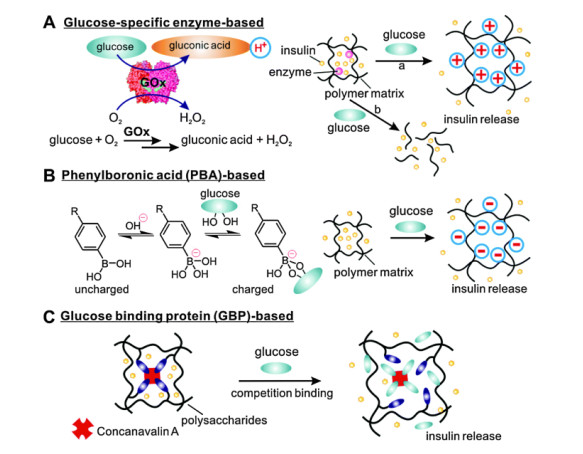
 图1
闭路智能胰岛素载药体系常见的三种葡萄糖分子响应机理
Figure1.
Three different mechanisms of the glucose-responsiveness in the classic closed-loop and smart insulin delivery systems
图1
闭路智能胰岛素载药体系常见的三种葡萄糖分子响应机理
Figure1.
Three different mechanisms of the glucose-responsiveness in the classic closed-loop and smart insulin delivery systems
引用本文:
李臻益, 胡晓玉, 强琚莉, 张冬梅, 肖守军, 林晨, 王乐勇. 闭路智能胰岛素载药体系的研究进展[J]. 有机化学,
2018, 38(1): 29-39.
doi:
10.6023/cjoc201708065

Citation: Li Zhenyi, Hu Xiaoyu, Jiang Juli, Zhang Dongmei, Xiao Shoujun, Lin Chen, Wang Leyong. Recent Advances in Closed-Loop and Smart Insulin Delivery Systems[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 29-39. doi: 10.6023/cjoc201708065
Citation: Li Zhenyi, Hu Xiaoyu, Jiang Juli, Zhang Dongmei, Xiao Shoujun, Lin Chen, Wang Leyong. Recent Advances in Closed-Loop and Smart Insulin Delivery Systems[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 29-39. doi: 10.6023/cjoc201708065
闭路智能胰岛素载药体系的研究进展
English
Recent Advances in Closed-Loop and Smart Insulin Delivery Systems
Abstract:
Insulin is a commonly prescribed drug for the treatment of type Ⅰ and type Ⅱ diabetes. As for the efficiency in controlling blood glucose level, insulin therapy is one of the most effective treatments for diabetes. The current administration route of insulin is mainly through subcutaneous injection, which leads to many undesirable side effects such as pain, local tissue necrosis or infection, and nerve damage. Recently, various closed-loop and smart insulin delivery systems have been developed based on the emerging nanotechnologies. Recent progress in the construction of closed-loop and smart insulin delivery system, which mainly focuses on the response mechanism, different strategies for fabricating the carrier matrix, and the regulation principle of the smart insulin release is described. Advantages and drawbacks of the current insulin delivery systems are also discussed, along with the opportunities and challenges in future.
-
Key words:
- diabetes
- / insulin
- / glucose-response
- / closed-loop delivery system
- / smart drug release
-
1 糖尿病概述及胰岛素治疗的给药方式
1.1 糖尿病概述
糖尿病是因胰岛素分泌缺陷或胰岛素敏感性降低, 而造成的以高血糖为特征的慢性代谢型疾病.长期存在的高血糖会导致人体各种组织(如眼、心脏、肾脏、血管、神经组织等)的慢性受损.空腹血糖浓度大于或等于7.0 mmol/L或餐后2 h血糖浓度大于或等于11.1 mmol/L即可确诊为糖尿病.常见的糖尿病主要分为Ⅰ型糖尿病和Ⅱ型糖尿病两种[1]. Ⅰ型糖尿病患病的年龄分布广泛, 主要病因是由于胰腺中负责胰岛素生成的β细胞在自身免疫反应中受损[2], 导致自身胰岛素分泌不足. Ⅰ型糖尿病受遗传因素的影响, 常存在家族发病倾向, 外界环境因素对发病也存在一定的影响[3]. Ⅱ型糖尿病则常见于中老年人, 肥胖者患病率高, 常常伴随有高血压、动脉硬化等其它疾病. Ⅱ型糖尿病的发病机制较为复杂, 但存在明显的遗传异质性[4].目前尚无根治糖尿病的有效方法, 但可以通过多种治疗手段来控制糖尿病患者的病情.目前糖尿病的治疗方法主要有以下几种:预防治疗、饮食治疗、运动治疗以及胰岛素治疗.轻微的糖尿病患者可以通过饮食及运动疗法来控制病情的进一步发展, 存在康复的可能性; 对于Ⅰ型糖尿病患者以及晚期Ⅱ型糖尿病患者胰岛素治疗的效果是最优的也是必不可少的[5].
1.2 胰岛素给药体系的主要类型
人工合成胰岛素的实现为糖尿病的胰岛素干预治疗提供了可行性[6].但是, 由于胰岛素是生物大分子, 其可渗透能力较弱, 难以穿透体内的各种生理屏障, 同时胰岛素的稳定性较差, 容易受到外界化学因素或生物酶降解作用的影响, 使其生物活性大大降低[7], 因此, 需要对胰岛素先进行包载及转运, 在到达病灶区域时再进行释放, 以维持其生物活性和功能.现阶段胰岛素给药体系主要分为以下几种类型[8]: (1)口服给药体系、(2)通过鼻腔的喷雾类给药体系、(3)通过口腔的吸入类给药体系、(4)经皮给药体系及(5)皮下穿刺给药体系.其中, 皮下穿刺注射胰岛素是最为常用的糖尿病治疗方法.糖尿病患者需要在就餐前注射短效针, 睡前注射长效针, 以调节因进食或睡眠引起的血糖浓度波动, 进而维持血糖浓度处于正常水平, 使生物机体能够正常运作.长期皮下注射不仅给患者带来穿刺的疼痛感还有可能造成局部组织麻痹或感染以及神经受损等毒副作用[9], 大大降低了糖尿病患者的生活质量.此外, 由于皮下穿刺注射胰岛素区域的不同, 深入肌肉组织的深度不同, 以及注射药量的不同都可能对胰岛素的有效吸收产生影响[6b].因此, 亟待开发更为有效且具有良好顺从性的胰岛素载药体系.随着纳米技术的日益成熟, 利用纳米技术合成策略构筑的载药体系受到人们的广泛关注[8b, 10], 闭路智能胰岛素载药体系随之应运而生并得以迅速发展.
闭路智能胰岛素载药体系是一类将葡萄糖浓度检测与胰岛素智能释放有机结合在一起的一类新型的载药体系.这类载药体系能够模拟人类胰腺的功能, 可以检测所处环境中血糖浓度的不同, 进而做出智能响应:当检测到外界血糖浓度超出正常范围时, 释放出体系中包载的胰岛素来降低血糖浓度; 而当外界血糖浓度处于正常范围时则不会出现响应, 从而避免了胰岛素释放过量所导致的低血糖等不良症状, 并在长时间内维持血糖浓度处于正常的范围, 有效防止血糖浓度出现大幅度波动.这类新型载药体系实现了从血糖检测到胰岛素释放再到血糖检测的闭回路, 因此这类集血糖检测与胰岛素智能释放于一体的新型载药体系被称之为“闭路智能胰岛素载药体系”[11].
2 闭路智能胰岛素载药体系的主要类型
闭路智能胰岛素载药体系的载运母体大部分都是由高分子生化材料构筑的, 具备良好的机械强度与生物相容性, 同时载药母体的结构中存在化学响应基团能够实现所包载胰岛素的可控释放.基于超分子自聚集构筑的胰岛素载运体系最近也有报道[12].人工合成胰岛素分子通常包裹在载运母体中.这些载体可以包载或自身含有对葡萄糖分子具有响应性的功能基团, 通过对葡萄糖分子响应机理的不同可将闭路智能胰岛素载药体系大致分为如图 1中所示的三类[13]: (1)基于葡萄糖氧化酶(GOx)催化机理的载药体系、(2)基于苯硼酸单元(PBA)与葡萄糖可逆结合作用的载药体系以及(3)基于糖结合蛋白质(GBP)与糖类物质竞争结合作用的载药体系.虽然上述三类载药体系对葡萄糖分子的响应机理存在差异, 但药物的释放原理不外乎载运母体发生形变或者载运母体被降解, 破坏了原有载药体系的结构从而使包载的胰岛素分子得以释放.因此, 基于胰岛素载药体系响应机制的不同, 我们将近年来闭路智能胰岛素载药体系分为上述三大类进行简要概述.
图1
闭路智能胰岛素载药体系常见的三种葡萄糖分子响应机理
Figure1.
Three different mechanisms of the glucose-responsiveness in the classic closed-loop and smart insulin delivery systems
2.1 基于葡萄糖氧化酶(GOx)催化机理的胰岛素载药体系
葡萄糖氧化酶(GOx)是一种葡萄糖专属催化酶, 它能够在消耗氧气将葡萄糖转化为葡萄糖酸的同时释放出过氧化氢[14]. GOx催化产生葡萄糖酸的速率与外界葡萄糖的浓度有密切关系, 因此GOx被广泛应用于葡萄糖检测以及具有葡萄糖响应性能的胰岛素载运系统中[15]. GOx对葡萄糖分子有高效灵敏的响应性, 在GOx的催化下, 葡萄糖转化为葡萄糖酸的同时也使得催化区域内的pH值出现下降, 因此GOx常常包裹在具有pH响应性能的胰岛素载运母体中, 载体由于催化区域内pH值的下降, 出现不同程度的形变或者水解作用, 原有的结构与性能被破坏, 从而释放出包载在其中的胰岛素分子[16].由此, 将两者有机整合在一起即可构筑闭路智能胰岛素载运体系.
Wu课题组[17]在2010年报道了纳米混杂生物有机膜用于胰岛素的载运及释放.通过交联聚合物p(NIPAM-MAA)微凝胶纳米颗粒、MnO2纳米颗粒、GOx和过氧化氢酶(CAT, 用于分解催化反应中产生的过氧化氢)形成纳米混杂生物有机膜, 随后进行胰岛素的包载.在外界高葡萄糖浓度下, 由于GOx的催化作用, 体系pH值降低引起聚合物p(NIPAM-MAA)微凝胶纳米颗粒的体积收缩, 使得生物有机膜呈现多孔状态, 包载的胰岛素得以有效释放. MnO2纳米颗粒不仅能够增强生物有机膜的机械强度, 同时也是过氧化氢的淬灭剂, 进一步提升了GOx的稳定性与催化效率.该纳米混杂生物有机膜体系能够使患Ⅰ型糖尿病小鼠的血糖浓度维持在正常水平长达4 d. Kim课题组[18]也有类似的工作报道.他们将GOx和CeNPs包载于多孔的硅纳米颗粒中, 再与药物分子一同包裹在具有pH响应的壳聚糖微凝胶中.当接触外界葡萄糖时, 由于GOx催化作用, 体系pH值下降引起凝胶体积膨胀, 从而使包载药物分子得以可控释放.
顾臻课题组[11a]于2013年报道了基于GOx催化机理的可注射纳米网络结构的新型胰岛素载药体系(图 2).他们首先利用双重乳液的封装方法将人工合成胰岛素分子、GOx和CAT包载进乙缩醛改性的糖苷中, 从而形成具有pH响应性能的纳米颗粒.在GOx的催化作用下, 葡萄糖转化形成葡萄糖酸时pH值降低引起改性糖苷分解为水溶性的右旋糖苷和小分子的丙酮与乙醇, 纳米颗粒随之被降解, 伴随着所包载胰岛素的释放.为了避免在纳米粒子降解过程中胰岛素的爆发式释放, 他们在纳米颗粒外层分别附上正电荷分子和负电荷分子, 然后将两者混合, 利用异种电荷间的相互吸引作用形成了纳米网络结构.该纳米网络具有多孔的三维立体结构, 为催化反应提供了许多高效的反应位点, 同时外界的葡萄糖分子及包载的胰岛素分子均能够有效地在纳米网络中进出.该载药体系在外界葡萄糖浓度处于高糖浓度(400 mg/dL)时能够迅速释放胰岛素来降低葡萄糖浓度, 在外界葡萄糖浓度处于正常水平(100 mg/dL)时则基本无释放, 避免了胰岛素过量释放引起的低血糖症状, 实现了体外高效智能的胰岛素释放调控.此外, 该纳米网络结构还具有类似可注射凝胶的性能, 这使得其在应用方面有了更大的发挥空间.在患Ⅰ型糖尿病小鼠体内注射一次能够维持其体内葡萄糖浓度在正常水平长达10 d之久, 具有更为良好的治疗效果.
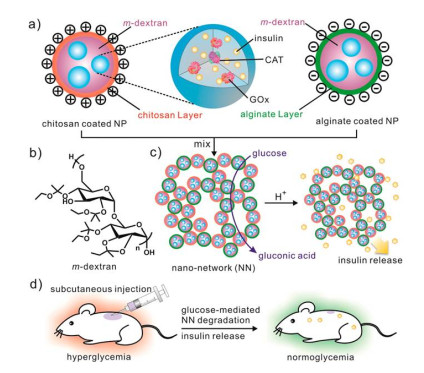 图2
可注射纳米网络结构胰岛素载运体系示意图
Figure2.
Schematic illustration of the construction and the glucose-responsive mechanism of the injectable nano-network
图2
可注射纳米网络结构胰岛素载运体系示意图
Figure2.
Schematic illustration of the construction and the glucose-responsive mechanism of the injectable nano-network
同年, 顾臻课题组[19]报道了利用羧化辅酶(TPP)作催化剂合成了以壳聚糖为主体的微型凝胶, 在微型凝胶中同时包载了GOx、催化剂(CAT)及人工合成胰岛素.壳聚糖分子中含有氨基官能团, 在催化作用发生时, 区域pH值下降, 氨基质子化, 链与链之间的排斥作用增强, 使得凝胶膨胀, 包载的胰岛素得以释放.
葡萄糖催化反应过程中存在区域环境的pH值下降, 构筑不同形态的pH响应载药母体(网状凝胶结构、纳米颗粒、囊泡结构等)就可以得到不同类型的pH响应性载药体系, 而缩酮结构单元是典型的酸敏感型响应基团.基于此原理, 顾臻课题组[20]在2014年进一步构筑了基于缩酮结构单元对pH具有响应性的囊泡结构, 用于胰岛素的载运和可控释放.他们利用尾端含有缩酮结构的两亲性分子PEG-p(Ser-Ketal)构筑了囊泡粒子, 同时将GOx、CAT以及人工合成胰岛素分子包载于囊泡内部.双亲分子自组装形成的囊泡膜能够有效地防止包载胰岛素的泄露[21], 同时由于葡萄糖分子是中性小分子, 能够自由地穿透囊泡膜进入囊泡内部[22].当外界葡萄糖浓度到达较高浓度时, 葡萄糖分子迅速扩散进入囊泡内部, 与GOx接触, 催化反应随之发生, 囊泡内区域环境的pH值由于葡萄糖酸的产生而下降, 两亲性分子的尾端的缩酮部分随之酸化水解, 使得整个分子由原来的两亲性分子转化为水溶性分子, 囊泡结构被破坏, 使得包载在囊泡内部的胰岛素得以释放.而当外界葡萄糖浓度处于正常水平时, 葡萄糖分子扩散缓慢, 包载的胰岛素基本无释放, 达到了应对外界葡萄糖不同浓度做出智能释放的效果.这类囊泡分子能够负载在温敏型可注射水凝胶的网状结构中, 通过皮下注射进入作用区域.在患Ⅰ型糖尿病的小鼠实验中能够长时间维持小鼠体内葡萄糖浓度处于正常水平, 具有良好的应用前景.
赵文茹课题组[16b]在2011年构筑了多层酶修饰的硅纳米颗粒, 用于葡萄糖响应的胰岛素包载与释放.利用硅纳米颗粒的多孔结构作为胰岛素的载运空腔, 在包载有胰岛素的硅纳米颗粒外表面通过戊二醛交联作用修饰上GOx与CAT的多层酶结构, 用作胰岛素释放的调节开关.当该体系处于不同的葡萄糖浓度时, 在GOx催化作用下葡萄糖转化为葡萄糖酸, 引起区域环境pH值不同程度的下降, 进而使多层酶结构产生不同程度的扩张, 包封在内层孔径中的胰岛素因而也得到不同程度的可控释放.
上述几篇报道对于GOx催化反应过程中产生的过氧化氢都采用了CAT分解的方法, CAT分解过氧化氢的反应过程中又会释放出氧气, 而GOx催化过程则需要消耗氧气, 理论上说这是一个良性循环[11].但不可避免在实际过程中存在部分催化产生的过氧化氢未能及时被分解, 残留的过氧化氢会降低GOx的活性, 进而影响体系整体对葡萄糖分子的响应和催化效率, 对正常细胞也存在一定的毒害作用[23], 同时CAT的包载会挤占包载空间, 使胰岛素药物分子的包载效率相对降低.另外, 由于pH响应较为缓慢, 不能迅速实现胰岛素的有效释放, 因此非pH响应的胰岛素快速释放体系受到人们的广泛关注与深入研究.
过氧化氢是常见的氧化剂, 具有中等强度的氧化能力.如果利用催化过程中产生的过氧化氢通过氧化还原反应来破坏载运母体的结构或性能, 既能达到药物释放的效果, 也能消除催化反应体系中生成的过氧化氢对载药体系带来的负作用.基于上述设计, 顾臻课题组[24]在2017年报道了基于过氧化氢响应的新型胰岛素载药体系(图 3).他们用在生理pH条件下对过氧化氢有高效响应性的两亲性分子mPEG-b-p(Ser-PBE)取代了对pH响应的两亲性分子PEG-p(Ser-Ketal).同样, 利用两亲性分子的结构特性构筑成囊泡结构并进行人工合成胰岛素与GOx的包载.当外界处于高葡萄糖浓度时, 葡萄糖分子迅速扩散进入囊泡中, 与GOx接触, 催化反应过程中产生过氧化氢, 此时两亲性分子在过氧化氢氧化作用下失去PBE侧链基团, 分解为水溶性的分子mPEG-b-polyserine, 囊泡由于分子亲疏水性质的转变随之破裂, 胰岛素得以高效快速地释放.该体系不同于上述几个基于对pH敏感单元构筑的载药体系, 因为在活体实验中, 生物体生理pH值存在着不可预知的迅速转变[13], 给胰岛素的释放带来了不可控的因素; 另外, 通过pH变化引起载运母体形变或者水解的速率相对缓慢[25], 不能达到高效快速释放胰岛素的要求, 因此限制了pH响应性胰岛素载药体系的进一步应用.而该体系是基于对过氧化氢响应的胰岛素载药体系, 生理pH值变化幅度内不会影响胰岛素的包载、转运以及智能释放.在临床应用方面, 用透明质酸制作成经皮微针贴纸中的微型针头[26], 该针头具有很好的生物相容性和机械强度.将包载胰岛素的囊泡负载于微型针头中, 当微针帖纸附着在皮肤表面时, 微针深入皮肤表面的毛细血管及淋巴血管, 若血管处于高浓度的葡萄糖环境时即可实现囊泡的破裂与胰岛素的释放[27].相较于现阶段常见的皮下穿刺, 能够明显减轻糖尿病人的痛苦感, 携带也更简便.
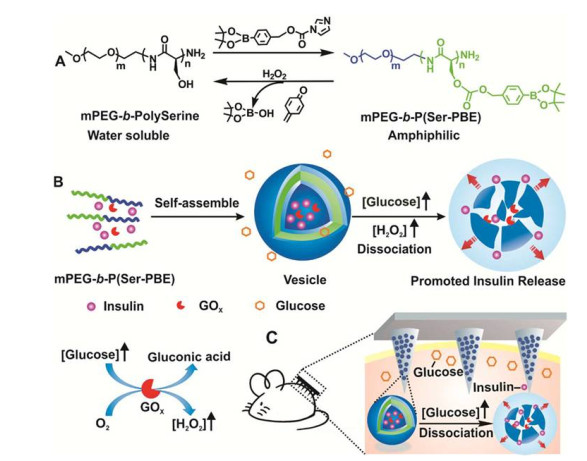 图3
基于过氧化氢响应的胰岛素载运体系的示意图
Figure3.
Schematic illustration of the H2O2-responsive mechanism for glucose-mediated insulin delivery system
图3
基于过氧化氢响应的胰岛素载运体系的示意图
Figure3.
Schematic illustration of the H2O2-responsive mechanism for glucose-mediated insulin delivery system
在GOx催化过程中除了pH值的下降, 过氧化氢的生成, 还有就是氧气的消耗.因此在催化反应过程中就形成了一个相对乏氧的区域. 2-硝基咪唑单元是疏水性的单元, 常用于癌症细胞组织的细胞显色[28].它在生物体乏氧环境中可以被硝基还原酶催化经过单电子还原形成亲水性的2-氨基咪唑单元[29].因此, 2015年顾臻课题组[30]利用上述2-硝基咪唑在乏氧环境下亲疏水性质转变的原理, 报道了一种基于乏氧环境响应的新型胰岛素载药体系.他们用2-硝基咪唑修饰的透明质酸作为两亲性分子构筑成囊泡, 并对GOx和人工合成胰岛素进行包载.该体系利用区域乏氧环境从无到有作为胰岛素释放的调控开关, 当外界环境处于高葡萄糖浓度时, 随着葡萄糖扩散进入囊泡中, 与GOx发生反应, 消耗区域内的氧气, 使得该区域出现相对乏氧环境, 从而引起2-硝基咪唑单元转化为2-氨基咪唑单元, 囊泡分子的亲疏水性质发生转变, 由原来的两亲性分子转变为水溶性分子, 囊泡随之破裂, 包载在囊泡中的胰岛素也随之得到释放.该囊泡同样能够负载于微针贴纸的微型针头中, 进行经皮的胰岛素释放与治疗.在患Ⅰ型糖尿病的小鼠实验中, 该体系能够对高葡萄糖浓度做出快速响应, 但对于正常葡萄糖浓度则基本无释放, 具备智能释放的性能, 有潜在的应用前景.
上述乏氧响应体系对葡萄糖分子有灵敏的响应速率, 生理环境下的pH波动对该体系也没有很大的影响, 但该体系并未对催化产生的过氧化氢做出一个合适的处理, 所以, 顾臻课题组[31]进一步改进了两亲性分子的结构, 他们在2017年进一步构筑了基于乏氧环境和过氧化氢双重响应的载药体系(图 4).在该体系中, 他们首先构筑了一个两亲性分子PEG-p(Ser-S-NI), 它不仅具备常见两亲性分子的特性, 能够自组装形成囊泡.而且, 该两亲性分子具有两段比较特殊的结构单元, 尾端疏水性的2-硝基咪唑单元, 在乏氧环境下可以被生物体内的硝基还原酶转变为亲水性的2-氨基咪唑单元, 从而实现亲疏水性质的转变; 中间是疏水性的硫醚单元, 可以被过氧化氢氧化形成亲水性的砜结构基团, 进一步加速亲疏水性质的转变.因此, 该囊泡在外界高浓度葡萄糖条件下, 随着葡萄糖扩散进入囊泡与GOx发生催化作用, 消耗区域内氧气生成区域乏氧环境的同时产生过氧化氢, 两者协调作用将原来两亲性的分子转变为水溶性的分子, 囊泡结构因此被破坏, 包载的胰岛素也随着得到有效的释放.在活体小鼠实验中也证实了其具有良好的糖尿病治疗效果.
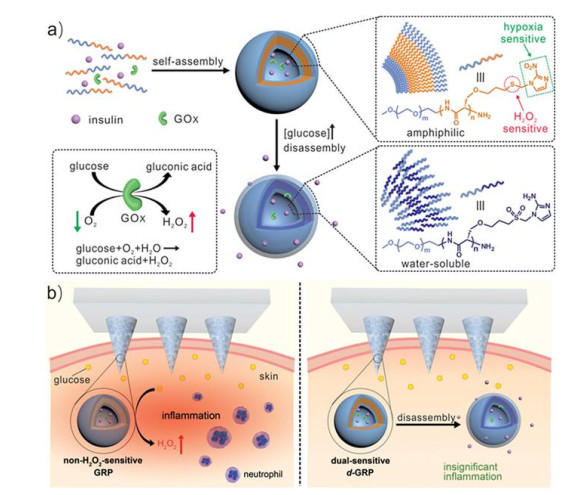 图4
基于乏氧环境和过氧化氢双重响应的胰岛素载运体系
Figure4.
Schematic illustration of the smart insulin delivery system using hypoxia and H2O2 dual-sensitive polymersome-based vesicles
图4
基于乏氧环境和过氧化氢双重响应的胰岛素载运体系
Figure4.
Schematic illustration of the smart insulin delivery system using hypoxia and H2O2 dual-sensitive polymersome-based vesicles
该体系是基于乏氧环境的产生来调控囊泡分子的亲疏水性质, 若采用CAT来淬灭产生的过氧化氢则会在淬灭的同时产生氧气和水, 使得区域乏氧环境被削减, 从而降低GOx的催化效率, 进而降低了胰岛素的释放速率与效率.硫醚键的引入既不会对GOx的催化效率造成影响, 又较使用CAT能提高体系胰岛素的包载率和葡萄糖响应速率; 另外硫醚键在消耗过氧化氢的同时进一步促进两亲性分子向亲水性分子的转变, 加速囊泡的破裂和胰岛素的释放; 过氧化氢的消耗使得体系对正常细胞的毒副作用大幅度降低, 在生物相容性能上有了较大的提升.综上可知, 此类基于乏氧环境和过氧化氢双重响应性的新型胰岛素载药体系涵盖了GOx催化机理的许多优点, 在活体实验中也取得了不错的效果, 具有很强的应用潜能与广阔的发展前景.
此外, 除了利用GOx的催化机制, 经过改性的GOx同样也能够应用于载药体系中.吴伟泰课题组[32]在2016年报道了一种葡萄糖专一响应的聚合物微凝胶, 其具有潜在的胰岛素载运与释放功能.该聚合物微凝胶是将apo-GOx掺杂在聚异丙基丙烯酰胺聚合物网络中形成半互穿结构的聚合物微凝胶. apo-GOx是一种去除辅酶处于未激活状态的葡萄糖氧化酶, 它与葡萄糖之间仍存在很强的结合能力与高选择性, 但不再具有催化作用. apo-GOx与葡萄糖可逆结合作用过程中存在构象变化, 进而能够迅速引起微凝胶体积的改变, 微凝胶体积随体系外界葡萄糖浓度的增加或降低产生相应地膨胀或收缩, 具备了智能调控的性能; 其次apo-GOx只对葡萄糖有响应, 而对于其它的糖类、多糖及糖蛋白都不存在结合作用, 因此, 具有专一选择性; 另外apo-GOx的自身荧光强度随葡萄糖浓度的增加而增加, 具备了初步可视化的功能; 最后apo-GOx对葡萄糖的敏感响应范围处在糖尿病临床相关的血糖浓度范围内, 为实现胰岛素载运提供了可能.
2.2 基于苯硼酸单元(PBA)与葡萄糖可逆结合作用的胰岛素载药体系
苯硼酸基团与含有顺式二醇链的糖类化合物的可逆结合作用被Lorand与Edwards发现以来[33], 已经被广泛应用于碳水化合物的检测与纯化及药物载运等方面[34].在这类胰岛素载药体系中, 载运母体的结构中通常都含有苯硼酸基团及其衍生物.苯硼酸单元既是糖类物质检测响应的功能位点, 又能够在与糖类物质可逆结合后转变为带有负电性的结构单元, 在分子间同种电荷相互排斥的作用下, 使聚合物网状载运母体结构出现不同程度的膨胀或破损[35], 进而使包载的药物相应地得以释放.苯硼酸单元具有较好的稳定性[15b], 为长期稳定载药提供了可能性; 同时由于硼化学的多样性, 通过化学改性, 使得基于苯硼酸衍生物的感应材料在较宽的葡萄糖浓度范围以及pH范围内都有很好的响应作用, 在临床医学及工业应用方面都有潜在的应用前景.但由于苯硼酸的相对选择性较差, 可能会与血液中的α-羟基酸或其他二醇链的糖类化合物结合[36], 使得载药的可控性与精确度降低.
血糖浓度的实时监测值是糖尿病临床医疗诊断与治疗的重要依据之一[37], 因此血糖浓度的可视化检测具有重要的临床意义与广阔的应用前景.葡萄糖浓度可视化是将葡萄糖浓度转化为其它可见形式的信号(如光学信号、电学信号等)来表达. Zhou课题组[38]在2010年报道了一种新型的具有可视化葡萄糖检测与胰岛素智能调控释放性能的多功能混合型纳米凝胶的载药体系.他们首先将高分子聚合物p(VPBA-b-DMAEA)包裹在银纳米颗粒的表面, 形成混合型纳米凝胶网络结构.这样的网络结构由于高分子聚合物链的存在能够提高体系的机械强度, 同时多孔网状的结构特性能够实现对人工合成胰岛素的高效包载.高分子聚合物链中苯硼酸单元(PBA)经过化学改性, pKa值下降至7左右, 能够在生理pH值和生理温度下对葡萄糖浓度(0~30 mmol/L)有高效率和高选择性的响应.该纳米凝胶的高分子外壳随外界葡萄糖浓度的升高或降低出现相应的膨胀与收缩.在纳米凝胶外壳体积膨胀时, 包载的药物被释放, 实现了包载胰岛素随纳米凝胶体积对外界葡萄糖浓度的响应变化而实现智能释放.纳米凝胶的内核是银纳米粒子, 作为该体系的荧光感应单元, 纳米凝胶外壳体积膨胀或收缩时, 银纳米颗粒的外围物化环境相应也发生变化, 从而使得其荧光强度发生改变, 即葡萄糖浓度影响凝胶外壳体积膨胀或收缩, 凝胶外壳体积膨胀或收缩影响银纳米粒子的荧光强度, 使得葡萄糖浓度通过荧光强度就可以得到可视化检测.但该体系也存在有一定缺陷, 因为葡萄糖浓度与荧光强度之间没有线性对应关系, 通过荧光强度大小只能在粗略地反应体系所处环境葡萄糖浓度的高低, 而不是相对精确地测定葡萄糖浓度的具体数值.
造成上述体系非线性对应关系的原因是凝胶体系内的苯硼酸单元与葡萄糖在低浓度条件下会形成2:1的络合物, 此时葡萄糖分子具有类似交联剂的作用, 使得凝胶体积缩小; 而在高浓度条件下两者则形成1:1的带负电荷的络合物, 由于同种电荷排斥作用, 使得凝胶体积膨胀, 因此产生了葡萄糖浓度与凝胶体积的非对应关系[39].
为克服上述问题, 研究发现1, 2-二醇类衍生物与苯硼酸的结合能力比1, 3-二醇类化合物的结合能力强[40], 因此在低温条件下先用1, 3-二醇类化合物将苯硼酸单元全部结合形成1:1络合物, 保证在其与葡萄糖等1, 2-二醇类衍生物结合时, 通过竞争结合只出现1:1络合物, 进而实现凝胶体积只出现膨胀现象, 这使得葡萄糖浓度与凝胶体积具有线性对应关系.由于1, 3-二醇类衍生物的存在使得凝胶体积只出现线性膨胀, 因此把1, 3-二醇类化合物称为体积调节剂. 2014年, Braun课题组[41]就报道了此类具有线性葡萄糖浓度可视化的水凝胶.他们首先将丙烯酰胺溶于结晶态聚苯乙烯胶体悬浮液中, 聚合形成结晶胶体阵列的光子晶体聚合物.随后, 其进一步将部分酰胺位点水解为羧酸位点, 再通过硼化反应产生硼酸单元, 最后加入体积调节剂聚乙烯醇, 最终制备了目标光子晶体水凝胶[42].光子晶体水凝胶的光学反射波长是水凝胶体积的函数, 测定凝胶的光学反射波长就可以测定出凝胶体积[43]; 由于体积调节剂的存在, 葡萄糖浓度与凝胶体积为对应线性关系.因此只要检测凝胶在所处环境下的光学反射波长, 经过数学演算即可将光学信号(反射波长)转化为具体的葡萄糖浓度数值, 实现了葡萄糖浓度的可视化检测.设想将这类光子晶体水凝胶应用于胰岛素载药体系, 光子晶体水凝胶既作为载运母体, 通过体积变化智能释放胰岛素分子, 同时又是葡萄糖浓度的检测器, 将可视化检测与智能药物释放有机结合, 相信在未来的胰岛素载药体系中有着广阔的发展前景.
以囊泡结构作为载运母体是胰岛素载药体系中常见的一种载药手段.史林启课题组[44]在2010年利用高分子链PEG-b-p(AA-co-APBA)自组装形成囊泡, 将人工合成胰岛素包载在囊泡中, 当外界葡萄糖扩散进入囊泡内部时, 苯硼酸单元与葡萄糖结合由原来的平面三角形结构转变为四面体结构, 降低了体系在中性pH环境下对葡萄糖的响应灵敏性, 只有当葡萄糖浓度达到5 g/L时才能彻底破坏囊泡结构, 释放包载的胰岛素.为解决灵敏性较差的问题, 2012年史林启课题组[45]构筑了一种基于苯硼酸单元与不同糖类分子竞争结合作用的新型载药体系.其将PEG-b-P(AA-co-APBA)分子与P(AA-co-AGA)分子混合自组装形成囊泡.该囊泡内核是由p(AA-co-APBA)分子中的苯硼酸单元与p(AA-co-AGA)分子中的糖类衍生物单元可逆结合形成, 囊泡外壳是则是水溶性的聚乙二醇链(PEG).这样的结构能够有效地防止囊泡进一步聚集, 囊泡结构更加稳定; 同时囊泡的纳米级尺寸有利于对葡萄糖小分子的响应速率; 相比糖类高分子衍生物, 葡萄糖与苯硼酸单元的结合作用更强, 因此也提高了体系对葡萄糖的响应效率.该体系初步解决了对葡萄糖响应速度缓慢及效率低下的问题, 但在生物相容性方面存在不足.因此, 2013年史林启课题组[46]基于上述构想, 进一步改进载药体系, 采用生物可降解的PEG-b-p(Asp-co-AspPBA)分子与p(AA-co-AGA)分子构筑混合囊泡作为胰岛素载运母体, 并在体外实验中取得了良好的效果.
杨晶课题组[47]在2012年构筑了基于竞争结合作用引起亲疏水性质转变的新型胰岛素载运体系.他们首先设计了mPEG5000-block-PBDEMA两亲性分子, 它能够在中性pH条件下自组装形成内腔亲水的囊泡结构.由于苯硼酸单元与葡萄糖分子的结合作用比与环二醇强, 因此, 囊泡处于外界高葡萄糖浓度时葡萄糖扩散进入囊泡空腔, 苯硼酸与环二醇解络合转与葡萄糖结合, 引起囊泡分子由原来的两亲性分子转变为亲水性分子, 囊泡结构随之解体, 包载的胰岛素从而得到有效释放.
目前, 闭路智能胰岛素载运体系中利用的囊泡结构大部分是由生物高分子材料构成, 具备了较好的生物相容性和良好的机械强度.而基于超分子自聚集构筑囊泡结构用于胰岛素载运的体系则相对少见.因此, 王乐勇课题组[12]在2017年报道了基于水溶性柱[5]芳烃与吡啶硼酸衍生物主客体作用构筑的超分子囊泡用于胰岛素的载运与可控释放(图 5).超分子主客体复合物是客体分子进入主体大环分子的空腔, 通过非共价键作用结合形成的, 基于主客体作用构筑的超分子两亲性分子由于其非共价键作用的本质, 可以进行结构、形貌的可逆转换, 也能够对外界刺激做出响应, 近年来被广泛应用于药物转运中[48].水溶性柱[5]芳烃是内部为疏水性空腔, 环端边缘为亲水基团的刚性大环主体分子, 它具有良好的生物相容性[49], 为其应用于载药体系提供了可能, 另外它与吡啶盐单元具有较强的主客体作用[50].因此, 我们设计合成了一个含有长烷基链作为疏水端的吡啶硼酸衍生物, 它能够与水溶性柱[5]芳烃通过主客体作用形成稳定的超分子两亲体, 进一步自组装形成超分子囊泡结构.该囊泡能够对重组胰岛素进行高效地包载.当囊泡处于高葡萄糖浓度环境时, 葡萄糖与吡啶硼酸衍生物结合成亲水性更强的复合物, 两亲性主客体平衡被打破, 主客体结构被破坏, 囊泡破裂, 胰岛素被释放.在体外实验中, 该体系能够对外界不同葡萄糖浓度做出智能响应.该体系为实现以小分子/超分子做载运母体用于胰岛素载药体系提供了新的思路与方法.
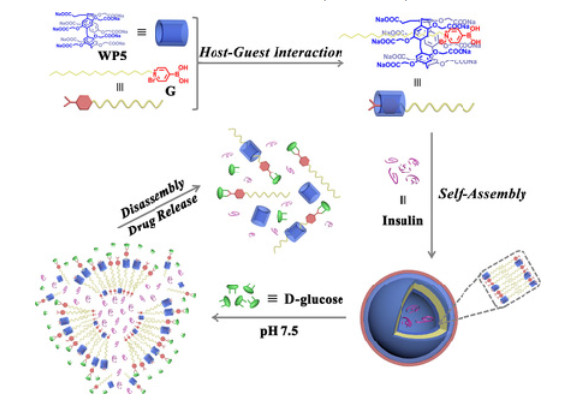 图5
基于水溶性柱[5]芳烃与吡啶硼酸衍生物主客体作用构筑的超分子囊泡用于胰岛素载运的示意图
Figure5.
Schematic illustration of the smart insulin delivery system using supramolecular vesicles
图5
基于水溶性柱[5]芳烃与吡啶硼酸衍生物主客体作用构筑的超分子囊泡用于胰岛素载运的示意图
Figure5.
Schematic illustration of the smart insulin delivery system using supramolecular vesicles
除了囊泡结构的药物载运体系, 含有苯硼酸单元的高分子聚合物构筑的纳米粒子也被广泛用于胰岛素的载运.张新歌课题组[51]多次报道了利用含有苯硼酸单元的聚合物单体(A)与含有氨基葡萄糖的聚合物单体(B)聚合形成纳米小球颗粒, 并包载胰岛素, 通过与外界葡萄糖的竞争结合作用或外界环境pH值的变化, 改变纳米小球颗粒的结构形态来实现葡萄糖响应的胰岛素智能释放.例如, 该课题组[52]在2014年通过RAFT聚合制备了二嵌段的聚合物分子p(AAPBA-b-GAMA).聚合物单体AAPBA中的苯硼酸单元作为对葡萄糖的响应单元, 聚合物单体GAMA中含有二醇链, 具有可再生性能, 同时能够降低聚合物分子整体的生物毒性.聚合物分子p(AAPBA-b-GAMA)通过分子内及分子间的结合作用自聚集形成聚合物纳米小球颗粒, 随其所处的环境中葡萄糖浓度的高低实现不同程度的膨胀, 从而达到包载胰岛素的智能响应与释放.
王柯敏课题组[53]在2015年报道了聚合物p(VPBA-DMAEA)修饰的多孔二氧化硅纳米粒子载运体系.该体系将有机高分子材料p(VPBA-DMAEA)与无机多孔二氧化硅纳米粒子通过化学改性相结合形成混合纳米粒子.该体系在中性或偏碱性的pH值环境时, 聚合物分子呈现堆叠坍塌状, 将二氧化硅纳米粒子的表面孔径堵塞住, 能够有效防止孔径内部包载药物的释放; 然而, 当体系处于偏酸性的pH值环境时, 由于聚合物链间的电荷作用以及增溶作用, 使得聚合物分子链之间相互排斥, 不再堆叠, 二氧化硅纳米粒子的表面孔径疏通, 包载的药物分子也随之得以释放.此外, 聚合物分子中含有的PBA单元, 是常见的葡萄糖识别单元, 又因为其与葡萄糖可逆结合后高分子链之间产生电荷排斥作用, 可以作为包载药物的释放开关, 具有潜在的胰岛素载运与可控释放功能.
介孔二氧化硅纳米粒子不论在体外还是在体内试验中都被证实具有非常好的生物相容性, 能够保证包载药物的零预释放[54].介孔材料不仅外表面拥有许多负载位点, 内部同样也可以负载药物, 成为双药载运体系的常用载体.环腺苷酸(cAMP)能够激活胰腺β细胞的钙离子通道, 从而进一步促进生物体自身胰岛素的分泌[55].环腺苷酸(cAMP)与胰岛素的共同释放有助于提高Ⅱ型糖尿病的理疗效果[55b, 56].因此, Lin课题组[57]在2009报道了葡萄糖响应的胰岛素与环腺苷酸(cAMP)双药智能释放的介孔二氧化硅纳米粒子载药体系(图 6).环腺苷酸(cAMP)自身难以穿透生物膜, 而包载于介孔二氧化硅纳米粒子内部孔径时, 可以通过细胞内吞作用随纳米粒子进入细胞内部.将介孔二氧化硅纳米粒子的外表面修饰上苯硼酸单元, 化学改性的胰岛素分子与苯硼酸作用, 结合在纳米粒子的外表面上, 达到胰岛素负载的效果, 环腺苷酸(cAMP)则包载于介孔二氧化硅纳米粒子的内部孔径中.体系在与葡萄糖接触时, 由于竞争结合作用, 苯硼酸单元转与葡萄糖结合, 胰岛素得以释放的同时, 打开了内部孔径通往外界的通道, 环腺苷酸(cAMP)也随之释放.该双药载运与智能释放体系的构筑为进一步实现复杂高效的载药体系提供了可能.
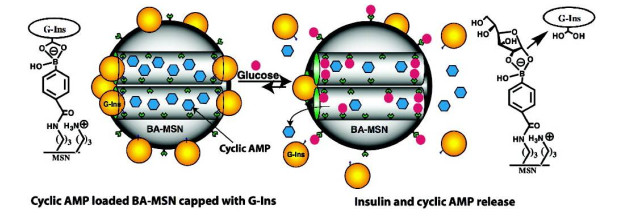 图6
胰岛素与环腺苷酸双药智能释放的介孔二氧化硅纳米粒子用于胰岛素载运的示意图
Figure6.
Schematic illustration of the glucose-responsive MSN-based delivery system for simultaneous release of insulin and cAMP
图6
胰岛素与环腺苷酸双药智能释放的介孔二氧化硅纳米粒子用于胰岛素载运的示意图
Figure6.
Schematic illustration of the glucose-responsive MSN-based delivery system for simultaneous release of insulin and cAMP
2.3 基于葡萄糖结合蛋白质(GBP)与糖类物质竞争结合作用的胰岛素载药体系
葡萄糖结合蛋白质(GBP)是一种天然的蛋白质, 具有与碳水化合物相结合的性能, 其主要为外源凝集素, 可用于细胞信号转导及细胞分子识别[58].伴刀豆凝集素A (Con A)就是最为常见的一种糖类结合蛋白质, 它与葡萄糖具有很强的结合能力, 因此在胰岛素载药体系中被广泛使用[59]. Con A具有四个能与糖类物质相结合的位点, 通过糖基化作用, 葡萄糖酸改性的胰岛素能够与Con A形成稳定的络合物.但当葡萄糖分子介入时, 由于Con A与葡萄糖的结合常数更大, 结合能力更强, 竞争结合作用使得Con A与葡萄糖酸改性胰岛素解络合, 转而与葡萄糖结合, 使得葡萄糖酸改性的胰岛素得以释放[60].但因为Con A糖类结合蛋白质的生物相容性不理想, 因此在一定程度上限制了其临床应用.寻找新型的糖结合蛋白质或改进现有的糖结合蛋白成为未来胰岛素载药体系的发展方向之一.
聂俊课题组[61]在2011年报道了Con A和糖类衍生物(GEMA)的复合物单体与甲基丙烯酸二甲胺乙酯(DMAEMA)单体通过光聚合作用制备的微型水凝胶胰岛素载药体系.由于GEMA的侧链端含有糖类结构单元, 与Con A糖结合蛋白存在结合作用, 而这种结合作用能够被葡萄糖的竞争结合作用所破坏, 凝胶网络结构间的交联作用减弱, 凝胶体积膨胀, 包载的胰岛素随之得以释放.此外, 聚合物单体DMAEMA中含有叔胺官能团, 在酸性条件下可被质子化, 并且具有很强的灵敏度, 即使对生理条件下微小的pH变化也能够做出有效的响应.因此, 当pH值下降时, 水凝胶内部叔胺质子化, 链与链之间的排斥作用增强, 使得凝胶体积膨胀, 实现包载胰岛素的释放.该体系具有葡萄糖与pH双重响应性, 在体外实验中也具有着良好的实验结果.
杜学忠课题组[62]于2013年报道了一个以Con A糖蛋白作调控开关, 甘露糖修饰的介孔二氧化硅纳米粒子为载体的载药体系.他们首先将甘露糖基团修饰在二氧化硅纳米颗粒表面, 当药物分子包载于纳米颗粒表面的孔洞中后利用Con A糖结合蛋白与甘露糖的结合作用可以将药物分子封锁在孔洞中, 从而实现药物的包载.在酸性条件下或者有葡萄糖分子存在时, Con A糖结合蛋白与甘露糖解络合, 包载的药物得以释放.虽然, 他们在实验中未使用胰岛素作为药物分子, 但其利用葡萄糖与Con A糖结合蛋白的特殊结合作用作为葡萄糖的响应与检测, 利用不同糖类分子与Con A糖结合蛋白的竞争结合作用作为包载药物释放的开关, 同时葡萄糖浓度高低与该系统药物的释放速率快慢相一致, 因此, 该体系具备了闭路智能载药体系的特征.
3 总结与展望
利用微米或纳米技术合成策略构筑的闭路胰岛素智能载药体系在实验和临床方面都取得了一定的进展, 并具有巨大的治疗潜能与应用前景.这些利用微米或纳米级别的载运母体所构筑的胰岛素载药体系能够克服传统胰岛素给药体系的缺点与局限性.尽可能减少皮下穿刺的次数, 延长一次穿刺治疗的有效作用时间或者通过其他给药途径来缓解多次穿刺造成的不良影响.尽管其存在多方面的理论优势, 但如果进一步实现临床层面上的应用, 还存在许多问题有待解决.
首先, 现阶段的胰岛素载药体系与传统皮下穿刺给药方式相比, 将血糖浓度控制在同一正常水平所需的胰岛素剂量要大很多.因为胰岛素也是一种生长激素, 人体长时间处于高胰岛素浓度下可能会引起内分泌系统的紊乱, 进而引发一些未知的生理疾病[63].因此, 在未来的闭路胰岛素智能载药体系的设计开发过程中必须进一步提高胰岛素作用效率, 用最少的药物剂量来实现血糖浓度的智能调控并且能够长时间维持机体处于正常血糖浓度.
其次, 精确的胰岛素的释放量还需要进一步优化.当胰岛素包裹在载运母体中进入人体, 由于生物壁垒的存在, 很难确定最终达到作用位点时释放的胰岛素精确值.载药体系必须具备有灵敏的葡萄糖响应位点或具有时序性药物释放的功能, 这样能够随时根据外界葡萄糖浓度做出合理的胰岛素释放.因此, 更加高效灵敏的葡萄糖分子响应检测器件和分步时序性释放载药体系有待进一步的研发.
再者, 能否维持胰岛素分子在载运母体中以及释放时的物化和生理性能是新的载药体系开发时必须考虑的关键问题.人工合成的胰岛素通过化学改性能够有效提高其稳定性, 例如唾液酸改性的胰岛素[64]、聚乙二醇改性的胰岛素等[65].但化学改性胰岛素稳定性提高的同时, 可能会引起患者体重降低等毒副作用.因此, 对新型胰岛素药物分子的研发是未来胰岛素载药体系发展的必经之路.
最后, 现阶段载药体系的载运母体材料多为利用微米或纳米技术合成策略制备的具有良好生物相容性的高分子材料或有机超分子材料.随着生物工程、干细胞工程等新兴生物技术的大力发展, 整合生物技术来协调构建生物相容性更好的载运母体是未来胰岛素载药体系进一步发展的方向.
-
-
[1]
(a) Stumvoll, M.; Goldstein, B. J.; van Haeften, T. W. Lancet 2005, 365, 1333.
(b) Atkinson, M. A.; Eisenbarth, G. S. Lancet 2001, 358, 221. -
[2]
(a) Bach, J. F. Endocr. Rev. 1994, 15, 516.
(b) Eisenbarth, G. S.; Flier, J. S.; Cahill, G. N. Engl. J. Med. 1986, 314, 1360.
(c) Tisch, R.; McDevitt, H. Cell 1996, 85, 291. -
[3]
(a) Concannon, P.; Rich, S. S.; Nepom, G. T. N. Engl. J. Med. 2009, 360, 1646.
(b) Daneman, D. Lancet 2006, 367, 847.
(c) Davies, J. L.; Kawaguchi, Y.; Bennett, S. T.; Copeman, J. B.; Cordell, H. J.; Pritchard, L. E.; Reed, P. W.; Gough, S. C. L.; Jenkins, S. C.; Palmer, S. M.; Balfour, K. M.; Rowe, B. R.; Farrall, M.; Barnett, A. H.; Bain, S. C.; Todd, J. A. Nature 1994, 371, 130. -
[4]
(a) Boden, G.; Shulman, G. I. Eur. J. Clin. Invest. 2002, 32, 14.
(b) Sturnvoll, M.; Goldstein, B. J.; van Haeften, T. W. Endocr. Res. 2007, 32, 19. -
[5]
(a) Hamaty, M. Cleve. Clin. J. Med. 2011, 78, 332.
(b) Hayward, R. A.; Manning, W. G.; Kaplan, S. H.; Wagner, E. H.; Greenfield, S. J. Am. Med. Assoc. 1997, 278, 1663. -
[6]
(a) Owens, D. R.; Zinman, B.; Bolli, G. B. Lancet 2001, 358, 739.
(b) Gualandi-Signorini, A. M.; Giorgi, G. Eur. Rev. Med. Pharmacol. Sci. 2001, 5, 73. -
[7]
Langguth, P.; Bohner, V.; Heizmann, J.; Merkle, H. P.; Wolffram, S.; Amidon, G. L.; Yamashita, S. J. Controlled Release 1997, 46, 39. doi: 10.1016/S0168-3659(96)01586-6
-
[8]
(a) Heinemann, L.; Pfutzner, A.; Heise, T. Curr. Pharm. Des. 2001, 7, 1327.
(b) Owens, D. R. Nat. Rev. Drug Discovery 2002, 1, 529. -
[9]
(a) Buzasi, K.; Sapi, Z.; Jermendy, G. Diabetes Res. Clin. Pract. 2011, 94, E34.
(b) Chantelau, E.; Spraul, M.; Muhlhauser, I.; Gause, R.; Berger, M. Diabetologia 1989, 32, 421.
(c) Richardson, T.; Kerr, D. Am. J. Clin. Dermatol. 2003, 4, 661. -
[10]
(a) Jeandidier, N.; Boivin, S. Adv. Drug Delivery Rev. 1999, 35, 179.
(b) Wu, W.; Zhou, S. Macromol. Biosci. 2013, 13, 1464.
(c) Bratlie, K. M.; York, R. L.; Invernale, M. A.; Langer, R.; Anderson, D. G. Adv. Healthcare Mater. 2012, 1, 267.
(d) Ravaine, V.; Ancla, C.; Catargi, B. J. Controlled Release 2008, 132, 2. -
[11]
(a) Gu, Z. ; Aimetti, A. A. ; Wang, Q. ; Dang, T. T. ; Zhang, Y. ; Veiseh, O. ; Cheng, H. ; Langer, R. S. ; Anderson, D. G. ACS Nano 2013, 7, 4194.
(b) Zhang, Y. Q. ; Yu, J. C. ; Shen, Q. D. ; Gu, Z. Prog. Chem. 2015, 27, 11(in Chinese).
(张宇琪, 俞计成, 沈群东, 顾臻, 化学进展, 2015, 27, 11. ) -
[12]
Gao, L.; Wang, T.; Jia, K.; Wu, X.; Yao, C.; Shao, W.; Zhang, D.; Hu, X.-Y.; Wang, L. Chem.-Eur. J. 2017, 23, 6605. doi: 10.1002/chem.v23.27
-
[13]
Mo, R.; Jiang, T.; Di, J.; Tai, W.; Gu, Z. Chem. Soc. Rev. 2014, 43, 3595. doi: 10.1039/c3cs60436e
-
[14]
Bankar, S. B.; Bule, M. V.; Singhal, R. S.; Ananthanarayan, L. Biotechnol. Adv. 2009, 27, 489. doi: 10.1016/j.biotechadv.2009.04.003
-
[15]
(a) Steiner, M.-S.; Duerkop, A.; Wolfbeis, O. S. Chem. Soc. Rev. 2011, 40, 4805.
(b) Wu, Q.; Wang, L.; Yu, H.; Wang, J.; Chen, Z. Chem. Rev. 2011, 111, 7855. -
[16]
(a) Gordijo, C. R.; Koulajian, K.; Shuhendler, A. J.; Bonifacio, L. D.; Huang, H. Y.; Chiang, S.; Ozin, G. A.; Giacca, A.; Wu, X. Y. Adv. Funct. Mater. 2011, 21, 73.
(b) Zhao, W.; Zhang, H.; He, Q.; Li, Y.; Gu, J.; Li, L.; Li, H.; Shi, J. Chem. Commun. 2011, 47, 9459. -
[17]
Gordijo, C. R.; Shuhendler, A. J.; Wu, X. Y. Adv. Funct. Mater. 2010, 20, 1404. doi: 10.1002/adfm.200901581
-
[18]
Kim, M. Y.; Kim, J. ACS Biomater. Sci Eng. 2017, 3, 572. doi: 10.1021/acsbiomaterials.6b00716
-
[19]
Gu, Z.; Dang, T. T.; Ma, M.; Tang, B. C.; Cheng, H.; Jiang, S.; Dong, Y.; Zhang, Y.; Anderson, D. G. ACS Nano 2013, 7, 6758. doi: 10.1021/nn401617u
-
[20]
Tai, W.; Mo, R.; Di, J.; Subramanian, V.; Gu, X.; Buse, J. B.; Gu, Z. Biomacromolecules 2014, 15, 3495. doi: 10.1021/bm500364a
-
[21]
Discher, D. E.; Ahmed, F. Annu. Rev. Biomed. Eng. 2006, 8, 323. doi: 10.1146/annurev.bioeng.8.061505.095838
-
[22]
Napoli, A.; Boerakker, M. J.; Tirelli, N.; Nolte, R. J. M.; Sommerdijk, N.; Hubbell, J. A. Langmuir 2004, 20, 3487. doi: 10.1021/la0357054
-
[23]
(a) Kohen, R. Biomed. Pharmacother. 1999, 53, 181.
(b) Liu, Y.; Du, J.; Yan, M.; Lau, M. Y.; Hu, J.; Han, H.; Yang, O. O.; Liang, S.; Wei, W.; Wang, H.; Li, J.; Zhu, X.; Shi, L.; Chen, W.; Ji, C.; Lu, Y. Nat. Nanotechnol. 2013, 8, 187. -
[24]
Hu, X.; Yu, J.; Qian, C.; Lu, Y.; Kahkoska, A. R.; Xie, Z.; Jing, X.; Buse, J. B.; Gu, Z. ACS Nano 2017, 11, 613. doi: 10.1021/acsnano.6b06892
-
[25]
Veiseh, O.; Tang, B. C.; Whitehead, K. A.; Anderson, D. G.; Langer, R. Nat. Rev. Drug Discovery 2015, 14, 45. http://www.ncbi.nlm.nih.gov/pubmed/25430866
-
[26]
(a) Donnelly, R. F.; Singh, T. R. R.; Woolfson, A. D. Drug Delivery 2010, 17, 187.
(b) Yang, S.; Wu, F.; Liu, J.; Fan, G.; Welsh, W.; Zhu, H.; Jin, T. Adv. Funct. Mater. 2015, 25, 4633. -
[27]
(a) Harvey, A. J.; Kaestner, S. A.; Sutter, D. E.; Harvey, N. G.; Mikszta, J. A.; Pettis, R. J. Pharm. Res. 2011, 28, 107.
(b) Heo, Y. J.; Shibata, H.; Okitsu, T.; Kawanishi, T.; Takeuchi, S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13399. -
[28]
(a) Krohn, K. A.; Link, J. M.; Mason, R. P. J. Nucl. Med. 2008, 49, 129S.
(b) Nunn, A.; Linder, K.; Strauss, H. W. Eur. J. Nucl. Med. 1995, 22, 265. -
[29]
(a) Edwards, D. I. J. Antimicrob. Chemother. 1993, 31, 9.
(b) Takasawa, M.; Moustafa, R. R.; Baron, J.-C. Stroke 2008, 39, 1629. -
[30]
Yu, J.; Zhang, Y.; Ye, Y.; DiSanto, R.; Sun, W.; Ranson, D.; Ligler, F. S.; Buse, J. B.; Gu, Z. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8260. doi: 10.1073/pnas.1505405112
-
[31]
Yu, J.; Qian, C.; Zhang, Y.; Cui, Z.; Zhu, Y.; Shen, Q.; Ligler, F. S.; Buse, J. B.; Gu, Z. Nano Lett. 2017, 17, 733. doi: 10.1021/acs.nanolett.6b03848
-
[32]
Ye, T.; Bai, X.; Jiang, X.; Wu, Q.; Chen, S.; Qu, A.; Huang, J.; Shen, J.; Wu, W. Polym. Chem. 2016, 7, 2847. doi: 10.1039/C6PY00179C
-
[33]
Lorand, J. P.; Edwards, J. O. J. Org. Chem. 1959, 24, 769. doi: 10.1021/jo01088a011
-
[34]
Preinerstorfer, B.; Laemmerhofer, M.; Lindner, W. J. Sep. Sci. 2009, 32, 1673. doi: 10.1002/jssc.v32:10
-
[35]
(a) Miyake, K.; Tanaka, T.; McNeil, P. L. PLoS One 2007, 2.
(b) Vaz, A. F. M.; Souza, M. P.; Vieira, L. D.; Aguiar, J. S.; Silva, T. G.; Medeiros, P. L.; Melo, A. M. M. A.; Silva-Lucca, R. A.; Santana, L. A.; Oliva, M. L. V.; Perez, K. R.; Cuccovia, I. M.; Coelho, L. C. B. B.; Correia, M. T. S. Radiat. Phys. Chem. 2013, 85, 218. -
[36]
Huang, Y.-J.; Ouyang, W.-J.; Wu, X.; Li, Z.; Fossey, J. S.; James, T. D.; Jiang, Y.-B. J. Am. Chem. Soc. 2013, 135, 1700. doi: 10.1021/ja311442x
-
[37]
(a) Van den Berghe, G.; Wilmer, A.; Hermans, G.; Meersseman, W.; Wouters, P. J.; Milants, I.; Van Wijngaerden, E.; Bobbaers, H.; Bouillon, R. N. Engl. J. Med. 2006, 354, 449.
(b) Van den Berghe, G.; Wouters, P.; Weekers, F.; Verwaest, C.; Bruyninckx, F.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P.; Bouillon, R. N. Engl. J. Med. 2001, 345, 1359. -
[38]
Wu, W.; Mitra, N.; Yan, E. C. Y.; Zhou, S. ACS Nano 2010, 4, 4831. doi: 10.1021/nn1008319
-
[39]
(a) Alexeev, V. L.; Sharma, A. C.; Goponenko, A. V.; Das, S.; Lednev, I. K.; Wilcox, C. S.; Finegold, D. N.; Asher, S. A. Anal. Chem. 2003, 75, 2316.
(b) Zhang, C.; Losego, M. D.; Braun, P. V. Chem. Mater. 2013, 25, 3239. -
[40]
Shiino, D.; Murata, Y.; Kataoka, K.; Koyama, Y.; Yokoyama, M.; Okano, T.; Sakurai, Y. Biomaterials 1994, 15, 121. doi: 10.1016/0142-9612(94)90261-5
-
[41]
Zhang, C.; Cano, G. G.; Braun, P. V. Adv. Mater. 2014, 26, 5678. doi: 10.1002/adma.201401710
-
[42]
Holtz, J. H.; Asher, S. A. Nature 1997, 389, 829. doi: 10.1038/39834
-
[43]
Goponenko, A. V.; Asher, S. A. J. Am. Chem. Soc. 2005, 127, 10753. doi: 10.1021/ja051456p
-
[44]
Wang, B.; Ma, R.; Liu, G.; Liu, X.; Gao, Y.; Shen, J.; An, Y.; Shi, L. Macromol. Rapid Commun. 2010, 31, 1628. doi: 10.1002/marc.201000164
-
[45]
Ma, R.; Yang, H.; Li, Z.; Liu, G.; Sun, X.; Liu, X.; An, Y.; Shi, L. Biomacromolecules 2012, 13, 3409. doi: 10.1021/bm3012715
-
[46]
Yang, H.; Sun, X.; Liu, G.; Ma, R.; Li, Z.; An, Y.; Shi, L. Soft Matter 2013, 9, 8589. doi: 10.1039/c3sm51538a
-
[47]
Yao, Y.; Zhao, L.; Yang, J.; Yang, J. Biomacromolecules 2012, 13, 1837. doi: 10.1021/bm3003286
-
[48]
(a) Cao, Y.; Zou, X.; Xiong, S.; Li, Y.; Shen, Y.; Hu, X.; Wang, L. Chin. J. Chem. 2015, 33, 329.
(b) Hu, X.-Y.; Jia, K.; Cao, Y.; Li, Y.; Qin, S.; Zhou, F.; Lin, C.; Zhang, D.; Wang, L. Chem.-Eur. J. 2015, 21, 1208.
(c) Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Chem. Soc. Rev. 2015, 44, 3568. -
[49]
(a) Chi, X.; Yu, G.; Ji, X.; Li, Y.; Tang, G.; Huang, F. ACS Macro Lett. 2015, 4, 996.
(b) Li, B.; Meng, Z.; Li, Q.; Huang, X.; Kang, Z.; Dong, H.; Chen, J.; Sun, J.; Dong, Y.; Li, J.; Jia, X.; Sessler, J. L.; Meng, Q.; Li, C. Chem. Sci. 2017, 8, 4458. -
[50]
(a) Li, C.; Shu, X.; Li, J.; Chen, S.; Han, K.; Xu, M.; Hu, B.; Yu, Y.; Jia, X. J. Org. Chem. 2011, 76, 8458.
(b) Li, C.; Xu, Q.; Li, J.; Yao, F.; Jia, X. Org. Biomol. Chem. 2010, 8, 1568. -
[51]
(a) Cheng, C.; Zhang, X.; Xiang, J.; Wang, Y.; Zheng, C.; Lu, Z.; Li, C. Soft Matter 2012, 8, 765.
(b) Wang, Y.; Zhang, X.; Han, Y.; Cheng, C.; Li, C. Carbohydr. Polym. 2012, 89, 124. -
[52]
Guo, Q.; Wu, Z.; Zhang, X.; Sun, L.; Li, C. Soft Matter 2014, 10, 911. doi: 10.1039/c3sm52485j
-
[53]
Chang, Y.-J.; Liu, X.-Z.; Zhao, Q.; Yang, X.-H.; Wang, K.-M.; Wang, Q.; Lin, M.; Yang, M. Chin. Chem. Lett. 2015, 26, 1203. doi: 10.1016/j.cclet.2015.08.005
-
[54]
(a) Lee, C.-H.; Cheng, S.-H.; Wang, Y.; Chen, Y.-C.; Chen, N.-T.; Souris, J.; Chen, C.-T.; Mou, C.-Y.; Yang, C.-S.; Lo, L.-W. Adv. Funct. Mater. 2009, 19, 215.
(b) Slowing, I. I.; Wu, C.-W.; Vivero-Escoto, J. L.; Lin, V. S. Y. Small 2009, 5, 57.
(c) Taylor, K. M. L.; Kim, J. S.; Rieter, W. J.; An, H.; Lin, W.; Lin, W. J. Am. Chem. Soc. 2008, 130, 2154. -
[55]
(a) Charles, M. A.; Fanska, R.; Schmid, F. G.; Forsham, P. H.; Grodsky, G. M. Science 1973, 179, 569.
(b) Dyachok, O.; Idevall-Hagren, O.; Sagetorp, J.; Tian, G.; Wuttke, A.; Arrieumerlou, C.; Akusjarvi, G.; Gylfe, E.; Tengholm, A. Cell Metabolism 2008, 8, 26. -
[56]
Tengholm, A. Upsala J. Med. Sci. 2012, 117, 355. doi: 10.3109/03009734.2012.724732
-
[57]
Zhao, Y.; Trewyn, B. G.; Slowing, I. I.; Lin, V. S. Y. J. Am. Chem. Soc. 2009, 131, 8398. doi: 10.1021/ja901831u
-
[58]
Geijtenbeek, T. B. H.; Gringhuis, S. I. Nat. Rev. Immunol. 2009, 9, 465. doi: 10.1038/nri2569
-
[59]
Sharon, N.; Lis, H. Science 1972, 177, 949. doi: 10.1126/science.177.4053.949
-
[60]
(a) Brownlee, M.; Cerami, A. Science 1979, 206, 1190.
(b) Seminoff, L. A.; Gleeson, J. M.; Zheng, J.; Olsen, G. B.; Holmberg, D.; Mohammad, S. F.; Wilson, D.; Kim, S. W. Int. J. Pharm. 1989, 54, 251. -
[61]
Yin, R.; Tong, Z.; Yang, D.; Nie, J. Int. J. Biol. Macromol. 2011, 49, 1137. doi: 10.1016/j.ijbiomac.2011.09.014
-
[62]
Wu, S.; Huang, X.; Du, X. Angew. Chem., Int. Ed. 2013, 52, 5580. doi: 10.1002/anie.201300958
-
[63]
(a) Brogden, R. N.; Heel, R. C. Drugs 1987, 34, 350.
(b) Reichard, P.; Nilsson, B. Y.; Rosenqvist, U. N. Engl. J. Med. 1993, 329, 304. -
[64]
Jain, S.; Hreczuk-Hirst, D. H.; McCormack, B.; Mital, M.; Epenetos, A.; Laing, P.; Gregoriadis, G. Biochim. Biophys. Acta, Gen. Subj. 2003, 1622, 42. doi: 10.1016/S0304-4165(03)00116-8
-
[65]
Bergenstal, R. M.; Rosenstock, J.; Arakaki, R. F.; Prince, M. J.; Qu, Y.; Sinha, V. P.; Howey, D. C.; Jacober, S. J. Diabetes Care 2012, 35, 2140. doi: 10.2337/dc12-0060
-
[1]
-

图 1 闭路智能胰岛素载药体系常见的三种葡萄糖分子响应机理
Figure 1 Three different mechanisms of the glucose-responsiveness in the classic closed-loop and smart insulin delivery systems
Reproduced from Ref. [13] with permission of The Royal Society of Chemistry. http://dx.doi.org/10.1039/C3CS60436E

图 2 可注射纳米网络结构胰岛素载运体系示意图
Figure 2 Schematic illustration of the construction and the glucose-responsive mechanism of the injectable nano-network
Reprinted with permission from Ref. [11a] Copyright 2013 American Chemical Society

图 3 基于过氧化氢响应的胰岛素载运体系的示意图
Figure 3 Schematic illustration of the H2O2-responsive mechanism for glucose-mediated insulin delivery system
Reprinted with permission from Ref. [11] Copyright 2017 American Chemical Society

图 4 基于乏氧环境和过氧化氢双重响应的胰岛素载运体系
Figure 4 Schematic illustration of the smart insulin delivery system using hypoxia and H2O2 dual-sensitive polymersome-based vesicles
Reprinted with permission from Ref. [29] Copyright 2017 American Chemical Society

图 5 基于水溶性柱[5]芳烃与吡啶硼酸衍生物主客体作用构筑的超分子囊泡用于胰岛素载运的示意图
Figure 5 Schematic illustration of the smart insulin delivery system using supramolecular vesicles
Reproduced with permission from Ref. [45] Copyright 2017 Wiley-VCH
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 6631
- HTML全文浏览量: 457
 下载:
下载:
