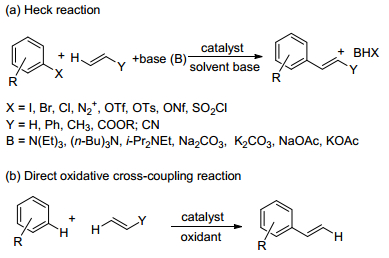
图式 1.
氧化偶联反应制备烯烃的两种方法
Scheme 1.
Two main methods of olefin formation by coupling reaction

不饱和脂肪族烃是重要的有机合成前体, 金属催化的氧化反应一直是化学工作者们研究的热点.不饱和脂肪族烃可以与金属催化剂配位得到活化, 从而更易于进行反应得到有用的有机化合物.在这些金属催化剂中, 钯占用重要的地位, 一方面是因为钯有容易相互转化的Pd(0), Pd(Ⅱ), Pd(Ⅳ)三种价态, 还因为易于通过改变配体、碱、溶剂、温度和添加剂等来优化反应条件.随着能源紧缺与环境污染问题的日益突出, 客观上要求化学工作者们发展出更为绿色、经济的反应, 提供原子利用率更高且对环境更为友好的合成策略[1], 此时氧化偶联反应便应运而生.钯是在催化氧化偶联反应中应用较多的过渡金属[2].目前对于催化不饱和脂肪族烃参与的氧化偶联反应的研究仍存在一定的局限性, 针对这种情况, 我们课题组报道了一个烯烃与苯醌在四价铈氧化催化下发生氧化偶联环化生成苯并四氢呋喃类衍生物的反应.
1967年, 日本化学家Fujiwara和Moritani首次采用苯乙烯氯化钯和苯反应得到了trans-二苯乙烯[3], 随后这种过渡金属催化的方法逐渐发展成为一种制备芳基烯烃的重要方法(Scheme 1).由于该方法所采用的底物无需预先官能化, 这不仅大大缩短了反应步骤, 并且反应过程中无卤素等官能化基团的脱除, 故能有效避免废盐等副产物的产生.因此, 该方法同Heck反应相比具有原子利用率高、原子经济性好等绿色化学的典型特征, 这使其日益受到人们的重视.
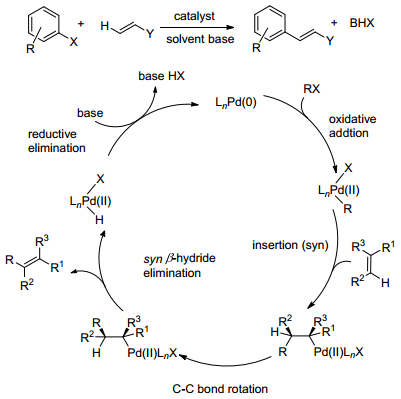
Heck反应作为构建芳基烯烃最有效的方法之一(Scheme 2), 其已经在合成领域的各个方面均有着十分广泛的应用以及出色的表现[4].但因其需要提前对底物进行预官能化, 步骤一般较为繁琐, 且在生成产物的同时产生了大量的有害物质受到了极大的限制, 与如今的绿色化学理念亦有冲突.
在过渡金属催化的作用下, 非官能团化的烯烃与芳烃直接进行氧化偶联从而构造出芳基烯烃.相比于Heck反应, 该构建芳基烯烃的方法因其原子利用率较高, 步骤简单等优势受到化学工作者的广泛关注.他们指明在该反应中乙酸是这种转化所必须的, 而这指导了随后几十年的后续研究, 即便是近几年, 使用酸性体系仍是直接构建芳基烯烃的主要方法.
接下来, Fujiwara课题组[5]使用催化量的Pd(OAc)2来催化反应进行进一步的拓展, 证实了各种杂环类芳烃(如噻吩和呋喃)亦可得到相应的取代烯烃产物[6, 7].紧接着, Itahara与他的同事将吡咯和吲哚也引入了这种类型的氧化偶联反应当中(Eq. 1).醌与尿嘧啶可以在该条件下直接进行芳基化或烯烃化, 但通常需要定量的钯催化剂且产率低下.
|
|
(1) |
在20世纪末, Fujiwara课题组[8]报道了一个芳烃与烯烃的氧化偶联反应, 该反应以叔丁基过氧化氢作为氧化剂并使用1 mol% Pd(OAc)2与10 mol%苯醌作为催化剂, 底物拓展表明, 烯烃以及芳基上连有不同取代基时, 均能以中等以上产率得到目标产物.特别注意的是, 该反应体系对于杂环芳烃具有良好的区域选择性和立体选择性, 如呋喃和吲哚等, 且主要生成反式烯烃(Scheme 3).
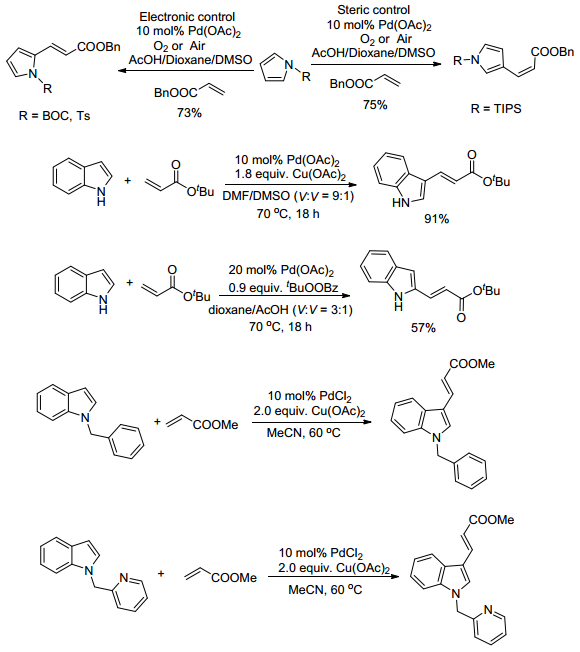
化学工作者们继续探索这一体系, 发现芳环烯基化的区域选择性亟待解决[9, 10].杂芳环的氧化偶联反应一般发生在3位, 随着研究的深入, 人们发现钯催化的吲哚烯基化可以通过控制溶剂的方式使氧化偶联反应发生在2位[11]. 2005年, Ricci课题组[12]就发现当反应在极性溶剂(如DMSO或DMF)中进行时, 丙烯酸正丁酯选择性地在吲哚的3位发生氧化偶联反应.而当反应在非极性溶剂(如CH2Cl2)中进行且以Ac2O作为助溶剂时, 丙烯酸正丁酯则选择性地在吲哚的2位发生氧化偶联反应.作者推测是这种酸性条件减缓了吲哚在钯作用下的去质子化, 从而使得Pd—C键从3位迁移到了2位, 进而生成了2位取代烯烃的产物.实验结果表明, 同样可以通过对于吲哚基的N原子上面取代不同的取代基来实现这种区域选择性, 比如将N上的取代基从2-吡啶甲基换为苄基, 则烯烃的取代从2位, 变为了3位实现了氧化偶联反应区域选择性.
在氧化C—H键进行烯烃化时大多数选择的芳烃都是富电子的, 因为电子与钯的空轨道进行配位往往是转化的关键.然而就在近期, Yu课题组报道了一个氧化偶联反应, 该反应是缺电子的芳烃进行烯烃化反应.当配体使用2, 6-二烷基吡啶时, 烯烃选择性的生成芳基间位和对位的取代产物, 并主要生成间位产物.使用这种特殊配体的具体原因目前还未有定论, 但其邻位上选择性的抑制可以将其归因于配体的空间位阻影响.
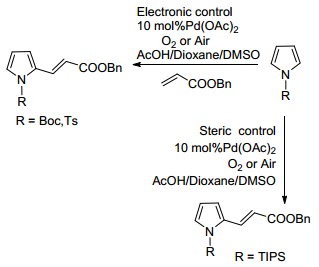
2006年, Gaunt课题组[13]研究发现, 在有氧存在的条件下, 对吡咯上的N使用不同保护基团进行保护也可以控制其与烯烃进行氧化偶联反应时的区域选择性(Scheme 4).在使用吸电性的保护基团例如N-Boc以及N-Ts保护时, 能够有效的降低吡咯的反应活性, 并能以相当不错的产率生成2位选择的取代烯烃的产物.当其他条件一致, 选择空间位阻较大的保护基团N-TIPS时, 其对2位有一定屏蔽的作用, 进而导致选择性的生成3位取代的产物.
缺电子的杂芳烃例如N上连有O的吡啶也能够作为底物与烯烃进行氧化偶联反应.这种底物能够在Pd(OAc)2作催化剂, 氧化剂选择Ag2CO3的条件下选择性的生成2位取代的产物[14].这与大多数C—H活化不同, 该反应在碱性条件下
添加了1.0 equiv.的吡啶作为添加剂.烯烃上取代缺电子基团或脂肪烃时均能得到中等以上的产率.其他的N上连有O的杂芳烃衍生物如吡嗪以及哒嗪等亦能适用.这种反应体系为吡啶2位的官能化提供了一种非常有效的选择(Eq. 2).
|
|
(2) |
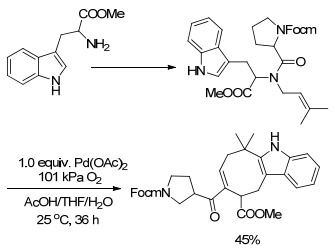
烯烃参与的氧化偶联反应也可在分子内进行. 2002年, Corey课题组[15]报道了一种分子内的烯烃氧化偶联反应, 用来合成天然产物.该反应首先以吲哚氨基化合物作为原料, 分别经过5步与6步反应得到最终的两种天然产物, 而其中由分子内烯烃氧化偶联反应得到的吲哚并环烯烃化合物正是这两种生物碱在全合成过程中的关键中间体.作者报道的该反应是吲哚烯烃类衍生物进行分子内的氧化偶联反应, 从而得到吲哚并八元环烯烃类衍生物.催化剂选择Pd(OAc)2, 氧化剂选择O2, 溶剂选择CH3COOH/H2O/THF的混合溶液(Scheme 5).对于取代苯环的烯基化, 使用导向基团来实现区域选择性则是目前一种通用的方法.在氧化偶联反应中使用乙酰氨基作为导向基团受到化学工作者的广泛关注.
而早在2002年, Van Leeuwen课题组[16]就第一次报道了这种氧化偶联反应体系, 是缺电子烯烃在氧化条件下进行的.在这个体系下仅能观察到邻位取代的产物, 表明了酰胺基团在反应中对于区域选择的重要性.该反应使用Pd(OAc)2作催化剂, 苯醌作为氧化剂, TsOH作为添加剂, 在室温条件下进行反应.实验结果表明, 苯醌在整个催化剂循环的过程中起到了对各种形式钯的稳定作用.作者发现富电子的芳烃反应更快, 这表明该反应是一个亲电过程.随后作者对反应的限速步骤进行了实验探究, 通过动力学同位素效应的实验, 证实了限速步骤的确为芳烃的C—H键的裂解.
随后, 在类似的反应条件下, 2005年Prasad课题组[17]引入卤代乙酰苯胺作为底物, 并使用氧气代替苯醌作氧化剂, 使用Pd(OAc)2作为催化剂, 在丙酮作溶剂的体系中进行反应, 也得到了相当不错的转化率.在2009年, Brown[18]和Liu小组[19]分别报道了使用阳离子钯催化剂催化这类反应.这种类型的反应还可以在水中进行, 其它的金属催化剂铑也开始被运用于该类反应之中(Eq. 3).
|
|
(3) |
同样是用钯作催化剂, 2008年Booker-Milburn课题组[20]报道了一个与之类似的反应.该反应是苯脲类化合物与双烯烃进行烯化环化得到目标产物苯并二氢吲哚类化合物.该反应在进行过程中, 烯烃π键首先与Pd进行配位, 其后进行一步亲核取代得到杂环化合物.体系中的氧化剂并没有将苯并二氢吲哚进一步氧化, 另外当R2或R3有一个是H时, 环化反应会继续进行而生成三环化合物(Eq. 4).
|
|
(4) |
在2008年, Yu课题组[21]进一步提出了在使用三氟甲基与羧基作导向基团时(Scheme 6), 对取代苯环进行烯基化的氧化偶联反应.当导向基团选择使用三氟甲基时, 能够生成邻位取代烯烃的产物, 表现出优秀的区域选择性.

2009年, Yu课题组[24]报道了钯催化缺电子芳烃的间位烯基化反应(Eq. 5).该反应中使用了一个新的吡啶配体Pyr, 能够有效地促进间位烯基化产物的生成.在Pyr的存在下, 含吸电子基团(例如三氟甲基、硝基、酯、酮)的芳烃能和钯催化剂发生协同作用, 并进行C(sp2)—H的碳氢键活化.氧化烯基化的区域选择性取决于吸电子基团所增强间位碳氢键的酸性.在该反应中Ac2O是必不可少的, 且不能被分子筛替代的, 这可能是由于该添加剂在有氧条件下有助于把H-Pd—OAc转换为Pd(OAc)2.
|
|
(5) |
2010年, Yu课题组[22]又研究了苯乙酸和烯烃氧化偶联反应(Eq. 6), 推测羧酸的C=O和Pd(OAc)2之间的弱相互作用, 可以促进邻位碳氧键的官能团化.反应体系可以在比较温和的反应条件下以较高收率得到苯乙酸衍生物, 同时具有很好的官能团兼容性.烯烃参与的氧化偶联反应除了区域选择性之外, sp3杂化的C—H活化也一向是化学工作者格外关注的热点问题, 也是催化领域难题之一.同年, 他们又发现氨基酸作为配体可以增强邻位烯基化反应的区域选择性和反应效率[23].在该条件下, 惰性底物如苄基乙酸也可以顺利地进行成烯反应.
|
|
(6) |
在Yu的邻位烯基化反应中, 配体通常可以加快反应速率和增强区域选择性, 同时也可以控制对映选择性(Eq. 7).在手性氨基酸的作用下, 二苯基羧酸盐可以通供了一个很有价值的手性羧酸, 但是该条件下带有α质子的产物更倾向于外消旋.
|
|
(7) |
近期, Yu课题组[25]报道了一个烷基烯化的反应.该反应使用多种金属盐作为添加剂, 使用Pd(OAc)2作为该反应的催化剂, 以丙烯酸苄酯作为偶联试剂进行氧化偶联反应.该反应为sp3的C—H键的直接进行活化偶联提供了一种可行性(Eq. 8).
|
|
(8) |
近期, Mhaske课题组[26]报道了一种钯催化的使用一步法使分子内的烯烃酰胺化/环化的方案, 用于合成8-氧代原花青素的基本框架, 从而进一步应用在合成8-氧肉桂酸、8-氧代四氢化他汀以及8-氧代异黄酮等天然产物骨架上, 对于大量合成类似天然产物有积极的推动作用(Eq. 9).该反应使用Cu(OAc)2以及NaOAc作为添加剂, 以O2为氧化剂, 并使用10 mol%的Pd(OAc)2作催化剂, 在DMSO与H2O以10:1比例混合的混合溶剂中进行反应.从底物拓展实验可以看到, 该反应体系对于不同的取代基都表现出不错的耐受性, 均能够以中等及以上的产率得到目标产物.
|
|
(9) |
全氟代芳烃的氧化偶联具有一定的难度, 因为碳氧键活化导致了一个携有Pd—Ar—F键中间体的生成, 该络合物对催化剂相当稳定[27]. 2010年, Zhang课题组[28]报道了钯催化的全氟代芳烃的成烯反应. Ag2CO3在反应中既作为碱又作为氧化剂, 其他的钯催化剂[如PdCl2, Pd(TFA)2, Pd(dba)3]和氧化剂[如Cu(OAc)2, Oxone, benzoquinone, PhI(OAc)2, O2]都是无效的.该脱氢交叉偶联适用于各种底物, 包括丙烯酸、丙烯酰胺和乙稀基酯, 包括吡啶这种路易斯碱也可以兼容(Eq. 10).当苯乙酸与烯烃发生氧化偶联反应时, 该反应同样能在钯作催化剂, 苯醌作氧化剂的条件下温和地进行, 并得到高区域选择性的产物[29].同时发现了药物酮洛芬、布洛芬和萘普生的合成适用于该体系, 并能够以较高的收率得到顺式的烯化产物(Eq. 11).当3-芳基丙酸和多取代苯乙酸作为反应底物时, 作者他们能够观察到配体所影响的反应性和选择性.当使用mono-N氨基酸作为配体时, 产物产率从8%激增至60%, 3-甲基-5-甲氧基苯乙酸的Ha和Hb的选择性也得到了一定改善, 不过效率并不高.随后, 有化学工作者使用Pd(OAc)2作催化剂, 氨基酸Ac-Val-OH作配体在该体系下得到了高产率的邻位双取代的产物[30].作者还对配体的影响进行了探索, 发现反应速率随着C—H键裂解速度的加快而加快.从而证实了C—H键的裂解为该反应的限速步骤[31].
|
|
(10) |
|
|
(11) |
近期, Sridharan课题组[32]报道了一个钯催化的烯烃参与的分子内的氧化偶联反应(Scheme 7).该反应方案是在室温的条件下, 反应物在PdCl2(MeCN)2或者PdCl2的催化作用下, 在乙腈或者四氢呋喃作溶剂的条件下, 使用4 equiv.水进行反应.以高产率、高选择性得到水合烯烃插入的反应产物, 即2, 3-二氢-1H-茚-1-酮, 该产物在生物学中有重要的应用价值.通过底物拓展实验观察到, 当内部亲核试剂为对甲苯磺酰胺, R1为氢时, R2不论是苯基还是取代苯基甚至是烷基均能以高产率得到对应的目标产物.而R1为甲氧基, 将R2替换为不同取代基时, 同样能得到相应产物并且产率相对较高.对于内部亲核试剂为羟基的反应, 总体产率略低于选择对甲苯磺酰胺做亲核试剂, 对于不同基团同样有不错的耐受性.作者对于最可能的反应机理进行了推测, 并尝试做了一系列的探究实验.最后推测可能的反应机理为:最开始二价钯与初始反应物的炔上电子进行配位, 接着在亲核试剂的辅助下进行区域选择性水合, 而后进行质子化得到烯醇结构, 最后进行迈克尔加成得到最终的产物.
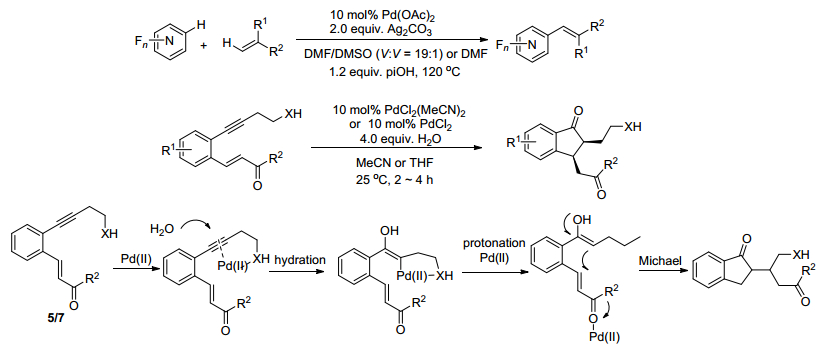
2016年, Shenvi及其同事[33]报道了一个烯烃与碘代芳基在镍-钴共催化作用下偶联反应, 该反应在5 mol% (dtbbpy)NiBr2与20 mol% Co(SaltBu, tBu)共同催化, 使用50 mol%含氟配体, 添加剂为2 equiv.的Ph(iPrO)SiH2, 在N, N-二甲基丙烯脲溶剂中, 220 ℃下反应得到相应的产物.
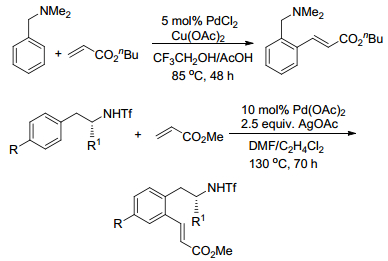
Shi课题组等[34]研究报道了当以N, N-二甲基苄胺作底物时, 其亦能与丙烯酸正丁酯反应得到邻位烯烃化产物, 收率86%.底物拓展实验表明:苄胺氮上取代基的种类及数目对底物活性具有极为显著的影响; 当N上的甲基被H原子取代时, 苄胺即失去反应活性; 当以苯环上卤代的N, N-二甲基苄胺作底物时, 产物中未发现Heck偶联产物形成; 当以α-甲基丙烯酸酯作底物时, 反应得到非共轭的苯基烯烃产物(Scheme 8).
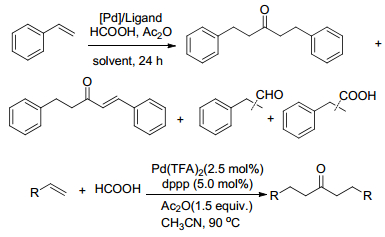
2017年, Chang课题组[35]报道了钯催化烯烃和HCOOH的氧化偶联反应, 可以生成酮类化合物, 该反应得到很高的反应产率, 高达90% (Scheme 9).在刚开始的实验研究中, 苯乙烯作为初步测试底物, Pd(OAc)2作催化剂, 作者还研究了Pd(TFA)2为催化剂. dppp为配体, 溶剂是乙腈时, 最后产物酮的产率可高达95%.
2017年, Liao课题组[36]报道了钯催化烯烃的氟烷基活性氧化偶联环化反应, 烯烃和现成的Rf—I键发生环氧化偶联反应, 可得到相应的氟烷基化2, 3-二氢苯并呋喃和二氢吲哚衍生物, 具有中等至优异的产率.这是一种用一步反应法构建C(sp3)—CF2和C—O/N键新颖有效的方法(Eq. 13).
|
|
(13) |
依然是在2017年的时候, Zang课题组[37]报道了钯催化酰胺分子内的氧化偶联反应, 可以生成螺二氢喹啉和八氢衍生品化合物(Scheme 10).分子内C(sp2)—H未活化的环烯烃的氧化芳基化的方法, 已经是获得螺二氢喹啉和八氢衍生品化合物的一个简单而有效的方法.用甲基丙烯酰胺作为导向基团, 通过烯基苯胺直接氧化芳基化, 可以获得优异收率的螺二氢喹啉类化合物.
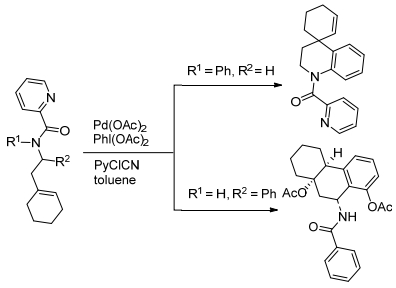
同年, Raju课题组[38]报道了通过使用亚氨基醌作为导向基团和内部氧化剂或辅助氧化剂的条件下, 用钯催化C—H键活化生成了二氢吩草素和苯并噻吩类化合物.钯催化的C—H键活化反应, 要经过氧化还原过程, 是为了制备二氢苯丙氨酸, 菲啶和咔唑衍生物是从联芳基亚氨基醌发展而来的.亚氨基醌被预先设计为一种导向基团进行邻位C—H活化和内部氧化剂或共氧化剂(Scheme 11).这个催化过程通过以下顺序进行: C—H键活化, 协调反应, 和活性烯烃的插入, β-氢化物消除, H转移插入, 和质子化或β-氢化物消除.除此之外, 可以使用这种方法有效地制备咔唑, 而不添加外部氧化剂.
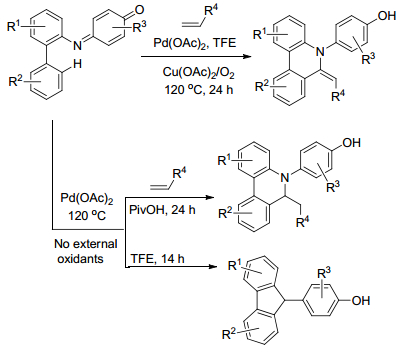
2013年, Cook课题组[39]报道钯催化下炔与非活化的烷基碘代物的插入、还原反应(Eq. 14), 以此来合成一系列三取代的烯烃.该反应具有反应条件温和、官能团容忍性好等特点.
|
|
(14) |
随后, 该课题组[40]报道了β-H存在下钯催化分子内碘迁移反应(Eq. 15).该反应以Pd(PPh3)4为催化剂, Cs2CO3为碱, 甲苯为溶剂, 50 ℃条件下, 一系列非环状的二级烷基碘代物能以较高的收率转变为含碘甲基的二奎烷类化合物.反应具有原子经济性高、官能团兼容性好、产物易于衍生化等特点.
|
|
(15) |
2015年, Gu课题组[41]报道了一种新颖的合成方法, 实现了邻位氨基化、原位炔基化的芳基官能团化的产物.该反应采用炔酸作为末端偶联试剂, 二级胺作为氨基源, 首先经过一个Catellani邻位的C—H氨化, 随后再发生脱羧炔基化反应.在反应体系中, 各种取代的炔酸都能够很好地进行反应.和先前报道的用末端炔作为偶联试剂相比, 该体系有三个明显的优势: (1)避免了分批加入; (2)大大缩短了反应的时间; (3)无论是芳基炔酸还是烷基炔酸, 都能有效地参与反应(Eq. 16).
|
|
(16) |
同年, Nevado课题组[42]报道了钯催化下端炔双官能团化反应.在PdCl2(PPh3)2催化下, K2CO3为碱, CH2Cl2和H2O作为溶剂, 芳基或烯基硼酸和全氟碘代物同时与端炔进行反应, 以较高区域选择性及立体选择性合成系列三取代的烯烃化合物(Eq. 17).
|
|
(17) |
2016年, Chaładaj课题组[43]报道了一种钯络合物催化下炔烃的烷基化反应(Eq. 18).一系列炔烃(端位和非端位)、芳基硼酸、全氟碘代物通过一锅法反应, 以较高的收率、高区域选择性、高立体选择性得到三取代或四取代的烯烃.
|
|
(18) |
Zhou课题组[44]发展了一种钯催化的杂芳烃与二、三级卤代烃的区域选择性烷基化方法(Eq. 19).在Pd(PPh3)4为催化剂、dppp为配体、Cs2CO3为碱的条件下, 1, 3-苯并噁唑、咪唑、吡咯、呋喃、噻吩等杂环化合物可以实现与二级、三级碘代烷烃的烷基化反应.添加NaI后, 溴代烷烃在上述条件下也可以很好的反应, 同时在体系中检测到了少量的碘代烷烃, 说明参与反应的可能是碘代烷烃.
|
|
(19) |
在2015年, Alexanian课题组[45]发展了一种钯催化非活化烷基卤代烃与芳烃的分子内环化反应(Eq. 20).以Pd(PPh3)4为催化剂, PMP为碱, 苯基叔丁烷为溶剂, 130 ℃反应48 h(以碘代烷烃为底物, 反应条件更为温和), 高产率地得到了一系列四氢萘、二氢茚、氮取代的吲哚啉、喹啉以及异喹啉等环化产物.
|
|
(20) |
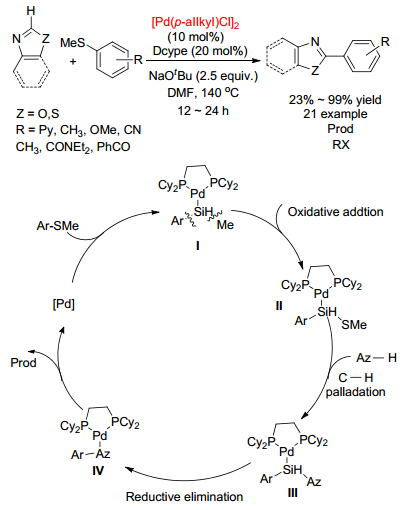
2016年, Yu课题组[46]报道了10 mol%的[Pd(π-allyl)Cl]2作为催化剂, 2.5 equiv.的NaOtBu为碱, DMF作为溶剂的条件下噻唑和芳基硫醚发生的氧化偶联反应(Scheme 12).反应12~24 h可以得到二(杂)芳基产物, 并且有很高的产率, 并对它的反应机理进行了探究.首先反应物Ⅰ经过氧化加成, 芳基硫醚的C—S键发生断裂, 形成中间体产物Ⅱ, 再经过一步钯催化得到产物3, 随后, CAr—CAz发生一步还原消除得到产物Ⅳ, 偶联产物同时释放再生钯催化剂, 使得Pd催化剂可以循环利用.
同年Wang课题组[47]报道了钯催化苯甲酸和酚类化合物发生的环氧化偶联, 得到生成二苯并吡喃类化合物.非常易得和廉价的原料被应用到这种新颖的方法中, 用来合成非常有价值和有用的二苯并吡喃酮支架.这种方法不仅适应的底物范围广, 而且操作也很简便.苯并吡喃又称为色烯, 它本身并不重要, 但它的某些衍生物却很重要.例如, 许多花的颜色物质, 叫作花色素, 是苯并吡喃盐的衍生物.苯并吡喃的羰基衍生物苯并-α-吡喃酮和苯并-γ-吡喃酮存在于许多天然化合物的结构中.经过初步的机械研究, 了解了这种化学作用, 并启发不同功能化芳烃的氧化偶联的新设计(Eq. 21).
|
|
(21) |
目前对于催化烯烃参与的氧化偶联反应的研究仍存在一定的局限性, 针对这种情况和前面工作基础上, 2011年, Shi课题组[48]报道了KOtBu促进的1, 1-二芳基苯乙烯的无过渡金属催化的芳基化反应(Eq. 22).使用30 mol%的1, 8-二苯菲作配体, 3 equiv.的KOtBu, 以苯作溶剂, 使得1, 1-二芳基苯乙烯和3-碘苯在110 ℃条件下进行氧化偶联反应得到芳基化产物, 产率达90%.该反应能够得到与Mizoroki-Heck反应同样的反应结果, 但并没有使用任何过渡金属催化剂.实验发现苯是反应效果最好的溶剂, 能够高度选择地生成相应的烯烃芳基化产物, 仅有微量的联芳基化合物作为副产物生成.通过不同的底物的拓展实验发现, 该反应对于不同官能团具有良好的耐受性, 已被用于多种药物中[49].
|
|
(22) |
Shi课题组在报道了芳基卤化物与烯烃的分子间的反应之后, 首次报道了两者分子内的氧化偶联反应(Eq. 23).但是环化产物并不是简单的偶联产物, 而是环化/异构化产物.同样是使用双氮化合物作为配体, 在KOtBu作添加剂, 以苯为溶剂, 使邻碘苯基烯丙基醚进行分子内的偶联反应, 并以良好的产率得到了一系列苯并呋喃的衍生物.这种转化为通过2-卤代苯酚和烯丙醇制备苯并呋喃提供了一种新颖的更为简单廉价的替代方法.
|
|
(23) |
苯并二氢呋喃是木脂素的重要结构单元, 因此对于该结构的合成一直受到广大合成化学工作者的关注.其广泛地存在于自然界的活性分子中.为了构建此类结构, 科研工作者们已经研究发展了多种合成方法[50].各种配体和5 mol% Pd(OAc)2, 2.0 equiv.的HCOOH, 1.0 equiv的Ac2O, 溶剂是正己烷, 90 ℃条件下反应24 h.然而苯并四氢呋喃作为苯并二氢呋喃的类似物, 因各种原因, 其合成工作一直没有太大进展, 处于一个亟待发展的阶段. 2013年Lei及其同事[51]报道了在HOTf催化下使苯醌与烯烃直接进行氧化偶联合成苯并四氢呋喃.即便如此, 探求构建苯并四氢呋喃的更为温和高效的合成方法仍然充满挑战.此外, 使用过渡金属催化的氧化偶联反应向来被化学工作者视为一种构建C—C键的高效的方法, 并有可能进一步获得复杂分子, 而合适的氧化剂则是获得这种有效的转化的强有力保障.
2017年我们课题组[52]报道了一个烯烃与苯醌在四价铈氧化催化下发生氧化偶联环化生成苯并四氢呋喃类衍生物的反应(Scheme 13).该反应使用廉价的过渡金属铈盐作为氧化促进剂, 使用无水硫酸镁作添加剂, 甲苯作溶剂, 在室温、空气氛围下进行.该反应能够普遍得到良好的产率, 并且具有良好的官能团耐受性, 特别对于酸性敏感的基团具有不错的效果.将其反应量扩大至克级时, 该反应也能以良好的产率得到目标产物.实验初期以对甲氧基苯酚和α-甲基苯乙烯作为底物, 欲探索出一种更为温和、高效、廉价的合成苯并二氢呋喃的方法.我们进一步探究发现对甲氧基苯酚在进行反应之前先一步被Ce(SO4)2•4H2O以95%以上的产率氧化为苯醌.因此接下来的工作则明确为苯醌与芳基烯烃在Ce(SO4)2•4H2O的促进下进行氧化偶联反应生成苯并四氢呋喃类衍生物的方法探究.
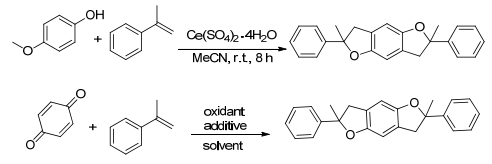
近些年来, 高价态的铈盐作为一种高效且廉价的氧化剂越来越多地受到化学工作者们的关注, 并被用于许多有机合成反应当中, 包括合成乙酰氨基苯酚、从烯烃制备羧酸酯以及将肟转化为醛和酮等.总之, 我们发现了一种通过四价铈盐氧化催化的由苯醌与烯烃合成苯并四氢呋喃的简单方法.该方法在室温条件下无需惰性气体保护就能得到一系列苯并四氢呋喃类化合物, 尤其是该反应对于酸敏感的基团同样有良好的耐受性.在有机合成反应中C—C键的构建是非常关键的一环.自从偶联反应被化学工作者发现以来, 该反应便已经在C—C键的形成领域占据了十分重要的地位, 且在过去的半个多世纪中取得了重大的进展, 进而成为化学工作者手中形成C—C键最有效也最直接的工具之一.随着能源紧缺与环境污染问题的日益突出, 客观上要求化学工作者们发展出更为绿色、经济的反应.
如今, “钯催化的偶联反应”被应用于许多物质的合成研究和工业化生产中.例如合成抗癌药物紫杉醇和抗炎症药物萘普生, 以及有机分子中一个体格特别巨大的成员——水螅毒素.科学家还尝试用这些方法改造一种抗生素——万古霉素的分子, 用来灭有超强抗药性的细菌.此外, 利用这些方法合成的一些有机材料能够发光, 可用于制造只有几毫米厚、像塑料薄膜一样的显示器.科学界一些人士表示, 依托“钯催化交叉偶联反应”, 一大批新药和工业新材料应运而生, 这些科学家的科研成果如今已经成为支撑制药、材料化学等现代工业文明的巨大力量.药物、塑料和许多其他的工业化学品都包含有大的碳基分子, 为了制造这些复杂的化学物质, 化学家需要将碳原子连接在一起.不过, 碳原子的稳定性很强, 碳原子之间并不能够轻易发生反应.有了钯作催化剂, 科学家能够在更低的温度下, 使用更少的溶剂、更小的成本来制造化合物, 而且反应产生的废物更少.
通常把钯催化的烯烃芳基化和烯基化偶联反应在药物合成方面有很多的应用, 尤其对于分子内的关环反应, 构建拥挤的季碳中心极为方便.因此偶联反应在苯(沙坦联苯)是很多治疗高血压用沙坦类药物的关键中间体.用Pd(OAc)2代替PdCl2, 在碱性条件方面用磷酸钾或钠代替醋酸钠, 以邻氯苯腈为原料, 经过偶联反应得到, 其收率达到95% (Eq. 24).
|
|
(24) |
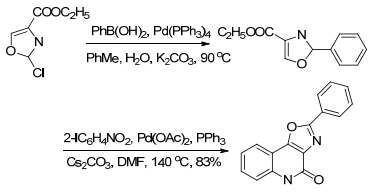
在众多的有关耐受菌药物的研究中, 神经系统药物2-芳基氧杂唑喹啉酮有抗精神抑郁、提高记忆力的作用.反应从简单的原料2-氯氧杂唑-4-醋酸酯开始, 经过偶联反应引入一芳环, 再与卤代建喹啉环得到目标产物[53] (Scheme 14).
Beraprost 3是前列腺素的一种衍生物[54], 具有口服抗血栓活性, 不但水溶性好, 而且稳定, 细胞毒性也小.以Pd(OAc)2为催化剂, 通过改变不同的有机胺、溶剂、温度、时间来寻求条件, 发现当用Pd(OAc)2, (n-Bu)3N, (n-C12H25)3N或(n-C8H17)3N, 溶剂用DMF或没有溶剂时, 收率都很高, 最高达到94%. (S)-Camptathecin是一种重要的抗癌药物先导化合物, 利用Heck反应合成该物质.此反应利用商业易得的原料2-氯甲基吡啶和2-氯喹啉, 经过多步反应可以合成抗癌药物[55].天然产物Resveratrol具有防癌抗癌、抗血小板凝聚及降血脂等作用[56].其合成引起化学家的广泛兴趣, 利用钯催化的脱羧Heck偶联反应.反应从廉价易得的5-二羟基苯甲酸开始, 经过简单的几步反应得到天然产物Resveratrol (Scheme 15).
三羟基二苯乙烯(又名白藜芦醇)具有良好的生物活性, 是一类重要的天然产物, 但在植物中含量极低, 要通过化学反应合成, Heck反应是关键的步骤, 催化剂是Pd(OAc)2 [57] (Scheme 16).
利用过渡金属作为催化剂进行氧化偶联反应具有很多优点[58, 59].不论从环境角度讲, 还是从实用角度讲都是一种绿色环保、价廉而极富吸引力的合成方法, 因此成为人们所研究的热点.在过渡金属催化条件下, 氧化偶联反应是合成多种天然产物和药物中间体杂环化合物的最重要的方法之一, 同时对具有官能化的材料和特殊性质的物质的合成提供了有力的帮助; 又由于此类反应具有反应条件温和、环境友好、原子经济型好等优点, 因此成为化学工作者们的一个研究热点.由于杂环化合物所特有的生理活性和药物活性, 以及其在医药、材料等物质合成中的普遍应用, 因此成为催化化学中一个重要的研究领域.目前已发现多种合成方法, 但由于一些条件的局限性, 如反应步骤复杂、产率低、底物的适应性不够广泛等, 探索新型杂环化合物的研发及合成的新策略成为化学工作者们的一项长期而艰巨的任务.除继续加大对更加高效、高选择性, 以及具有更广泛的底物适用性的钯等催化体系的研究之外, 亦应加大对其它等价廉易得的金属催化剂体系的研究, 以降低反应的成本, 使该反应朝着实用性和易用性方面发展; 进一步拓展在天然产物全合成及具有生理、生物活性化合物合成中的应用.
近年来偶联反应的研究发展较迅速, 在基础研究以及应用方面均得到了有价值的研究结果, 但以钯为催化剂具有成本高、催化剂的重复利用性差、不符合工业化生产等缺点, 因此, 进一步寻求价格便宜、催化活性高的催化剂仍是偶联反应今后的研究重点.
Alberico, D.; Scott, M.-E.; Lautens, M. Chem. Rev. 2007, 107, 174. doi: 10.1021/cr0509760
Boudier, A.; Bromm, L.-O.; Lotz, M. Angew. Chem., Int. Ed. 2000, 39, 4414. doi: 10.1002/(ISSN)1521-3773
Fujiwara, Y.; Jia, C. Pure Appl. Chem. 2001, 73, 319. doi: 10.1351/pac200173020319
Moritanl, I.; Fujiwara, Y. Tetrahedron Lett. 1967, 8, 119. doi: 10.1016/S0040-4039(00)90498-2
Maruyama, O.; Yoshidomi, M.; Fujiwara, Y. Chem Lett. 1979, 10, 1229.
Fujiwara, Y.; Maruyama, O.; Yoshidomi, M. J. Org. Chem. 1981, 46, 851. doi: 10.1021/jo00318a005
Itahara, T.; Kawasaki, K.; Ouseto, F. Bull. Chem. Soc. Jpn. 1984, 57, 3488. doi: 10.1246/bcsj.57.3488
Jia, C.-G.; Lu, W.-J.; Kitamura, T.; Fujiwara, Y. Tetrahedron Lett. 2016, 57, 243. doi: 10.1016/j.tetlet.2015.12.057
Itahara, T. J. Org. Chem. 1985, 50, 5546. doi: 10.1021/jo00350a023
Hirota, K.; Isobe, Y.; Kitade, Y. Synthesis 1987, 495. https://es.scribd.com/doc/92685461/Materials-Science-and-Technology
Grimster, N.-P.; Gauntlett, C. Angew. Chem., Int. Ed. 2005, 117, 3185. doi: 10.1002/(ISSN)1521-3757
Capito, E.; Brown, J.-M.; Ricci. Chem. Commun. 2005, 854. doi: 10.1039/b417035k
Beck, E.-M.; Grimster, N.-P.; Hatley, R.; Gaunt, M.-J. J. J. Am. Chem. Soc. 2006, 128, 2528. doi: 10.1021/ja058141u
Cho, S.-H; Hwang, S.-J.; Chang, S. J. Am. Chem. Soc. 2008, 130, 9254. doi: 10.1021/ja8026295
Baran, P.-S.; Corey, E.-J. J. Am. Chem. Soc. 2002, 124, 7904. doi: 10.1021/ja026663t
Boele, M.-D.; van Strijdonck, G.-P.; de Vries, A.-H.; Kamer, P.-C.; de Vries, J.-G.; van Leeuwen, P.-W. J. Am. Chem. Soc. 2002, 124, 1586. doi: 10.1021/ja0176907
Lee, G.-T.; Jiang, X.; Prasad, K. Adv. Synth. Catal. 2005, 347, 1921 doi: 10.1002/(ISSN)1615-4169
Rauf, W.; Thompson, A.-L.; Brown, J.-M. Chem. Commun. 2009, 26, 3874. https://www.inspq.qc.ca/file/11058/download?token=XH72jfI_
Wang, J.-R.; Yang, C.-T.; Liu, L. Tetrahedron lett. 2007, 48, 5449. doi: 10.1016/j.tetlet.2007.06.001
Houlden, C.-E.; Bailey, C.-D.; Ford, J.-G.; Gagné, M.-R.; Lloyd-Jones, G.-C.; Booker-Milburn, K.-I. J. Am. Chem. Soc. 2008, 130, 10066. doi: 10.1021/ja803397y
Li, J.-J.; Mei, T.-S.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 120, 6552. doi: 10.1002/ange.v120:34
Wang, D.-H.; Engle, K.-M.; Shi, P.-F.; Yu, J.-Q. Science 2010, 327, 315. doi: 10.1126/science.1182512
Engle, K.-M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2010, 49, 6169. doi: 10.1002/anie.201002077
Zhang, Y.-H.; Shi, B.-F.; Yu, J.-Q. ChemInform 2009, 131, 72. doi: 10.1080/00032719.2013.845899?src=recsys
Wasa, M.; Engle, K.-M.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 3680. doi: 10.1021/ja1010866
Garad, D.-N.; Mhaske, S.-B. Org. Lett. 2016, 18, 3862. doi: 10.1021/acs.orglett.6b01868
(a) Lafrance, M. ; Shore, A. ; Fagnou, K. ChemInform 2006, 5097.
(b) Do, H. -Q. ; Daugulis, O. ChemInform 2008, 130, 1128.
(c) Nakao, Y. ; Kashihara, N. ; Kanyiva, K. -S. J. Am. Chem. Soc. 2008, 130, 170.
Zhang, X.; Fan, S.; He, C.-Y. J. Am. Chem. Soc. 2010, 132, 506. https://www.sciencedirect.com/science/article/pii/S0956566316312106
Wang, D.-H.; Engle, K.-M.; Shi, B.-F. Science 2010, 327, 315-319. doi: 10.1126/science.1182512
Engle, K.-M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2010, 49, 6169-6173. doi: 10.1002/anie.201002077
Engle, K.-M.; Wang, D.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 14137. doi: 10.1021/ja105044s
Vinoth, P.; Nagarajan, S.; Maheswari, C. U.; Sudalai, A.; Pace§, V.; Sridharan, V. Org. Lett. 2006, 18, 3442. http://www.ncbi.nlm.nih.gov/pubmed/27355446
Green, S.-A.; Matos, J.-L.; Yagi, A.; Shenvi, R.-A. J. Am. Chem. Soc. 2016, 138, 12779. doi: 10.1021/jacs.6b08507
Cai, G.; Fu, Y.; Shi, Z. J. Am. Chem. Soc. 2007, 129, 7666. doi: 10.1021/ja070588a
Chang, W.; Dai, J.; Li, J. Org. Chem. Front. 2017. http://www.ncbi.nlm.nih.gov/pubmed/20165796
Liao, J.; Fan, L.; Guo, W. Org. Lett. 2017, 19, 1008. doi: 10.1021/acs.orglett.6b03865
Zang, Z.-L.; Karnakanti, S.; Zhao, S. Org. Lett. 2017, 19, 6. doi: 10.1021/acs.orglett.6b02819
Raju, S.; Annamalai, P.; Chen, P.-L. Org. Lett. 2017. http://www.ncbi.nlm.nih.gov/pubmed/28718655
Fruchey, E. R.; Monks, B.-M.; Patterson, A.-M.; Cook, S.-P. Org. Lett. 2013, 15, 4362. doi: 10.1021/ol4018694
Monks, B.-M.; Cook, S.-P. Angew. Chem., Int. Ed. 2013, 52, 14214. doi: 10.1002/anie.201308534
Sun, F.; Gu, Z. Org. Lett. 2015, 17, 2222. doi: 10.1021/acs.orglett.5b00830
Li, Z.; García-Domínguez, A.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 11610. doi: 10.1021/jacs.5b07432
Domański, S.; Chaładaj, W. ACS Catal. 2016, 6, 3452. doi: 10.1021/acscatal.6b00777
Wu, X.-J.; See, J.-W.; Xu, K.; Hirao, H.-J.; Roger, J.-L.; Hierso, J.-C.; Zhou, H. J.-R. Angew. Chem., Int. Ed. 2014, 53, 13573. doi: 10.1002/anie.201408355
Venning, A. R. O.; Bohan, P.-T.; Alexanian, E.-J. J. Am. Chem. Soc. 2015, 137, 3731. doi: 10.1021/jacs.5b01365
Yang, Y.-M.; Dang, Z.-M.; Yu, H.-Z. Org. Biomol. Chem. 2016, 14, 4499. doi: 10.1039/C6OB00607H
Wang, Y.; Gu, J.-Y.; Shi, Z.-J. Org. Lett. 2017. doi: 10.1007/s00214-017-2174-z
Sun, C.-L.; Gu, Y.-F.; Wang, B. Chemistry 2011, 17, 10844. doi: 10.1002/chem.v17.39
(a) Van, M. -S. ; Van, D. -S. ; Schmidt, T. -J. Bioorg. Med. Chem. Lett. 2005, 13, 661.
(b) Chu, G. -H. ; Gu, M. ; Cassel, J. -A. Bioorg. Med. Chem. Lett. 2005, 15, 5114.
(c) Nguyen, R. -V. ; Li, C. -J. Synlett 2008, 1897.
(d) Moreno. ; Clavijo, E. ; Vargas, A. -J. ; Kieffer, R. Org. Lett. 2011, 13, 6244.
(a) Fries, P. ; Halter, D. ; Kleinschek, A. J. Am. Chem. Soc. 2011, 133, 3906.
(b) Taleb, A. ; Lahrech, M. ; Hacini, S. Synlett 2009, 1597.
(c) Guo, X. ; Yu, R. ; Li, H. J. Am. Chem. Soc. 2009, 131, 17387.
(d) Chen, P. ; Wang, J. ; Liu, K. J. Org. Chem. 2008, 73, 339.
(e) Trost, B. -M. ; McClory, A. Angew. Chem., Int. Ed. 2007, 119, 2120.
(f) Tadd, A. -C. ; Fielding, M. -R. ; Willis, M. -C. Tetrahedron Lett. 2007, 48, 7578.
(g) Nagamochi, M. ; Fang, Y. -Q. ; Lautens, M. -A. Org. Lett. 2007, 9, 2955.
(h) Felluga, F. ; Ghelfi, F. ; Pagnoni, U. -M. Synthesis 2007, 1882.
(i) Bryans, J. -S. ; Chessum, N. E. A. ; Huther, N. Tetrahedron 2003, 59, 6221.
Meng, L.-K.; Zhang, G.-H.; Liu, C.; Lei, A.-W. Angew. Chem., Int. Ed. 2013, 125, 10385. doi: 10.1002/ange.201305885
Fang, J.-Z.; Xu, L.-G.; Fang, B. Synth. Commun. 2017, 28, 1491.
Diederich, F. ; Stang, P. -J. New York, Wiley-VCH, 1998, pp. 203~210.
Kazuhiro, H.; Kazuyuki, S.; Hisanori, N. Org. Lett. 2003, 5, 3703. doi: 10.1021/ol035371+
Daniel, L.-C.; Jason, M.-N. Org. Lett. 2001, 3, 4255. doi: 10.1021/ol0169271
Merrit, B.-A.; Jing, L.; Erik, L.-M. Tetrahedron Lett. 2003, 44, 4819. doi: 10.1016/S0040-4039(03)01131-6
Marcella, G.; Carolina, M.; Angela, F. Tetrahedron Lett. 2003, 44, 9107. doi: 10.1016/j.tetlet.2003.10.060
(a) Punniyamurthy, T. ; Velusamy, S. Chem. Rev. 2005, 105, 2329.
(b) Nugent, R. -A. ; Murphy, M. J. Org. Chem. 1987, 52, 2206.
(a) Chuang, G. -J. ; Wang, W. J. Am. Chem. Soc. 2011, 13, 1760.
(b) Du, F. -T. ; Ji, J. -X. Chem. Sci. 2012, 3, 460.
(c) Wei, W. ; Hu, X. -Y. ; Yan, X. -W. Chem. Commun. 2012, 48, 305.

图式 1 氧化偶联反应制备烯烃的两种方法
Scheme 1 Two main methods of olefin formation by coupling reaction

图式 2 Heck反应的芳基烯烃氧化偶联反应
Scheme 2 Heck reaction of arylene oxide oxidation coupling reaction

图式 4 不同保护基下的吡咯与烯烃的氧化偶联反应
Scheme 4 Oxidative coupling reaction of pyrrole with olefins under different protecting groups

图式 5 分子内的烯烃氧化偶联反应用来合成天然产物5, 6, 7, 8, 9, 10, 11, 11-八氢-5H-环辛吲哚化合物
Scheme 5 Intramolecular olefin oxidation coupling reaction used to synthesize natural products 5, 6, 7, 8, 9, 10, 11, 11-octahy-dro-5H-cycloocta indole

图式 6 苯环上进行烯基化的氧化偶联反应
Scheme 6 Benzene ring on the alkenylation of the oxidative coupling reaction

图式 7 钯催化的烯烃参与的分子内的氧化偶联反应
Scheme 7 Palladium-catalyzed olefins participate in intramolecular oxidative coupling reactions

图式 8 钯催化N, N-二甲基苄胺(N-苯乙基磺酰胺)与丙烯酸酯的偶联反应
Scheme 8 Palladium catalyzed coupling reaction of N, N-dieth-ylbenzylamine (N-phenethyl sulfonamides) and acrylates

图式 9 钯催化烯烃和甲酸的氧化偶联反应
Scheme 9 Palladium-catalyzed oxidation coupling reaction of olefins and formic acids

图式 10 钯催化酰胺分子内的C(sp2)—H未活化的环烯烃的氧化偶联反应
Scheme 10 Palladium catalyzes the oxidative coupling reaction of C(sp2)—H unactivated cyclic olefins within the amide molecule

图式 11 钯催化C—H键活化生成了二氢吩草素和苯并噻吩类化合物的反应
Scheme 11 Palladium catalyzed activation of the C—H bond resulted in the reaction of dihydrogaphene and benzothiophene compounds

图式 12 钯催化偶氮或噻唑与芳基发生的氧化偶联反应
Scheme 12 Palladium-catalyzed oxidative coupling of azo or thiazole with aryl

图式 13 硫酸铈催化烯烃与苯醌的氧化偶联环化反应
Scheme 13 Catalytic oxidation coupling of olefins with benzoquinone catalyzed by cerium sulfate

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:























 下载:
下载:



