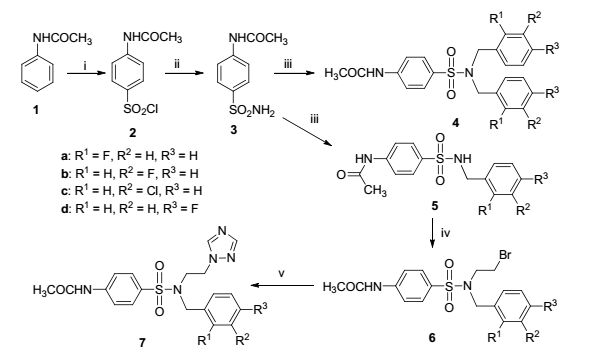
 图式 1
磺胺1, 2, 4-三唑类新化合物的合成
Scheme1.
Synthesis of novel sulfonamide 1, 2, 4-triazoles
图式 1
磺胺1, 2, 4-三唑类新化合物的合成
Scheme1.
Synthesis of novel sulfonamide 1, 2, 4-triazoles
引用本文:
刘庆龙, 房鹏金, 赵志龙, 张慧珍, 周成合. 新型磺胺1, 2, 4-三唑类化合物的设计合成、抗微生物活性及与小牛胸腺DNA相互作用研究[J]. 有机化学,
2017, 37(12): 3146-3154.
doi:
10.6023/cjoc201708010

Citation: Liu Qinglong, Fang Pengjin, Zhao Zhilong, Zhang Huizhen, Zhou Chenghe. Design, Synthesis, and Biological Evaluation of Novel Sulfonamide 1, 2, 4-Triazoles and Their Interaction with Calf Thymus DNA[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3146-3154. doi: 10.6023/cjoc201708010
Citation: Liu Qinglong, Fang Pengjin, Zhao Zhilong, Zhang Huizhen, Zhou Chenghe. Design, Synthesis, and Biological Evaluation of Novel Sulfonamide 1, 2, 4-Triazoles and Their Interaction with Calf Thymus DNA[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3146-3154. doi: 10.6023/cjoc201708010
新型磺胺1, 2, 4-三唑类化合物的设计合成、抗微生物活性及与小牛胸腺DNA相互作用研究
摘要:
磺胺类药物是重要的化学治疗药物,设计合成新型的磺胺类药物是近年来研究的热点领域之一.以乙酰苯胺为起始原料,经过磺酰化、氨解、N-烷化等多步反应合成了一系列新型磺胺1,2,4-三唑类化合物,其结构经IR、1H NMR、13C NMR、MS和HRMS证实.体外考察了合成的磺胺三唑类化合物的抗细菌和抗真菌活性,结果表明所合成化合物的活性优于磺胺,且化合物普遍给出较好的抗大肠杆菌活性,特别是N-(4-(N-(2-(1H-1,2,4-三唑-1-基)乙基)-N-(3-氟苄基)磺酰氨基)苯基)乙酰胺(7b)显示较优的抗大肠杆菌活性(MIC=16 μg/mL).此外,为了初步探讨化合物可能的抗微生物作用机制,利用紫外光谱法初步研究了强活性化合物7b与小牛胸腺DNA的相互作用.研究表明化合物7b可能通过嵌入的方式与DNA形成复合物进而发挥抗微生物效力.
-
关键词:
- 磺胺
- / 1, 2, 4-三唑
- / 合成
- / 抗微生物
- / 小牛胸腺DNA
English
Design, Synthesis, and Biological Evaluation of Novel Sulfonamide 1, 2, 4-Triazoles and Their Interaction with Calf Thymus DNA
Abstract:
Sulfonamides as an important type of chemotherapeutic drugs have been one of the research topics recently. A series of sulfonamide 1, 2, 4-triazoles were successfully synthesized starting from commercial acetanilide via a multi-step sequence of sulfonylation, aminolysis and N-alkylation, and were confirmed by IR, 1H NMR, 13C NMR, MS as well as HRMS spectra. All the synthesized new compounds were evaluated for their in vitro antibacterial and antifungal activities. The bioactive assay showed that most of the synthesized compounds exhibited better inhibitory potency than sulfanilamide against all tested bacterial strains, and most of the compounds gave good anti-Escherichia coli activity in comparison with other microorganisms. Especially, N-(4-(N-(2-(1H-1, 2, 4-triazol-1-yl)ethyl)-N-(3-fluorobenzyl)sulfamoyl)phenyl) acetamide (7b) bearing m-fluorobenzyl group exhibited excellent antibacterial activities against Escherichia coli with minimal inhibition concentration (MIC) value of 16 mg/mL. Preliminary research revealed that compound 7b could effectively intercalate into calf thymus DNA to form compound 7b-DNA complex which might block DNA replication and thus exert antimicrobial activities.
-
Key words:
- sulfonamide
- / 1, 2, 4-triazole
- / synthesis
- / antimicrobial
- / calf thymus DNA
-
抗感染药物是目前世界上使用量最大、最为广泛的临床药物, 在保障人类健康、延长人类寿命方面发挥了重要作用.然而, 日益频发的严重的药物耐药性、顽固的致病性微生物和频繁新出现的有害微生物已使众多临床广泛使用的抗生素药物如β-内酰胺类(青霉素类、头孢类)、大环内酯类、氨基糖苷类、四环素类等抗生素和人工合成的抗菌药如磺胺类、喹诺酮类、噁唑烷酮类等抗菌药的治疗作用越来越有限[1], 迫使众多的研究者致力于新型高效抗感染药物的开发, 特别是不同于现有临床药物的新结构多靶点的药物由于其能有效解决耐药性问题而成为全球共同关注的重大课题[2].
具有对氨基苯磺酰胺结构的磺胺(Sulfonamide, SA)类药物是第一类人工合成抗菌药, 用于临床已有80余年.该类药物在细菌二氢叶酸的生物合成中和对氨基苯甲酸(p-aminobenzoic acid, PABA)竞争性地与酶结合, 影响核酸前体物的合成, 从而抑制细菌生长和繁殖[3].早期研究认为, 苯氨基和磺酰胺基必须处于苯环对位, 苯环的其他部位不能修饰, 且磺酰胺基氮原子必须单取代或未取代的磺胺类结构才具有较好的生物活性[4].随着研究的深入, 发现含有对氨基苯磺酰胺经典结构的磺胺类化合物不仅具有强的抗菌能力, 而且修饰后的衍生物还有抗真菌、抗病毒、抗癌、抗寄生虫、消炎镇痛、抗糖尿病、抗癫痫、利尿等多种生物活性[5].随着超分子药物化学的诞生, 磺胺类超分子药物也得到了一定的发展[6], 如作为治疗局部烫伤感染的抗菌药磺胺嘧啶银, 它的抗菌活性明显高于磺胺嘧啶和硝酸银.迄今已有大量的工作在SO2NH基团上引入含氮杂环如嘧啶、哒嗪、噻唑、异噁唑等, 可有效增强其抗微生物活性.目前, 此领域已取得了众多杰出成果, 现已有较多的药物已成功用于临床[7], 如抗菌药物磺胺嘧啶、磺胺氯哒嗪、磺胺噻唑、磺胺异噁唑等[8].因此, 高效低毒的新型磺胺衍生物的合成与开发已吸引了众多目光[4].
叔胺作为活性化合物中的重要结构片段, 易形成氢键、与金属离子配位以及接受质子季铵化等, 不仅能够调节目标分子的理化性质, 而且能够与生物体内的酶、受体、DNA发生多种非共价键协同超分子相互作用.近年来, 研究发现叔胺类化合物作为新型抗微生物化合物显示出巨大的潜力[9]. 1, 2, 4-三唑环的独特五元芳香氮杂环结构使其易发生多种非共价键相互作用, 易与生物体内多种酶和受体结合, 表现出多种生物活性[10].将三唑药效团引入到磺胺结构中, 有利于调节目标分子的理化性质, 增强抗微生物活性, 拓宽抗菌谱.烷基链的引入可有效增强分子的柔韧性, 影响电荷分布, 调节理化性质.此外, 将不同卤苄基引入到磺胺结构中进行了一系列修饰, 以考察不同基团对生物活性的影响, 并期望得到广谱高效的磺胺类衍生物[11].
因此, 本工作设计合成了一系列磺胺1, 2, 4-三唑类化合物(Scheme 1), 并考察了所合成中间体和磺胺1, 2, 4-三唑类化合物的体外抗细菌抗真菌活性, 为了初步探讨化合物可能的抗微生物作用机制, 利用紫外光谱法初步研究了强活性化合物与小牛胸腺DNA的相互作用.
图式 1
磺胺1, 2, 4-三唑类新化合物的合成
Scheme1.
Synthesis of novel sulfonamide 1, 2, 4-triazoles
1 结果与讨论
1.1 化学合成
以乙酰苯胺为起始原料, 经过与氯磺酸反应得到化合物2, 氨解合成化合物3, 化合物3进一步与取代苄卤反应合成双苄基取代化合物4a~4d (30.1%~33.4%)和单苄基取代化合物5a~5d (20.8%~31.7%), 化合物5a~5d分别与1, 2-二溴乙烷反应得到化合物6a~6d, 产率为71.3%~83.6%;继而与1, 2, 4-三唑反应合成新型磺胺1, 2, 4-三唑类化合物7a~7d (72.8%~81.5%).目标化合物合成路线见Scheme 1.其中, 单苄基磺胺化合物5a~5d的制备是整个反应体系较难控制的环节.因为苄基为给电子基, 使得磺酰氨基碱性增强, 更容易继续与氯苄发生反应得到双取代化合物, 故反应时间的选择对该反应较为重要.通过实验发现, 当反应时间为12 h时, 化合物产率较高.
1.2 结构表征
所合成的重要中间体和目标化合物经IR, 1H NMR, 13C NMR, MS和HRMS进行了结构确认, 其中化合物5a经过X射线单晶衍射仪表征.
1.2.1 1H NMR数据分析
磺胺化合物4~7的波谱数据中, 磺胺结构的苯环氢的化学位移在δ 7.87~7.65范围内.乙酰基中甲基氢在δ 2.16~2.07处出现一组单峰, 而化学位移δ 10.43~10.37处的单峰显示出氮原子所连的氢的存在.而化合物5中, 磺酰胺基氮原子上的氢在δ 8.08~8.03范围内出现一组单峰.由于苯环的强拉电子作用, 化合物苄基上的质子在δ 4.40~3.98范围内出现强吸收单峰.磺胺三唑化合物7a~7d波谱数据中的δ 7.90~7.85和8.34~8.26范围内的两组单峰分别归属为三唑环3位和5位的质子.
1.2.2 13C NMR数据分析
在碳谱的化学位移数据中, δ 169.6~169.4范围内的弱吸收显示了乙酰基中羰基碳的存在.苯环上有氟原子取代时, 碳谱的信号峰会出现裂分, 并且出峰组数会以氟原子个数的2倍呈现.所有的碳原子均在合适的化学位移值处出现吸收峰.
1.2.3 化合物5a的单晶数据分析
化合物5a溶于CHCl3和CH3OH的混合溶液中, 其单晶是在室温下随着溶剂缓慢蒸发而生长的, 图 1是化合物的晶体结构.化合物分子结构通过氢键和π-π堆积作用形成的空间堆积图(图 2). 表 1中的数据显示了化合物5a分子结构中的氢键参数(表 1).
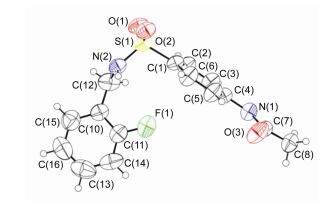 图1
化合物5a的单晶结构图
Figure1.
Crystal structure of compound 5a
图1
化合物5a的单晶结构图
Figure1.
Crystal structure of compound 5a
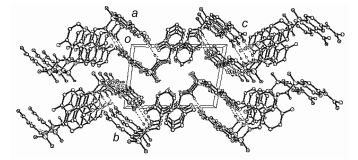 图2
化合物5a的空间堆积图
Figure2.
Packing diagram of compound 5a
图2
化合物5a的空间堆积图
Figure2.
Packing diagram of compound 5a
 表 1
化合物5a分子结构的氢键参数(Å, °)a
Table 1.
Hydrogen-bond geometry (Å, °) of compound 5a
表 1
化合物5a分子结构的氢键参数(Å, °)a
Table 1.
Hydrogen-bond geometry (Å, °) of compound 5a
D—H…A D—H H…A D…A D—H…A N(1)—H(1)…O(2)i 0.77(3) 2.26(3) 3.000(3) 162(3) N(2)—H(2A)…O(3)ii 0.80(3) 2.08(3) 2.873(3) 178(3) ai, (x-1, y, z); ii, (-x, -y, -z). 1.3 生物活性
1.3.1 抗细菌抗真菌活性
化合物4~7的体外抗细菌、抗真菌数据见表 2.测试的细菌有:金黄色葡萄球菌、大肠杆菌、蕈状芽孢杆菌、耻垢分支杆菌4种, 和白色念珠菌、红色酵母菌以及黄曲霉菌3种真菌.活性测试中分别选用磺胺、依诺沙星和氟康唑作为参考药物.
表 2
磺胺类新化合物4~7的抗细菌和抗真菌活性(MIC, µg/mL)
Table 2.
Antibacterial and antifungal data as MIC (μg/mL) for sulfonamide compounds 4~7
化合物 细菌 真菌 金黄色葡萄球菌 大肠杆菌 蕈状芽孢杆菌 耻垢分支杆菌 白色念珠菌 红色酵母菌 黄曲霉菌 4a 512 128 512 512 256 512 512 4b 512 128 256 512 512 256 512 4c 512 256 512 512 512 512 512 4d 512 128 512 512 512 512 512 5a 512 128 256 256 256 256 256 5b 256 64 512 512 256 256 256 5c 128 256 256 256 256 512 128 5d 256 128 256 512 512 256 256 6a 128 128 128 128 256 128 128 6b 64 64 128 64 128 128 64 6c 64 128 64 128 128 128 128 6d 256 128 256 256 256 128 128 7a 256 32 256 256 128 128 128 7b 64 16 128 64 64 64 32 7c 256 128 128 128 128 64 64 7d 256 64 256 128 128 128 128 磺胺 512 512 >512 >512 >512 >512 >512 依诺沙星 8 16 8 16 — — — 氟康唑 — — — — 8 8 256 体外抗细菌活性结果表明:磺胺叔胺化合物6a~6d和7a~7d对所测试细菌表现出了一定的抑制作用, 而且相对于中间体4和5, 目标化合物的抗细菌能力有了一定的提高; 值得注意的是, 与磺胺相比, 它们的抗细菌能力得到了一定的改善, 为磺胺的2~32倍.相对于其他测试菌株, 磺胺类化合物对大肠杆菌表现出较好的抑制活性(MIC=16~256 μg/mL).特别是3-氟苄基取代的磺胺1, 2, 4-三唑7b显示出与依诺沙星相当的抗大肠杆菌活性(MIC=16 μg/mL), 为磺胺(MIC=512 μg/mL)的32倍, 可作为抗菌药物值得进一步研究开发.而且对于其他测试菌株金黄色葡萄球菌、蕈状芽孢杆菌、耻垢分支杆菌, 化合物7b也显示出中等的抑制活性(MIC=64~128 μg/mL).此外, 2-氟苄基化合物7a对大肠杆菌也显示出较好的抑制活性(MIC=32 μg/mL).因此, 在磺胺母体中引入苄基、烷基和1, 2, 4-三唑环等片段能够在一定程度上改善化合物的抗微生物活性.
抗真菌活性结构显示, 中间体4和5给出较弱的抗真菌活性.与参考药物氟康唑相比, 磺胺叔胺化合物6a~6d和7a~7d对所测试真菌表现出弱到中等(MIC=32~128 μg/mL)的抑制活性; 而对耐氟康唑的黄曲霉菌的抑制活性普遍强于氟康唑(MIC=256 μg/mL), 尤其是化合物7b给出相对较强的抗黄曲霉菌活性(MIC=32 μg/mL), 是氟康唑的8倍.可见磺胺类化合物在抗曲霉菌领域的研究值得进一步探索.
1.3.2 化合物7b与小牛胸腺DNA的相互作用研究
DNA是生物体中重要的一类生物大分子, 对生命遗传密码的翻译、转录、复制起着非常重要的作用, 是大多数抗菌、抗癌和抗病毒类药物在体内的主要靶分子, 在生物医学领域引起了广泛的关注.小分子化合物与DNA的相互作用研究对新型高效DNA靶标药物的合理设计具有非常重要的指导意义, 为临床药物的使用提供了有效的理论指导.小牛胸腺DNA由于其价格低廉和简单易用等属性, 而被广泛选择作为研究的DNA模型.为了从分子水平上初步探讨化合物的抗微生物作用机制, 利用紫外-可见光谱法研究了强活性化合物7b与小牛胸腺DNA的相互作用[12].
通常情况下, 将DNA吸收光谱的减色红移现象视为小分子化合物与DNA发生了嵌插作用, 小分子的芳香发色团与DNA的碱基对发生强烈的堆积, 形成化合物-DNA复合物导致; 而认为增色性是由于DNA双链的二级结构被破坏导致[13].如图 3所示, 在紫外-可见吸收光谱中, 随着化合物7b浓度的增加, DNA在260 nm处的吸光度比例增大.同时, 化合物7b和DNA体系的吸光度值稍小于相应浓度的游离化合物和DNA的吸光度总和, 表明DNA吸收光谱存在减色效应(图 4), 可能是由于化合物7b嵌插入DNA双螺旋结构, 通过π-π堆积的静电作用力形成大的共轭体系导致[14].
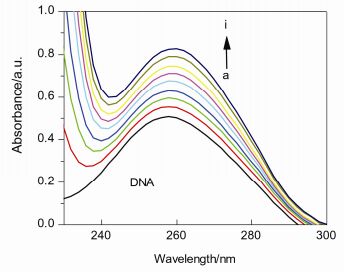 图3
DNA在不同浓度的化合物7b存在下的紫外吸收光谱(pH=7.4, 室温)
Figure3.
UV absorption spectra of DNA with different concentrations of compound 7b (pH=7.4, room temperature)
图3
DNA在不同浓度的化合物7b存在下的紫外吸收光谱(pH=7.4, 室温)
Figure3.
UV absorption spectra of DNA with different concentrations of compound 7b (pH=7.4, room temperature)
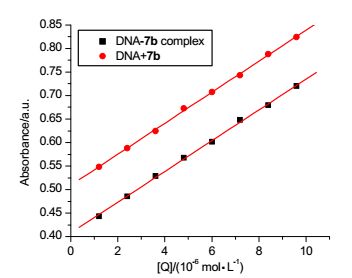 图4
260 nm处7b-DNA络合物的吸光度以及游离的DNA与游离化合物7b的吸光度之和
Figure4.
Comparison of absorption at 260 nm between the 7b-DNA complex and the sum values of free DNA and free compound 7b
图4
260 nm处7b-DNA络合物的吸光度以及游离的DNA与游离化合物7b的吸光度之和
Figure4.
Comparison of absorption at 260 nm between the 7b-DNA complex and the sum values of free DNA and free compound 7b
基于DNA吸收光谱在化合物7b存在时的变化, 可用Eq. 1分析计算DNA与化合物的结合常数K.
A0和A分别代表没有和有化合物7b存在下的DNA吸光度值, ξC和ξD-C表示化合物7b和7b-DNA体系的吸收系数, 根据方程和吸收滴定拟合曲线图(图 5), 可计算出相应的结合常数K=8.16×104 L/mol, R=0.999, SD=0.13 (R代表相关性系数, SD是标准偏差).
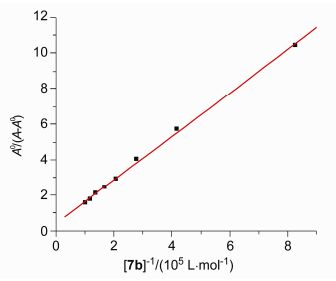 图5
A0/(A-A0)与1/[7b]的标准曲线
Figure5.
Plot of A0/(A-A0) versus 1/[7b]
图5
A0/(A-A0)与1/[7b]的标准曲线
Figure5.
Plot of A0/(A-A0) versus 1/[7b]
中性红(NR)作为吩嗪类平面染料, 可与DNA发生嵌插作用, 而且NR本身毒性很低, 水溶液可长时间放置.因此, 选用中性红作为光谱探针研究化合物7b与DNA的相互结合模式. 图 6显示了NR染料在DNA加入过程中的吸收光谱变化.结果表明, 随着DNA浓度的增加, NR在460 nm附近的吸收峰逐渐减小, 而在530 nm处出现一个新的吸收峰, 这归因于新的DNA-NR复合物的形成, 在504 nm处的等色点为DNA-NR复合物的形成提供了证据[15].
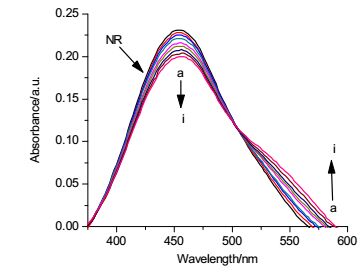 图6
中性红在不同浓度DNA下的紫外吸收光谱(pH=7.4, 室温)
Figure6.
UV absorption spectra of NR in the presence of DNA at pH 7.4 and room temperature
图6
中性红在不同浓度DNA下的紫外吸收光谱(pH=7.4, 室温)
Figure6.
UV absorption spectra of NR in the presence of DNA at pH 7.4 and room temperature
图 7显示了中性红和化合物7b与DNA竞争结合的吸收光谱.随着化合物7b浓度的增加, 最大吸收峰在530 nm的DNA-NR复合物的吸收下降, 而在460 nm处的吸收略微增强, 与随着DNA浓度增加时游离NR在460 nm左右的吸收相比(图 6), 图 7(插图)中的吸收光谱显示出相反的过程.结果表明, 化合物7b插入到DNA的双螺旋结构, 代替NR, 与DNA发生络合, 形成了7b-DNA复合物.
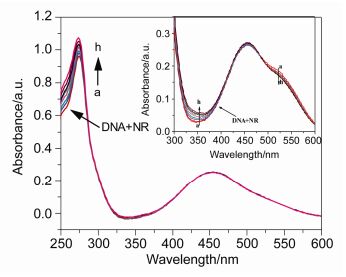 图7
DNA在中性红与化合物7b竞争下的紫外吸收光谱
Figure7.
UV Absorption spectra of the competitive reaction between 7b and neutral red with DNA
图7
DNA在中性红与化合物7b竞争下的紫外吸收光谱
Figure7.
UV Absorption spectra of the competitive reaction between 7b and neutral red with DNA
2 结论
以乙酰苯胺和氯磺酸为起始原料经磺酰化、氨解、及1, 2, 4-三唑的N-烷化反应合成了一系列的新型磺胺1, 2, 4-三唑类化合物, 化合物结构经IR、1H NMR、13C NMR、MS和HRMS证实.合成的化合物对所测试的细菌和真菌呈现出了不同程度的抑制作用, 普遍优于前体磺胺, 且化合物普遍给出较好的抗大肠杆菌活性, 特别是化合物7b显示较优的抗大肠杆菌活性(MIC=16 μg/mL).因此, 磺胺1, 2, 4-三唑类化合物作为抗微生物药物具有开发潜力, 值得进一步研究.此外, 利用紫外光谱法初步研究了强活性化合物7b与小牛胸腺DNA的相互作用, 结果表明化合物7b可能通过嵌入的方式与DNA形成复合物进而发挥抗微生物效力.
3 实验部分
3.1 仪器与试剂
熔点仪: X-6型精密显微熔点测定仪(温度未校正); 红外光谱仪: Bio-Rad FTS-185, Bruker RFS100/S (KBr压片, 测试范围400~4000 cm−1); 核磁共振仪: Bruker AV 400核磁共振仪(TMS内标); 质谱仪: LCMS-2010A (TOF电离源); 高分辨质谱仪: Bruker Daltonics; 电热鼓风干燥箱:郑州长城科工贸有限公司; 台式恒温振荡器:太仓市实验设备厂; 立式压力蒸汽灭菌器: LDZX-50KB蒸汽灭菌器, 上海申安医疗机械厂; 紫外可见分光光度计: TU-1901型.
硅胶:青岛海洋化工厂; 乙酰苯胺、氯磺酸、苄卤、烷基二溴化物、1, 2, 4-三唑及其他所需试剂均为市售分析纯; 氟康唑:富阳金伯士化工有限公司; 依诺沙星:重庆科瑞制药有限责任公司; 磺胺:富阳金伯士化工有限公司; 牛肉膏、蛋白胨、RMPI 1640 (Gibco BRL)、吗啡啉丙磺酸、琼脂、葡萄糖、酵母浸膏等:北京奥博星生物技术有限公司.
3.2 实验方法
3.2.1 对乙酰氨基苯磺酰胺3的制备
在冰水浴条件下, 向150 mL圆底烧瓶中加入化合物1 (5.040 g, 37.3 mmol), 置于磁力搅拌器上, 逐滴加入氯磺酸(12.5 mL, 188.1 mmol), 过程中有大量雾状物产生, 用带有干燥管的尾气吸收装置将其导入氢氧化钠水溶液.待反应稍缓和后摇动圆底烧瓶使固体完全溶解, 体系为浅棕色稠状液体. 60 ℃温水浴中搅拌反应2 h, 冷却至室温.在通风橱中、充分搅拌下缓慢将反应液倒入盛有约75 g碎冰块的250 mL烧杯中, 用玻璃棒将大块固体捣碎使成均匀的白色小颗粒, 抽滤, 以少量水多次洗涤, 晾干, 得到淡粉色固体.将得到的固体转入250 mL烧杯中, 在搅拌下缓慢滴加17.5 mL浓氨水, 滴加完毕后搅拌反应3 h.加入适量冰水、抽滤, 用冰水洗涤除去过量的氨.干燥得到3.840 g白色固体, 产率为48.24%. m.p. 216~218 ℃(文献[8]: m.p. 217~219 ℃).
3.2.2 N-(4-(N, N-双(4-氟苄基)磺酰氨基)苯基)乙酰胺(4a)的制备
将化合物3 (5.997 g, 25.8 mmol)和邻氟苄氯(3.996 g, 27.6 mmol)同时加入到盛有140 mL丙酮的250 mL圆底烧瓶中.将250 mL的圆底烧瓶放入油浴锅, 在50 ℃条件下搅拌反应20 min后, 再投入碳酸钾(4.600 g, 32.5 mmol)作碱, 搅拌反应12 h后停止反应.期间薄层色谱(TLC)跟踪(展开剂:二氯甲烷).将反应体系趁热过滤, 洗涤.经硅胶柱色谱纯化(洗脱剂:二氯甲烷), 得白色固体4a, 产率31.3%; m.p.: 134~136 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.16 (s, 3H, COCH3), 4.40 (s, 4H, ArFCH2), 7.08~7.00 (m, 4H, ArFH), 7.30~7.22 (m, 4H, ArFH), 7.85 (br, 4H, ArH), 10.42 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 46.1, 115.3, 119.2, 123.6, 124.6, 128.8, 130.1, 130.8, 132.5, 143.8, 160.6, 169.6; IR (KBr) ν: 3365 (N—H), 2925, 2856 (CH2), 1703 (C=O), 1592, 1529, 1493 (aromatic frame), 1327, 1154, 783, 621 cm-1; MS m/z: 431 [M+H]+; HRMS (TOF) calcd for C22H21F2N2O3S [M+H]+ 431.1235; found 431.1241.
3.2.3 N-(4-(N, N-双(3-氟苄基)磺酰氨基)苯基)乙酰胺(4b)的制备
化合物4b的合成参照化合物4a的制备方法, 得白色固体4b, 产率30.1%; m.p. 112~114 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.15 (s, 3H, COCH3), 4.37 (s, 4H, ArFCH2), 7.11~7.03 (m, 6H, ArFH), 7.38~7.32 (m, 2H, ArFH), 7.87 (br, 4H, ArH), 10.40 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 45.9, 114.3, 114.6, 119.0, 123.9, 128.1, 130.6, 134.7, 141.3, 143.2, 162.4, 169.5; IR (KBr) ν: 3357 (N—H), 2937, 2848 (CH2), 1707 (C=O), 1594, 1527, 1489 (aromatic frame), 1334, 1157, 776, 611 cm-1; MS m/z: 431 [M+H]+; HRMS (TOF) calcd for C22H21F2N2O3S [M+H]+ 431.1235; found 431.1239.
3.2.4 N-(4-(N, N-双(3-氯苄基)磺酰氨基)苯基)乙酰胺(4c)的制备
化合物4c的合成参照化合物4a的制备方法, 得白色固体4c, 产率33.4%; m.p. 156~158 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.12 (s, 3H, COCH3), 4.30 (s, 4H, ArClCH2), 7.09~7.07 (m, 4H, ArClH), 7.22~7.21 (m, 4H, ArClH), 7.83 (br, 4H, ArH), 10.40 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.7, 52.1, 119.3, 127.3, 127.7, 128.5, 128.7, 130.4, 132.8, 133.3, 139.6, 144.0, 169.6; IR (KBr) ν: 3363 (N—H), 2935, 2851 (CH2), 1714 (C=O), 1598, 1531, 1492 (aromatic frame), 1341, 1162, 778, 613 cm-1; MS m/z: 464 [M+H]+; HRMS (TOF) calcd for C22H21Cl2N2O3S [M+H]+ 463.0644; found 463.0648.
3.2.5 N-(4-(N, N-双(4-氟苄基)磺酰氨基)苯基)乙酰胺(4d)的制备
化合物4d的合成参照化合物4a的制备方法, 得白色固体4d, 产率32.2%; m.p. 117~119 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.11 (s, 3H, COCH3), 4.30 (s, 4H, ArFCH2), 7.08~7.06 (m, 4H, ArFH), 7.21~7.20 (m, 4H, ArFH), 7.83 (br, 4H, ArH), 10.39 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.7, 52.1, 119.3, 127.3, 129.5, 130.3, 132.8, 133.3, 139.6, 144.0, 169.6; IR (KBr) ν: 3341 (N—H), 2925, 2843 (CH2), 1721 (C=O), 1592, 1524, 1493 (aromatic frame), 1336, 1153, 765, 604 cm-1; MS m/z: 431 [M+H]+; HRMS (TOF) calcd for C22H21F2N2O3S [M+H]+ 431.1235; found 431.1241.
3.2.6 N-(4-(N-(2-氟苄基)磺酰氨基)苯基)乙酰胺(5a)的制备
化合物5a的合成参照化合物4a的制备方法, 得白色固体5a, 产率25.4%; m.p.: 121~123 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.09 (s, 3H, COCH3), 4.00 (s, 2H, ArFCH2), 7.34~7.12 (m, 4H, ArFH), 7.73 (br, 4H, ArH), 8.03 (s, H, SO2NH), 10.31 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 46.0, 115.5, 119.0, 124.7, 125.0, 128.1, 129.9, 130.9, 134.5, 143.2, 160.6, 169.5; IR (KBr) ν: 3442, 3336 (N—H), 2923, 2845 (CH2), 1715 (C=O), 1593, 1527, 1497 (aromatic frame), 1341, 1162, 771, 612 cm-1; MS m/z: 323 [M+H]+; HRMS (TOF) calcd for C15H16FN2O3S [M+H]+ 323.0860; found 323.0868.
3.2.7 N-(4-(N-(3-氟苄基)磺酰氨基)苯基)乙酰胺(5b)的制备
化合物5b的合成参照化合物4a的制备方法, 得白色固体5b, 产率20.8%; m.p. 127~129 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.09 (s, 3H, COCH3), 4.00 (s, 2H, ArFCH2), 7.09~7.02 (m, 3H, ArFH), 7.35~7.29 (m, 1H, ArFH), 7.72 (d, J=8 Hz, 2H, ArH), 7.73 (d, J=8 Hz, 2H, ArH), 8.07 (s, H, SO2NH), 10.30 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 45.9, 114.3, 114.6, 119.0, 123.9, 128.1, 130.58, 134.7, 141.3, 143.2, 162.4, 169.4; IR (KBr) ν: 3451, 3329 (N—H), 2919, 2857 (CH2), 1721 (C=O), 1599, 1518, 1490 (aromatic frame), 1331, 1157, 773, 617 cm-1; MS m/z: 323 [M+H]+; HRMS (TOF) calcd for C15H16FN2O3S [M+H]+ 323.0860; found 323.0867.
3.2.8 N-(4-(N-(3-氯苄基)磺酰氨基)苯基)乙酰胺(5c)的制备
化合物5c的合成参照化合物4a的制备方法, 得白色固体5c, 产率31.7%; m.p. 145~147 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.09 (s, 3H, COCH3), 3.98 (s, 2H, ArClCH2), 7.33~7.20 (m, 4H, ArClH), 7.72 (d, J=4.0 Hz, 2H, ArH), 7.74 (d, J=4.0 Hz, 2H, ArH), 8.08 (s, H, SO2NH), 10.30 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 45.9, 119.1, 126.6, 127.5, 127.8, 128.1, 130.5, 133.4, 134.8, 140.9, 143.3, 169.5; IR (KBr) ν: 3459, 3323 (N—H), 2927, 2863 (CH2), 1728 (C=O), 1589, 1511, 1493 (aromatic frame), 1322, 1165, 784, 623 cm-1; MS m/z: 340 [M+H]+; HRMS (TOF) calcd for C15H16Cl-N2O3S [M+H]+ 339.0565; found 339.0572.
3.2.9 N-(4-(N-(4-氟苄基)磺酰氨基)苯基)乙酰胺(5d)的制备
化合物5d的合成参照化合物4a的制备方法, 得白色固体5d, 产率25.4%; m.p. 121~123 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.07 (s, 3H, COCH3), 3.90 (s, 2H, ArFCH2), 7.19~7.08 (m, 4H, ArFH), 7.75 (d, J=4.0 Hz, 2H, ArH), 7.78 (d, J=4.0 Hz, 2H, ArH), 8.04 (s, H, SO2NH), 10.27 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 29.21, 50.86, 123.77, 130.93, 132.08, 132.55, 134.64, 138.90, 144.90, 147.88, 174.05; IR (KBr) ν: 3443, 3327 (N—H), 2921, 2875 (CH2), 1712 (C=O), 1593, 1503, 1487 (aromatic frame), 1314, 1154, 771, 612 cm-1; MS m/z: 323 [M+H]+; HRMS (TOF) calcd for C15H16FN2O3S [M+H]+ 323.0860; found 323.0867.
3.2.10 N-(4-(N-(2-溴乙基)-N-(2-氟苄基)磺酰氨基)苯基)乙酰胺(6a)的制备
向盛有50 mL丙酮的100 mL圆底烧瓶中加入化合物5a (0.395 g, 1.4 mmol), 1, 2-二溴乙烷(1.258 g, 6.7 mmol)和碳酸钾(1.130 g, 8.2 mmol), 置于50 ℃下搅拌反应后(TLC跟踪反应, 展开剂:二氯甲烷), 冷却至室温, 减压蒸馏回收丙酮, 水溶解反应体系中的固体, 二氯甲烷(50 mL×3)萃取, 合并有机相, 无水Na2SO4干燥, 浓缩, 经硅胶柱色谱纯化(洗脱剂:二氯甲烷), 得白色固体6a, 产率71.3%; m.p. 131~133 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.10 (s, 3H, COCH3), 3.33~3.29 (t, J=6.0 Hz, 2H, NCH2CH2), 3.45 (t, J=8.0 Hz, 2H, NCH2CH2), 4.40 (s, 2H, ArFCH2), 7.44~7.15 (m, 4H, ArFH), 7.78 (br, 4H, ArH), 10.38 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 30.5, 46.4, 50.3, 115.8, 119.2, 124.1, 125.0, 128.8, 130.5, 131.2, 132.4, 143.9, 160.6, 169.6; IR (KBr) ν: 3335 (N—H), 2935, 2891 (CH2), 1732 (C=O), 1587, 1515, 1492 (aromatic frame), 1324, 1159, 778, 623 cm-1; MS m/z: 430 [M+H]+; HRMS (TOF) calcd for C17H19BrFN2O3S [M+H]+ 429.0278; found 429.0282.
3.2.11 N-(4-(N-(2-溴乙基)-N-(3-氟苄基)磺酰氨基)苯基)乙酰胺(6b)的制备
化合物6b的合成参照化合物6a的制备方法, 得白色固体6b, 产率75.1%; m.p. 119~121 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.11 (s, 3H, COCH3), 3.31 (t, J=6.0 Hz, 2H, NCH2CH2), 3.46 (t, J=6.0 Hz, 2H, NCH2CH2), 4.36 (s, 2H, ArFCH2), 7.21~7.08 (m, 3H, ArFH), 7.43~7.37 (m, 1H, ArFH), 7.83 (br, 4H, ArH), 10.40 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 30.5, 50.3, 51.7, 114.9, 115.2, 119.3, 124.5, 128.7, 130.9, 132.5, 140.3, 143.9, 162.6, 169.6; IR (KBr) ν: 3341 (N—H), 2932, 2893 (CH2), 1721 (C=O), 1593, 1524, 1489 (aromatic frame), 1327, 1165, 769, 614 cm-1; MS m/z: 430 [M+H]+; HRMS (TOF) calcd for C17H19BrFN2O3S [M+H]+ 429.0278; found 429.0284.
3.2.12 N-(4-(N-(2-溴乙基)-N-(3-氯苄基)磺酰氨基)苯基)乙酰胺(6c)的制备
化合物6c的合成参照化合物6a的制备方法, 得白色固体6c, 产率83.6%; m.p. 157~159 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.10 (s, 3H, COCH3), 3.32 (t, J=8.0 Hz, 2H, NCH2CH2), 3.46 (t, J=6.0 Hz, 2H, NCH2CH2), 4.35 (s, 2H, ArClCH2), 7.40~7.29 (m, 4H, ArClH), 7.82 (br, 4H, ArH), 10.39 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 30.5, 50.3, 51.5, 119.3, 127.1, 128.0, 128.3, 128.7, 130.8, 132.5, 133.5, 140.0, 144.0, 169.5; IR (KBr) ν: 3359 (N—H), 2941, 2897 (CH2), 1719 (C=O), 1597, 1516, 1493 (aromatic frame), 1326, 1171, 773, 620 cm-1; MS m/z: 447 [M+H]+; HRMS (TOF) calcd for C17H19BrClN2O3S [M+H]+ 444.9983; found 444.9991.
3.2.13 N-(4-(N-(2-溴乙基)-N-(4-氟苄基)磺酰氨基)苯基)乙酰胺(6d)的制备
化合物6d的合成参照化合物6a的制备方法, 得白色固体6d, 产率77.3%; m.p. 124~126 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.10 (s, 3H, COCH3), 3.25 (t, J=8.0 Hz, 2H, NCH2CH2), 3.41 (t, J=8.0 Hz, 2H, NCH2-CH2), 4.31 (s, 2H, ArClCH2), 7.18 (t, J=8.4 Hz, 2H, ArFH), 7.45~7.28 (m, 2H, ArFH), 7.81 (br, 4H, ArH), 10.37 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 30.4, 49.1, 50.0, 115.8, 119.3, 128.7, 130.7, 130.8, 132.7, 143.9, 161.4, 169.5; IR (KBr) ν: 3337 (N—H), 2929, 2887 (CH2), 1715 (C=O), 1595, 1529, 1493 (aromatic frame), 1319, 1158, 773, 622 cm-1; MS m/z: 430 [M+H]+; HRMS (TOF) calcd for C17H19BrFN2O3S [M+H]+ 429.0278; found 429.0280.
3.2.14 N-(4-(N-(2-(1H-1, 2, 4-三唑-1-基)乙基)-N-(2-氟苄基)磺酰氨基)苯基)乙酰胺(7a)的制备
在60 ℃下, 向装有20 mL乙腈的100 mL圆底烧瓶中加入1, 2, 4-三唑(0.074 g, 1 mmol)和碳酸钾(0.147 g, 1 mmol), 搅拌反应30 min后, 冷却至室温.将化合物6a (0.250 g, 0.6 mmol)加入反应体系, 继续在70 ℃的搅拌反应结束后(TLC跟踪反应, 展开剂:二氯甲烷/丙酮, V:V=5:1), 冷却至室温, 减压蒸馏回收乙腈, 水溶解反应体系中的固体, 二氯甲烷(50 mL×3)萃取, 合并有机相, 无水Na2SO4干燥, 浓缩, 经硅胶柱色谱纯化(洗脱剂:二氯甲烷/丙酮, V:V=20:3), 得白色固体7a, 产率72.8%; m.p. 109~111 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.15 (s, 3H, COCH3), 3.56 (t, J=8.0 Hz, 2H, NCH2CH2), 4.26 (t, J=6.0 Hz, 2H, NCH2CH2), 4.37 (s, 2H, ArFCH2), 7.40~7.17 (m, 4H, ArFH), 7.80 (d, J=4.0 Hz, 2H, ArH), 7.82 (d, J=4.0 Hz, 2H, ArH), 7.90 (s, 1H, triazole-3-CH), 8.34 (s, 1H, triazol-5-CH), 10.43 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 46.4, 48.0, 48.2, 115.8, 119.2, 123.6, 125.0, 128.8, 130.4, 131.0, 132.1, 143.9, 144.7, 151.9, 160.6, 169.6; IR (KBr) ν: 3345 (N—H), 2993, 2896 (CH2), 1721 (C=O), 1589, 1516, 1487 (aromatic frame), 1323, 1161, 769, 614 cm-1; MS m/z: 418 [M+H]+; HRMS (TOF) calcd for C19H21F-N5O3S [M+H]+ 418.1344; found 418.1349.
3.2.15 N-(4-(N-(2-(1H-1, 2, 4-三唑-1-基)乙基)-N-(3-氟苄基)磺酰氨基)苯基)乙酰胺(7b)的制备
化合物7b的合成参照化合物7a的制备方法, 得白色固体7b, 产率75.1%; m.p. 97~99 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.11 (s, 3H, COCH3), 3.51 (t, J=6.0 Hz, 2H, NCH2CH2), 4.20 (t, J=6.0 Hz, 2H, NCH2CH2), 4.26 (s, 2H, ArFCH2), 7.12~6.69 (m, 4H, ArFH), 7.79 (d, J=8.0 Hz, 2H, ArH), 7.80 (d, J=8.0 Hz, 2H, ArH), 7.85 (s, 1H, triazole-3-CH), 8.26 (s, 1H, triazole-5-CH), 10.39 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 47.9, 48.1, 51.8, 114.9, 115.1, 119.2, 124.4, 128.8, 130.8, 132.2, 139.9, 143.9, 144.6, 151.8, 162.6, 169.5; IR (KBr) ν: 3334 (N−H), 2987, 2891 (CH2), 1716 (C=O), 1594, 1524, 1496 (aromatic frame), 1335, 1172, 773, 623 cm-1; MS m/z: 418 [M+H]+; HRMS (TOF) calcd for C19H21FN5O3S [M+H]+ 418.1344; found 418.1356.
3.2.16 N-(4-(N-(2-(1H-1, 2, 4-三唑-1-基)乙基)-N-(3-氯苄基)磺酰氨基)苯基)乙酰胺(7c)的制备
化合物7c的合成参照化合物7a的制备方法, 得白色固体7c, 产率81.5%; m.p.: 134~136 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.11 (s, 3H, COCH3), 3.52 (t, J=6.0 Hz, 2H, NCH2CH2), 4.21 (t, J=6.0 Hz, 2H, NCH2-CH2), 4.26 (s, 2H, ArClCH2), 7.19~7.17 (m, 2H, ArClH), 7.39~7.33 (m, 2H, ArClH), 7.78 (d, J=8.0 Hz, 2H, ArH), 7.80 (d, J=8.0 Hz, 2H, ArH), 7.86 (s, 1H, triazol-3-CH), 8.27 (s, 1H, triazole-5-CH), 10.39 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 47.9, 48.1, 51.7, 119.2, 127.0, 128.0, 128.2, 128.7, 128.9, 130.7, 132.2, 133.6, 139.5, 144.0, 144.6, 144.7, 151.8, 169.5; IR (KBr) ν: 3347 (N—H), 2999, 2883 (CH2), 1728 (C=O), 1598, 1517, 1488 (aromatic frame), 1326, 1168, 775, 631 cm-1; MS m/z: 444 [M+H]+; HRMS (TOF) calcd for C19H21ClN5O3S [M+H]+ 434.1048; found 434.1058.
3.2.17 N-(4-(N-(2-(1H-1, 2, 4-三唑-1-基)乙基)-N-(4-氟苄基)磺酰氨基)苯基)乙酰胺(7d)的制备
化合物7d的合成参照化合物7a的制备方法, 得白色固体7d, 产率77.3%; m.p. 113~115 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 2.10 (s, 3H, COCH3), 3.45 (t, J=6.0 Hz, 2H, NCH2CH2), 4.14 (t, J=6.0 Hz, 2H, NCH2CH2), 4.22 (s, 2H, ArFCH2), 7.13 (d, J=8.0 Hz, 2H, ArFH), 7.26 (d, J=8.0 Hz, 2H, ArFH), 7.76 (d, J=8.0 Hz, 2H, ArH), 7.79 (d, J=8.0 Hz, 2H, ArH), 7.85 (s, 1H, triazole-3-CH), 8.24 (s, 1H, triazole-5-CH), 10.37 (s, 1H, NHCOCH3); 13C NMR (100 MHz, DMSO-d6) δ: 24.9, 48.0, 49.1, 51.7, 115.7, 115.8, 119.3, 128.7, 130.6, 132.4, 143.9, 144.7, 151.9, 161.3, 169.5; IR (KBr) ν: 3341 (N—H), 2992, 2886 (CH2), 1723 (C=O), 1587, 1531, 1485 (aromatic frame), 1347, 1164, 766, 624 cm-1; MS m/z: 418 [M+H]+; HRMS (TOF) calcd for C19H21FN5O3S [M+H]+ 418.1344; found 418.1346.
3.3 活性测试
化合物的活性测试采用美国国家实验室标准委员会(NCCLS)[16]推荐的药敏实验方法.化合物和参考药物均用二甲基亚砜(DMSO)溶解, 配成12.8 μg•mL−1溶液.取无菌96孔板, 在每一孔中加入100 μL菌液.取100 μL用无菌肉汤稀释好的受试药液于1号孔中, 混匀后取100 μL于2号孔中, 依次倍比稀释至11号孔, 使各孔中的药物浓度分别为512, 256, 128, 64, 32, 16, 8, 4, 2, 1和0.5 μg• mL−1, 各孔中DMSO含量均低于1%, 目的是为了排除溶剂DMSO对体外抗细菌抗真菌活性测试的影响, 然后在11号孔中取100 μL去掉, 12号孔不含药物, 作阳性对照, 各药敏板在37 ℃下于恒温摇床上培养24 h.
辅助材料(Supporting Information) 代表性中间体和目标产物的核磁共振氢谱、碳谱和高分辨谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) Brown, E. D.; Wright, G. D. Nature 2016, 529, 336.
(b) Baym, M.; Stone, L. K.; Kishony, R. Science 2016, 351, 40.
(c) Brown, D. Nat. Rev. Drug Discovery 2015, 14, 821. -
[2]
(a) Gadakh, B.; Van Aerschot, A. Curr. Med. Chem. 2015, 22, 2140.
(b) Gale, R. T.; Brown, E. D. Curr. Opin. Microbiol. 2015, 27, 69.
(c) Staker, B. L.; Buchko, G. W.; Myler, P. J. Curr. Opin. Microbiol. 2015, 27, 133.
(d) Oktar, F. N.; Yetmez, M.; Ficai, D.; Ficai, A.; Dumitru, F.; Pica, A. Curr. Top. Med. Chem. 2015, 15, 1583. -
[3]
(a) Zhang, H. Z.; He, S. C.; Peng, Y. J.; Zhang, H. J.; Gopala, L.; Tangadanchu, V. K. R.; Gan, L. L.; Zhou, C. H. Eur. J. Med. Chem. 2017, 136, 165.
(b) Zhang, H. Z.; Jeyakkumar, P.; Kumar, K. V.; Zhou, C. H. New J. Chem. 2015, 39, 5776. -
[4]
何世超, Jeyakkumar, P., Avula, S. R., 王宪龙, 张慧珍, 周成合.中国科学:化学, 2016, 46, 823.He, S. C.; Jeyakkumar, P.; Avula, S. R.; Wang, X. L.; Zhang, H. Z.; Zhou, C. H. Sci. Sin.:Chim. 2016, 46, 823(in Chinese).
-
[5]
(a) Zessel, K.; Mohring, S.; Hamscher, G.; Kietzmann, M.; Stahl, J. Environ. Toxicol. Chem. 2005, 24, 771.
(b) Supuran, C. T. Nat. Rev. Drug. Discovery 2008, 7, 168.
(c) Gawin, R.; Clercq, E. D.; Naesens, L.; Koszytkowska-Stawińska, M. Bioorg. Med. Chem. 2008, 16, 8379.
(d) Bouissane, L.; Kazzouli, S. E.; Léonce, S.; Pfeiffer, B.; Rakib, E. M.; Khouili, M.; Guillaumet, G. Bioorg. Med. Chem. 2006, 14, 1078. -
[6]
(a) Marques, L. L.; Oliveira, G. M.; Lang, E. S.; Campos, M. M.; Gris, L. R. Inorg. Chem. Commun. 2007, 10, 1083.
(b) Chohan, Z. H.; Shad, H. A. J. Enzyme Inhib. Med. Chem. 2012, 27, 403.
(c) Herole, R. A.; Velingkar, V. S. Int. J. Pharm. Chem. 2011, 1, 45. -
[7]
(a) Alanazi, A. M.; El-Azab, A. S.; Al-Suwaidan, I. A.; ElTahir, K. E.; Asiri, Y. A.; Abdel-Aziz, N. I.; Abdel-Aziz, A. A. Eur. J. Med. Chem. 2015, 92, 115.
(b) Laev, S. S.; Salakhutdinov, N. F. Bioorg. Med. Chem. 2015, 23, 3059. -
[8]
王宪龙, 甘淋玲, 闫聪彦, 周成合, 中国科学:化学, 2011, 41, 451.Wang, X. L.; Gan, L. L.; Yan, C. Y.; Zhou, C. H. Sci. Sin.:Chim. 2011, 41, 451(in Chinese).
-
[9]
(a) Zhang, H. Z.; Damu, G. L. V.; Cai, G. X.; Zhou, C. H. Eur. J. Med. Chem. 2013, 64, 329.
(b) Fang, B.; Zhou, C. H.; Rao, X. C. Eur. J. Med. Chem. 2010, 45, 4388.
(c) Dai, L. L.; Zhang, H. Z.; Nagarajan, S.; Rasheed, S.; Zhou, C. H. Med. Chem. Commun. 2015, 6, 147. -
[10]
(a) Zhang, H. Z. ; Gan, L. L. ; Wang, H. ; Zhou, C. H. Mini-Rev. Med. Chem. 2017, 17, 122.
(b) Cheng, Y. ; Wang, H. ; Addla, D. ; Zhou, C. H. Chin. J. Org. Chem. 2016, 36, 1 (in Chinese).
(程宇, 王辉, Addla, D., 周成合, 有机化学, 2016, 36, 1. )
(c) Zhou, C. H. ; Wang, Y. Curr. Med. Chem. 2012, 19, 239. -
[11]
(a) Jeyakkumar, P.; Zhang, L.; Avula, S. R.; Zhou, C. H. Eur. J. Med. Chem. 2016, 122, 205.
(b) Zhang, H. Z.; Lin, J. M.; Rasheed, S.; Zhou, C. H. Sci. China, Chem. 2014, 57, 807. -
[12]
Berdis, A. J. Biochemistry 2008, 47, 8253. doi: 10.1021/bi801179f
-
[13]
Rahban, M.; Divsalar, A.; Saboury, A. A.; Golestani, A. J. Phys. Chem. C 2010, 114, 5798. doi: 10.1021/jp910656g
-
[14]
(a) Li, X. L.; Hu, Y. J.; Wang, H.; Yu, B. Q.; Yue, H. L. Biomacromolecules 2012, 13, 873.
(b) Zhang, G. W.; Fu, P.; Wang, L.; Hu, M. M. J. Agric. Food Chem. 2011, 59, 8944. -
[15]
Ni, Y.; Dua, S.; Kokot, S. Anal. Chim. Acta 2007, 584, 19. doi: 10.1016/j.aca.2006.11.006
-
[16]
Kadi, A. A.; El-Brollosy, N. R.; Al-Deeb, O. A; Habib, E. E.; Ibrahim, T. M.; El-Emam, A. A. Eur. J. Med. Chem. 2007, 42, 235. doi: 10.1016/j.ejmech.2006.10.003
-
[1]
-

图式 1 磺胺1, 2, 4-三唑类新化合物的合成
Scheme 1 Synthesis of novel sulfonamide 1, 2, 4-triazoles
Reagents and conditions: (ⅰ) HSO3Cl, 0 ℃; (ⅱ) NH3•H2O, 0 ℃; (ⅲ) halobenzyl halide, K2CO3, acetone, 50 ℃; (ⅳ) 1, 2-dibromoethane, K2CO3, acetone, 50 ℃; (ⅴ) 1, 2, 4-triazole, K2CO3, CH3CN, 65 ℃.

图 3 DNA在不同浓度的化合物7b存在下的紫外吸收光谱(pH=7.4, 室温)
Figure 3 UV absorption spectra of DNA with different concentrations of compound 7b (pH=7.4, room temperature)

图 4 260 nm处7b-DNA络合物的吸光度以及游离的DNA与游离化合物7b的吸光度之和
Figure 4 Comparison of absorption at 260 nm between the 7b-DNA complex and the sum values of free DNA and free compound 7b
c(DNA)=4.8×10-5 mol•L-1, and c(7b)=0~9.6×10-6 mol•L-1 for curves a~i respectively at increment 1.2×10-6

图 6 中性红在不同浓度DNA下的紫外吸收光谱(pH=7.4, 室温)
Figure 6 UV absorption spectra of NR in the presence of DNA at pH 7.4 and room temperature
c(NR)=2×10-5 mol/L, and c(DNA)=0~3.84×10-5 mol/L for curves a~i respectively at increment of 0.48×10-5

图 7 DNA在中性红与化合物7b竞争下的紫外吸收光谱
Figure 7 UV Absorption spectra of the competitive reaction between 7b and neutral red with DNA
c(DNA)=4.8×10-5 mol/L, c(NR)=2×10-5 mol/L, and c(7b)=0~8.4×10-6 mol/L for curves a~h respectively at increment 1.2×10-6. Inset: Absorption spectra of the system with the increasing concentration of 7b in the wavelength range of 250~300 nm absorption spectra of competitive reaction between compound 7b and NR with DNA
表 1 化合物5a分子结构的氢键参数(Å, °)a
Table 1. Hydrogen-bond geometry (Å, °) of compound 5a
D—H…A D—H H…A D…A D—H…A N(1)—H(1)…O(2)i 0.77(3) 2.26(3) 3.000(3) 162(3) N(2)—H(2A)…O(3)ii 0.80(3) 2.08(3) 2.873(3) 178(3) ai, (x-1, y, z); ii, (-x, -y, -z).  下载: 导出CSV
下载: 导出CSV
表 2 磺胺类新化合物4~7的抗细菌和抗真菌活性(MIC, µg/mL)
Table 2. Antibacterial and antifungal data as MIC (μg/mL) for sulfonamide compounds 4~7
化合物 细菌 真菌 金黄色葡萄球菌 大肠杆菌 蕈状芽孢杆菌 耻垢分支杆菌 白色念珠菌 红色酵母菌 黄曲霉菌 4a 512 128 512 512 256 512 512 4b 512 128 256 512 512 256 512 4c 512 256 512 512 512 512 512 4d 512 128 512 512 512 512 512 5a 512 128 256 256 256 256 256 5b 256 64 512 512 256 256 256 5c 128 256 256 256 256 512 128 5d 256 128 256 512 512 256 256 6a 128 128 128 128 256 128 128 6b 64 64 128 64 128 128 64 6c 64 128 64 128 128 128 128 6d 256 128 256 256 256 128 128 7a 256 32 256 256 128 128 128 7b 64 16 128 64 64 64 32 7c 256 128 128 128 128 64 64 7d 256 64 256 128 128 128 128 磺胺 512 512 >512 >512 >512 >512 >512 依诺沙星 8 16 8 16 — — — 氟康唑 — — — — 8 8 256
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 8
- 文章访问数: 4199
- HTML全文浏览量: 628
 下载:
下载:
