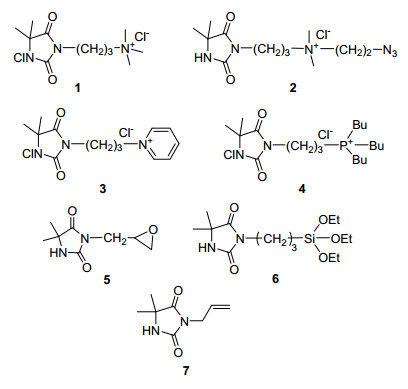
图 1.
部分已报道的DMH氯胺(前体)衍生物结构
Figure 1.
Structure of representative DMH N-chloramine (precursor) derivatives

氯胺是指含一个或多个N-Cl键的有机化合物, 是一类稳定长效、抗菌功能可再生、最为理想的绿色广谱抗菌剂之一[1~3].依靠结构中强氧化性氯, 氯胺可高效杀死绝大部分葡萄球菌、大肠杆菌和绿脓假单胞菌等常见病菌, 甚至包括某些病毒:从而使细菌产生耐药性的可能性甚微[2~4].然而氯胺一旦由胺制得后(N—H→ N—Cl), 亲水性降低, 这非常不利于其触杀进程[5].鉴于此, Li等将季铵盐[6, 7]、吡啶盐[8]和季膦盐[9]等阳离子结构引入到氯胺结构中制备了图 1所示离子型5, 5-二甲基海因(DMH)类氯胺(前体)分子1~4.研究表明, 阳离子结构的引入改善氯胺分子亲水性的同时, 显著提升了氯胺结构的杀菌活性[6~9]; 而且进一步研究发现3有比1更优的抗菌活性[8].尽管前体2通过“click”化学反应被固载到了棉纤维和聚对苯甲二酸乙二醇酯(PET)表面(预先接枝端炔基)[5], 制备了相应的非溶出型抗菌材料, 但这一方法颇显复杂, 直接将1、3和4的氯胺前体固载于惰性材料表面来制备相应抗菌材料还是相当有挑战性的研究课题.
将氯胺前体共价固载于材料表面主要依赖于基质表面的亲电/亲核反应和自由基引发的表面接枝聚合反应.含有键和基团(如硅氧烷基、环氧乙烷基和烯丙基等)的氯胺前体5~6(图 1)曾被分别接枝固载于棉纤维表面[10, 11], 但实现在惰性表面的固载则仍较为困难.虽然7可通过表面接枝聚合反应修饰于PET和聚丙烯(PP)等惰性材料表面[12], 但这一反应体系相对复杂, 丙烯基结构也存在自阻聚问题, 常需与其它单体共聚才能完成有效接枝聚合.
全氟苯基叠氮化合物(PFPA)由于其制备简单, 稳定性较好, 是近年来惰性材料表面修饰研究中较多采用的偶联剂之一[13]. PFPA在紫外光照时下产生氮卡宾, 它可与材料表面N—H/C—H键或C=C键发生插入反应进而实现与表面的共价偶联.采用两步PFPA偶联策略, Li等将肝素(heparin)在不经任何预处理前提下直接以共价键的形式固载到了聚氨酯(PU)膜表面, 而且固载后的肝素保留了相当好的抗凝血栓活性[14].
本文以羟基吡啶为链接, 将PFPA基团引入到吡啶盐-氯胺3的前体结构中, 制备了可固载型氯胺前体分子8~9, 合成方案见Scheme 1.选用在医疗卫生领域常用的聚氨酯(PU膜)材料[15]为模板, 在254 nm光照条件下将前体分子8~9共价固载于PU表面.对表面修饰进行了表征, 初步考察了前体分子结构对表面修饰固载和抗菌性能影响.
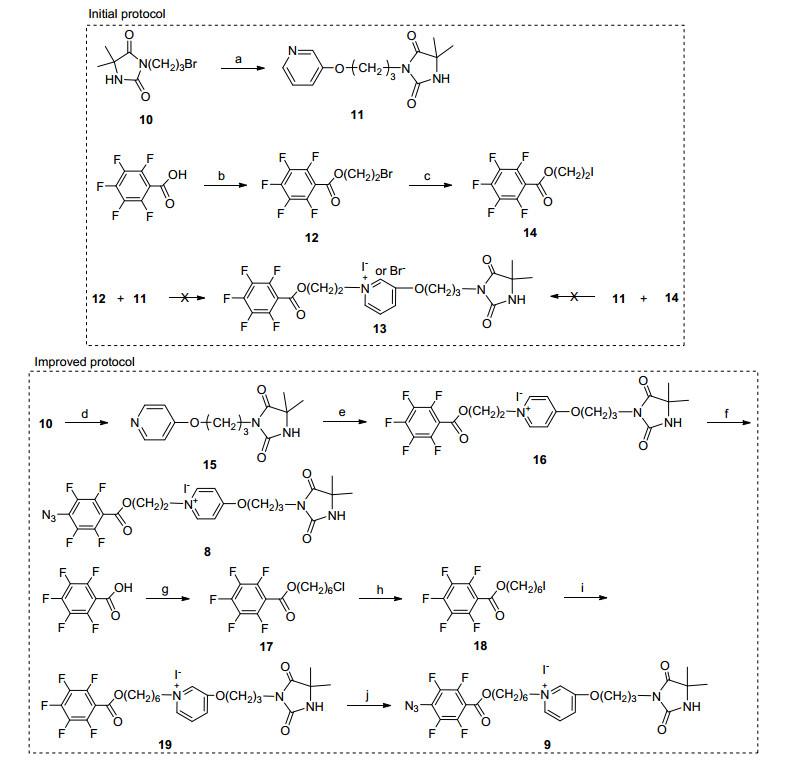
Reagents and conditions: (a) 3-hydroxypyridine, K2CO3, CH3CN, reflux, 2 h, 70.0%; (b) BrCH2CH2OH, dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), CH2Cl2, r.t., 24 h, 94.6%; (c) NaI, acetone, reflux, 8 h, 98.5%; (d) 4-hydroxypyridine, K2CO3, CH3CN, reflux, 35.2%; (e) 14, CH3CN, reflux, 42.0%; (f) NaN3, acetone-H2O, reflux, overnight, 91.0%; (g) Br(CH2)6OH, DCC, DMAP, CH2Cl2, r.t., overnight, 91.1%; (h) NaI, acetone, reflux, 8 h, 92.0%; (i) 11, CH3CN, reflux, overnight, 63.9%; (j) NaN3, acetone-H2O, reflux, overnight, 90.5%.
受本组已有工作启发[16], 最初选用间羟基吡啶为链接单元尝试将DMH和PFPA通过拟形成的吡啶盐单元结合起来.如Scheme 1所示, 间羟基吡啶在碱性条件下烷基化可将DMH单元与吡啶环链接起来制得了中间体11, 随后以五氟苯甲酸为原料制备了溴乙基酯12.然而, 将12与中间体11置于乙腈中回流反应12 h后, 薄层色谱(TLC)检测发现并未有预期产品13的生成.推测可能至少与两方面因素有关: (1)间羟基吡啶结构中羟基为邻对位定位基团, 使得间位吡啶N原子上的电子云密度相对较少, 亲核性也相对较弱, 因此间羟基吡啶N原子上的季铵化反应较为困难; (2) 11与12同为芳香性环结构, 芳环之间π-π stacking作用可能使11和12处于某种定向排布状态, 而12结构中的溴原子与氟代苯环之间的链长(2个CH2)又较短, 不利于12中BrCH2进攻另一芳环间羟基吡啶N原子发生季铵化反应.所以, 我们首先参照文献[17], 将12进行卤素交换反应制备了对应碘代物14, 进一步尝试表明14与11依然无法进行季铵化反应来制备目标分子13, 说明中间体14中亲电活性较强的ICH2并未有效克服本步季铵化反应的瓶颈.
基于上述探索, 我们尝试以对羟基吡啶为原料来制备了中间体15.随后将其与14进行季铵化反应, 成功制得了吡啶盐16, 这显然是相对中间体11而言15中N原子亲核性更强的缘故.此外, 从五氟苯甲酸出发, 经酯化反应和卤素交换反应制备了中间体18, 随后尝试其与11的季铵化制备了吡啶季铵盐19, 这可能是增大了18中碘原子与氟代苯环之间的距离, ICH2有足够的空间许可(6个CH2)进攻11中吡啶环N原子并完成季铵化反应的缘故.最后参考文献方法[18], 在氟代苯环对位引入叠氮基团成功制备了目标分子8和9.
成功制得前体8/9后, 采用文献所述光照偶联实验条件[13, 14]尝试将其共价接枝到PU膜表面(图 2).起初采用FT-IR ATR技术鉴定表面修饰, 然而数据表明, 光固载前后PU膜的ATR谱图几乎没有差异, 这可能是PU材料本身和所合成前体分子均含有相同的官能团(如酰胺基和酯基等)所致.随后借鉴文献思路[14], 采用酸性橙染色法对光照修饰的膜材料PU-9进行了初步验证.如图 2所示, PU-9膜表面呈现棕黄色, 而PU膜略微呈淡黄色.颜色的差异表明PU-9表面阳离子吡啶盐单元的存在, 同时也表明采用PFPA光偶联策略已将前体9经成功固载于PU膜表面.
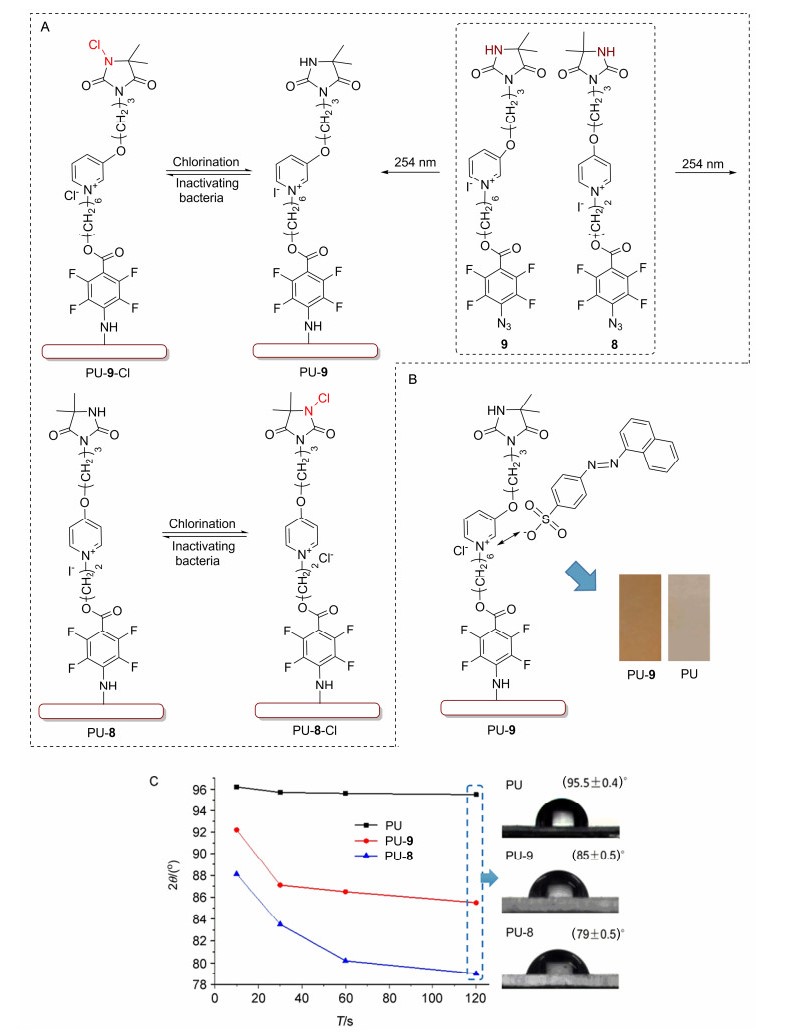
(A) Schematic illustration; (B) visualization of orange sodium salt staining; (C) contact angel data
PU膜表面吡啶盐结构的引入势必会使表面亲水性增加, 因此对PU-8/PU-9进行了接触角测试.考虑到修饰膜表面可能的亲水性, 水滴与PU膜接触会有一个稳定过程, 因此分别测定10、30、60和120 s的静态接触角.如图 2所示, 接触角变化趋势在60 s至120 s基本趋于稳定. PU膜的接触角为(95.5±0.4)°, 呈现疏水性; PU-9的接触角为(85.0±0.5)°, 亲水性略增; 而PU-8的接触角为(79.0±0.5)°, 较PU-9而言亲水性增强更为明显.光照修饰后PU膜亲水性不同程度的增大也表明前体8/9在紫外光照条件下已被成功接枝固载于膜材料表面.
为进一步对膜表面改性进行表征, 采用X射线光电子能谱分析了改性PU膜的表面元素.改性后PU膜的X射线光电子能谱宽扫描及F1s高分辨扫描数据如图 3所示: PU-8及PU-9在685.7 eV结合能出现F1s峰, 表明8/9被成功接枝固载到PU膜表面, 因为基质PU材料中并不含有F元素[14].同时可以看出, PU-9表面F含量(1.07%)低于PU-8表面F含量(1.52%), 这意味着8在PU表面有较高的固载率.这一现象与接触角数据一定程度上相符.前体8本身分子结构较小, 可能在表面的堆砌密度较大, 从而固载较多; 光照后PU-8表面较多的吡啶盐单元可能使得表面亲水性相对较好, 掩盖了表面F原子引入可能带来的疏水性倾向[19].当然, 仅仅依靠前体8/9的结构差别很难准确定位前体结构与固载效率及表面亲水性变化之间的关系, 我们将在今后的工作中深入研究这部分内容.
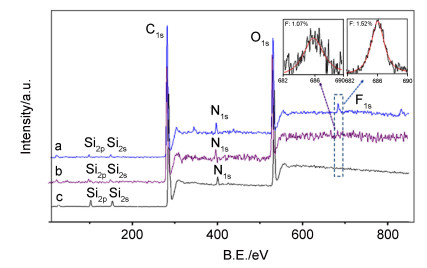
(a) PU-9; (b) PU-8; (c) PU
经稀漂白水处理后得到了表面携有氯胺基团的抗菌材料PU-8-Cl和PU-9-Cl.采用标准碘量/硫代硫酸钠滴定法测定了膜表面活性氯的含量分别为(1.17±0.04)和(1.52±0.05) μg/cm2. PU-8-Cl较低的表面活性氯含量可能和8在PU表面较高的固载率相关, 这点与XPS的数据分析是相吻合的.值得说明的是, 此处滴定所得表面活性氯的差别并不一定完全对等于图 3中XPS所显示F含量的差别, 这是因为前体分子8/9极有可能在PU表面实现多层分子固载[20], 而氯化过程中被氯化前体的可能仅仅位于多层固载层最外面可及的区域, 当然这种假设需要今后更多的实验数据来支持.
采用大肠杆菌(ATCC 25922)和金黄色葡萄球菌(ATCC 25923)为模式菌株, 对PU-8-Cl/PU-9-Cl进行了抗菌性能测试.数据如表 1所示, PU-9-Cl/PU-8-Cl在15 min内分别可灭活(17.74±4.84)%、(53.23±4.85)%的大肠杆菌以及(48.63±5.92)%、(72.52±4.23)%金黄色葡萄球菌, PU-8-Cl相对于PU-9-Cl杀菌能力更强.首先, 杀菌能力的体现本身就是PU表面氯胺结构单元成功引入的验证, 其次PU-8-Cl较优的杀菌能力与其表面较高的活性氯含量和较好的亲水性有关, 这两方面因素都可能使得细菌细胞更易在材料表面吸附, 更易完成表面氯胺结构的触杀进程[6].
 下载:
导出CSV
下载:
导出CSV
| 样品 | E. colia | S. aureusb | |||
| 减少值c/% | 减少量/log | 减少值c/% | 减少量/log | ||
| PU-9-Cl | 17.74±4.84 | 0.09±0.02 | 48.63±5.92 | 0.29±0.02 | |
| PU-8-Cl | 53.23±4.85 | 0.33±0.04 | 72.52±4.23 | 0.56±0.06 | |
| a大肠杆菌浓度为2.31×107 CFU/mL; b金黄色葡萄球菌浓度为1.46×107 CFU/mL; c此处细菌减少量计算是以相同抗菌条件下未改性PU膜为对照. | |||||
本文合成了两种携有PFPA结构的吡啶盐-氯胺前体8和9, 并在254 nm光照条件下将其固载到PU表面, 采用酸性橙染料染色、静态接触角和XPS等方法进行表面修饰的表征.氯化后的修饰PU膜展现了良好的抗菌性能, 尤以PU-8-Cl杀菌能力更优.此外本研究表明, 前体结构对光固载效率和膜材料抗菌能力都有一定的影响, 更多前体分子的合成和固载正在进展中, 用以深入考察这一构效关系.总之, 本研究在制备非溶出型氯胺抗菌材料的同时, 也提供了一种惰性材料表面固载氯胺前体的普适方法.
NMR分析使用AVANCE Ⅲ HD-500MHz核磁共振仪, 以氘代氯仿和氘代水为溶剂; 高分辨质谱(HRMS)分析采用Q-Tof MS spectrometer (Micromass, Manchester, England)质谱仪; 表面红外分析采用Nicolet iN10 MX & iS10 (ThermoFisher, America)红外光谱仪; X射线光电子能谱仪为Thermo Fisher公司产品ESCALAB™ 250Xi.
本文所用到的药品购自阿拉丁和麦克林分析纯试剂.柱层析用硅胶及薄层层析硅胶板均系山东省烟台市芝罘黄务硅胶开发试验厂产品, 薄层层析板采用碘缸显色或于紫外灯(254 nm)下观察.化合物熔点经北京泰克仪器有限公司产品X-4型显微熔点测定仪(数显)测试获得.聚氨酯膜购自烟台万华聚氨酯股份有限公司; 测试菌株大肠杆菌(ATCC 25922)和金黄色葡萄球菌(ATCC 25923)均为自大连医科大学宁安红女士友赠.
溴丙基海因10的合成参见文献[21], 所得产品为白色固体(m.p. 103~104 ℃), NMR数据与文献完全一致[21].
将3-羟基吡啶(1.52 g, 16.0 mmol)溶于乙腈中, 再加入无水碳酸钾(6.63 g, 48.0 mmol), 回流反应1 h, 然后将化合物10 (4.98 g, 20.0 mmol)加入体系中, 继续回流1 h, 停止反应.过滤除去无机盐, 浓缩后经柱层析[V(甲醇):V(二氯甲烷)=6:100为洗脱剂]纯化即得到白色固体11 2.95 g, 产率70.0%. m.p. 119~120 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.30 (dd, J=2.7, 1.0 Hz, 1H), 8.21 (dd, J=4.2, 1.8 Hz, 1H), 7.26~7.13 (m, 2H), 4.06 (t, J=6.0 Hz, 2H), 3.73 (t, J=6.8 Hz, 2H), 2.23~2.09 (m, 2H), 1.43 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 177.1, 156.0, 154.9, 142.3, 138.1, 123.8, 121.2, 66.1, 58.7, 36.1, 27.9, 25.1; HRMS calcd for C13H18N3O3 [M+H]+: 264.1348, found 264.1343.
将五氟苯甲酸(2.16 g, 10.2 mmol)溶于70 mL二氯甲烷中, 再将6-氯-1-己醇(2.05 g, 15.0 mmol)、N, N'-二环己基碳二亚胺(2.10 g, 10.2 mmol)及4-二甲氨基吡啶(0.06 g, 0.5 mmol)分别加入体系中.室温下反应24 h后过滤除去固体, 将滤液浓缩后经柱层析纯化[(乙酸乙酯):V(石油醚)=5:95为洗脱剂]得到无色油状透明液体17 3.01 g, 产率91.1%. 1H NMR (500 MHz, CDCl3) δ: 4.40 (t, J=6.5 Hz, 2H), 3.55 (t, J=6.6 Hz, 2H), 1.84~1.75 (m, 4H), 1.58~1.39 (m, 4H); 13C NMR (126 MHz, CDCl3) δ: 159.1, 146.3 (d, J=6.3 Hz), 144.2 (m), 142.1 (d, J=13.2 Hz), 138.7 (t, J=13.2 Hz), 136.7 (d, J=12.5 Hz), 108.4 (m), 66.7, 44.8, 32.4, 28.3, 26.4, 25.1; HRMS calcd for C13H12ClF5O2Na [M+Na]+ 353.0344, found 353.0352.
将化合物17 (3.01 g, 9.1 mmol)溶于30 mL丙酮中, 再将碘化钠(2.99 g, 20 mmol)加入体系中, 搅拌溶解后, 回流反应8 h.过滤除去固体, 将滤液浓缩后经柱层析纯化[(乙酸乙酯):V(石油醚)=5:95为洗脱剂]后得到无色油状透明液体18 3.53 g, 产率92.0%. 1H NMR (500 MHz, CDCl3) δ: 4.41 (t, J=6.5 Hz, 2H), 3.22 (t, J=7.0 Hz, 2H), 1.93~1.72 (m, 4H), 1.55~1.41 (m, 4H); 13C NMR (126 MHz, CDCl3) δ: 159.1, 146.3 (m), 144.2 (m), 142.1 (m), 138.7 (m), 136.8 (m), 108.4 (m), 66.7, 33.2, 30.0, 28.7, 24.8, 6.1.
将化合物11 (0.79 g, 3.0 mmol)溶于20 mL乙腈中, 再将化合物18 (2.50 g, 5.9 mmol)加入体系中, 回流过夜.停止反应浓缩后经柱层析纯化[V(甲醇):V(二氯甲烷)=10:100为洗脱剂]得到淡黄色固体19: 1.31 g, 产率63.9%. m.p. 73~74 ℃; 1H NMR (500 MHz, D2O) δ: 8.46 (t, J=1.8 Hz, 1H), 8.38 (d, J=5.9 Hz, 1H), 8.00~7.98 (m, 1H), 7.87~7.84 (m, 1H), 4.50 (t, J=7.1 Hz, 2H), 4.32 (t, J=6.3 Hz, 2H), 4.15 (t, J=5.6 Hz, 2H), 3.61 (t, J=6.4 Hz, 2H), 2.06~2.09 (m, 2H), 1.94~1.97 (m, 2H), 1.65~1.69 (m, 2H), 1.37~1.40 (m, 2H), 1.27~1.31 (m, 2H), 1.28 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 177.8, 159.1, 158.1, 156.0, 146.35, 144.3 (d, J=13.8 Hz), 142.1, 138.7, 137.3, 136.6, 132.1, 131.8, 128.8, 108.2 (d, J=17.6 Hz), 69.5, 66.6, 62.0, 58.9, 35.3, 31.8, 28.0, 27.5, 25.4, 25.2, 25.0; HRMS calcd for C26H29F5N3O5 [M-I]+ 558.2022, found 558.2021.
将化合物19 (1.31 g, 2.0 mmol)溶于丙酮-水(18 mL~3 mL)混合溶剂中, 然后加入叠氮化钠(0.16 g, 2.4 mmol), 加热回流过夜后停止反应.浓缩后经柱层析纯化[V(甲醇):V(二氯甲烷)=10:100为洗脱剂]得到白色固体9 1.18 g, 产率90.5%. m.p. 78~79 ℃; 1H NMR (500 MHz, D2O) δ: 9.35 (d, J=2.3 Hz, 1H), 8.60 (d, J=5.8 Hz, 1H), 7.98 (J=1.5 Hz, 1H), 7.94 (d, J=5.7 Hz, 1H), 5.03 (t, J=7.6 Hz, 2H), 4.47 (t, J=5.5 Hz, 2H), 4.37 (dt, J=9.3, 6.4 Hz, 2H), 3.75 (t, J=6.3 Hz, 2H), 2.19~2.23 (m, 2H), 2.10~2.14 (m, 2H), 1.78~1.82 (m, 2H), 1.56~1.60 (m, 2H), 1.51~1.55 (m, 2H), 1.49 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 177.8, 159.4, 158.1, 156.0, 146.3, 144.2, 141.4 (d, J=14.1 Hz), 139.4 (d, J=14.1 Hz), 137.3, 132.1, 131.8, 128.8, 123.3 (d, J=11.6 Hz), 107.8 (t, J=15.4 Hz), 69.5, 66.4, 62.0, 58.9, 35.3, 31.8, 28.1, 27.5, 25.4, 25.2, 25.1; HRMS calcd for C26H29F4N6O5 [M-I]+ 581.2130, found 581.2139.
将4-羟基吡啶(0.76 g, 8.0 mmol)置于60 mL乙腈中, 搅拌溶解后, 将无水碳酸钾(3.32 g, 24.0 mmol)加入体系中加热回流1 h, 再加入化合物10 (2.49 g, 10.0 mmol), 反应过夜后, 过滤除去无机盐, 将滤液浓缩后经柱层析纯化[V(甲醇):V(二氯甲烷)=6:100为洗脱剂]得到淡黄色粉末12 0.74 g, 产率35.2%. m.p. 203~204 ℃; 1H NMR (500 MHz, D2O) δ: 7.84~7.78 (m, 1H), 6.53~6.46 (m, 1H), 4.02 (t, J=6.9 Hz, 1H), 3.47 (t, J=6.6 Hz, 1H), 2.12 (p, J=6.7 Hz, 1H), 1.28 (s, 3H); 13C NMR (126 MHz, D2O) δ: 180.6, 179.4, 157.2, 142.5, 117.4, 59.0, 55.0, 35.8, 27.9, 23.4. HRMS calcd for C13H18N3O3 [M+H]+264.1348, found 264.1338.
将五氟苯甲酸(2.12 g, 10.0 mmol)溶于70 mL二氯甲烷中, 再将溴乙醇(2.07 g, 0.82 mL, 16.5 mmol)加入体系中, 然后加入N, N'-二环己基碳二亚胺(2.06 g, 10.0 mmol)、4-二甲氨基吡啶(0.05 g, 0.4 mmol), 室温下反应24 h后过滤除去固体杂质, 将滤液浓缩后经柱层析纯化[V(乙酸乙酯):V(石油醚)=5:95为洗脱剂]得到淡黄色透明液体12 3.02 g, 产率94.6%; 1H NMR (500 MHz, CDCl3) δ: 4.70~4.66 (m, 2H), 3.62 (td, J=6.1, 1.9 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ: 158.6, 146.6 (m), 144.6 (m), 142.6 (m), 138.6 (m), 136.6 (m), 107.5 (t, J=15.5 Hz), 65.7, 27.5; HRMS calcd for C9H4BrF5O2Na [M+Na]+ 340.9207, found 340.9211.
将化合物12 (1.17 g, 3.7 mmol)、碘化钠(1.10 g, 7.3 mmol)置于20 mL丙酮中, 搅拌溶解后, 回流反应5 h.浓缩后经柱层析纯化[(乙酸乙酯):V(石油醚)=5:95为洗脱剂]得到淡红色透明液体, 即为化合物14 1.32 g, 产率98.5%; 1H NMR (500 MHz, CDCl3) δ: 4.65 (t, J=6.9 Hz, 2H), 3.41 (t, J=6.9 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ: 160.3, 146.4 (t, J=5.0 Hz), 144.4, 140. 6 (t, J=16.5, Hz), 138.6 (m), 136.5, 109.6 (t, J=15.6 Hz), 68.5, 0.40.
将化合物12 (0.27 g, 1 mmol)溶于20 mL乙腈中, 再将化合物15 (0.74 g, 2 mmol)加入体系中, 回流过夜.浓缩后经柱层析纯化[V(甲醇):V(二氯甲烷)=10:100为洗脱剂]得到白色固体16 0.26 g, 产率42.0%. m.p. 68~69 ℃; 1H NMR (500 MHz, D2O) δ: 8.65~8.50 (m, 2H), 7.56~7.33 (m, 2H), 4.85~4.72 (m, 2H), 4.72~4.69 (m, 2H), 4.40 (t, J=7.1 Hz, 2H), 3.47 (t, J=6.6 Hz, 2H), 2.22 (t, J=6.9 Hz, 2H), 1.33 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 178.3, 169.8, 158.8, 156.4, 146.7, 144.6 (d, J=11.2 Hz), 142.4, 138.7, 136.7 (d, J=11.2 Hz), 132.4, 114.5, 107.3 (d, J=15.0 Hz), 69.1, 63.6, 59.4, 57.8, 35.4, 30.4, 25.0. HRMS calcd for C22H21F5N3O5 [M-I]+ 502.1396, found 502.1405.
将化合物16 (0.26 g, 0.4 mmol)溶于丙酮与水(6 mL/1 mL)混合溶剂中, 加入叠氮化钠(0.06 g, 0.9 mmol), 加热回流过夜后, 停止反应.浓缩后经柱层析纯化[V(甲醇):V(二氯甲烷)=10:100为洗脱剂]得到白色固体8 0.24 g, 产率91.0%. m.p. 73~74 ℃; 1H NMR (500 MHz, CDCl3) δ: 9.12 (m, 2H), 7.57 (m, 2H), 7.12 (s, 1H), 4.74~4.67 (m, 4H), 3.53 (t, J=5.9 Hz, 2H), 3.38 (m, J=3.8 Hz, 2H), 2.34~2.28 (m, 2H), 1.40 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 178.3, 169.8, 160.4 (t, J=3.2 Hz), 155.9, 146.7, 144.6 (d, J=34.9 Hz), 141.7 (d, J=13.6 Hz), 139.73 (d, J=16.7 Hz), 123.5, 122.43, 114.47, 110.7 (d, J=17.3 Hz), 69.1, 63.4, 59.2, 57.6, 35.0, 30.4, 25.1; HRMS calcd for C22H21F4N6O5 [M-I]+ 525.1504, found 525.1504.
分别将前体8/9的乙醇溶液(7 mg/mL)涂覆到PU膜(3 cm×3 cm)表面, 置于烘箱(60 ℃) 20 min, 取出后进一步室温晾干.然后将膜置于紫外交联仪(254 nm)中照射100 s (膜距离紫外灯管5 cm, 接受总能量为1.01 J/cm2), 最后用乙醇将PU膜充分漂洗3次, 所得修饰材料记为PU-8和PU-9.随后将其置于饱和氯化钠溶液中恒温振荡30 min, 取出后用去离子水冲洗干净, 再浸于稀释10倍的家用漂白水中, 恒温振荡120 min后取出用去离子水充分漂洗3次, 晾干后室温避光保存备用, 所得抗菌膜材料记为PU-8-Cl和PU-9-Cl.
将PU-9和未改性PU膜一同浸入2 wt%酸性橙染液中, 室温持续振荡16 h后取出.用去离子水充分洗涤直至洗涤水中颜色消失, 拍照比较二者对染料的吸附情况.
采用标准碘量/Na2S2O3滴定法测定改性抗菌PU膜的活性氯含量.分别称量0.08 g (1.5×1.5 cm2)样品(PU-8-Cl/PU-9-Cl), 剪碎后置于盛有50 mL (含10%乙醇和0.05 g Tritonx-100)水中, 加入0.4 g KI并调节pH=4.0, 置于暗处搅拌5 h.以1%的淀粉溶液为指示剂, 使用0.001 mol/L的Na2S2O3溶液(由碘酸钾标准溶液标定)滴定至终点(三组平行样, 并取平均值), 活性氯含量计算公式为:
Cl+=(C×V×35.45)/(2×S)×106
其中, Cl+活性氯的重量百分比, C和V是Na2S2O3溶液的浓度(mol/L)和体积(L), S是PU膜的面积.
采用“Sandwich Test”方法测定抗菌PU膜的抗菌性能.将1片膜材料(以PU-9-Cl为例, 3 cm×3 cm)置于直身瓶中(V=300 mL, d=7.6 cm), 取10 μL细菌溶液(约107 CFU/mL)均匀滴加到膜表面并涂匀, 随后将另外完全相同的一片PU-9-Cl膜置于其上, 用灭菌小烧杯(V=20 mL)压紧两片PU膜以保证二者与菌液的充分接触. 15 min后, 向瓶中加入20 mL 0.03% Na2S2O3溶液以中和膜材料中可能残存的活性氯.随之将直接瓶旋涡振荡2 min以除去黏附膜表面的细菌, 进而取出100 μL悬浮液进行连续稀释, 并取100 μL置于固体培养基平板上(3个平行样), 于37 ℃恒温箱中培养24 h, 采用平板计数法确定菌落数来计算存活细菌数.采用同样的方法对未改性PU膜做测试作为对照实验.测试样品的抗菌活性使用细菌减少百分数(%)及Log值减少量来判断, 计算公式如下:
细菌减少量(%)=(A-B)/A×100
Log减少量=Log (A/B) (B>0)或Log A (B=0)
其中, A为对照试验中菌落数目菌液浓度(CFU/mL), B为依据抗菌材料测试后剩余菌落数计算出的菌液浓度(CFU/mL).
辅助材料(Supporting Information) 目标化合物及中间体的核磁共振数据.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
亢真真, 焦玉超, 张冰, 梁杰, 上海师范大学学报(自然科学版), 2012, 41, 540. doi: 10.3969/j.issn.1000-5137.2012.05.016Kang, Z.; Jiao, Y.; Zhang, B.; Liang, J. J. Shanghai Normal Univ. (Nat. Sci. Ed.) 2012, 41, 540(in Chinese). doi: 10.3969/j.issn.1000-5137.2012.05.016
Hui, F.; Debiemme-Chouvy, C. Biomacromolecules 2013, 14, 585. doi: 10.1021/bm301980q
Dong, A.; Wang, Y.-J.; Gao, Y.; Gao, T.; Gao G. Chem. Rev. 2017, 117, 4806. doi: 10.1021/acs.chemrev.6b00687
Gottardi, W.; Debabov, D.; Nagl, M. Antimicrob. Agents Chemother. 2013, 57, 1107. doi: 10.1128/AAC.02132-12
Jie, Z.; Yan, X.; Zhao, L.; Worley, S. D.; Liang. J. RSC Adv. 2014, 4, 6048. doi: 10.1039/c3ra47147k
Li, L.; Pu, T.; Zhanel, G.; Zhao, N.; Ens, W.; Liu, S. Adv. Healthcare Mater. 2012, 1, 609. doi: 10.1002/adhm.201200018
Ning, C.; Li, L.; Logsetty, S.; Ghanbar, S.; Guo, M.; Ensf, W.; Liu, S. RSC Adv. 2015, 5, 93877. doi: 10.1039/C5RA15714E
Li, L.; Zhao, Y.; Zhou, H.; Ning, A.; Zhang, F.; Zhao, Z. Tetrahedron Lett. 2017, 58, 321. doi: 10.1016/j.tetlet.2016.12.021
Li, L.; Zhou, H.; Gai, F.; Chi, X.; Zhang, F.; ZhaoZ. RSC Adv. 2017, 7, 13244. doi: 10.1039/C6RA24954J
Liang, J.; Chen, Y.; Ren, X. Ind. Eng. Chem. Res. 2007, 46, 6425. doi: 10.1021/ie0707568
Wu, L.; Liu A.; Li, Z. Fibers Polym. 2015, 16, 550. doi: 10.1007/s12221-015-0550-7
Sun, Y. Y.; Sun, G. J. Appl. Polym. Sci. 2001, 81, 1517. doi: 10.1002/(ISSN)1097-4628
Liu, L.-H.; Yan, M. Acc. Chem. Res. 2010, 43, 1434. doi: 10.1021/ar100066t
Li, L.; Li, J.; Kulkarni, A.; Liu, S. J. Mater. Chem., 2013, 1, 571. doi: 10.1039/C2TB00248E
Williamson, M. R.; Black, R.; Kielty, C. Biomaterials 2006, 27, 3608.
Li, L.; Chi, X.; Gai, F.; Zhou, H.; Zhang, F.; Zhao, Z. J. Appl. Polym. Sci. 2017, 134, 45323. doi: 10.1002/app.45323
Dastgir, S.; Coleman, K. S.; Green, M. L. H. Dalton Trans. 2011, 40, 661. doi: 10.1039/C0DT00760A
Keana, J. F. W.; Cai, S. X. J. Org. Chem. 1990, 55, 3640. doi: 10.1021/jo00298a048
Xiong, J.; Xia, L.; Shentu, B.; Weng, Z. J. Appl. Polym. Sci. 2014, 131, 39812.
Renaudie, L.; Narvor, C. L.; Lepleux, E.; Roger, P. Biomacromolecules 2007, 8, 679. doi: 10.1021/bm060961r
Li, L.; Zhao, N.; Liu, S. Polymer 2012, 53, 67. doi: 10.1016/j.polymer.2011.11.036

图 1 部分已报道的DMH氯胺(前体)衍生物结构
Figure 1 Structure of representative DMH N-chloramine (precursor) derivatives

Scheme 1 氯胺前体8~9的合成路线
Scheme 1 Synthesis route of N-chloramine precursors 8~9
Reagents and conditions: (a) 3-hydroxypyridine, K2CO3, CH3CN, reflux, 2 h, 70.0%; (b) BrCH2CH2OH, dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), CH2Cl2, r.t., 24 h, 94.6%; (c) NaI, acetone, reflux, 8 h, 98.5%; (d) 4-hydroxypyridine, K2CO3, CH3CN, reflux, 35.2%; (e) 14, CH3CN, reflux, 42.0%; (f) NaN3, acetone-H2O, reflux, overnight, 91.0%; (g) Br(CH2)6OH, DCC, DMAP, CH2Cl2, r.t., overnight, 91.1%; (h) NaI, acetone, reflux, 8 h, 92.0%; (i) 11, CH3CN, reflux, overnight, 63.9%; (j) NaN3, acetone-H2O, reflux, overnight, 90.5%.

图 2 氯胺前体在PU表面的固载
Figure 2 Immobilization of N-chloramine precursors on PU film
(A) Schematic illustration; (B) visualization of orange sodium salt staining; (C) contact angel data

图 3 改性PU膜的XPS谱图
Figure 3 XPS spectra of surface modified PU film
(a) PU-9; (b) PU-8; (c) PU
表 1 改性PU膜的抗菌活性
Table 1. Antibacterial activity of surface modified PU film
| 样品 | E. colia | S. aureusb | |||
| 减少值c/% | 减少量/log | 减少值c/% | 减少量/log | ||
| PU-9-Cl | 17.74±4.84 | 0.09±0.02 | 48.63±5.92 | 0.29±0.02 | |
| PU-8-Cl | 53.23±4.85 | 0.33±0.04 | 72.52±4.23 | 0.56±0.06 | |
| a大肠杆菌浓度为2.31×107 CFU/mL; b金黄色葡萄球菌浓度为1.46×107 CFU/mL; c此处细菌减少量计算是以相同抗菌条件下未改性PU膜为对照. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们