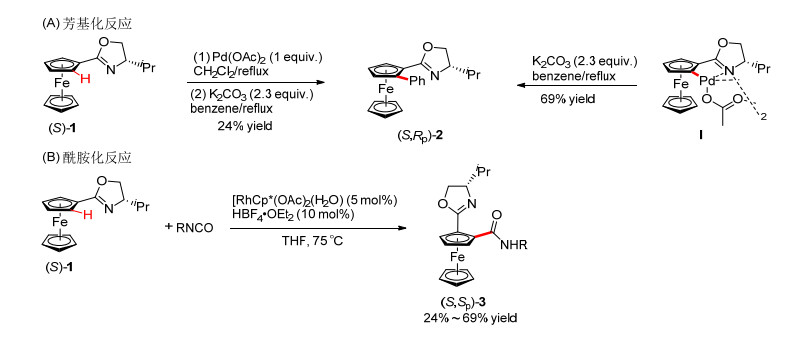
 图式 1
手性噁唑啉导向的非对映选择性碳氢键芳基化和酰胺化反应
Scheme1.
Diastereoselective C—H arylation and amidation reactions by using chiral oxazoline as a directing group
图式 1
手性噁唑啉导向的非对映选择性碳氢键芳基化和酰胺化反应
Scheme1.
Diastereoselective C—H arylation and amidation reactions by using chiral oxazoline as a directing group
引用本文:
黄家翩, 顾庆, 游书力. 过渡金属催化的碳氢键官能团化反应合成平面手性二茂铁化合物[J]. 有机化学,
2018, 38(1): 51-61.
doi:
10.6023/cjoc201708030

Citation: Huang Jiapian, Gu Qing, You Shuli. Synthesis of Planar Chiral Ferrocenes via Transition-Metal-Catalyzed Direct C-H Bond Functionalization[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 51-61. doi: 10.6023/cjoc201708030
Citation: Huang Jiapian, Gu Qing, You Shuli. Synthesis of Planar Chiral Ferrocenes via Transition-Metal-Catalyzed Direct C-H Bond Functionalization[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 51-61. doi: 10.6023/cjoc201708030
过渡金属催化的碳氢键官能团化反应合成平面手性二茂铁化合物
English
Synthesis of Planar Chiral Ferrocenes via Transition-Metal-Catalyzed Direct C-H Bond Functionalization
Abstract:
Ferrocenes bearing planar chirality have been demonstrated to be highly efficient ligands or catalysts in asymmetric catalysis. In view of their atom and step economies, direct asymmetric C—H bond functionalization is the most concise and powerful method for the construction of planar chiral ferrocenes compared with traditional approaches. This review summarizes recent progress on the development of novel methods to synthesize planar chiral compounds via transition-metal (Cu-, Pd-, Ir-, Rh-, Au-, Pt-) catalyzed asymmetric C—H bond functionalization. Preparation of a variety of new planar chiral ferrocene-based catalysts and ligands by utilizing these methods and their application in catalytic asymmetric reactions are also discussed.
-
二茂铁化合物具有热稳定、化学稳定和易衍生等特点, 在材料化学、合成化学和药物化学等领域中有着广泛的应用[1].尤其是含有平面手性的二茂铁化合物, 在不对称催化反应中作为手性配体或者手性催化剂得到了广泛的研究[2], 其中一些已经在工业上实现了大规模应用, 例如用(R, Sp)-xyliphos作为手性配体, 金属铱催化的亚胺不对称氢化反应来合成除草剂金都尔的关键中间体[3].因此, 化学家们发展了众多方法将平面手性引入二茂铁骨架.到目前为止, 最为常用的策略包括手性辅基导向的非对映选择性邻位金属化[4a~4c, 4e, 4g~4h]、外加手性试剂的不对称邻位锂化[4d, 4f, 4i]、消旋体的手性拆分[4j]、烯烃复分解[4m]或偶联[4k~4l]等去对称化反应.然而这些方法具有明显的局限性, 如需要预先引入手性导向基团, 需要使用当量的手性试剂, 手性拆分效率偏低等等.
过渡金属催化的不对称碳氢键官能团化反应由于原子经济性好、合成效率高等特点而受到广泛关注, 逐渐成为不对称合成的重要方法[5].通过不对称碳氢键活化策略来合成平面手性二茂铁化合物无疑是最简洁高效的方法, 其挑战是如何在相对温和的反应条件下实现碳氢键的对映选择性活化.可喜的是, 在过去几年中, 不对称碳氢键官能团化反应构建平面手性二茂铁化合物已取得了不少重要的研究成果[6].因此, 本文将对这一领域的进展进行简要的综述, 根据反应类型分为非对映选择性反应(手性辅基导向)和对映选择性反应(手性催化), 而后者则根据过渡金属类型分为铜、钯、铱/铑和金/铂催化的碳氢键官能团化反应进行综述.
1 非对映选择性反应合成平面手性二茂铁
非对映选择性碳氢键官能团反应的手性控制是通过底物中的手性基团来实现的, 因而关键在于找到一个合适的手性辅基, 通过分子内诱导取得优秀的立体选择性控制. 2007年, 游书力课题组[7]发展了手性噁唑啉取代的二茂铁(S)-1与简单芳烃的交叉偶联反应合成了平面手性二茂铁(S, Rp)-2 (Scheme 1A).值得注意的是, 将预先制备好的二聚体钯络合物I在溶剂苯中回流也可以得到相同产物(S, Rp)-2. 2012年, Shibata课题组[8]使用相同的手性辅基实现了铑催化的二茂铁和异氰酸脂的酰胺化反应(Scheme 1B).
图式 1
手性噁唑啉导向的非对映选择性碳氢键芳基化和酰胺化反应
Scheme1.
Diastereoselective C—H arylation and amidation reactions by using chiral oxazoline as a directing group
2 对映选择性反应合成平面手性二茂铁
2.1 铜催化的碳氢键官能团化反应
与手性辅基策略相比, 使用不对称催化的方法来引入平面手性无疑更加令人关注. 1997年, Schmalz小组[9]开创性地使用CuOTf/双噁唑啉(L1)作为催化剂, 实现了二茂铁碳氢键不对称卡宾插入反应, 以较好的收率和对映选择性获得相应环化产物(Scheme 2).
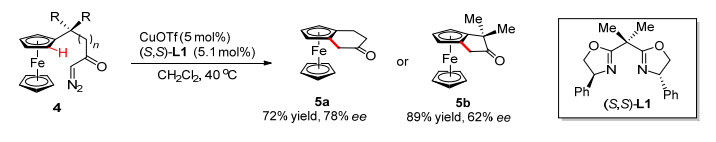 图式 2
铜催化二茂铁碳氢键不对称卡宾插入反应
Scheme2.
Cu-catalyzed enantioselective insertion of carbenoid into C—H bond of ferrocene
图式 2
铜催化二茂铁碳氢键不对称卡宾插入反应
Scheme2.
Cu-catalyzed enantioselective insertion of carbenoid into C—H bond of ferrocene
2.2 二价钯催化的碳氢键官能团化反应
早在1978年, Sokolov小组[10]使用当量的手性氨基酸, 实现了二甲氨甲基二茂铁不对称环钯化反应, 获到了光学活性的二茂铁氯化钯二聚体, 再用于下一步转化.由于光学活性的钯络合物需要预先制备, 而且需要使用当量的手性配体, 因此这一方法缺乏足够的实用价值.在钯催化不对称碳氢键官能团化反应领域, 余金权小组[11]首次使用催化量的手性氨基酸作为配体, 发展了一系列不对称碳氢键官能团化反应, 实现了中心手性的构建.受这些工作的启发, 2013年, 游书力小组[12]实现了二甲氨甲基二茂铁的催化不对称碳氢键芳基化反应, 使用Boc-L-Val-OH为手性配体, 空气为氧化剂, 在温和的反应条件下, 以良好的收率和优秀的对映选择性得到了芳基化产物7 (Scheme 3), 但该反应对于脂肪硼酸的结果不佳.
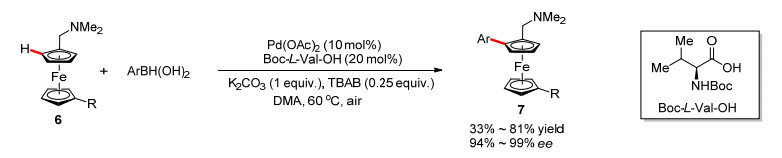 图式 3
钯催化二茂铁和芳基硼酸的不对称碳氢键芳基化反应
Scheme3.
Pd-catalyzed asymmetric C—H bond arylation of ferrocenes with arylboronic acids
图式 3
钯催化二茂铁和芳基硼酸的不对称碳氢键芳基化反应
Scheme3.
Pd-catalyzed asymmetric C—H bond arylation of ferrocenes with arylboronic acids
该反应可能的催化循环如图 1所示, 在配体Boc-L-Val-OH作用下, 经协同钯化-去质子化过程, 对映选择性地切断二茂铁碳氢键, 生成手性二价钯中间体A, 接着与芳基硼酸发生转金属化生成中间体B, 最后还原消除生成芳基化产物(Sa)-7a, 释放零价钯并氧化生成二价钯, 完成催化循环[13].其次, μ-羧基桥联的二聚体也被认为可能是活性催化物种[14].
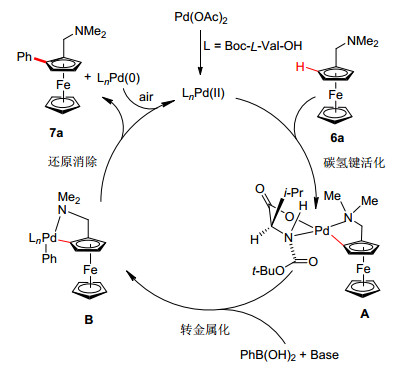 图1
可能的催化循环
Figure1.
Plausible catalytic cycle
图1
可能的催化循环
Figure1.
Plausible catalytic cycle
几乎同时, 崔秀灵和吴养洁课题组[15]报道了以Boc-L-Phe-OH作为最优配体催化的不对称氧化Heck反应, 该反应同样首先生成平面手性二价钯中间体, 然后烯烃插入碳钯键, β-氢消除, 实现了平面手性烯基取代二茂铁的高效合成(Scheme 4A).该反应具有很好的底物普适性, 丙烯酸酯、取代的苯乙烯和丙烯酰胺等都可以取得良好的收率和优秀的对映选择性控制.随后, 游书力课题组[16]报道了钯催化二茂铁和双芳基炔烃的不对称环化反应(Scheme 4B), 该工作是受崔秀灵和吴养洁小组报道[17]的消旋反应启发而开展的.值得注意的是, 从产物(Sp)-9a出发, 可以方便地合成P, N-双齿配体(Sp)-L2, 将该配体应用于催化不对称烯丙基取代反应, 可以获得44% ee的对映选择性控制.
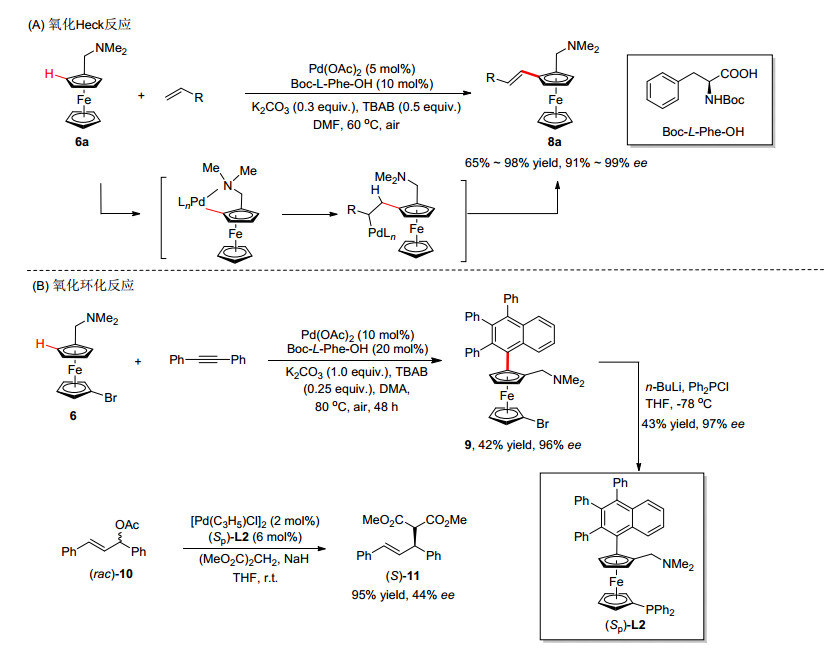 图式 4
二价钯催化的氧化Heck和环化反应
Scheme4.
Pd(Ⅱ)-catalyzed oxidative Heck and annulation reactions
图式 4
二价钯催化的氧化Heck和环化反应
Scheme4.
Pd(Ⅱ)-catalyzed oxidative Heck and annulation reactions
对于手性联芳基化合物的合成, 通过双碳氢键活化来实现不对称氧化偶联反应是最直接高效的方法. 2016年, 游书力课题组[18]使用Pd(OAc)2和Boc-L-Ile-OH催化体系, 实现了二茂铁化合物6与富电子杂芳烃12的不对称氧化交叉偶联反应(Scheme 5).该反应可能的催化循环与前面所述芳基硼酸的偶联反应类似, 唯一的区别在于中间体A与苯并呋喃发生亲电取代反应而不是和芳基硼酸的转金属化反应.
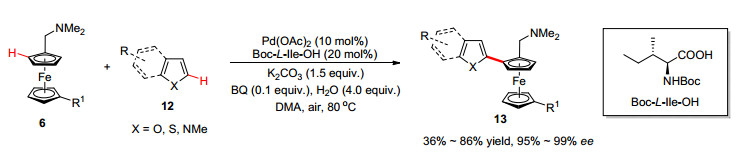 图式 5
不对称双碳氢键氧化交叉偶联反应
Scheme5.
An enantioselective oxidative twofold C—H cross-coupling reaction
图式 5
不对称双碳氢键氧化交叉偶联反应
Scheme5.
An enantioselective oxidative twofold C—H cross-coupling reaction
除了钯催化二茂铁碳氢键直接芳基化反应、氧化Heck反应和环化反应, 其他的反应类型相对较少. 2014年, 崔秀灵和吴养洁课题组[19]使用Pd(OAc)2和Ac-L-Phe-OH催化体系, 芳基二酮为酰基化试剂, 以令人满意的收率和优秀的对映选择性实现了二茂铁的不对称碳氢键酰基化反应(Scheme 6).对于反应机理初步研究发现, 自由基捕获剂四甲基哌啶氧化物(TEMPO)可以抑制该反应, 这表明很有可能经历的是自由基历程.可能的反应机理如图 2所示, 首先二茂铁底物发生不对称碳氢键活化生成手性环钯中间体A, 芳基二酮在过氧叔丁醇的作用下产生苯甲酰基自由基, 该自由基与中间体A反应生成三价钯或四价钯中间体B, 最后还原消除生成目标产物, 释放二价钯, 完成催化循环.
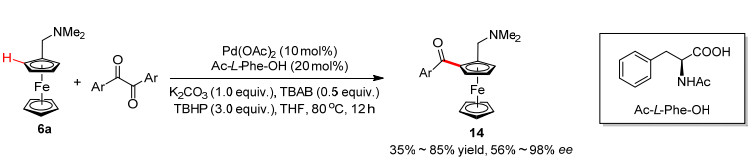 图式 6
二茂铁化合物催化不对称碳氢键酰基化反应
Scheme6.
Catalytic enantioselective C—H acylation of ferrocenes
图式 6
二茂铁化合物催化不对称碳氢键酰基化反应
Scheme6.
Catalytic enantioselective C—H acylation of ferrocenes
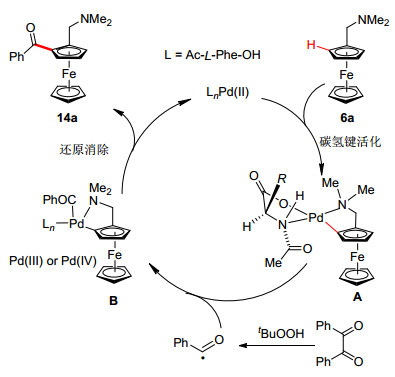 图2
可能的反应机理
Figure2.
Proposed reaction mechanism
图2
可能的反应机理
Figure2.
Proposed reaction mechanism
2.3 零价钯催化的碳氢键官能团化反应
二价钯催化的不对称碳氢键官能团化反应已经成为合成平面手性二茂铁的一个重要方法, 然而在这类反应中, 催化剂用量依然较高(5~10 mol%), 反应的效率偏低, 而且必须使用外加氧化剂才能实现催化循环. 2014年, 游书力课题组[20]使用Pd(OAc)2/BINAP催化体系, 实现了零价钯催化的分子内碳氢键芳基化反应, 以非常高的效率构建了一系列平面手性二茂铁并环化合物(Scheme 7).与之前氧化性的碳氢键芳基化反应(PdⅡ/Pd0催化)相比, 该氧化还原中性反应(Pd0/PdⅡ催化)无需使用外加氧化剂, 避免了底物和配体的氧化, 同时加速了催化循环, 显著提高了催化效率.即使催化剂用量降低至0.5 mol%, 反应依然能够获得99%收率和96% ee对映选择性.利用该方法, 从产物Rp-16a出发, 通过4步转化合成了P, N-手性配体(S, Rp)-20, 该配体在钯催化不对称烯丙基化及胺化反应中显示良好的手性控制(Scheme 8)[21].
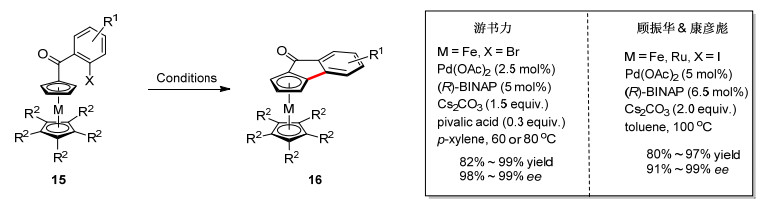 图式 7
零价钯催化的分子内不对称碳氢键芳基化反应
Scheme7.
Pd(0)-catalyzed enantioselective intramolecular C—H arylation
图式 7
零价钯催化的分子内不对称碳氢键芳基化反应
Scheme7.
Pd(0)-catalyzed enantioselective intramolecular C—H arylation
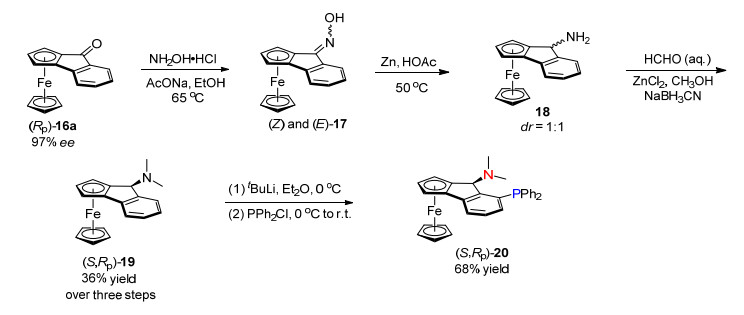 图式 8
高效合成平面手性P, N-配体(S, Rp)-20
Scheme8.
Efficient synthesis of planar chrial P, N-ligand (S, Rp)-20
图式 8
高效合成平面手性P, N-配体(S, Rp)-20
Scheme8.
Efficient synthesis of planar chrial P, N-ligand (S, Rp)-20
几乎同时, 顾振华和康彦彪小组[22]也报道了使用二茂铁衍生的芳基碘底物的工作, 除了二茂铁, 二茂钌底物同样可以实现该反应, 都表现出了良好的官能团兼容性.此外, 段伟良和叶金星小组[23]使用手性磷酸作为手性源和非手性膦配体, 同样实现了钯催化的二茂铁分子内不对称碳氢键芳基化反应, 对映选择性最高可达83% ee.作者推测手性磷酸在催化循环中起到了立体选择性辅助拔氢的作用.
受上述工作启发, Guiry课题组[24]从C2对称的二茂铁化合物ent-16i出发, 合成了一系列手性二茂铁二醇化合物, 研究了其催化的hetero-Diels-Alder反应, 最高可以以61%收率和59% ee对映选择性得到相应环化产物(Scheme 9).
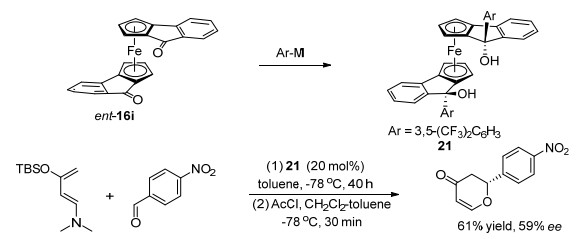 图式 9
手性二醇21的高效合成及不对称hetero-Diels-Alder反应
Scheme9.
Efficient synthesis of diol 21 and asymmetric hetero-Diels-Alder reaction
图式 9
手性二醇21的高效合成及不对称hetero-Diels-Alder反应
Scheme9.
Efficient synthesis of diol 21 and asymmetric hetero-Diels-Alder reaction
顾振华课题组[25a]、刘澜涛和赵文献课题组[25b]以酰胺链接的二茂铁22为反应底物, 分别以(R, Sa)-O-PINAP和(R, R)-L3作为手性配体, 也实现了钯催化的分子内不对称碳氢键芳基化反应, 以优秀的收率和良好的对映选择性构建了手性喹啉酮并二茂铁化合物(Scheme 10).
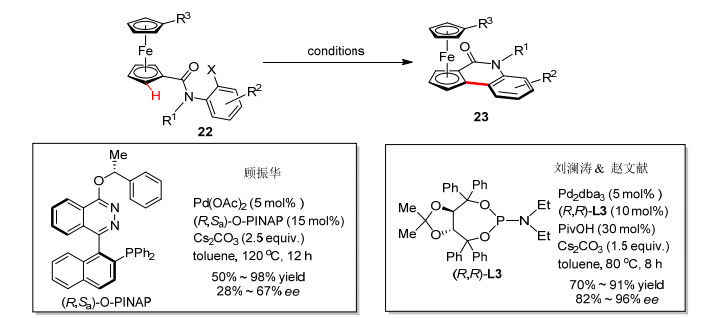 图式 10
二茂铁酰胺化合物分子内不对称碳氢键芳基化反应
Scheme10.
Enantioselective intramolecualr C—H arylation of ferrocenecarboxamides
图式 10
二茂铁酰胺化合物分子内不对称碳氢键芳基化反应
Scheme10.
Enantioselective intramolecualr C—H arylation of ferrocenecarboxamides
含有吡啶或吡啶氮氧片段的手性二茂铁化合物是一类非常高效的亲核性或路易斯碱催化剂, 然而其以往合成通常需要使用手性制备液相色谱对消旋体进行分离. 2015年, 游书力课题组[26]以Pd(OAc)2/(R)-BINAP作为催化剂, 实现了钯催化的分子内不对称碳氢键吡啶基化反应, 高效合成了一系列含吡啶的平面手性二茂铁化合物, 例如手性DMAP和PPY类似物[DMAP=4-(二甲氨基)吡啶, PPY=4-(1-吡咯烷基)-吡啶](Scheme 11).将底物进一步氧化合成了手性氮氧化合物26, 在其催化环氧化合物27的不对称开环反应中, 可以95%收率及66% ee对映选择性得到相应产物.
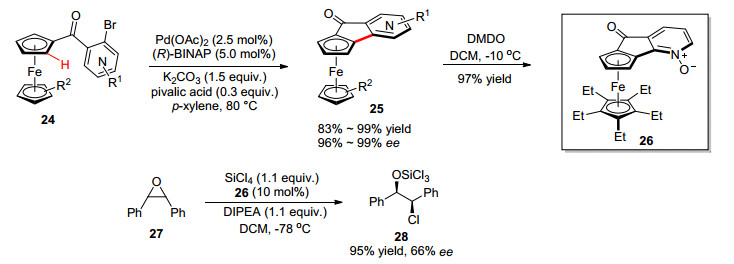 图式 11
高对映选择性合成手性平面二茂铁吡啶衍生物及其在环氧化合物不对称开环反应中的应用
Scheme11.
Highly enantioselective synthesis of planar chiral ferrocenylpyridine derivatives and their application in asymmetric opening of meso-epoxide
图式 11
高对映选择性合成手性平面二茂铁吡啶衍生物及其在环氧化合物不对称开环反应中的应用
Scheme11.
Highly enantioselective synthesis of planar chiral ferrocenylpyridine derivatives and their application in asymmetric opening of meso-epoxide
可能的反应机理如图 3a所示, 催化剂和底物首先发生氧化加成生成中间体A, 在碱的存在下发生配体交换得到中间体B, 经协同钯化-选择性去质子化过程, 生成中间体C, 最后还原消除得到产物, 同时释放出催化剂, 完成催化循环.还通过密度泛函(DFT)计算阐明了该反应取得高对映选择性的原因.计算发现生成S构型产物的过渡态(TS-CMD-S)的吉布斯自由能比R构型产物的过渡态(TS-CMD-R)的吉布斯自由能高出8.8 kcal/mol.象限分析TS-CMD-S过渡态, 在象限Ⅱ中, 茂环和(R)-BINAP的苯基之间存在显著的空间相互作用, 而对于TS-CMD-R过渡态, 茂环则位于开放的象限Ⅰ从而避免了相互作用(图 3b, 3c).
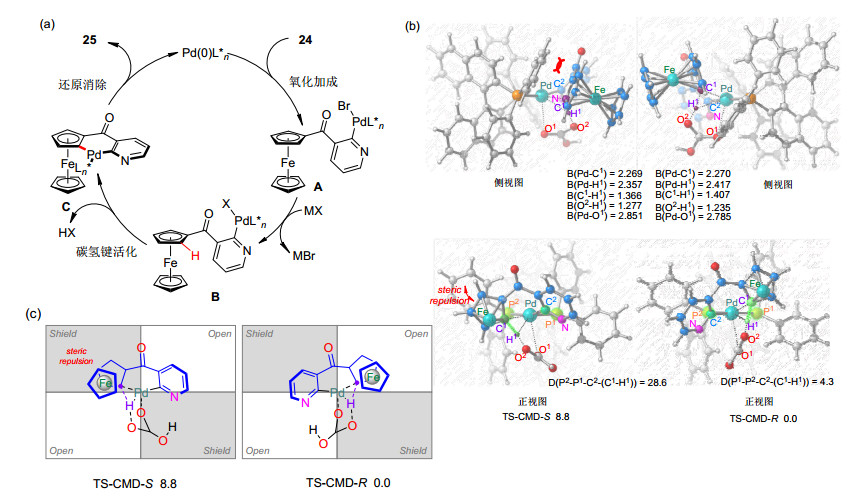 图3
(a) 可能的反应机理、(b)两种过渡态TS-CMD-R和TS-CMD-S的结构以及(c)两种过渡态象限分析
Figure3.
(a) Proposed reaction mechanism, (b) the structures of the two transition states TS-CMD-R and TS-CMD-S, and (c) the quadrant analysis of the two transition states
图3
(a) 可能的反应机理、(b)两种过渡态TS-CMD-R和TS-CMD-S的结构以及(c)两种过渡态象限分析
Figure3.
(a) Proposed reaction mechanism, (b) the structures of the two transition states TS-CMD-R and TS-CMD-S, and (c) the quadrant analysis of the two transition states
相对于二茂铁的不对称芳基化反应, 不对称分子内烯基化反应研究较少.游书力等[27]利用碳氢键官能团化的策略首次实现了二茂铁、二茂钌分子内不对称烯基化反应(Scheme 12).该催化体系具有非常好的底物适用范围, 可以兼容多种官能团.
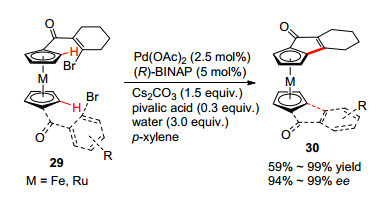 图式 12
零价钯催化碳氢键烯基化反应
Scheme12.
Pd(0)-catalyzed C—H alkenylation
图式 12
零价钯催化碳氢键烯基化反应
Scheme12.
Pd(0)-catalyzed C—H alkenylation
2.4 铱/铑催化的碳氢键官能团化反应
和钯催化不对称碳氢键官能团化相比, 铑和铱催化反应发展相对比较缓慢, 尤其是催化合成平面手性二茂铁化合物. 2014年, Shibata等[28a]使用[Ir(coe)2Cl]2/手性双烯L4为催化体系, 首次实现了2-异喹啉二茂铁与烯烃的不对称碳氢键烷基化反应, 使用2-异喹啉作为导向基团, 可以显著抑制二次烷基化过程.烷基化试剂包括烯丙基苯、1-辛烯、甲基丙烯酸甲酯、冰片烯等烯烃; 当使用苯乙烯作为烷基化试剂时, 反应存在区域选择性, 但仍以直链产物为主(Scheme 13).可能的反应机理如图 4所示[28b], 首先[Ir(coe)2Cl]2与手性双烯配体, NaBArF反应, 生成阳离子铱/手性双烯催化剂, 然后与底物发生不对称碳氢键活化生成铱氢物种A, 烯烃插入生成B, 最后还原消除生成产物, 同时释放出催化剂, 完成催化循环.
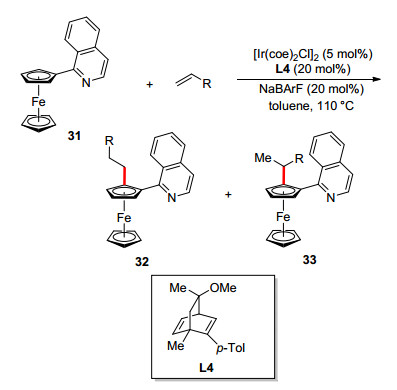 图式 13
铱催化二茂铁不对称碳氢键烷基化反应
Scheme13.
Ir-catalyzed enantioselective C—H alkylation of ferrocene
图式 13
铱催化二茂铁不对称碳氢键烷基化反应
Scheme13.
Ir-catalyzed enantioselective C—H alkylation of ferrocene
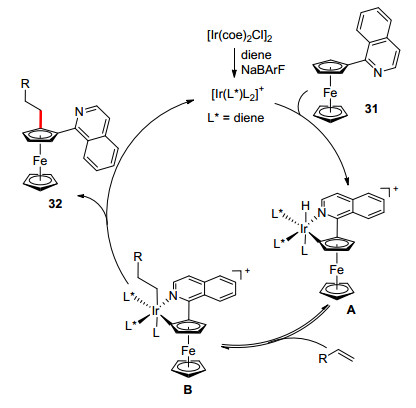 图4
可能的反应机理
Figure4.
Proposed reaction mechanism
图4
可能的反应机理
Figure4.
Proposed reaction mechanism
鉴于有机硅化合物的重要性, 如何有效构建碳硅键, 特别是碳氢键直接硅基化反应具有重要意义. 2015年, 三个课题组[29]几乎同时报道了铑催化二茂铁分子内不对称碳氢键硅基化反应.其中, Shibata课题组[29a]使用手性双烯配体L5实现了脱氢偶联反应, 何伟课题组[29b]和Murai, Takai课题组[29c]则分别使用手性双膦配体(S)-L6和(R)-DTBM-Segphos也实现脱氢偶联反应, 反应条件温和, 对映选择性优秀(Scheme 14).
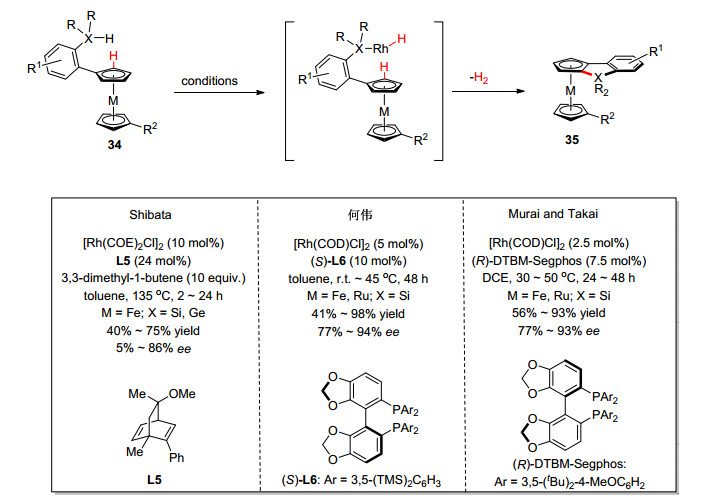 图式 14
铑催化分子内不对称碳氢键硅基化反应
Scheme14.
Rh-catalyzed intramolecular asymmetric C—H silylation
图式 14
铑催化分子内不对称碳氢键硅基化反应
Scheme14.
Rh-catalyzed intramolecular asymmetric C—H silylation
2016年, 游书力等[30]采用[RhCp*Cl2]2作为催化剂, 实现了N-甲氧基二茂铁甲酰胺与炔烃的环化反应, 合成[30]一系列二茂铁并吡啶酮类化合物, 该反应体系不需要额外氧化剂.使用手性(Ra)-Rh1催化剂, 能初步实现平面手性产物的构建(46% ee) (Scheme 15).可能的反应机理图 5所示[31], 在氧化条件下生成Rh(Ⅲ)催化剂, 和底物发生不对称碳氢键活化形成五元环铑中间体A, 接着炔烃插入Rh—C键, 最后经历C—N键形成/C—O键切断得到最终产物38.
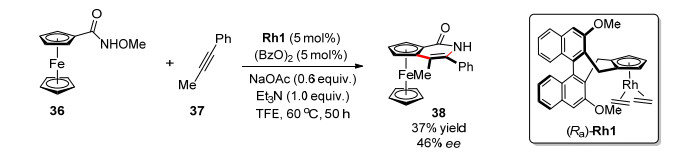 图式 15
铑催化不对称环化反应
Scheme15.
Rh(Ⅲ)-catalyzed asymmetric annulation reaction
图式 15
铑催化不对称环化反应
Scheme15.
Rh(Ⅲ)-catalyzed asymmetric annulation reaction
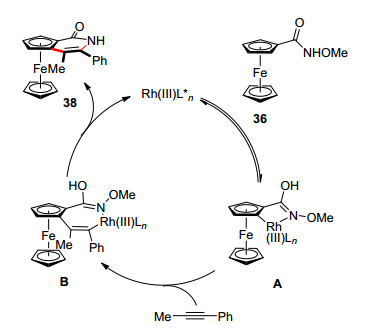 图5
可能的反应机理
Figure5.
Proposed reaction mechanism
图5
可能的反应机理
Figure5.
Proposed reaction mechanism
2.5 金/铂催化的碳氢键官能团化反应
金催化的分子内环异构化反应是合成菲及螺旋化合物重要方法.然而, 对映选择性的环异构化反应却鲜有报道. Urbano和Carre o等[32]发展了金催化的不对称环异构化反应合成平面手性二茂铁三环化合物, 该反应具有良好的底物普适性和优秀的对映选择性控制.随后, Shibata课题组[33]报道了几乎相同的反应, 使用金属铂作为催化剂(Scheme 16).可能的反应机理如图 6所示[34], 首先底物炔基和催化剂配位形成η2-金属络合物A, 接着发生6-endo环化反应, 生成产物40.
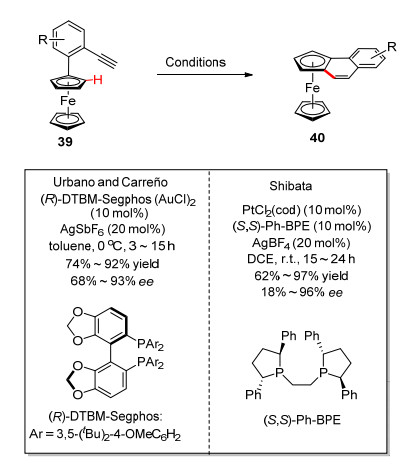 图式 16
Au/Pt催化的不对称环异构化反应
Scheme16.
Au/Pt-catalyzed cycloisomerization
图式 16
Au/Pt催化的不对称环异构化反应
Scheme16.
Au/Pt-catalyzed cycloisomerization
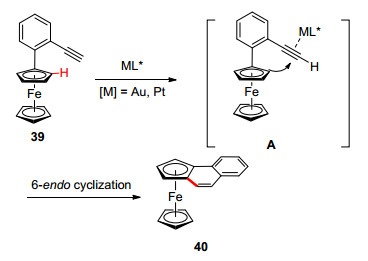 图6
可能的反应机理
Figure6.
Proposed reaction mechanism
图6
可能的反应机理
Figure6.
Proposed reaction mechanism
3 结论与展望
主要总结了过渡金属(铜、钯、铱、铑、金和铂)催化的不对称碳氢键直接官能团化反应合成结构多样的平面手性二茂铁化合物的最新研究成果, 与传统方法相比, 这些策略构建平面手性二茂铁更加简洁高效.在二价钯催化的反应中, 催化剂和二茂铁底物首先发生不对称碳氢键活化, 生成关键的手性钯中间体, 然后与不同偶联组分反应, 生成结构多样的产物.对于零价钯催化分子内不对称碳氢键芳基化反应, 催化剂用量低, 反应的对映选择性控制优秀, 产物也可容易转化为手性配体或催化剂, 并且实现其不对称催化反应.另外, 铱、铑、金和铂催化反应也提供了结构多样的平面手性二茂铁化合物.虽然在这一领域已取得了不少重要的进展, 但其发展仍处于起步阶段, 催化体系及反应类型还比较局限.例如, 除了碳-碳键之外, 构建其他碳-杂原子键(例如C—P、C—N、C—S键等)的相关工作还未见报道; 催化效率还不够高, 特别是在二价钯催化反应中, 这一定程度上限制了其实用性.其次, 导向基团的使用使得反应的适用性受限制, 发展可直接利用的导向基团非常有必要.总体来说, 不对称碳氢键官能团化构建平面手性二茂铁化合物已经取得很大进展, 发展更加高效的新催化体系, 新催化反应来构建平面二茂铁化合物仍然是这一领域的热门课题.
-
-
[1]
(a) Hayashi, T.; Togni, A. Ferrocenes, VCH, Weinheim, Germany, 1995.
(b) Togni, A.; Haltermann, R. L. Metallocenes, VCH, Weinheim, Germany, 1998.
(c) Štěpnička, P. Ferrocenes, Wiley, Chichester, 2008.
(d) Dai, L.-X.; Hou, X.-L. Chiral Ferrocenes in Asymmetric Catalysis, Wiley, Weinheim, 2010. -
[2]
(a) Fu, G. C. Acc. Chem. Res. 2000, 33, 412.
(b) Wu, Y. -J. ; Huo, S. -Q. ; Gong, J. -F. ; Cui, X. -L. ; Ding, L. ; Ding, K. -L. ; Du, C. -X. ; Liu, Y. -H. ; Song, M. -P. J. Organomet. Chem. 2001, 637, 27.
(c) Dai, L. -X. ; Tu, T. ; You, S. -L. ; Deng, W. -P. ; Hou, X. -L. Acc. Chem. Res. 2003, 36, 659.
(d) Fu, G. C. Acc. Chem. Res. 2004, 37, 542.
(e) Atkinson, R. C. J. ; Gibson, V. C. ; Long, N. J. Chem. Soc. Rev. 2004, 33, 313.
(f) Hou, X. -L. ; You, S. -L. ; Tu, T. ; Deng, W. -P. ; Wu, X. -W. ; Li, M. ; Yuan, K. ; Zhang, T. -Z. ; Dai, L. -X. Top. Catal. 2005, 35, 87.
(g) Arrayás, R. -G. ; Adrio, J. ; Carretero, J. -C. Angew. Chem. , Int. Ed. 2006, 45, 7674.
(h) Song, Q. -B. ; Dong, Y. Chin. J. Org. Chem. 2007, 27, 66(in Chinese).
(宋庆宝, 东宇, 有机化学, 2007, 27, 66. )
(i) Wu, Y. -J. ; Yang, F. ; Zhang, J. -L. ; Cui, X. -L. ; Gong, J. -F. ; Song, M. -P. ; Li, T. -S. Chin. Sci. Bull. 2010, 55, 2784.
(j) No l, T. ; Van der Eycken, J. Green Process. Synth. 2013, 2, 297.
(k) Schaarschmidt, D. ; Lang, H. Organometallics 2013, 32, 5668. -
[3]
(a) Blaser, H.-U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Top. Catal. 2002, 19, 3.
(b) Blaser, H.-U.; Pugin, B.; Spindler, F. J. Mol. Catal. A 2005, 231, 1. -
[4]
(a) Battelle, L. F.; Bau, R.; Gokel, G. W.; Oyakawa, R. T.; Ugi, I. K. J. Am. Chem. Soc. 1973, 95, 482.
(b) Rebière, F.; Riant, O.; Ricard, L.; Kagan, H. B. Angew. Chem., Int. Ed. 1993, 32, 568.
(c) Richards, C. J.; Damalidis, T.; Hibbs, D. E.; Hursthouse, M. B. Synlett 1995, 74.
(d) Tsukazaki, M.; Tinkl, M.; Roglans, A.; Chapell, B. J.; Taylor, N. J.; Snieckus, V. J. Am. Chem. Soc. 1996, 118, 685.
(e) Enders, D.; Peters, R.; Lochtman, R.; Raabe, G. Angew. Chem., Int. Ed. 1999, 38, 2421.
(f) Laufer, R. S.; Veith, U.; Taylor, N. J.; Snieckus, V. Org. Lett. 2000, 2, 629.
(g) Bolm, C.; Kesselgruber, M.; Muñiz, K.; Raabe, G. Organometallics 2000, 19, 1648.
(h) Bolm, C.; Kesselgruber, M.; Raabe, G. Organometallics 2002, 21, 707.
(i) Genet, C.; Canipa, S. J.; O'Brein, P.; Taylor, S. J. Am. Chem. Soc. 2006, 128, 9336.
For a review on kinetic resolution:(j) Alba, A.-N.; Rios, R. Molecules 2009, 14, 4747.
(k) Mercier, A.; Yeo, W. C.; Chou, J.; Chaudhuri, P. D.; Bernardinelli, G.; Kündig, E. P. Chem. Commun. 2009, 5227.
(l) Mercier, A.; Urbaneja, X.; Yeo, W. C.; Chaudhuri, P. D.; Cumming, G. R.; House, D.; Bernardinelli, G.; Kündig, E. P. Chem.-Eur. J. 2010, 16, 6285.
(m) Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Chem.-Eur. J. 2013, 19, 4151. -
[5]
For reviews and book, see:
(a) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242.
(b) Peng, H. M.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2010, 49, 5826.
(c) Wencel-Delord, J.; Colobert, F. Chem.-Eur. J. 2013, 19, 14010.
(d) Engle, K. M.; Yu, J.-Q. J. Org. Chem. 2013, 78, 8927.
(e) Zheng, C.; You, S.-L. RSC Adv. 2014, 4, 6173.
(f) Ye, B.; Cramer, N. Acc. Chem. Res. 2015, 48, 1308.
(g) You, S.-L. Asymmetric Functionalization of C-H Bonds, RSC, Cambridge, U.K., 2015.
(h) Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351.
(i) Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017, 117, 8908. -
[6]
(a) Wang, Y. ; Zhang, A. ; Liu, L. ; Kang, J. ; Zhang, F. ; Ma, W. Chin. J. Org. Chem. 2015, 35, 1399(in Chinese).
(王艳芳, 张安安, 刘澜涛, 康建勋, 张富强, 马文瑾, 有机化学, 2015, 35, 1399. )
(b) Arae, S. ; Ogasawara, M. Tetrahedron Lett. 2015, 6, 1751.
(c) López, L. A. ; López, E. Dalton. Trans. 2015, 44, 10128.
(d) Zhu, D. -Y. ; Chen, P. ; Xia, J. -B. ChemCatChem 2016, 8, 68. -
[7]
Xia, J.-B.; You, S.-L. Organometallics 2007, 26, 4869. doi: 10.1021/om700806e
-
[8]
Takebayashi, S.; Shizuno, T.; Otani, T.; Shibata, T. Beilstein J. Org. Chem. 2012, 8, 1844. doi: 10.3762/bjoc.8.212
-
[9]
Siegel, S.; Schmalz, H.-G. Angew. Chem., Int. Ed. 1997, 36, 2456. doi: 10.1002/(ISSN)1521-3773
-
[10]
(a) Sokolov, V. I.; Troitskaya, L. L. Chimia 1978, 32, 122.
(b) Sokolov, V. I.; Troitskaya, L. L.; Reutov, O. A. J. Organomet. Chem. 1979, 182, 537.
(c) Günay, M. E.; Richards, C. J. Organometallics 2009, 28, 5833. -
[11]
(a) Shi, B.-F.; Maugel, N.; Zhang, Y.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 4882.
(b) Shi, B.-F.; Zhang, Y.-H.; Lam, J. K.; Wang, D.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 460.
(c) Wasa, M.; Engle, K. M.; Lin, D. W.; Yoo, E. J.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 19598.
(d) Musaev, D. G.; Kaledin, A.; Shi, B.-F.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 1690.
(e) Xiao, K.-J.; Lin, D. W.; Miura, M.; Zhu, R.-Y.; Gong, W.; Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 8138.
(f) Chu, L.; Xiao, K.-J.; Yu, J.-Q. Science 2014, 346, 451.
(g) Chan, K. S. L.; Fu, H.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 2042.
(h) Laforteza, B. N.; Chan, K. S. L.; Yu, J.-Q. Angew. Chem., Int. Ed. 2015, 54, 11143.
(i) Xiao, K.-J.; Chu, L.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 2856.
(j) Xiao, K.-J.; Chu, L.; Chen, G.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 7796. -
[12]
Gao, D.-W.; Shi, Y.-C.; Gu, Q.; Zhao, Z.-L.; You, S.-L. J. Am. Chem. Soc. 2013, 135, 86. doi: 10.1021/ja311082u
-
[13]
Cheng, G.-J.; Chen, P.; Sun, T.-Y.; Zhang, X.; Yu, J.-Q.; Wu, Y.-D. Chem. Eur. J. 2015, 21, 11180. doi: 10.1002/chem.v21.31
-
[14]
Gair, J.-J.; Haines, B.-E.; Filatov, A.-S.; Musaev, D.-G.; Lewis, J.-C. Chem. Sci. 2017, 8, 5746. doi: 10.1039/C7SC01674C
-
[15]
Pi, C.; Li, Y.; Cui, X.-L.; Zhang, H.; Han, Y.-B.; Wu, Y.-J. Chem. Sci. 2013, 4, 2675. doi: 10.1039/c3sc50577d
-
[16]
Shi, Y.-C.; Yang, R.-F.; Gao, D.-W.; You, S.-L. Beilstein J. Org. Chem. 2013, 9, 1891. doi: 10.3762/bjoc.9.222
-
[17]
Zhang, H.; Cui, X.-L.; Yao, X.-Y.; Wang, H.; Zhang, J.-Y.; Wu, Y.-J. Org. Lett. 2012, 14, 3012. doi: 10.1021/ol301063k
-
[18]
Gao, D.-W.; Gu, Q.; You, S.-L. J. Am. Chem. Soc. 2016, 138, 2544. doi: 10.1021/jacs.6b00127
-
[19]
Pi, C.; Cui, X.-L.; Liu, X.-Y.; Guo, M.X.; Zhang, H.-Y.; Wu, Y.-J. Org. Lett. 2014, 16, 5164. doi: 10.1021/ol502509f
-
[20]
Gao, D.-W.; Yin, Q.; Gu, Q.; You, S.-L. J. Am. Chem. Soc. 2014, 136, 4841. doi: 10.1021/ja500444v
-
[21]
Fukuzawa, S.-I.; Yamamoto, M.; Hosaka, M.; Kikuchi, S. Eur. J. Org. Chem. 2007, 5540. doi: 10.1002/ejoc.200700470
-
[22]
Deng, R.; Huang, Y.; Ma, X.; Li, G.; Zhu, R.; Wang, B.; Kang, Y.-B.; Gu, Z. J. Am. Chem. Soc. 2014, 136, 4472. doi: 10.1021/ja500699x
-
[23]
张松, 陆俊筑, 叶金星, 段伟良, 有机化学, 2016, 36, 752. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201604009&dbname=CJFD&dbcode=CJFQZhang, S.; Lu, J.; Ye, J.; Duan, W.-L. Chin. J. Org. Chem. 2016, 36, 752(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201604009&dbname=CJFD&dbcode=CJFQ
-
[24]
(a) Nottingham, C.; Müller-Bunz, H.; Guiry, P. J. Angew. Chem., Int. Ed. 2016, 55, 11115.
(b) Nottingham, C.; Müller-Bunz, H.; McGlinchey, M. J.; Guiry, P. J. Eur. J. Org. Chem. 2017, 2848. -
[25]
(a) Ma, X.; Gu, Z. RSC. Adv. 2014, 4, 36241.
(b) Liu, L.; Zhang, A.-A.; Zhao, R.-J.; Li, F.; Meng, T.-J.; Ishida, N.; Murakami, M.; Zhao, W.-X. Org. Lett. 2014, 16, 5336. -
[26]
Gao, D.-W.; Zheng, C.; Gu, Q.; You, S.-L. Organometallics 2015, 34, 4618. doi: 10.1021/acs.organomet.5b00730
-
[27]
Gao, D.-W.; Gu, Y.; Wang, S.-B.; Gu, Q.; You, S.-L. Organometallics 2016, 35, 3227. doi: 10.1021/acs.organomet.6b00569
-
[28]
(a) Shibata, T.; Shizuno, T. Angew. Chem., Int. Ed. 2014, 53, 5410.
(b) Takebayashi, S.; Shibata, T. Organometallics 2012, 31, 4114. -
[29]
(a) Shibata, T.; Shizuno, T.; Sasaki, T. Chem. Commun. 2015, 51, 7802.
(b) Zhang, Q.-W.; An, K.; Liu, L.-C.; Yue, Y.; He, W. Angew. Chem., Int. Ed. 2015, 54, 6918.
(c) Murai, M.; Matsumoto, K.; Takeuchi, Y.; Takai, K. Org. Lett. 2015, 17, 3102. -
[30]
Wang, S.-B.; Zheng, J.; You, S.-L. Organometallics 2016, 35, 1420. doi: 10.1021/acs.organomet.6b00020
-
[31]
Guimond, N.; Gouliaras, C.; Fagnou, K. J. Am. Chem. Soc. 2010, 132, 6908. doi: 10.1021/ja102571b
-
[32]
Urbano, A.; Hernández-Torres, G.; del Hoyo, A. M.; Martínez-Carrión, A.; Carre o, M. C. Chem. Commun. 2016, 52, 6419. doi: 10.1039/C6CC02624A
-
[33]
Shibata, T.; Uno, N.; Sasaki, T.; Kanyiva, K. S. J. Org. Chem. 2016, 81, 6266. doi: 10.1021/acs.joc.6b00825
-
[34]
Fürstner, A.; Mamane, V. J. Org. Chem. 2002, 67, 6264. doi: 10.1021/jo025962y
-
[1]
-

图式 1 手性噁唑啉导向的非对映选择性碳氢键芳基化和酰胺化反应
Scheme 1 Diastereoselective C—H arylation and amidation reactions by using chiral oxazoline as a directing group

图式 2 铜催化二茂铁碳氢键不对称卡宾插入反应
Scheme 2 Cu-catalyzed enantioselective insertion of carbenoid into C—H bond of ferrocene

图式 3 钯催化二茂铁和芳基硼酸的不对称碳氢键芳基化反应
Scheme 3 Pd-catalyzed asymmetric C—H bond arylation of ferrocenes with arylboronic acids

图式 4 二价钯催化的氧化Heck和环化反应
Scheme 4 Pd(Ⅱ)-catalyzed oxidative Heck and annulation reactions

图式 5 不对称双碳氢键氧化交叉偶联反应
Scheme 5 An enantioselective oxidative twofold C—H cross-coupling reaction

图式 6 二茂铁化合物催化不对称碳氢键酰基化反应
Scheme 6 Catalytic enantioselective C—H acylation of ferrocenes

图式 7 零价钯催化的分子内不对称碳氢键芳基化反应
Scheme 7 Pd(0)-catalyzed enantioselective intramolecular C—H arylation

图式 8 高效合成平面手性P, N-配体(S, Rp)-20
Scheme 8 Efficient synthesis of planar chrial P, N-ligand (S, Rp)-20

图式 9 手性二醇21的高效合成及不对称hetero-Diels-Alder反应
Scheme 9 Efficient synthesis of diol 21 and asymmetric hetero-Diels-Alder reaction

图式 10 二茂铁酰胺化合物分子内不对称碳氢键芳基化反应
Scheme 10 Enantioselective intramolecualr C—H arylation of ferrocenecarboxamides

图式 11 高对映选择性合成手性平面二茂铁吡啶衍生物及其在环氧化合物不对称开环反应中的应用
Scheme 11 Highly enantioselective synthesis of planar chiral ferrocenylpyridine derivatives and their application in asymmetric opening of meso-epoxide

图 3 (a) 可能的反应机理、(b)两种过渡态TS-CMD-R和TS-CMD-S的结构以及(c)两种过渡态象限分析
Figure 3 (a) Proposed reaction mechanism, (b) the structures of the two transition states TS-CMD-R and TS-CMD-S, and (c) the quadrant analysis of the two transition states

图式 13 铱催化二茂铁不对称碳氢键烷基化反应
Scheme 13 Ir-catalyzed enantioselective C—H alkylation of ferrocene

图式 14 铑催化分子内不对称碳氢键硅基化反应
Scheme 14 Rh-catalyzed intramolecular asymmetric C—H silylation
-


 下载:
下载:





















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 57
- 文章访问数: 7242
- HTML全文浏览量: 894
 下载:
下载:
