 图1
结合雌激素主要成分
Figure1.
Components of conjugated estrogens
图1
结合雌激素主要成分
Figure1.
Components of conjugated estrogens
引用本文:
郑修文, 马海燕, 丁凯. 马烯雌酮及其衍生物的高效合成[J]. 有机化学,
2018, 38(2): 464-470.
doi:
10.6023/cjoc201709005

Citation: Zheng Xiuwen, Ma Haiyan, Ding Kai. Practical Syntheses of Equilin and Its Derivatives[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 464-470. doi: 10.6023/cjoc201709005
Citation: Zheng Xiuwen, Ma Haiyan, Ding Kai. Practical Syntheses of Equilin and Its Derivatives[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 464-470. doi: 10.6023/cjoc201709005
马烯雌酮及其衍生物的高效合成
English
Practical Syntheses of Equilin and Its Derivatives
Abstract:
Equilin and its derivatives are important active ingredients of conjugated estrogens, extracted from pregnant horse urine. However, they are still challenge synthetic targets due to the presence of an unstable non-conjugated double bond. Equilin (7), 17β-dihydroequilin (8) and 17α-dihydroequilin (9) were efficiently synthesized from commercially available 19-hydroxyandrost-4-ene-3, 17-dione via retro-aldol aromatization with the overall yields of 32%, 37% and 25%, respectively.
-
Key words:
- equilin
- / conjugated estrogen
- / aromatization
- / steroid
-
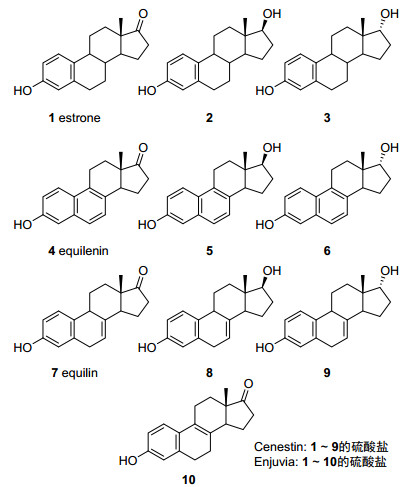
倍美力(Premarin)是惠氏公司1941年上市的药物——结合雌激素(Conjugated estrogens)的商品名, 目前为辉瑞制药所有.上市多年来, 一直是雌激素替代治疗(Hormone replacement therapy, HRT)的首选药物, 用于绝经后妇女补充雌激素以缓解其更年期症状[1].结合雌激素是多种甾体雌激素的硫酸盐的混合物, 提取于孕母马尿中.主要成分为雌酚酮(1, estrogen)、马萘雌酮(4, equilenin)和马烯雌酮(7, equilin)及其衍生物的硫酸盐(图 1)[2].
图1
结合雌激素主要成分
Figure1.
Components of conjugated estrogens
多年以来, 倍美力等结合雌激素药物一直通过从孕马尿中提取进行生产.不同批次的孕马尿有效物质含量并不稳定, 同时众多无需进行质量控制的低含量雌激素物质也会对药效有影响.通过合成或分离得到主要成分的纯品后, 按一定比例混合制得的药物称为合成结合雌激素, 具有产品质量稳定、副作用小等特点.目前上市的合成结合雌激素有Cenestin(合成结合雌激素A)和Enjuvia(合成结合雌激素B), 分别于1999年和2004年批准上市, 由Barr旗下的Duramed公司生产(2008年被Teva公司收购).其中Cenestin主要成分为9种甾体雌激素(图 1, 硫酸盐), Enjuvia主要成分为10种[3].
目前仅有雌酚酮可通过化学合成法从易得的甾体原料大规模生产.马萘雌酮(4)和马烯雌酮(7)仍然依靠从孕马尿中提取获得.最近, 我们发展了以易得的19-羟基-4-烯-3, 17-二酮雄甾(10)为原料, 利用retro-aldol芳构化反应合成甾体雌激素的方法[4].基于此方法, 完成了马萘雌酮(4)及其衍生物5和6的合成[5].
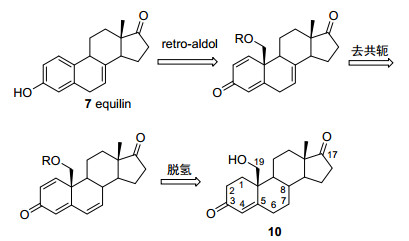
马烯雌酮合成的难点在于其B环上有一个非共轭的双键, 一方面容易异构化为更加稳定的共轭双键, 另一方面容易氧化, 导致B环芳构化生成马萘雌酮.因此, 需要避免高温、强酸、强碱和强氧化条件, 这使合成非常困难. 1958年, Zderic等[6a, 6b]利用19去甲基的诺龙衍生物为原料, 4, 6-二烯-3-酮中6, 7位双键的迁移构建7, 8位双键, 发酵法1, 2位脱氢芳构化A环, 完成了首个马烯雌酮的半合成. 1964年, Bagli等[6c]使用类似的策略构建7, 8位双键, 但改用化学法1, 2位脱氢, retro-aldol芳构化构建A环合成马烯雌酮, 但多个关键步骤产率非常低, 无合成价值. 1966年, Stein等[6d, 6e]报道了使用Δ-8-雌酚酮为原料合成马烯雌酮, 其中利用环氧-烯丙醇重排反应构建了8位羟基, 消除得到7, 8位双键.该方法选择性好、产率较高, 但是原料难得, 需要用全合成的方法合成. Bailey和Deghengh在1967年和1969年分别报道了使用马萘雌酮为原料, 通过Birch还原构建7, 8位双键合成马烯雌酮的方法.该方法路线短, 但是原料昂贵, 产率低[6f, 6g].目前为止, 与结合雌激素药物中马烯雌酮结构相关的三个化合物7, 8和9还没有实用的合成方法.本工作中, 我们以易得的19-羟基-4-烯-3, 17-二酮雄甾(10)为原料, 利用Saegusa-Ito反应成功构建了1, 2位双键.再通过强碱作用下烯酮去共轭构建了7, 8位双键, 最终利用retro-aldol芳构化反应合成了马烯雌酮(7)及其衍生物8和9 (Scheme 1).该路线中, 原料为商品化的药物中间体(6000 RMB/kg), 使用的试剂便宜易得, 合成路线短, 产率高, 为结合雌激素药物的化学法生产和进一步的生理活性研究提供了实用的化学合成方法.
 图式 1
反合成分析
Scheme1.
Retrosynthetic analysis
图式 1
反合成分析
Scheme1.
Retrosynthetic analysis
1 结果与讨论
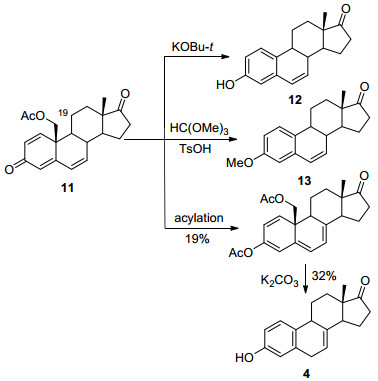
马烯雌酮合成的关键是B环非共轭的7, 8位双键的构建, 可以利用4, 6-二烯-3-酮中6, 7位双键的迁移获得.先前在研究retro-aldol芳构化反应时发现, 起始原料10中的19位羟基只有用酯基保护时, 才能2, 3-二氯-5, 6-二氰对苯醌(DDQ)氧化获得脱氢产物11[4].用甲基或甲氧甲基(MOM)保护时, 1, 2位脱氢无法进行.在使用中间体11为前体尝试合成马烯雌酮时(Scheme 2), 发现用碱烯醇化, 直接获得异构体12, 说明芳构化比烯醇化快得多.进一步尝试在酸性条件下与原酸酯反应合成烯醇醚, 却直接获得3-醚化的雌激素13[4, 7].与乙酸酐、乙酰氯和吡啶混合物反应可获得烯醇酯, 但收率不到20%, 在碱的作用下芳构化后虽然可获得目标产物马烯雌酮(7), 但产率也很低, 无实用价值[4b].
 图式 2
前期研究工作
Scheme2.
Our previous studies
图式 2
前期研究工作
Scheme2.
Our previous studies
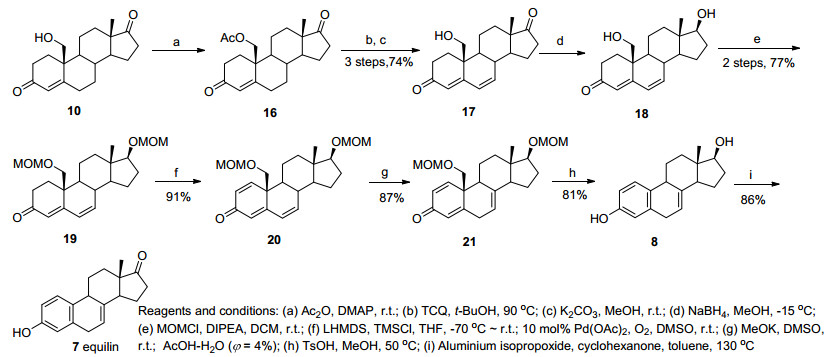
由此可见, 对于1, 4, 6-三烯-3-酮, 烯醇化需要强碱性条件, 19位羟基必须使用对碱稳定的保护基团.我们使用MOM保护的14为底物, 重新探索了1, 2脱氢条件(表 1). DDQ, 2-碘酰基苯甲酸(IBX)等经典脱氢试剂[4, 8]对底物完全不反应.也尝试了2位溴代后消除的方法[9], 但是对于底物14, 溴代和消除都得到复杂的混合物, 无合成意义.最终发现使用两步的Saegusa-Ito反应可以高产率获得目标产物15.常规的Saegusa-Ito反应需要使用亚化学计量的钯催化剂, 按照Larock发展的催化的Saegusa-Ito反应条件[10], 也可以获得满意的结果, 反应规模放大到10 mmol也可顺利进行, 产率不变.
 表 1
1, 2-脱氢反应的优化
Table 1.
Optimization of 1, 2-dehydrogenation
表 1
1, 2-脱氢反应的优化
Table 1.
Optimization of 1, 2-dehydrogenation

Condition Result 1 IBX, DMSO, toluene, 85 ℃ No reaction 2 DDQ, dioxane, 110 ℃ No reaction 3 DDQ, TBSCl, dioxane, r.t. No reaction 4 Bromination and elimination
(various conditions)Complex 5 (ⅰ) LHMDS, TMSCl, -78 ℃
(ⅱ) 5 mol% Pd(OAc)2, O2, DMSO88% yield 解决了1, 2位脱氢的问题后, 我们开始合成马烯雌酮. 19-羟基雄甾-4-烯-3, 17-二酮(10)与四氯苯醌反应可以直接获得B环脱氢产物17, 但是同时生成部分产物极性一致的未知副产物, 只能通过结晶去除, 分离产率非常低(<60%).我们发现乙酰化产物16脱氢没有副产物形成, 改用三步法从10合成17, 路线虽然较长, 但产率较高, 后处理方便.二烯酮17可被选择性还原获得二醇18, 两个羟基MOM保护后获得中间体19.使用前面优化过的Saegusa-Ito反应条件顺利得到1, 2脱氢产物20.烯酮20使用经典的叔丁醇钾条件烯醇化[11]未获成功, 完全回收原料.改用位阻更小的KOMe烯醇化, 稀醋酸淬灭后顺利得到非共轭烯酮21.中间体21在酸性条件下去除保护基顺利发生retro-aldol芳构化, 以81%产率获得了17β-二氢马烯雌酮(8).值得注意的是, 反应需要在严格的无氧条件下进行, 否则产物在酸性条件下被空气氧化, 生成B环芳构化的副产物17β-二氢马萘雌酮5.由于化合物8具有容易被氧化的A环酚羟基和B环双键, 氧化17位羟基遇到困难.我们筛选了DMP, IBX, Swern氧化和多种铬的氧化剂都得到复杂的产物, 最后使用Oppenauer氧化获得了成功.最终以九步反应, 收率32%获得了马烯雌酮(7) (Scheme 3).
 图式 3
马烯雌酮与17β-二氢马烯雌酮的合成路线
Scheme3.
Synthesis of equilin and 17β-dihydroequilin
图式 3
马烯雌酮与17β-二氢马烯雌酮的合成路线
Scheme3.
Synthesis of equilin and 17β-dihydroequilin
甾体17位羰基的还原立体专一性地生成17β-羟基, 因此17α-羟基甾体化合物不能通过17-酮的还原制备, 一般都通过17β-羟基化合物的翻转获得.由于甾体化合物中17位位阻较大, 羟基翻转的条件比较剧烈, 对于稳定性差的17β-二氢马烯雌酮(8), 常规方法难以获得目标产物9.为完成这一转化, 我们最近发展了一种温和条件下羟基翻转的方法[12]. 17β-二氢马烯雌酮3位苯甲酰基保护后, 17位羟基用全氟丁基磺酰氟[13]活化, 生成的磺酸酯23与甲酸在非常温和的条件下发生亲核取代, 以81%收率获得17α-羟基产物24, 未发现双键氧化或迁移的副产物形成.最后在碱性条件下脱除苯甲酰基和甲酰基保护, 得到17α-二氢马烯雌酮9 (Scheme 4).
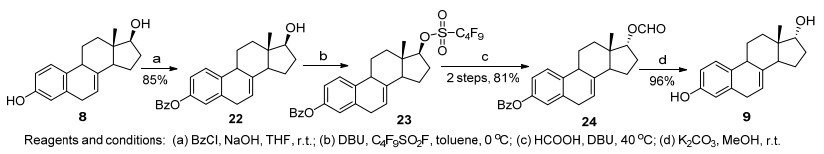 图式 4
17α-二氢马烯雌酮的合成路线
Scheme4.
Synthesis of equilin and 17α-dihydroequilin
图式 4
17α-二氢马烯雌酮的合成路线
Scheme4.
Synthesis of equilin and 17α-dihydroequilin
2 结论
以易得的19-羟基-4-烯-3, 17-二酮雄甾为原料, 通过Saegusa-Ito反应和四氯苯醌脱氢分别引入1, 2位和6, 7位双键, 碱性条件下烯醇化双键迁移获得7, 8位双键, 再以retro-aldol芳构化为关键反应合成了17β-二氢马烯雌酮(8)和马烯雌酮(7).在此基础上, 利用先前发展的17位羟基翻转反应, 合成了17α-二氢马烯雌酮(9).
3 实验部分
3.1 仪器与试剂
1H NMR和13C NMR由Bruker-AM 400型或Bruker-AM 300型核磁共振仪测定, 若无特别注明, 均以CDCl3作溶剂, 残留的未氘代溶剂为内标(δ 7.26 1H NMR, δ 77.0 13C NMR); EI-HRMS由Waters Micromass GCT Premier质谱仪测定; ESI-HRMS由Thermo Fisher Scientific LTQ FT Ultra质谱仪测定; 旋光是由PERKIN ELMER Polarimeter 1030旋光仪测定; 固体化合物熔点由RY-1G熔点仪测定, 所测熔点均未经温度计校正.试剂纯化参照Purification of Laboratory Chemicals (6th edition); 薄层色谱(TLC)采用GF254高效板, 显色剂为紫外灯照射、碘缸显色或10%磷钼酸乙醇溶液; 柱层析用硅胶H (100~200目和200~300目)作为固定相.
3.2 实验方法
3.2.1 19-羟基雄甾-4, 6-二烯-3, 17-二酮(17)的合成
向化合物10 (29 mmol, 8.76 g)中依次加入乙酸酐(145 mmol, 14.8 g)和4-二甲氨基吡啶(3 mmol, 0.367 g), 室温搅拌15 min, 体系由悬浊液变澄清, TLC显示原料反应完全.浓缩, 加入二氯甲烷稀释, 加入饱和碳酸氢钠溶液至体系中性, 分液, 水洗, 饱和氯化钠溶液洗, 无水Na2SO4干燥, 100~200目硅胶过滤[V(石油醚):V(乙酸乙酯)=1:1洗涤], 抽干得到白色固体9.98 g.
将9.98 g固体溶解于干燥的叔丁醇中, 加入四氯苯醌(32 mmol, 7.87 g), 90 ℃反应约30 min, 黄色固体消失, 核磁跟踪反应, 显示原料转化完全.浓缩, 加入约100 mL二氯甲烷, 抽滤, 向滤液中加入40 mL 1 mol/L氢氧化钠水溶液形成黑绿色悬浮液, 抽滤, 分出有机相, 水洗, 饱和氯化钠溶液洗, 无水Na2SO4干燥, 浓缩后用适量中性氧化铝快速柱层析, 得到7.53 g淡黄色油状物16.
7.53 g化合物16溶解于66 mL甲醇中, 加入碳酸钾(44 mmol, 4.36 g), 室温搅拌30 min, 有大量固体析出, TLC显示原料转化完全.旋去甲醇, 加二氯甲烷溶解后滤去无机物, 快速柱层析得到6.47 g淡黄色固体17, 三步收率74%. m.p. 172~174 ℃; [α]D30 +121.2 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 6.17 (s, 2H), 5.75 (s, 1H), 3.85 (dd, J=11.6, 5.6 Hz, 1H), 3.74 (dd, J=11.6, 3.6 Hz, 1H), 0.93 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 219.9, 200.1, 159.4, 139.3, 128.9, 125.7, 63.6, 50.6, 49.0, 48.3, 41.3, 37.4, 35.5, 34.1, 31.7, 29.8, 21.3, 20.9, 13.6; HRMS calcd for C19H24O3 300.1725, found 300.1723.
3.2.2 17, 19-二(甲氧基甲醚)雄甾-4, 6-二烯-3-酮(19)的合成
将化合物17(2 mmol, 600 mg)溶于36 mL无水甲醇, 置于-15 ℃低温浴中, 向其中滴加硼氢化钠(2 mmol, 75.6 mg)的甲醇溶液(4 mL), 持续1 min, 滴加完毕后继续反应约10 min, 大量固体析出, 向反应液滴加饱和氯化铵溶液淬灭反应.稍浓缩后抽滤, 水洗滤渣两次, 收集滤渣并对滤液用二氯甲烷萃取, 水洗两次, 用无水Na2SO4干燥, 有机相与滤渣合并, 抽干.得到含有14%过度还原产物(NMR确定)的白色固体18 592 mg.
将粗品18溶解于20 mL干燥的二氯甲烷溶液中, 依次加入二异丙基乙基胺(12 mmol, 2.10 mL)和氯甲基甲醚(8 mmol, 0.61 mL), 室温反应, 体系由浑浊逐渐澄清, TLC监控约1 h反应完全.加入20 mL二氯甲烷稀释, 2 mol/L盐酸10 mL, 搅拌10 min后, 分液, 水洗, 无水Na2SO4干燥, 柱层析纯化后得到600 mg白色固体19, 两步收率77%. m.p. 77~79 ℃; [α]D30 +54.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 6.14 (dd, J=10.0, 2.0 Hz, 1H), 6.09 (d, J=10.0 Hz, 1H), 5.78 (s, 1H), 4.66~4.57 (m, 3H), 4.54 (d, J=6.4 Hz, 1H), 3.75 (d, J=10.0 Hz, 1H), 3.59 (d, J=10.0 Hz, 1H), 3.55 (t, J=8.4 Hz, 1H), 3.35 (s, 3H), 3.33 (s, 3H), 2.75~2.65 (m, 1H), 2.51~2.29 (m, 3H), 2.12~2.04 (m, 1H), 1.93~1.88 (m, 1H), 1.83~1.57 (m, 5H), 1.52~1.42 (m, 1H), 1.33~1.26 (m, 1H), 1.19~1.06 (m, 2H), 0.86 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 199.9, 159.3, 140.6, 128.4, 125.7, 96.9, 96.1, 86.03, 69.0, 55.6, 55.3, 50.9, 48.7, 43.6, 40.3, 38.0, 37.4, 34.4, 30.6, 28.0, 23.0, 21.5, 11.7; HRMS calcd for C23H35O5 [M+H]+ 391.2479, found 391.2476.
3.2.3 17, 19-二(甲氧基甲醚)雄甾-1, 4, 6-三烯-3-酮(20)的合成
化合物19 (7 mmol, 2.73 g)溶解于35 mL干燥的四氢呋喃, 置于干冰-丙酮浴, 滴加六甲基二硅基氨基锂(21 mL 1 mol/L的四氢呋喃溶液), 30 min后滴加四甲基氯硅烷(21 mmol, 2.7 mL), 逐渐升温至室温, TLC监控原料反应完全, 抽干.加入饱和碳酸氢钠溶液和乙酸乙酯, 分液, 水洗, 无水Na2SO4干燥, 抽干, 得到黄色油状物粗品.
向中间体粗品中加入10 mol%醋酸钯, 将体系内气体置换为氧气(气球储气, 1.1 kPa), 注入63 mL二甲基亚砜, 室温下剧烈搅拌, 1 h后TLC发现原料转化完全.加入少量NaHCO3后, 减压蒸馏除去大部分二甲基亚砜, 加乙酸乙酯稀释, 硅胶滤除固体.滤液水洗两次, 无水Na2SO4干燥, 柱层析纯化.两步以收率91%得2.47 g淡黄色油状物20. [α]D30 -46.3 (c 0.55, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 7.16 (d, J=10.0 Hz, 1H), 6.38 (d, J=9.6 Hz, 1H), 6.25 (d, J=10.0 Hz, 1H), 6.07 (s, 1H), 6.01 (d, J=9.6 Hz, 1H), 4.62 (d, J=6.4 Hz, 1H), 4.59 (d, J=6.4 Hz, 1H), 4.49 (d, J=6.8 Hz, 1H), 4.45 (d, J=6.8 Hz, 1H), 3.84 (d, J=9.2 Hz, 1H), 3.59 (d, J=9.2 Hz, 1H), 3.52 (t, J=8.4 Hz, 1H), 3.33 (s, 3H), 3.26 (s, 3H), 2.39 (t, J=10.8 Hz, 1H), 2.12~2.02 (m, 1H), 1.97~1.83 (m, 3H), 1.82~1.40 (m, 4H), 1.19~1.10 (m, 2H), 0.87 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 186.5, 158.1, 149.6, 137.7, 130.6, 127.7, 125.8, 96.6, 96.0, 85.8, 69.7, 55.4, 55.2, 48.9, 48.7, 45.8, 43.0, 38.1, 37.0, 27.9, 22.8, 22.4, 11.7; HRMS calcd for C23H32O5 388.2250, found 388.2256.
3.2.4 17, 19-二(甲氧基甲醚)雄甾-1, 4, 7-三烯-3-酮(21)的合成
在25 mL Schlenk管中加入化合物20 (1 mmol, 388 mg)和甲醇钾(3 mmol, 210 mg), 氩气保护下注入2 mL干燥的二甲基亚砜, 室温反应1 h后, 缓慢滴加通氩气除氧后的乙酸水溶液(φ=4%) 6 mL淬灭反应(前1 mL用5 s/滴速度, 之后稍快; 体系温度不能太高, 根据环境温度判断是否需要冷水浴).加入乙酸乙酯和水溶解固体, 分液, 水洗两次后, 无水Na2SO4干燥, 柱层析纯化.得到336 mg黄色油状物21, 收率87%. [α]D30 -29.6 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 7.15 (d, J=10.0 Hz, 1H), 6.35 (dd, J=10.0, 1.6 Hz, 1H), 6.21 (s, 1H), 5.34~5.26 (m, 1H), 4.63 (d, J=6.4 Hz, 1H), 4.60 (d, J=6.4 Hz, 1H), 4.53~4.47 (m, 2H), 3.90 (d, J=9.2 Hz, 1H), 3.84 (d, J=9.2 Hz, 1H), 3.67~3.57 (m, 1H), 3.34 (s, 3H), 3.26 (s, 3H), 3.20 (d, J=18.8 Hz, 1H), 2.88~2.79 (m, 1H), 0.70 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 186.3, 162.8, 152.4, 137.1, 130.1, 125.6, 117.1, 96.6, 96.0, 86.2, 68.9, 55.6, 55.2, 49.1, 48.5, 47.2, 43.4, 36.7, 32.7, 27.6, 23.1, 22.0, 12.0; HRMS calcd for C23H32O5 [M+]: 388.2250, found 388.2243.
3.2.5 17β-二氢马烯雌酮(8)的合成
氩气保护下, 化合物21(0.4 mmol, 155 mg)与通氩气除氧后的1 mL 0.1 mol/L对甲苯磺酸的甲醇溶液混合, 50 ℃反应过夜. TLC监控反应至中间体基本转化.浓缩溶剂, 用乙酸乙酯稀释, 加入饱和碳酸氢钠至中性, 分液, 水洗, 饱和氯化钠洗, 无水Na2SO4干燥, 柱层析纯化, 得到88.0 mg白色固体, 收率81%. m.p. 172~175 ℃(dec.); [α]D30 +191.5 (c 0.50, THF); 1H NMR (Methanol-d4, 400 MHz) δ: 7.01 (d, J=8.4 Hz, 1H), 6.60 (dd, J=8.4, 2.8 Hz, 1H), 6.50 (d, J=2.8 Hz, 1H), 5.37~5.32 (m, 1H), 3.76 (t, J=8.4 Hz, 1H), 3.43~3.21 (m, 2H), 3.03~2.94 (m, 1H), 2.10 (m, 2H), 1.98~1.87 (m, 2H), 1.79~1.68 (m, 1H), 1.56~1.38 (m, 4H), 0.61 (s, 3H); 13C NMR (Methanol-d4, 101 MHz) δ: 156.0, 138.6, 135.3, 130.3, 129.7, 115.1, 115.0, 114.7, 82.7, 51.4, 46.4, 41.9, 38.7, 33.9, 30.7, 30.7, 21.7, 11.8; HRMS calcd for C18H22O2 270.1620, found 270.1626.
3.2.6 马烯雌酮(7)的合成
氩气保护下, 化合物8 (0.5 mmol, 135 mg)和异丙醇铝(0.5 mmol, 120 mg)的混合物中加入5 mL干燥的甲苯和环己酮(20 mmol, 2.0 mL)中, 130 ℃反应1.25 h, TLC监控至绝大部分原料已转化, 降至室温后加入1 mL水搅拌10 min后, 加入乙酸乙酯和水稀释, 分液, 饱和氯化钠溶液洗, 无水Na2SO4干燥, 柱层析纯化, 得到115 mg白色固体7, 收率86%. m.p. 232~236 ℃ (dec.); [α]D30 +286.0 (c 0.50, THF); 1H NMR (DMSO-d6, 400 MHz) δ: 9.10 (s, 1H), 7.04 (d, J=8.4 Hz, 1H), 6.59 (d, J=8.4 Hz, 1H), 6.49 (s, 1H), 5.47 (s, 1H), 3.40 (d, J=22.8 Hz, 1H), 3.29 (d, J=22.8 Hz, 1H), 3.12~2.97 (m, 1H), 2.49~2.40 (m, 1H), 2.32 (m, 1H), 2.21~2.12 (m, 2H), 1.93~1.76 (m, 2H), 1.76~1.68 (m, 1H), 1.55 (td, J=13.2, 4.0 Hz, 1H), 1.34 (qd, J=13.0, 4.0 Hz, 1H), 0.68 (s, 3H); 13C NMR (DMSO-d6, 101 MHz) δ: 219.1, 155.1, 135.9, 133.5, 128.5, 127.6, 115.4, 113.9, 113.9, 49.7, 48.8, 39.6, 35.3, 31.8, 31.7, 29.2, 19.3, 13.4; HRMS calcd for C18H20O2 268.1463, found 268.1457.
3.2.7 3-苯甲酰基-17β-二氢马烯雌酮(22)的合成
氩气保护下, 化合物8(1 mmol, 270 mg)溶解于1 mL THF(四氢呋喃), 加入NaOH (20 mmol, 800 mg)的水溶液(5 mL), 40 ℃反应30 min, 置于冰浴, 大量固体析出, 滴加苯甲酰氯(5 mmol, 0.58 mL), 补加2 mL THF, 缓慢升至室温. TLC监控反应, 原料全部反应后, 浓缩, 加入乙酸乙酯和水稀释, 分液, 水洗, 无水Na2SO4干燥, 快速柱层析, 以85%收率得到318 mg白色固体22. m.p. 172~173 ℃; [α]D30 +148.8 (c 0.50, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 8.20 (d, J=7.2 Hz, 2H), 7.63 (t, J=7.2 Hz, 1H), 7.50 (t, J=7.2 Hz, 2H), 7.28 (d, J=8.4 Hz, 1H), 7.02 (dd, J=8.4, 2.0 Hz, 1H), 6.97 (d, J=2.0 Hz, 1H), 5.41 (s, 1H), 3.86 (t, J=8.4 Hz, 1H), 3.53 (d, J=22.0 Hz, 1H), 3.43 (d, J=22.0 Hz, 1H), 3.19~3.12 (m, 1H), 2.25~2.15 (m, 2H), 2.04~1.91 (m, 2H), 1.82~1.71 (m, 1H), 1.66~1.43 (m, 5H), 0.67 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 165.4, 148.7, 136.7, 135.5, 134.8, 133.5, 130.1, 129.4, 128.8, 128.5, 120.9, 119.4, 114.0, 82.0, 50.2, 45.2, 40.7, 37.5, 32.4, 30.5, 29.7, 20.8, 11.2; HRMS calcd for C25H26O3 374.1882, found 374.1877.
3.2.8 3-苯甲酰基-17α-甲酰基二氢马烯雌酮(24)的合成
氩气保护下, 在冰浴中向化合物22 (0.5 mmol, 187 mg)加入甲苯(4 mL)、1, 8-二氮杂二环十一碳-7-烯(1.2 mmol, 182 mg)和全氟丁基磺酰氟(0.6 mmol, 181 mg), 维持冰浴反应约10 min后, TLC监控反应原料转化完全.用少量粗硅胶快速过滤(洗脱液为甲苯), 抽干得到白色固体粗品23.
氩气保护下, 向23粗品中加入1, 8-二氮杂二环十一碳-7-烯(2 mmol, 304 mg)和甲酸(2 mmol, 92 mg)的THF (4 mL)混合溶液, 40 ℃反应30 h.浓缩, 加乙酸乙酯和水, 分液, 有机相水洗两次, 无水Na2SO4干燥, 柱层析纯化, 得到182 mg白色固体24, 两步分离收率81%. m.p. 166~167 ℃; [α]D30 +136.4 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 8.20 (d, J=7.6 Hz, 2H), 8.12 (s, 1H), 7.63 (t, J=7.6 Hz, 1H), 7.51 (t, J=7.6 Hz, 2H), 7.29 (d, J=8.4 Hz, 1H), 7.03 (dd, J=8.4, 2.4 Hz, 1H), 6.98 (d, J=2.4 Hz, 1H), 5.47 (s, 1H), 5.05 (d, J=6.0 Hz, 1H), 3.54 (d, J=22.0 Hz, 1H), 3.44 (d, J=22.0 Hz, 1H), 3.20 (dd, J=11.2, 5.6 Hz, 1H), 2.46~2.38 (m, 1H), 2.36~2.29 (m, 1H), 2.24~2.16 (m, 1H), 1.86 (td, J=14.0, 13.6, 4.0 Hz, 1H), 1.79~1.56 (m, 5H), 0.70 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ: 165.4, 160.7, 148.8, 136.5, 135.4, 134.7, 133.5, 130.1, 129.6, 128.9, 128.5, 120.9, 119.4, 114.4, 81.3, 49.4, 46.4, 40.5, 32.7, 32.0, 30.2, 29.8, 22.0, 16.5; HRMS calcd for C26H26O4 402.1831, found 402.1839.
3.2.9 17α-二氢马烯雌酮(9)的合成
氩气保护下, 向化合物24 (0.2 mmol, 80.4 mg)的甲醇溶液(2 mL)中加入KCO3 (0.3 mmol, 41.5 mg), 室温反应30 min, 体系由浑浊转为澄清, TLC发现原料反应完全.浓缩, 加入乙酸乙酯和水溶解固体, 分液, 有机相饱和碳酸氢钠洗, 水洗, 无水Na2SO4干燥, 柱层析纯化, 得到51.9 mg微黄的油状物9, 收率96%. [α]D30 +178.6 (c 0.75, THF); 1H NMR (Acetone-d6, 400 MHz) δ: 7.05 (d, J=8.4 Hz, 1H), 6.63 (dd, J=8.4, 2.4 Hz, 1H), 6.53 (d, J=2.4 Hz, 1H), 5.37 (s, 1H), 3.75 (d, J=6.0 Hz, 1H), 3.36 (d, J=21.6 Hz, 1H), 3.25 (d, J=21.6 Hz, 1H), 3.02~2.98 (m, 1H), 2.45~2.41 (m, 1H), 2.20~2.09 (m, 2H), 2.06~2.00 (m, 2H), 1.63~1.38 (m, 4H), 0.52 (s, 3H); 13C NMR (Methanol-d4, 101 MHz) δ: 155.9, 139.2, 135.4, 130.4, 129.8, 115.0, 114.9, 114.7, 80.2, 49.4, 48.5, 41.9, 33.8, 33.6, 32.9, 30.8, 23.0, 17.5; HRMS calcd for C18H22O2 270.1620, found 270.1618.
辅助材料(Supporting Information) 中间体和产物的1H NMR和13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) Grodin, J. M. ; Siiteri, P. K. ; Macdonald, P. C. J. Clin. Endocrinol. Metab. 1973, 36, 207.
(b) Longcope, C. Am. J. Obstet. Gynecol. 1971, 111, 778.
(c) Purohit, A. ; Tutill, H. J. ; Day, J. M. ; Chander, S. K. ; Lawrence, H. R. ; Allan, G. M. ; Fischer, D. S. ; Vicker, N. ; Newman, S. P. ; Potter, B. V. L. ; Reed, M. J. Mol. Cell. Endocrinol. 2006, 248, 199. -
[2]
Bhavnani, B. R.; Stanczyk, F. Z. J. Steroid Biochem. Mol. Biol. 2014, 142, 16. doi: 10.1016/j.jsbmb.2013.10.011
-
[3]
(a) Jing, Y. ; Xu, C. -G. ; Ding, K. ; Lin, J. -R. ; Jin, R. -H. ; Tian, W. -S. Tetrahedron Lett. 2010, 51, 3242.
(b) Jing, Y. M. S. Thesis, Shanghai Normal University, Shanghai, 2010 (in Chinese).
(景羽, 硕士论文, 上海师范大学, 上海, 2010. ) -
[4]
Yue, T.; Li, H. -P.; Ding, K. Tetrahedron Lett. 2016, 57, 4850. doi: 10.1016/j.tetlet.2016.09.062
-
[5]
(a) Zderic, J. A. ; Bowers, A. ; Carpio, H. ; Djerassi, C. J. Am. Chem. Soc. 1958, 80, 2596.
(b) Zderic, J. A. ; Carpio, H. ; Bowers, A. ; Djerassi, C. Steroids 1963, 1, 233.
(c) Bagli, J. F. ; Morand, P. F. ; Wiesner, K. ; Gaudry, R. Tetrahedron Lett. 1964, 5, 387.
(d) Stein, R. P. ; Buzby, G. C. ; Smith, H. Tetrahedron Lett. 1966, (41), 5015.
(e) Stein, R. P. ; Buzby, G. C. ; Smith, H. Tetrahedron 1970, 26, 1917.
(f) Bailey, E. J. ; Gale, A. ; Phillipp, G. ; Siddons, P. T. ; Smith, G., Chem. Commun. 1967, 1253.
(g) Marshall, D. J. ; Deghengh, R. Can. J. Chem. 1969, 47, 3127. -
[6]
Zheng, D. -Q.; Jing, Y.; Zheng, B. -Y.; Ye, Y. -F.; Xu, S.; Tian, W. -S.; Ma, H. -Y.; Ding, K. Tetrahedron 2016, 72, 2164. doi: 10.1016/j.tet.2016.03.002
-
[7]
(a) Nicolaou, K. C. ; Zhong, Y. L. ; Baran, P. S. J. Am. Chem. Soc. 2000, 122, 7596.
(b) Nicolaou, K. C. ; Montagnon, T. ; Baran, P. S. ; Zhong, Y. L. J. Am. Chem. Soc. 2002, 124, 2245.
(c) Chen, K. ; Liu, C. ; Deng, L. ; Xu, G. Steroids 2010, 75, 513. -
[8]
(a) Hogg, J. A. ; Lincoln, F. H. ; Nathan, A. H. ; Hanze, A. R. ; Schneider, W. P. ; Beal, P. F. ; Korman, J. J. Am. Chem. Soc. 1955, 77, 4438.
(b) Allen, G. R. ; Weiss, M. J. J. Am. Chem. Soc. 1959, 81, 4968.
(c) Schaub, R. E. ; Allen, G. R. ; Weiss, M. J., J. Am. Chem. Soc. 1959, 81, 4962. -
[9]
Larock, R. C.; Hightower, T. R.; Kraus, G. A.; Hahn, P.; Zheng, D. Tetrahedron Lett. 1995, 36, 2423. doi: 10.1016/0040-4039(95)00306-W
-
[10]
Fryszkowska, A.; Peterson, J.; Davies, N. L. Org. Process Res. Dev. 2016, 20, 1520. doi: 10.1021/acs.oprd.6b00215
-
[11]
Guo, P. -P.; Ding, K. Tetrahedron Lett. 2015, 56, 4096. doi: 10.1016/j.tetlet.2015.05.026
-
[12]
(a) Wang, W. -Y. ; Jin, H. -A. ; Yan, Z. -H. ; He, M. -C. ; Lin, S. ; Tian, W. -S. Tetrahedron Lett. 2017, 58, 3489.
(b) Yan, Z. -H. ; Jin, H. -A. ; Yu, X. -Q. ; Wang, W. -Y. ; Tian, W. -S. Chin. J. Org. Chem. 2017, 37, 196(in Chinese).
(严兆华, 金红爱, 余信权, 王汪阳, 田伟生, 有机化学, 2017, 37, 196. )
(c) Yan, Z. -H. ; Tian, H. ; Zhao, D. -D. ; Jin, H. -A. ; Tian, W. -S. Chin. Chem. Lett. 2016, 27, 96.
(d) Yan, Z. -H. ; Zhao, D. -D. ; Hu, W. ; Tian, H. ; Li, X. -S. ; Tian, W. -S. Chem. J. Chin. Univ. 2015, 36, 2441(in Chinese).
(严兆华, 赵冬冬, 胡伟, 田欢, 李小松, 田伟生, 高等学校化学学报, 2015, 36, 2441. )
-
[1]
-
表 1 1, 2-脱氢反应的优化
Table 1. Optimization of 1, 2-dehydrogenation
Condition Result 1 IBX, DMSO, toluene, 85 ℃ No reaction 2 DDQ, dioxane, 110 ℃ No reaction 3 DDQ, TBSCl, dioxane, r.t. No reaction 4 Bromination and elimination
(various conditions)Complex 5 (ⅰ) LHMDS, TMSCl, -78 ℃
(ⅱ) 5 mol% Pd(OAc)2, O2, DMSO88% yield  下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 24
- 文章访问数: 3740
- HTML全文浏览量: 885
 下载:
下载:
