(HOSE, Harmonic oscillator stabilization energy)[79], 磁共振能(MRE, Magnetic resonance energies)[80, 81], 多中心键级[82, 83]等, 在此不做叙述.需要强调的是, 由于各种判据自身的局限性, 一般不能仅通过一种判据判断某个未知体系的芳香性, 而要综合多种方法来考察.
3 芳香化合物的分类
芳香化合物根据不同的分类标准, 可以有多种芳香性分类方式.例如(1)根据原子种类, 可以分为全碳体系的芳香性、杂原子参与的芳香性、金属参与的芳香性等; (2)根据分子结构, 可以分为平面芳香性、Möbius带扭曲型芳香性、球芳香性等; (3)根据现代的分子轨道节点(面)数和电子数, 又可以分为经典Hückel-π芳香性、Möbius-π芳香性、σ芳香性等.本文以第三种分类方式, 结合前两种的内容, 对芳香性化合物及相对应的非芳香、反芳香化合物进行举例介绍, 并简述部分“芳香性”判据在这些典型体系的相关应用.
3.1 Hückel-π芳香性
Hückel芳香性是指Hückel最早提出的可依据(4n+2)规则判断的芳香性.如上所述, 该类芳香化合物通常应具有共平面、单环、(4n+2)个共轭π电子等特征[9].后来经过发展, 将(4n+2)规则进行了进一步拓展, 下面就这些体系进行介绍.
3.1.1 经典全碳体系
经典全碳体系是最早被研究的芳香体系, 具有共平面、单双键交替、π电子完全参与离域与共轭等典型特征.从Faraday分离得到化合物苯开始[1], 这项研究就已经拉开了序幕.
3.1.1.1 苯及苯的同系物
苯及苯的同系物(这里指广义的同系物)属于最经典的全碳芳香化合物.结合晶体数据的键长平均化情况来简要说明(图 10)[84~88], 单晶X射线衍射法表明其共平面性, 并且其键长不属于一般定域的单、双键, 而是相对平均化.在ASE的甲基-亚甲基法下, 苯环的ASE=33.2 kcal/mol, 萘为52 kcal/mol左右(多种构筑方式), 均显示其具有较高的芳香稳定化能[62].
 图10
苯及其同系物的键长(单位: Å)
Figure10.
Bond lengths of benzene and its homologues (Unit: Å)
图10
苯及其同系物的键长(单位: Å)
Figure10.
Bond lengths of benzene and its homologues (Unit: Å)
此外, Schleyer等[22]计算了部分苯及其同系物的NICS(0), 大部分为较大的负值(图 11), 为芳香性较强的化合物.除NICS以外, 上述判据中的AICD图像也可以很好地反应苯环的这一特点, 这里不再重复.
 图11
苯及其同系物的NICS(0)(单位: ppm)
Figure11.
NICS(0) values of benzene and its homologues (Unit: ppm)
图11
苯及其同系物的NICS(0)(单位: ppm)
Figure11.
NICS(0) values of benzene and its homologues (Unit: ppm)
3.1.1.2 轮烯
轮烯, 又称为[n]轮烯([n]annulene, 图 12), 最初由Sondheimer命名[89], 是指单双键交替的全碳单环分子(实际上除了图示中[10]轮烯、[12]轮烯可能有异构体, 其他更大的环系也可能存在多种异构体, 这里不再列出).值得注意的是, 大于8元环后的每个经典的全顺(All-cis)结构, 一般都会受到碳键角的张力约束而发生异构.
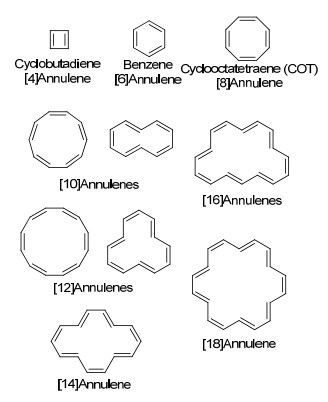 图12
部分轮烯的结构
Figure12.
Part of structures of annulenes
图12
部分轮烯的结构
Figure12.
Part of structures of annulenes
因为完全单双键交替的体系通常拥有偶数个电子, 符合Hückel规则中芳香或者反芳香体系的电子数情况(4n+2或4n).但结构的扭曲与否, 往往会产生非芳香体系的情况. Schleyer对于4n电子数轮烯的ASE情况研究, 反映了此现象, 上述ASE判据中已进行叙述[63]. Haley等[90]在2006年曾经撰写过一篇相关综述总结轮烯体系的相关研究.同年, Herges[91]关于设计拓扑Möbius带结构分子的综述中也有提及.
环丁二烯和苯可视为最简单的两种轮烯, 即[4]轮烯和[6]轮烯. [4]轮烯因为4电子平面的构型, 是经典的反芳香化合物, 其单独存在时非常不稳定; 苯环则是作为芳香化合物的母体分子.
[8]轮烯即环辛四烯, 早在1911年就被分离获得[92].如果其为平面构型, 按照[4n+2]规则, 是具有8电子的反芳香结构.因此, 该分子倾向于通过结构扭转降低反芳香性引发的去稳定化作用.实验数据表明环辛四烯分子是碗状结构, 并且通过构象翻转能实现越过一个平面型反芳香结构的过渡态的能垒[31].
[10]轮烯有多种因为双键顺反而导致的异构体, 于1969年被Masamune等[93]首次合成, 并且在同一篇文章中报道了两种异构体的光照转换, 均在加热条件下形成两个六元环以获得较好的键角(Scheme 7).
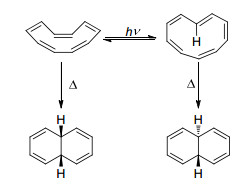 图式 7
[10]轮烯及其异构化
Scheme7a.
[10]Annulenes and their isomerization
图式 7
[10]轮烯及其异构化
Scheme7a.
[10]Annulenes and their isomerization
令人感到惊讶的是, 按照休克尔芳香性理论, [10]轮烯如果为平面结构, 是10电子的芳香体系, 但它实际却是扭曲的分子结构.人们将其解释为, 当按照全顺(All-cis)构型存在时, 每个键角为144°, 张力过大; 而当形成图 12所示的第二种结构时, 向内的两个H原子因为位阻会导致环平面的扭曲.这表明[10]轮烯的芳构化释放的能量, 不足以克服其键角张力和原子排斥力.值得注意的是, 理论计算表明当其中部分双键被替换成三键, 因为张力的释放, 该含三键分子应当是具有芳香性的[94].
[12]轮烯的首次分离在1970年由Oth等[95]报道.低温核磁表明其明显扭曲的分子结构, 被归因于图 12中[12]轮烯右侧结构内部相互排斥的三个氢原子, 但其因为异构体众多, 也有不少研究其中最稳定构型的计算化学论文[96, 97].如同[10]轮烯一样, [12]轮烯也会发生环的异构, 形成八并六元环结构, 这里不再细述. Castro和Karney等[98]的计算工作进一步地表明, Oth等给出的异构过程(Scheme 8)实际上经历了一个拓扑结构为Möbius带的过渡态. Möbius带拓扑结构是数学上的一种几何概念, 可以表述为将一张纸条的两端正反相接, 有关Mobius带的背景故事可以参看Herges等[91]的综述, 关于Mobius芳香性可以参见下文.
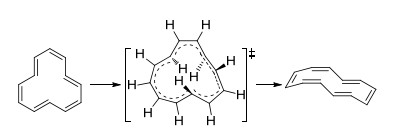 图式 8
[12]轮烯的异构
Scheme8.
Isomerization of [12]annulene
图式 8
[12]轮烯的异构
Scheme8.
Isomerization of [12]annulene
[14]轮烯由Sondheimer和Gaoni在1960年第一次报道合成[99].尽管晶体数据表明其结构为非平面, 但是许多理论和实验研究都表明其为芳香分子, 具体研究可以参看这一系列文献[100].其非平面性被归因于环内氢原子的相互排斥作用(图 13).研究发现在[14]轮烯的互变异构中最具贡献度的是图中两种具有四个反式双键的构型, 并且还有一种三个反式双键的构型也有贡献(8%).研究者也通过控温核磁研究了其衍生物的异构情况, 发现确实是四个反式双键的构型为主要产物(屏蔽区域的氢信号及去屏蔽区域的氢信号为4:10).
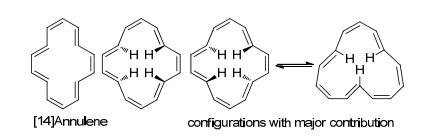 图13
[14]轮烯及其主要构型
Figure13.
[14]Annulene and its main configurations
图13
[14]轮烯及其主要构型
Figure13.
[14]Annulene and its main configurations
[16]轮烯在1961年的首次合成被报道, 仍旧归功于Sondheimer和Gaoni[101].此后更多学者进行了计算、X射线衍射等方面的研究[102]. Oth等[100]研究表明, 该16π电子结构的主要贡献是通过高度扭曲以克服其反芳香性(图 14).并且, 其通过温度调节发现, 当温度降低时, 分子更趋向于平面结构, 而且该结构的反芳香性更强.而值得注意的是, Schleyer等[63]的ASE计算结果表明, [16]轮烯的不稳定性很小(Scheme 7).
 图14
[16]轮烯及其主要构型
Figure14.
[16]Annulene and its main configurations
图14
[16]轮烯及其主要构型
Figure14.
[16]Annulene and its main configurations
近年来, 有关于计算的一篇文献预测了几种[16]轮烯可能存在的构型, 有一些具有Möbius拓扑结构[103].此外, 2003年Nature上报道的Möbius芳香性[16]轮烯的合成将在下述Möbius芳香性小节中提及[104].
[18]轮烯是准平面的芳香性结构(图 15), 其在1962年被Sondheimer及其合作者[105]报道合成. Longuet-Higgins和Salem预测随着环的增大, (4n+2)体系轮烯的芳构化提供的稳定性随之下降, 因而单双键交替会变得更为明显[106].对于大环轮烯结构及其芳香性的理论研究从[10]轮烯到[30]轮烯均有报道[93, 107~109].然而, [18]轮烯不同于上述(4n+2)类型轮烯的特点, 其环的大小使得内部的H原子的相互排斥较弱, 从而使得分子共平面的结构相对其他结构更为稳定, 这与Mislow对10碳以上的大环轮烯的“最小平面轮烯为[30]轮烯”的猜测相违背[105, 108].晶体结构和低温核磁均表明, [18]轮烯确实是近似共平面构型, 环中的H原子之间排斥较小, 只有微弱的上下翘起[110, 111].其他轮烯及其衍生物的相关研究可以参看综述报道[90].
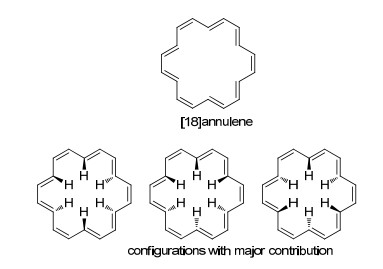 图15
[18]轮烯及其主要构型
Figure15.
[18]Annulene and its main configurations
图15
[18]轮烯及其主要构型
Figure15.
[18]Annulene and its main configurations
3.1.1.3 含奇数原子碳环的正负离子
除了中性结构, 平面环还可以通过得失π电子来满足芳香性规则, 以获得更好的稳定性.含奇数个碳原子的环系可以通过特殊的方法如得失电子使得其中所有碳原子成为sp2杂化, 从而实现与环上碳原子的轨道交盖.如环戊二烯负离子和环庚三烯正离子(图 16), 经过结构表征证明其确实是平面、键长均一化构型[112, 113], 而相应的中性化合物都是非芳香体系.这两个体系均是通过得失电子使得体系成为6电子的芳香结构.
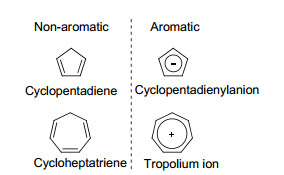 图16
环戊二烯和环庚三烯及其对应芳香离子
Figure16.
Cyclopentadiene and cycloheptatriene and their corresponding aromatic ions
图16
环戊二烯和环庚三烯及其对应芳香离子
Figure16.
Cyclopentadiene and cycloheptatriene and their corresponding aromatic ions
3.1.1.4 并环结构
除了苯的同系物, 还可以通过各种含不同碳原子数的环构成并环结构.通常认为, 这些结构为了使得整体能量最低, 往往会平衡各个环的电子数以获得芳香性.例如上述环戊二烯负离子和环庚三烯正离子的五并七元环结构薁(azulene, 也叫甘菊环)(图 17)[114, 115].体系总电荷数为0, 通过自身电子分散, 使得五元环成为带负电荷的环戊二烯负离子结构, 而七元环成为带正电荷的环庚三烯正离子结构, 导致两个环都为芳香结构, 比原本两个非芳香的中性结构更为稳定.其晶体结构、核磁共振和理论计算均表明了这一点.除了五并七的薁以外, 还有其他类似的体系, 此处不再逐一列举.
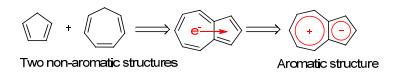 图17
薁通过自身内部的芳构化实现稳定化
Figure17.
Azulene stabilized itself by intramolecular aromatization
图17
薁通过自身内部的芳构化实现稳定化
Figure17.
Azulene stabilized itself by intramolecular aromatization
3.1.2 含杂原子芳香体系
含杂原子芳香体系(Heteroaromatics)的研究始于呋喃、吡咯、吡啶等稳定、简单的芳香化合物[9], 其芳香性源自于氧、氮原子提供π电子数不唯一的特异性(图 18上).这里采用简单体系为例.例如, 呋喃为五中心体系, 但由于氧原子可以提供一对孤对电子(lone pair)参与离域, 使得π电子数为6电子, 满足Hückel规则.吡咯也是如此.然而, 当氮杂环从五元环扩大为六元环时, 氮虽仍采取sp2杂化, 但其孤对电子存在于平面的sp2轨道上, 而非垂直于平面的p轨道上, 因此其仍然是6电子结构.关于杂环的Hückel芳香性研究很多, 目前相关研究已拓展至含各种杂原子(包括金属)、含多个杂原子甚至是全杂原子环系的研究[116~118].
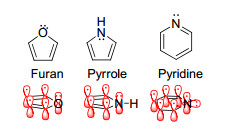 图18
一些简单的芳香性杂环化合物(上)以及π轨道示意图(下)
Figure18.
Some simple sturcuture of heteroaromatics (top) and π-orbitals (bottom)
图18
一些简单的芳香性杂环化合物(上)以及π轨道示意图(下)
Figure18.
Some simple sturcuture of heteroaromatics (top) and π-orbitals (bottom)
3.1.3 同芳香性体系
同芳香性(Homoaromaticity)是Winstein[119]在1959年提出的概念.与一般芳香性体系不同, 同芳香性体系并非是由全不饱和原子组成, 而是在全sp2体系的环中引入了一个或多个饱和碳原子.饱和碳原子左右连接的不饱和原子通过“跨越”它的方式参与环的电子离域, 使得环的离域不受饱和碳原子的影响, 使饱和碳原子如同“不存在”于环系中一般, 因此称为“同芳香性”.早在1956年, Doering等[120]就已提出了“假芳香性”(pseudo-aromaticity)一词用于描述环庚三烯衍生物(Buchner Acid)的同芳香性. Doering给出了图 19所示的异构原理, 表明环庚三烯衍生物倾向于形成离域结构的6电子芳香体系, 从而降低自身的能量.而忽略饱和碳原子的结构中6个sp2碳确实具有共平面性, 印证了其同芳香性的存在.此外, 在ASE、反应性等相应的研究也充分验证其芳香性, 可以参看2001年Williams[121]撰写的综述.
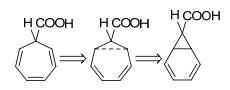 图19
环庚三烯衍生物(毕希纳酸)的同芳香性与异构化示意图
Figure19.
Homoaromaticity of a derivative of cyclopentatriene (buchner acid) and its isomerization
图19
环庚三烯衍生物(毕希纳酸)的同芳香性与异构化示意图
Figure19.
Homoaromaticity of a derivative of cyclopentatriene (buchner acid) and its isomerization
近期, 北京大学席振峰-张文雄团队[122]合成的氮杂半瞬烯(2, 6-Diazasemibullvalene, NSBV), 是同芳香性研究领域的重大突破.
3.1.4 富勒烯等球面的芳香性——球芳香性体系
类似于C60等一系列的富勒烯(Fullerenes)看似不稳定, 事实上却具有特殊的稳定性来稳定其弯曲的sp2碳原子面结构.这种特殊稳定性质即球芳香性.上述MO方法中提及的富勒烯C6010+离子, 就是典型的球芳香性结构[54]. Chen等[123]在2005年撰写了球芳香性相关综述对富勒烯、硼烷多面体(Polyhedral boranes)结构等进行了总结.
3.2 Möbius-π芳香性
Möbius芳香性是Craig在1958年首先提出(当时并未使用Möbius一词), 表述为“当一个共轭环系中掺杂入d轨道后, 会因为d轨道本身产生的相位翻转(图 20), 使得分子轨道整体全同相位的芳香性转变为有一个节点的芳香性”.这种利用原子d轨道相位翻转的Möbius芳香性, 现在被称为Craig模型(Craig Type).这个模型被用于描述一种轨道有奇数个节点、平面的Möbius芳香性的化合物模型[14].
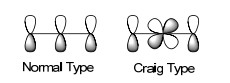 图20
d轨道参与共轭的Craig模型
Figure20.
Craig type conjugation with a d-orbital included
图20
d轨道参与共轭的Craig模型
Figure20.
Craig type conjugation with a d-orbital included
1964年, Heilbronner[15]在一篇关于轮烯的文章中提出, 当一个环具有Möbius拓扑结构时(图 21), 含有4n个π电子的化合物会具有芳香性, 即Möbius芳香性.这种芳香性的规则和Hückel的[4n+2]规则相反, 被认为是具有Möbius拓扑结构的Möbius芳香性概念的提出.事实上, 这更符合目前所用的“Möbius”一词的原意.
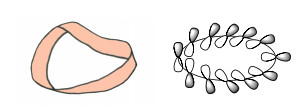 图21
Möbius带和Möbius拓扑结构的芳香性分子轨道示意图
Figure21.
Möbius strip and the MO sketch of aromaticity with Möbius-topology
图21
Möbius带和Möbius拓扑结构的芳香性分子轨道示意图
Figure21.
Möbius strip and the MO sketch of aromaticity with Möbius-topology
此外, Jiao和Schleyer[124, 125]在20世纪90年代曾研究过渡态的Möbius芳香性.关于Möbius芳香性, Rzepa[48]曾在2001年曾撰写相关综述报道, 该综述在展示Craig模型分子时并没有展示8电子的平面结构, 而是将分子轨道中的Möbius成分和Hückel成分加以区分, 说明体系含有Möbius芳香性的成分.下面将通过举例来介绍一下非过渡态的两类Möbius芳香性各自的特点.
3.2.1 扭曲结构
自1964年Heilbronner预测具有扭曲拓扑结构的Möbius芳香性以来, 许多研究者都尝试实现这一猜想, 但均因为扭曲结构具有较大的张力而不稳定, 且因为扭曲的p轨道中的π电子共轭被削弱而未能实现实验上的验证.因此, 这类物质虽然理论上认为应该存在, 但实际却很难合成. 2003年, Herges等[126]报道了一例[16]轮烯的衍生物, 并证明其Möbius芳香性, 实现了Heilbronner的预测.其合成策略如图 22所示, 巧妙地利用了常规的π体系与面内的π体系相结合, 通过光照和加热实现分子前线轨道不同的顺旋-顺旋、顺旋-对旋的匹配, 从而实现了具有Möbius结构轮烯的制备.
 图22
稳定Möbius芳香性轮烯结构的策略
Figure22.
Strategy to stabilize the Möbius structure of annulenes
图22
稳定Möbius芳香性轮烯结构的策略
Figure22.
Strategy to stabilize the Möbius structure of annulenes
Herges等[126]还利用理论计算研究了他们设计的[16]轮烯各异构体的结构并计算了相对能量, 并与Castro和Schleyer[103]计算的常规[16]轮烯结构进行了比照.他们发现不同于常规[16]轮烯(最稳定的为2种Hückel拓扑结构).他们设计的最稳定的前7种结构中, 有6种均为Möbius构型, 只有一种为Hückel构型(相对能量位于第六位).最终, 他们通过改进路线获得了2种Möbius结构和1种Hückel结构的化合物.晶体结构表明, 其中一种具有C2对称性的Möbius构型的离域性很好, 而Hückel构型的分子则键长平均程度更低.该Möbius构型的HOMA值为0.35, 与C60结构中的苯环HOMA值相仿, 比菲结构的中间环的HOMA值(0.4)略小; 而Hückel构型分子的HOMA值仅为0.16.茚-异茚法ASE计算结果表明, C2对称性的Möbius结构为4.04 kcal/mol, 而Hückel构型分子为-2.22 kcal/mol.因此他们认为, 该分子的Hückel构型为非芳香的, 而Möbius构型则有一定的芳香性.
由于全碳体系无法使用d轨道参与相位翻转, 所以必须通过碳环的扭转来实现这一点, 因此上述研究也是全碳体系Möbius芳香性的一个新突破.关于碳氢化合物的Möbius芳香性, Oda等[127]在2004年曾撰写相关综述讨论.
3.2.2 平面结构
根据上述Craig的描述, 只要是能够利用d轨道参与两边的原子(通常指碳)形成dπ-pπ键的体系, 均可能实现平面构型的Craig-Möbius芳香性, 例如S、P以及含有空的d轨道的金属原子等[14].并且Rzepa在其综述中提及了含有金属钒、钽等的配合物等过渡金属化合物同样具有这样的能力[48].
Mauksch和Tsogoeva[55]在2010年的报道中提及了金属杂环中的Craig-Möbius芳香性的证明.其中一例给出了如图 23所示化合物的π分子轨道图像.该分子一共是4个π轨道, 也就是8电子的平面结构, 其NICS(0)和NICS(1)zz的数值分别为-4.2和-23.0, ASE为15.5 kcal/mol, 并且优化结构中的键长较为平均.如果一定要将其中的轨道分成Hückel类型和Möbius类型的话, 也可以说其具有2个Möbius轨道的成分.因此, 我们可以认为其含有较好的Craig-Möbius芳香性.
 图23
[Fe(CH)6H2]的结构及其π分子轨道图像
Figure23.
The structure of [Fe(CH)6H2] and its π-MOs
图23
[Fe(CH)6H2]的结构及其π分子轨道图像
Figure23.
The structure of [Fe(CH)6H2] and its π-MOs
夏海平课题组与朱军课题组[26]在2013年报道了一类新型的Craig-Möbius芳香性金属有机化合物-金属杂戊搭炔(Metallapentalyne)(图 24).这种化合物的有机母体戊搭炔(Pentalyne)应当是Hückel反芳香性的.然而, 当桥头碳替换为金属锇时, 化合物体现出很强的芳香性.
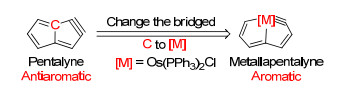 图24
金属杂戊搭炔的结构示意
Figure24.
Simplified structure of metallapentalyne
图24
金属杂戊搭炔的结构示意
Figure24.
Simplified structure of metallapentalyne
文中列举了一系列证据证明金属杂并双五元环结分子轨道(图 25)显示其为八电子共轭体系, 并且无论从构的芳香性特征, 包括上面提及的MO、ASE、NICS等.总电子数(8电子)的角度来看, 还是从分子轨道中是否含有Möbius轨道(C2对称性轨道)对芳香性贡献大小的角度来看, 这一例分子都具有Möbius芳香性.此外, 其还对NICS(0)zz的值做了计算, 在A环和B环上的值分别为-11.1和-10.8, 与苯环的-14.5相近, 却与戊搭炔母体的+45.0和+60.3大相径庭.此外, ASE值(约为20 kcal/mol)也证明了体系的芳香性.因此, 该研究表明通过替换原子的方式可以实现从反芳香性到芳香性的突变.
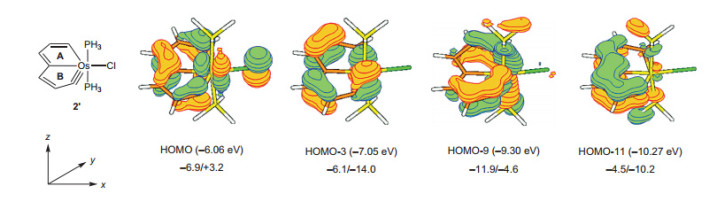 图25
金属杂戊搭炔的π分子轨道(图下数值分别对应A和B环的NICS(0)zz)
Figure25.
The π-MOs of metallapentalyne(The values below correspond to NICS(0)zz of A and B rings)
图25
金属杂戊搭炔的π分子轨道(图下数值分别对应A和B环的NICS(0)zz)
Figure25.
The π-MOs of metallapentalyne(The values below correspond to NICS(0)zz of A and B rings)
3.3 其他芳香性
除了上述的三大类, 还有一些独特的芳香性, 其在规则、内容上可能与上述有所交叉或重叠, 如σ芳香性[128~134]、γ芳香性[135]、δ芳香性[136]等, 近年来过渡态的芳香性也备受关注[137].值得注意的是, 席振峰课题组[27]成功合成了金属螺芳香化合物, 实现了Rzepa等[138]在2002年提出的理论模型的实验化.下文对这几种重要的芳香性原理进行简单的介绍.
3.3.1 σ芳香性
Dewar[128]认为, 对于仅含σ键的饱和碳原子体系, 例如环丙烷, 由于其键角(60°)显著偏离sp3碳原子的常规键角(109.5°), 本应具有较大的环张力却能稳定存在, 因此将其稳定性归因于体系的σ芳香性.这种芳香性与普通由π电子离域的芳香体系不同, 而是以σ电子离域为典型特征(图 26).例如, 环丙烷中三个成环的碳原子具有六个离域的σ电子, 被认为与6π电子的苯环“等共轭”(Isoconjugated). Dewar也在文献中描述了H3+的σ芳香性.
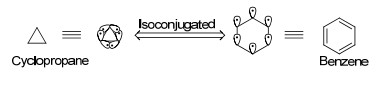 图26
环丙烷与苯环等共轭
Figure26.
Cyclopropane isoconjugated with benzene
图26
环丙烷与苯环等共轭
Figure26.
Cyclopropane isoconjugated with benzene
AICD研究表明环丙烷和环丁烷的σ电子离域情况相对于五元环及更大的环系更好.但σ芳香性这一概念在环丙烷体系的使用仍存在争议.近年来, 吴玮等[132]利用价键理论深入研究了环丙烷的σ芳香性, 认为“σ芳香性”在环丙烷和环丁烷中对体系稳定性的贡献均很小, 因此无法通过σ芳香性来解释环丙烷和环丁烷虽然角张力不同, 却拥有相似的稳定性.但是, 他们的研究却支持H3+体系确实存在σ芳香性.
3.3.2 γ芳香性
1972年, Gund[135]在经典芳香性定义的基础上, 首次提出了非环状的交叉共轭体系也具有芳香性——γ芳香性(又称为Y芳香性).这类具有“Y”形状的分子与苯等环状芳香分子类似, 也具有易发生取代反应的特点.例如, 胍离子等化合物(图 27)都具有一定的稳定性[139].胍具有极强的碱性(pKb=-2), 极易夺取体系中的质子形成阳离子.胍阳离子非常稳定, 被认为是“γ芳香性”的体现.
 图27
一些具有γ芳香性的化合物
Figure27.
Some γ-aromatic compounds
图27
一些具有γ芳香性的化合物
Figure27.
Some γ-aromatic compounds
3.3.3 δ芳香性
2007年, Boldyrev等提出δ体系也存在芳香性[136].他们通过理论计算, 研究[Ta3O3]-阴离子的芳香性, 认为这是一种δ键参与的新型芳香性.
3.3.4 螺芳香性
螺芳香性(Spiro-aromaticity)最早被Horada等用于描述“以一个sp3碳原子连接的两个独立的芳香环”体系[140]. 2002年, Rzepa等[138]首次将该词用于描述具有整体芳香性的螺环.与经典芳香体系不同, 通过共用单个原子的螺环结构也具有整体芳香性.近期, 席振峰课题组[27]报道了首例螺芳香体系(图 28).与Rzape的计算研究不同, 该螺芳香化合物的两个环处在同一个平面, 借助平面上下的锂原子的配位, 进一步稳定了这两个共用钯原子的五元环.该体系的芳香性也通过DFT计算的ISE值(-21.6 kcal/mol)和NICS(1)zz值(δ-15.7)进一步论证.
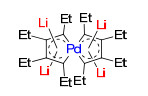 图28
平面螺芳香性
Figure28.
Planar spiro-aromaticity
图28
平面螺芳香性
Figure28.
Planar spiro-aromaticity
4 结语
芳香体系具有特殊的稳定性, 从而使得众多芳构化的反应往往能获得额外的芳香驱动力.利用这一特征可以用于特定产物结构的设计, 并探索实现绿色合成.值得注意的是, 各种化合物的芳香性判据仍然存在较大争议, 目前并没有一个明确的界限可以严格区分具有芳香性、非芳香性和反芳香性的三类化合物.此外, 每一种芳香性判据往往具有一定的局限性与应用的前提条件.因此, 对于新体系的芳香性研究, 不能简单地凭借单一的判据进行分析, 而应该结合实验数据, 选择合适的理论判据, 多种渠道进行小心验证.
尽管距离Faraday首次分离出芳香族母体分子苯已近200年, 但全新的芳香体系仍然不断涌现, 有关芳香性的新科学问题仍不断出现.有关芳香性的各种理论不断被完善和发展的同时, 众多理论学家、实验学家在“芳香性”相关主题上的争论从未停止, 这也正是芳香化学魅力之所在.
-
-
[1]
Faraday, M. Philos. Trans. R. Soc. London 1825, 115, 440. doi: 10.1098/rstl.1825.0022
-
[2]
Schleyer, P. v. R. Pure Appl. Chem. 1996, 68, 209. doi: 10.1351/pac199668020209
-
[3]
封继康, 分子科学学报, 2005, 21, 1. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=fzkb200504002&dbname=CJFD&dbcode=CJFQFeng, J. K. J. Mol. Sci. 2005, 21, 1(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=fzkb200504002&dbname=CJFD&dbcode=CJFQ
-
[4]
Lloyd, D. J. Chem. Inf. Comput. Sci. 1996, 36, 442. doi: 10.1021/ci950158g
-
[5]
(a) Kekulé, A. Bull. Soc. Chim. Paris 1865, 3, 98.
(b) Kekulé, A. Ann. 1866, 137, 129.
(c) Kekulé, A. Ann. 1872, 162, 77. -
[6]
Erlenmeyer, E. Ann. 1866, 137, 327.
-
[7]
Pascal, P. Ann. Chim. Phys. 1910, 19, 5.
-
[8]
Armit, J. W.; Robinson, R. J. Chem. Soc. 1925, 127, 1604. doi: 10.1039/CT9252701604
-
[9]
Hückel, E. Z. Phys. 1931, 70, 204. doi: 10.1007/BF01339530
-
[10]
Pauling, L. J. Chem. Phys. 1936, 4, 673. doi: 10.1063/1.1749766
-
[11]
Wheland, G. W. Resonance in Organic Chemistry, Wiley, New York, 1955.
-
[12]
London, F. J. Phys. Radium 1937, 8, 397. doi: 10.1051/jphysrad:01937008010039700
-
[13]
Pople, J. A. J. Chem. Phys. 1956, 24, 1111. doi: 10.1063/1.1742701
-
[14]
Craig, D. P. Nature 1958, 181, 1052. doi: 10.1038/1811052a0
-
[15]
Heilbronner, E. Tetrahedron Lett. 1964, 5, 1923. doi: 10.1016/S0040-4039(01)89474-0
-
[16]
Dewar, M. J. S.; Gleicher, G. J. J. Am. Chem. Soc. 1965, 87, 685. doi: 10.1021/ja01082a001
-
[17]
(a) Dauben, Jr., H. J.; Wilson, J. D.; Laity, J. L. J. Am. Chem. Soc. 1968, 90, 811.
(b) Dauben, Jr., H. J.; Wilson, J. D.; Laity, J. L. J. Am. Chem. Soc. 1969, 91, 1991.
(c) Dauben, Jr., H. J.; Wilson, J. D.; Laity, J. L. In Non-Benzenoid Aromatics, Ed.:Snyder, J. P., Vol. 2, Academic Press, New York, 1971. -
[18]
(a) Benson, R. C.; Flygare, W. H. J. Am. Chem Soc. 1970, 92, 7523.
(b) Schmalz, T. G.; Norris, C. L.; Flygare, W. H. J. Am. Chem Soc. 1973, 95, 7961.
(c) Schmalz, T. G.; Gierke, T. D.; Beak, P.; Flygare, W. H. Tetrahedron Lett. 1974, 33, 2885.
(d) Palmer, M. H.; Findlay, R. H. Tetrahedron Lett. 1974, 33, 253.
(e) Sutter, D. H.; Flygare, W. H. Top. Curr. Chem. 1976, 63, 89. -
[19]
Thorn, D. L.; Hoffmann, R. Nouv. J. Chim. 1979, 3, 39. http://www.researchgate.net/publication/284379561_Delocalization_in_metallocycles
-
[20]
(a) Kutzelnigg, W. Isr. J. Chem. 1980, 19, 193.
(b) Kutzelnigg, W.; Fleischer, U.; Schindler, M. NMR, Basic Principles and Progress, Vol. 23, Springer Verlag, Berlin, 1990, p. 165. -
[21]
Elliott, G. P.; Roper, W. R.; Waters, J. M. J. Chem. Soc., Chem. Commun. 1982, 14, 811.
-
[22]
Schleyer, P. v. R.; Maerker, C; Dransfeld, A. Jiao, H. J. Am. Chem. Soc. 1996, 118, 6317. doi: 10.1021/ja960582d
-
[23]
Wallenborn, E.-U.; Haldimann, R. F.; Klarner, F.-G.; Diederich, F. Chem. Eur. J. 1998, 4, 2258. doi: 10.1002/(ISSN)1521-3765
-
[24]
Wen, T. B.; Zhou, Z. Y.; Jia, G. Angew. Chem., Int. Ed. 2001, 42, 5954. http://www.ncbi.nlm.nih.gov/pubmed/11385683
-
[25]
Ajami, D.; Oeckler, O.; Simon & Amp, A.; Herges, R. Nature 2003, 426, 819. doi: 10.1038/nature02224
-
[26]
Zhu, C.; Li, S.; Luo, M.; Zhou, X.; Niu, Y.; Lin, M.; Zhu, J.; Cao, Z.; Lu, X.; Wen, T.; Xie, Z.; Schleyer, P. V. R.; Xia, H. Nat. Chem. 2013, 5, 698. doi: 10.1038/nchem.1690
-
[27]
Zhang, Y.; Wei, J.; Chi, Y, ; Zhang, X.; Zhang, W. X.; Xi, Z. J. Am. Chem. Soc. 2017, 139, 5039. doi: 10.1021/jacs.7b02039
-
[28]
Krygowski, T. M.; Cyranski M. K. Chem. Rev. 2001, 101, 1385. doi: 10.1021/cr990326u
-
[29]
Breslow, R. Chem. Eng. News 1965, 43, 90. doi: 10.1021/cen-v043n040.p090
-
[30]
Breslow, R. Acc. Chem. Res. 1973, 6, 393. doi: 10.1021/ar50072a001
-
[31]
Anet, F. A. L.; Bourn, A. J. R.; Lin, Y. S. J. Am. Chem. Soc. 1964, 86, 3576. doi: 10.1021/ja01071a046
-
[32]
Chen, Z.; King, R. B. Chem. Rev. 2005, 105, 3613. doi: 10.1021/cr0300892
-
[33]
Stock, A.; Pohland, E. Eur J. Inorg. Chem. 1926, 59, 2215.
-
[34]
Wiberg, E. Naturwissenschaften 1948, 35, 182. doi: 10.1007/BF00627385
-
[35]
Dixon, W. T. Tetrahedron 1962, 18, 875. doi: 10.1016/S0040-4020(01)92738-3
-
[36]
Avram, M.; Marica, E.; Nenitzescu, C. D. Eur. J. Inorg. Chem. 1959, 92, 1088.
-
[37]
Hirsch, A. The Chemistry of Fullerenes. Thieme, Stuttgart, 1994.
-
[38]
Taylor, R. C. R. Chim. 2006, 9, 982. doi: 10.1016/j.crci.2006.01.004
-
[39]
Rubin, Y. In Fullerenes and Related Structures, Ed.:Hirsch, A., Springer, Berlin & Heidelberg, 1999, 199, p. 67.
-
[40]
Sondheimer, F.; Wolovsky, R.; Amiel, Y. J. Am. Chem. Soc. 1962, 84, 274. doi: 10.1021/ja00861a030
-
[41]
Jackman, L. M.; Sondheimer, F.; Amiel, Y.; Ben-Efraim, D. A.; Gaoni, Y.; Wolovsky, R.; Bothner-By, A. A. J. Am. Chem. Soc. 1962, 84, 4307. doi: 10.1021/ja00881a022
-
[42]
Karplus, M. J. Chem. Phys. 1960, 33, 1842. doi: 10.1063/1.1731515
-
[43]
Nowakowski, J. Theor. Chim. Acta 1968, 10, 79. doi: 10.1007/BF00529046
-
[44]
Nyulászi, L. Chem. Rev. 2001, 101, 1229. doi: 10.1021/cr990321x
-
[45]
Randić, M. Chem. Rev. 2003, 103, 3449. doi: 10.1021/cr9903656
-
[46]
Lee, V. Y.; Sekiguchi, A. Angew. Chem., Int. Ed. 2007, 46, 6596. doi: 10.1002/(ISSN)1521-3773
-
[47]
Boldyrev, A. I.; Wang, L. S. Chem. Rev. 2005, 105, 3716. doi: 10.1021/cr030091t
-
[48]
Rzepa, H. S. Chem. Rev. 2005, 105, 3697. doi: 10.1021/cr030092l
-
[49]
Borden, W. T. Modern Molecular Orbital Theory for Organic Chemists, Prentice Hall, Englewood Cliffs, NJ, 1975.
-
[50]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
-
[51]
Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
-
[52]
Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200. doi: 10.1016/0009-2614(89)87234-3
-
[53]
Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5, Semichem Inc., Shawnee Mission KS, 2009.
-
[54]
Hirsch, A.; Chen, Z.; Jiao, H. Angew. Chem., Int. Ed. 2000, 39, 3915. doi: 10.1002/(ISSN)1521-3773
-
[55]
Mauksch, M.; Tsogoeva, S. B. Chem.-Eur. J. 2010, 16, 7843. doi: 10.1002/chem.201000396
-
[56]
Bleeke, J. R. Chem. Rev. 2001, 101, 1205. doi: 10.1021/cr990337n
-
[57]
Pauling, L.; Sherman, J. J. Chem. Phys. 1933, 1, 606. doi: 10.1063/1.1749335
-
[58]
Minkin, V. I.; Glukhovtsev, M. N.; Simkin, B. Y. Aromaticity and Antiaromaticity, Electronic and Structural Aspects, J. Wiley, New York, 1994.
-
[59]
George, P.; Trachtman, M.; Bock, C. W.; Brett, A. M. Theor. Chim. Acta 1975, 38, 121. doi: 10.1007/BF00581469
-
[60]
Hehre, W. J.; Ditchfield, R.; Radom, L.; Pople, J. A. J. Am. Chem. Soc. 1970, 92, 4796. doi: 10.1021/ja00719a006
-
[61]
Gordon, M. S. Modern Electronic Structure Theory, Part I, World Scientific, Singapore, 1995.
-
[62]
Schleyer, P. V. R.; Pühlhofer, F. Org. Lett. 2002, 4, 2873. doi: 10.1021/ol0261332
-
[63]
Wannere, C. S.; Moran, D.; Allinger, N. L.; Hess, B. A.; Schaad, L. J.; Schleyer, P. V. R. Org. Lett. 2003, 5, 2983. doi: 10.1021/ol034979f
-
[64]
Suresh, C. H.; Koga, N. Chem. Phys. Lett. 2006, 419, 550. doi: 10.1016/j.cplett.2005.12.028
-
[65]
Fallah-Bagher-Shaidaei, H.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. V. R. Org. Lett. 2006, 8, 863. doi: 10.1021/ol0529546
-
[66]
Islas, R.; Martinez-Guajardo, G.; Jemenez-Halla, J. O. C.; Sola, M; Merino, G. J. Chem. Theory Comput. 2010, 6, 1131. doi: 10.1021/ct100098c
-
[67]
Herges, R.; Geuenich, D. J. Phys. Chem. A 2001, 105, 3214. doi: 10.1021/jp0034426
-
[68]
Geuenich, D.; Hess. K.; Köhler, F.; Herges, R. Chem. Rev. 2005, 105, 3758. doi: 10.1021/cr0300901
-
[69]
Steiner, E.; Fowler, P. W. Int. J. Quantum Chem. 1996, 60, 609. doi: 10.1002/(ISSN)1097-461X
-
[70]
Kruszewski, J.; Krygowski, T. M. Tetrahedron Lett. 1972, 13, 3839. doi: 10.1016/S0040-4039(01)94175-9
-
[71]
Andrzejak, M.; Kubisiak, P. Struct. Chem. 2013, 24, 1171. doi: 10.1007/s11224-012-0148-2
-
[72]
Becke, A. D.; Edgecombe, K. E. J. Chem. Phys. 1990, 92, 5397. doi: 10.1063/1.458517
-
[73]
Savin, A.; Nesper, R.; Wengert, S.; F ssler, T. F. Angew. Chem., Int. Ed. 1997, 36, 1808. doi: 10.1002/(ISSN)1521-3773
-
[74]
Santos, J. C.; Tiznado, W.; Contreras, R.; Fuentealba, P. J. Chem. Phys. 2004, 120, 1670. doi: 10.1063/1.1635799
-
[75]
Zubarev, D. Y.; Boldyrev, A. I. Phys. Chem. Chem. Phys. 2008, 10, 5207. doi: 10.1039/b804083d
-
[76]
Juse, J.; Sundholm, D. Phys. Chem. Chem. Phys. 1999, 1, 3429. doi: 10.1039/a903847g
-
[77]
Bird, C. W. Tetrahedron 1985, 41, 1409. doi: 10.1016/S0040-4020(01)96543-3
-
[78]
Kotelevskii, S. I.; Prezhdo, O. V. Tetrahedron 2001, 57, 5715. doi: 10.1016/S0040-4020(01)00485-9
-
[79]
Cyrański, M. K. Chem. Rev. 2005, 105, 3773. doi: 10.1021/cr0300845
-
[80]
Aihara, J. I. J. Am. Chem. Soc. 2006, 128, 2873. doi: 10.1021/ja056430c
-
[81]
Aihara, J.; Kanno, H.; Ishida, T. J. Phys. Chem. A 2007, 111, 8873. doi: 10.1021/jp0733567
-
[82]
Giambiagi, M.; de Giambiagi, M. S.; dos Santos Silva, C. D., de Figueiredo, A. P. Phys. Chem. Chem. Phys. 2000, 2, 3381. doi: 10.1039/b002009p
-
[83]
Ponec, R.; Bultinck, P.; Saliner, A. G. J. Phys. Chem. A 2005, 109, 6606. doi: 10.1021/jp052179b
-
[84]
Piermarini, G. J.; Mighell, A. D.; Weir, C. E.; Block, S. Science 1969, 165, 1250. doi: 10.1126/science.165.3899.1250
-
[85]
Abrahams, S. C.; Robertson, J. M.; White, J. G. Acta Crystallogr. 1949, 2, 233. doi: 10.1107/S0365110X49000618
-
[86]
Mason, R. Acta Crystallogr. 1964, 17, 547. doi: 10.1107/S0365110X64001281
-
[87]
Petříček, V.; Císařová, I.; Hummel, L.; Kroupa, J.; Březina, B. Acta Crystallogr., Sect. B:Struct. Sci. 1990, 46, 830. doi: 10.1107/S0108768190007510
-
[88]
Frampton, C. S.; Knight, K. S.; Shankland, N.; Shankland, K. J. Mol. Struct. 2000, 520, 29. doi: 10.1016/S0022-2860(99)00280-X
-
[89]
Sondheimer, F.; Wolovsky, R. J. Am. Chem. Soc. 1962, 84, 260. doi: 10.1021/ja00861a028
-
[90]
Spitler, E. L.; Johnson Ⅱ, C. A.; Haley, M. M. Chem. Rev. 2006, 106, 5344. doi: 10.1021/cr050541c
-
[91]
Herges, R. Chem. Rev. 2006, 106, 4820. doi: 10.1021/cr0505425
-
[92]
Willstatter, R.; Waser, E. Eur. J. Inorg. Chem. 1911, 44, 3423. doi: 10.1002/cber.191104403216/full
-
[93]
Masamune, S.; Seidner, R. T. J. Chem. Soc. D:Chem. Commun. 1969, 542. doi: 10.1039/0577-6171/1969
-
[94]
Yavari, I.; Norouzi-Arasi, H. J. Mol. Struct.:THEOCHEM 2002, 593, 199. doi: 10.1016/S0166-1280(02)00318-4
-
[95]
Oth, J. F. M.; Rottele, H.; Schroder, G. Tetrahedron Lett. 1970, 11, 61. doi: 10.1016/S0040-4039(01)87565-1
-
[96]
Wiberg, K. B. Chem. Rev. 2001, 101, 1317. doi: 10.1021/cr990367q
-
[97]
Allinger, N. L.; Sprague, J. T. J. Am. Chem. Soc. 1973, 95, 3893. doi: 10.1021/ja00793a012
-
[98]
Castro, C.; Karney, W. L.; Valencia, M. A.; Vu, C. M. H.; Pemberton, R. P. J. Am. Chem. Soc. 2005, 127, 9704. doi: 10.1021/ja052447j
-
[99]
Sondheimer, F.; Gaoni, Y. J. Am. Chem. Soc. 1960, 82, 5765. doi: 10.1021/ja01506a061
-
[100]
(a) Oth, J. F. M. Pure Appl. Chem. 1971, 25, 573.
(b) Jug, K.; Fasold, E. J. Am. Chem. Soc. 1987, 109, 2263.
(c) Baumann, H.; Oth, J. F. M. Helv. Chim. Acta 1995, 78, 679.
(d) Choi, C. H.; Kertesz, M. J. Am. Chem. Soc. 1997, 119, 11994.
(e) Baumann, H.; Bunzli, J. J. Chem. Soc., Faraday Trans. 1998, 94, 2695.
(f) Oda, M.; Sakamoto, Y.; Kajioka, T.; Uchiyama, T.; Miyatake, R.; Kuroda, S. Angew. Chem., Int. Ed. 2001, 40, 2660.
(g) Vogel, E.; Engels, H.; Huber, W.; Lex, J.; Mullen, K. J. Am. Chem. Soc. 1982, 104, 3729. -
[101]
Sondheimer, F.; Gaoni, Y. J. Am. Chem. Soc. 1961, 83, 4863. doi: 10.1021/ja01484a042
-
[102]
(a) Martin-Santamaria, S.; Lavan, B.; Rzepa, H. S. J. Chem. Soc., Perkin Trans. 2 2000, 0, 1415.
(b) Oth, J. F. M.; Gilles, J.-M. Tetrahedron Lett. 1968, 6259.
(c) Johnson, S. M.; Paul, I. C.; King, G. S. D. J. Chem. Soc. (B) 1970, 643. -
[103]
Castro, C.; Isborn, C. M.; Karney, W. L.; Mauksch, M.; Schleyer, P. V. R. Org. Lett. 2002, 4, 3431. doi: 10.1021/ol026610g
-
[104]
Ajami, D.; Oeckler, O.; Simon, A.; Herges, R. Nature 2003, 426, 819. doi: 10.1038/nature02224
-
[105]
Sondheimer, F.; Wolovsky, R.; Amiel, Y. J. Am. Chem. Soc. 1962 84, 274. doi: 10.1021/ja00861a030
-
[106]
Longuet-Higgins, H. C.; Salem, L. Proc. R. Soc. London, Ser. A 1959, 251, 172. doi: 10.1098/rspa.1959.0100
-
[107]
Yoshizawa, K.; Kato, T.; Yamabe, T. J. Phys. Chem. 1996, 100, 5697. doi: 10.1021/jp953420x
-
[108]
Mislow, K. J. Chem. Phys. 1952, 20, 1489. doi: 10.1063/1.1700789
-
[109]
Coulson, C. A.; Golebiewski, A. Tetrahedron 1960, 11, 125. doi: 10.1016/0040-4020(60)89014-X
-
[110]
Bregman, J. Hirshfeld, F. L. Rabinovich, D. Schmidt, G. M. J. Acta Crystallogr. 1965, 19, 227. doi: 10.1107/S0365110X65003158
-
[111]
Stevenson, C. D.; Kurth, T. L. J. Am. Chem. Soc. 2000, 122, 722. doi: 10.1021/ja993604f
-
[112]
Reetz, M. T.; Hütte, S.; Goddard, R. Z. Naturforsch., B:J. Chem. Sci. 1995, 50, 415.
-
[113]
Gould, E. S. Acta Crystallogr. 1955, 8, 657. doi: 10.1107/S0365110X55002053
-
[114]
Robertson, J. M.; Shearer, H. M. M.; Sim, G. A.; Watson, D. G. Acta Crystallogr. 1962, 15, 1. doi: 10.1107/S0365110X62000018
-
[115]
Pople, J. A. Mol. Phys. 1958, 1, 175. doi: 10.1080/00268975800100211
-
[116]
Pozharskii, A. F. Chem. Heterocycl. Com. 1985, 21, 717. doi: 10.1007/BF00519137
-
[117]
(a) Bird, C. W. Tetrahedron 1992, 48, 335.
(b) Bird, C. W. Tetrahedron 1990, 46, 5697. -
[118]
Alkorta, I; Elguero, J. Struct. Chem. 2003, 14, 377. doi: 10.1023/A:1024402027760
-
[119]
Winstein, S. J. Am. Chem. Soc. 1959, 81, 6524. doi: 10.1021/ja01533a052
-
[120]
Doering, W. v. E.; Laber, G.; Vonderwahl, R.; Chamberlain, N. F.; Williams, R. B. J. Am. Chem. Soc. 1956, 78, 5448. doi: 10.1021/ja01601a080
-
[121]
Williams, R. V. Chem. Rev. 2001, 101, 1185. doi: 10.1021/cr9903149
-
[122]
Zhang, S.; Wei, J.; Zhan, M.; Luo, Q.; Wang, C.; Zhang, W. X.; Xi, Z. J. Am. Chem. Soc. 2012, 134, 11964. doi: 10.1021/ja305581f
-
[123]
Chen, Z.; King, R. B. Chem. Rev. 2005, 105, 3613. doi: 10.1021/cr0300892
-
[124]
Jiao, H.; Schleyer, P. v. R. Angew. Chem., Int. Ed. 1993, 32, 1763. doi: 10.1002/(ISSN)1521-3773
-
[125]
Jiao, H.; Schleyer, P. v. R. J. Chem. Soc., Perkin Trans. 2 1994, 407. doi: 10.1039%2Fp29940000407
-
[126]
Ajami, D.; Oeckler, O.; Simon, A.; Herges, R. Nature 2003, 426, 819. doi: 10.1038/nature02224
-
[127]
Kawase, T.; Oda, M. Angew. Chem., Int. Ed. 2004, 43, 4396. doi: 10.1002/(ISSN)1521-3773
-
[128]
Dewar, M. J. S. Bull. Soc. Chim. Belg. 1979, 88, 957. http://www.researchgate.net/publication/231451404_chemical_implications_of_.sigma._conjugation
-
[129]
Baeyer, A. Chem. Ber. 1885, 18, 2269. doi: 10.1002/(ISSN)1099-0682
-
[130]
Schleyer, P. V. R. In Substituent Effects in Radical Chemistry, Eds.:Viehe, H. G.; Janousek, Z.; Merenyi, R., Reidel, Dordrecht, 1986, pp. 69~81.
-
[131]
Benson, S. W.; Cruickshank, F. R.; Golden, D. M.; Haugen, G. R.; O'Neal, H. E.; Rodgers, A. S.; Shaw, R.; Walsh, R. Chem. Rev. 1969, 69, 279. doi: 10.1021/cr60259a002
-
[132]
Wu, W.; Ma, B.; Schleyer, P. v. R.; Mo, Y. Chem. Eur. J. 2009, 15, 9730. doi: 10.1002/chem.v15:38
-
[133]
Havenith, R. W. A.; De Proft, F.; Fowler, P. W. Geerlings, P. Chem. Phys. Lett. 2005, 407, 391. doi: 10.1016/j.cplett.2005.03.099
-
[134]
Zhu, C.; Zhou, X.; Xing, H.; An, K.; Zhu, J.; Xia, H. Angew. Chem., Int. Ed. 2015, 54, 3102. doi: 10.1002/anie.201411220
-
[135]
Gund, P. J. Chem. Edu. 1972, 49, 100. doi: 10.1021/ed049p100
-
[136]
Zhai, H. J.; Averkiev, B. B.; Zubarev, D. Y.; Wang, L. S.; Boldyrev, A. I. Angew. Chem., Int. Ed. 2007, 46, 4277. doi: 10.1002/(ISSN)1521-3773
-
[137]
Schleyer, P. V. R.; Wu, J. I.; Cossío, F. P.; Fernández, I. Chem. Soc. Rev. 2014, 43, 4909. doi: 10.1039/C4CS00012A
-
[138]
Rzepa, H. S.; Taylor, K. R. J. Chem. Soc., Perkin Trans. 2 2002, 1499. doi: 10.1039/b108727b
-
[139]
Sommerfeld, T. J. Am. Chem. Soc. 2002, 124, 1119. doi: 10.1021/ja012013y
-
[140]
Harada, N.; Ono, H.; Nishiwaki, T.; Uda, H. J. Chem. Soc., Chem. Commun. 1991, 24, 1753. doi: 10.1021%2Fja00010a029
-
[1]
-

图式 1 1, 3, 5, 7-环辛四烯的构象转换
Scheme 1 Ttransformation of conformations of 1, 3, 5, 7-cyclooctatetraene

图式 2 一般双键和芳香环双键的反应特性
Scheme 2 Reaction performances of a common C—C double and one on aromatic ring


图 5 计算得到的C6010+离子的π分子轨道
Figure 5 The calculated π-molecular orbitals of C6010+
Shell symbols in parenthesis, indicating s, p, d, f, and g shells. (Ref. [54], Copyright © 2000 WILEY-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany)

图式 5 使用甲基-亚甲基法研究吡啶的ASE
Scheme 5 Studying the ASE of pyridine by methyl-methylene method

图式 7 使用茚-异茚法研究[4n]轮烯的ASE
Scheme 7 Studying the ASE of [4n]annulenes by indene-iso-indene method

图 6 一系列五元环化合物(C4H4X)的NICS(0)和ASE的对照
Figure 6 NICS(0) and ASE values of a series of five-membered ring heterocycles C4H4X
(Reprinted with permission from Ref. [22]. Copyright © 1996 American Chemical Society.)

图 7 “左手定则”(上)及氢原子的自身感应电流密度和苯环的离域感应电流密度(下)
Figure 7 "Left-hand rule" (up); a local current density of hydrogen atom and a delocalized one of benzene (down)
(Reprinted with permission from Ref. [68]. Copyright © 2005 American Chemical Society.)

图 8 苯环的AICD图像:总贡献、σ贡献及π贡献
Figure 8 AICD isosurfaces of benzene: total, σ, and π contributions.
(Reprinted with permission from Ref. [68]. Copyright © 2005 American Chemical Society.)

图 9 苯(上)和萘(下)的等值面(0.3, 0.7和ELF点)
Figure 9 Isovalues of benzene (top) and naphthalene (bottom) (0.3, 0.7 and ELF points)
(Ref. [74]. Copyright © 2004, Rights managed by AIP Publishing LLC.)

图 11 苯及其同系物的NICS(0)(单位: ppm)
Figure 11 NICS(0) values of benzene and its homologues (Unit: ppm)

图 16 环戊二烯和环庚三烯及其对应芳香离子
Figure 16 Cyclopentadiene and cycloheptatriene and their corresponding aromatic ions

图 17 薁通过自身内部的芳构化实现稳定化
Figure 17 Azulene stabilized itself by intramolecular aromatization

图 18 一些简单的芳香性杂环化合物(上)以及π轨道示意图(下)
Figure 18 Some simple sturcuture of heteroaromatics (top) and π-orbitals (bottom)

图 19 环庚三烯衍生物(毕希纳酸)的同芳香性与异构化示意图
Figure 19 Homoaromaticity of a derivative of cyclopentatriene (buchner acid) and its isomerization

图 21 Möbius带和Möbius拓扑结构的芳香性分子轨道示意图
Figure 21 Möbius strip and the MO sketch of aromaticity with Möbius-topology

图 22 稳定Möbius芳香性轮烯结构的策略
Figure 22 Strategy to stabilize the Möbius structure of annulenes
(Ref. [126]. Copyright © 2003, Right Managed by Nature Publishing Group)

图 23 [Fe(CH)6H2]的结构及其π分子轨道图像
Figure 23 The structure of [Fe(CH)6H2] and its π-MOs
(Ref. [55]. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim)

图 25 金属杂戊搭炔的π分子轨道(图下数值分别对应A和B环的NICS(0)zz)
Figure 25 The π-MOs of metallapentalyne(The values below correspond to NICS(0)zz of A and B rings)
(Ref. [26]. Copyright © 2013, Rights Managed by Nature Publishing Group)
表 1 芳香性简史
Table 1. A brief history of aromaticity
年份/年 事件 关键词 1825前 人们将一类具有芳香气味的化合物称为“芳香化合物” 芳香化合物 1825 Faraday分离出一种具有芳香气味的化合物——苯[1] 苯 1865前 不饱和的、具有高C/H比的稳定化合物称为“芳香性化合物” 稳定性 1865 Kekulé提出著名的苯环的“Kekulé式”, 认为苯环是平面、环状分子, 并且具有3个C=C双键和3个C—C单键交替构成[5] Kekulé式 1866 Erlenmeyer提出了芳香性的化学判据:在实验上, 芳香化合物的取代反应比加成反应更易发生[6] 取代反应 1910 Pascal提出芳香性化合物的抗磁磁化率升高, 即该类化合物具有极强的抗磁性[7] 抗磁磁化率 1925 Armit和Robinson提出苯环为“六电子体(Electron Sextet)”, 认为苯环的性质与其电子有关, 并提出了杂原子参与的芳香性[8] 六电子体杂环芳香性 1931 Hückel提出著名的[4n+2]规则:在sp2杂化碳原子平面单环体系中, 含有(4n+2)个π电子的化合物具有芳香性[9] [4n+2]规则 1936 Pauling提出环电流理论:在一定条件下离域π电子可在苯环及同系稠环上形成环电流[10] 环电流 1937 Wheland根据大量实验数据总结提出了各种芳香烃共振能(ERE)的估算值[11] 芳香烃共振能(ERE) London提出了London抗磁性, 通过LCAO分子轨道法计算了π电子环电流对磁化率的贡献[12] London抗磁性 1956 Pople提出环流对核磁共振(NMR)化学位移的影响[13] NMR 1958 Craig首次提出了具有Craig模型的芳香性, 即如今的平面Möbius芳香性[14] Craig-Möbius芳香性 1964 Heilbronner首次提出:环状4n电子轮烯可能具有Möbius带扭曲结构的Möbius芳香性[15] 扭曲结构的Möbius芳香性 1965 Dewar提出基于原子化热的共振能(EDRE)作为芳香性的能量判据[16] Dewar芳香烃共振能 1969 Dauben进行抗磁磁化率的近代研究, 认为芳香化合物有较大的抗磁磁化率升高[17] 抗磁磁化率 1970 Flygare研究了芳香化合物磁化率的各向异性[18] 磁化率各向异性 1979 Hoffmann等预测了金属苯的芳香性[19] 金属苯 1980 Kutzelnigg通过IGLO量子化学计算了芳香化合物的磁性质:化学位移、磁化率、磁化率的各向异性等[20] 芳香化合物的磁性质 1982 Roper组报道首例金属苯的合成与表征, 并研究其芳香性[21] 金属苯 1996 Schleyer提出使用核独立化学位移(NICS)作为芳香性的判据[22] NICS Wallenborn首次将诱导电流密度的各向异性(AICD)用于芳香性领域, 并研究了富勒烯衍生物过渡态的芳香性[23] AICD 2001 贾国成组报道首例金属苯炔, 并研究了其芳香性[24] 金属苯炔 2003 Herges组首次合成了具有Möbius拓扑结构的Möbius芳香性化合物[25] 扭曲结构的Möbius芳香性 2013 夏海平组首次制备了金属杂戊搭炔, 并与朱军组合作研究了其芳香性, 发现可以通过掺杂金属实现分子骨架由反芳香到芳香的反转[26] 芳香性反转 2017 席振峰组报道首例螺芳香性化合物, 并研究其芳香性[27] 螺芳香性  下载: 导出CSV
下载: 导出CSV
表 2 传统芳香性化合物的特征
Table 2. Features of traditional aromatics
特征 芳香化合物 反芳香化合物 非芳香化合物 平面性 环共平面趋势 环平面扭曲趋势 一般为非平面 键长交替 平均化 单双键交替明显 与一般化合物类似 稳定性 稳定化 去稳定化 中等 环外1H NMR 去屏蔽 屏蔽 中等 反应性 相对更易取代、加成 相对更易加成 都可能发生 外加磁场 产生抗磁环电流 产生顺磁环电流 产生多向电流
下载: 导出CSV
-






































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1076
- 文章访问数: 36607
- HTML全文浏览量: 8641
 下载:
下载:
