 Figure1.
4H-3, 1-Benzoxazin-4-ones with bioactivities
Figure1.
4H-3, 1-Benzoxazin-4-ones with bioactivities
Citation:
Zhang Xiaoxiang, Lü Chang, Li Ping, Yong Wanxiong, Li Jing, Zhu Xinbao. Rapid Access to 4H-3, 1-Benzoxazin-4-ones via Gold-Catalyzed One-Pot Oxidative Rearrangement of 2-Alkynyl Arylazides[J]. Chinese Journal of Organic Chemistry,
2018, 38(1): 208-214.
doi:
10.6023/cjoc201706030

金催化2-炔基芳基叠氮氧化重排一锅法快速合成苯并噁嗪-4-酮
English
Rapid Access to 4H-3, 1-Benzoxazin-4-ones via Gold-Catalyzed One-Pot Oxidative Rearrangement of 2-Alkynyl Arylazides
Abstract:
The one-pot one-step synthetic method of 4H-3, 1-benzoxazin-4-ones via the gold-catalyzed oxidative rearrangement of 2-alkynyl arylazides has been developed. The desired products were obtained in moderate to excellent yields under mild reaction conditions. In most cases, using acetic acid as solvent the reactions were shown to proceed very fast within 1 h.
-
Key words:
- gold-catalyzed
- / oxidative rearrangement
- / one-pot
- / 2-alkynyl arylazides
-
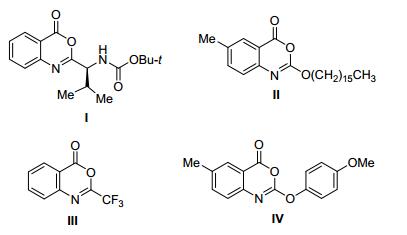
4H-3, 1-Benzoxazin-4-ones are very important core structures in organic chemistry due to their presence in a myriad of bioactive compounds and pharmaceutical drugs, [1]such as inhibitor of human leukocyte (Ⅰ), [2] cetillistat (Ⅱ), [3] chymotrypsin inactivator (Ⅲ)[4] and HSV-1 protease inhibitor (Ⅳ)[5] (Figure 1). They are also utilized as valuable synthetic intermediates in organic chemistry.[6] Because of those, an immense number of convenient and efficient methodologies for the construction of 4H-3, 1-benzoxazin-4-ones 3 have been established over the years.[7] Those have hitherto included the palladium catalyzed transformtion of 2-alkynyl arylazides 1 in the presence of oxone via aminopalladation/oxidative rearrangement, which afforded the corresponding 4H-3, 1-benzo-xazin-4-ones 3 (Scheme 1).[7h] While this synthetic method was shown to be reliable and suitable rout to the heterocycles, the reactions were reported to rely on the combination of a base, heating and lightless requirement, which led to the formation of 4H-3, 1-benzoxazin-4-ones in low yields after long reaction time. In this regard, the establishing of mild and efficient synthetic methods to get substituted 4H-3, 1-benzoxazin-4-ones from 2-alkynyl arylazides is desirable.
Figure1.
4H-3, 1-Benzoxazin-4-ones with bioactivities
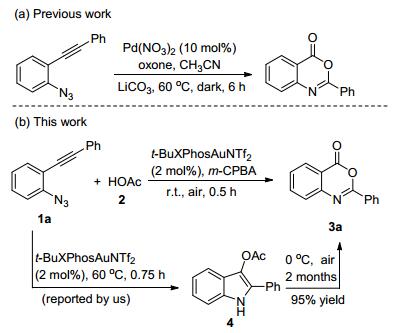 Scheme1.
Design for gold-catalyzed oxidation rearrangement of 2-alkynyl arylazides 1
Scheme1.
Design for gold-catalyzed oxidation rearrangement of 2-alkynyl arylazides 1
Gold-catalyzed tandem reactions[8] of 2-alkynyl arylazides have become one of the most versatile and efficient method for the access to N-containing heterocycles via α-imino gold carbenes intermediate generated in situ.[9]As part of an ongoing program exploring the gold-catalyzed transformation of 2-alkynyl arylazides, [10] we recently reported intermolecular trapping of α-imino gold carbenes by carboxylic acids for the synthesis of 1H-indol-3-yl esters.[10d] Very interestingly, the 2-phenyl-1H-indol-3-yl (4) stored in fridge was slowly converted to the oxidative compound 2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3a), which was isolated in 95% yield after two months. On the basis of these earlier studies, we reasoned that a novel synthetic method to 4H-benzo[d][1, 3]oxazin-4-ones (3) could be achieved through the use of suitable oxidants. Herein, we report a one-pot, one-step tBuXPhosAuNTf2 catalyzed oxidative rearrangement at room temperature in the presence of acetic acid and m-CPBA (Scheme 1). In most cases, the desired 4H-3, 1-benzoxazin-4-one products were obtained in moderate to excellent yields within 1 h.
1 Results and discussion
Initially, we chose to focus our attention on the reaction of 1-azido-2-(phenylethynyl)benzene (1a) and acetic acid (2) by different oxidants to establish the reaction conditions. This revealed treating a solution of reaction containing 1a (1 equiv.), m-CPBA (2 equiv.) and acetic acid (1 mL) with 2 mol% tBuXPhosAuNTf2 at room temperature for 0.5 h gave the best result (Table 1, Entry 1). Under these conditions, 4H-3, 1-benzoxazin-4-one (2a) was provided in 86% yield. On the other hand, a similar product yield was observed when the reaction temperature was increased to 60 ℃ (Table 1, Entry 2). In contrast, while the use of oxone and TBHP as the oxidants afforded 2a in markedly lower yields of 40%~67%, the expected 2a was not obtained employing air (Table 1, Entries 3~5). In this latter case, the reaction was found to result in the formation of 2-phenyl-1H-indol-3-yl acetate (4) at 60 ℃ after 24 h, which was provided in 85% yield. Lower product yield was observed when the reaction was conducted in MeCN, PhCl or (CH2Cl)2 in place of HOAc as the solvent (Table 1, Entries 6~8). Additional control experiment with HOAc at 40% of concentration led to lower product yield (Table 1, Entry 9). 69% yield of 3a was obtained when 1.5 equiv. of m-CPBA was used (Table 1, Entry 10). Only starting material was left when the reaction was conducted without catalyst, which was confirmed by TLC and crude 1H NMR analysis (Table 1, Entry 11).
 Table 1.
Optimization of reaction conditionsa
Table 1.
Optimization of reaction conditionsa

Entry Oxidant Solvent Time/h Yieldb/% 1 m-CPBA HOAc 0.5 86 2c m-CPBA HOAc 0.5 85 3c, d Air HOAc 24 — 4e oxone HOAc 0.5 67 5 TBHP HOAc 0.5 40 6f m-CPBA MeCN 0.5 49 7f m-CPBA PhCl 0.5 39 8f m-CPBA (CH2Cl)2 0.5 54 9g m-CPBA (CH2Cl)2 0.5 34 10h m-CPBA HOAc 0.5 69 11i m-CPBA HOAc 0.5 NR aUnless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with tBuXPhosAuNTf2:1a:oxidant ratio = 0.02:1:2. bIsolated yield. c At 60 ℃.d 85% of 4a obtained.e12% of 4a was obtained. f0.4 mL of HOAc was used.g Without HOAc.h1.5 equiv of m-CPBA was used.iWithout catalyst. NR=no reaction To define the scope of the present procedure, we next turned our attention to the reactions of a variety of 2-alkynyl arylazides 1 with acetic acid (Table 2). Reactions of 2-alkynyl arylazides with a pendant electron-donating or electron-withdrawing group on the alkyne carbon with acetic acid gave the desired 4H-3, 1-benzoxazin-4-ones in good to excellent yields (Table 2, 3a~3f). We also tested the effect of substituted group R1 on the aromatic ring and found that for substrates R1 = substituted halogen, CF3, or Me, excellent yields were obtained (Table 2, 3g~3k, 3n~3o). Substrate with R1=R2=Cl was also provided the desired product in 85% yield (Table 2, 3l). In contrast, ester, di-substituted methyl, and ether functional groups on the aromatic ring were also examined, which were afforded the corresponding compounds in 52%~56% yields (Table 2, 3m, 3p~3q).
Table 2.
Synthesis of 4H-3, 1-benzoxazin-4-ones 3a, b


















aUnless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with t-BuXPhosAuNTf2:1a:m-CPBA ratio=0.02:1:2. bIsolated yield. On the other hand, reactions of 2-alkynyl arylazides with aliphatic substituted group on the alkyne carbon were also tested under the standard reaction conditions. Interestingly, the reactions of substrates with R2=n-C4H9, n-C5H11, cyclopropyl, and t-But were found to proceed well and provide the anthranilic acid products 5 in excellent yields, which were due to the hydrolysis of corresponding 4H-3, 1-benzoxazin-4-ones (Table 3, 5a~5d).
Table 3.
Synthesis of anthranilic acids 5a, b





a Unless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with t-BuXPhosAuNTf2:1a:m-CPBA ratio=0.02:1:2. bNMR yield (mixed with 3-chlorobenzoic acid). Based on the above results, we tentatively propose the tBuXPhosAuNTf2-catalyzed 4H-3, 1-benzoxazin-4-ones forming reaction to proceed by the mechanism outlined in Scheme 2, although it is highly speculative. This could involve the activation of the 1-azido-2-(phenylethynyl)-benzene (1a) through coordination of the gold catalyst with alkyne bond, which was then attacked by azide to deliver the intermediate A. After the releasing of N2, α-imino gold carbene species B was afforded, which was then trapped by acetic acid to deliver the complex C. Subsequent protondemetalation step would then give the intermediate 2-phenyl-1H-indol-3-yl acetate 4, which was oxidized by m-CPBA to form intermediate D. Rearrangement of D led to form E, which could be oxidized by air to provide the desired product 4H-3, 1-benzoxazin-4-one 3a.
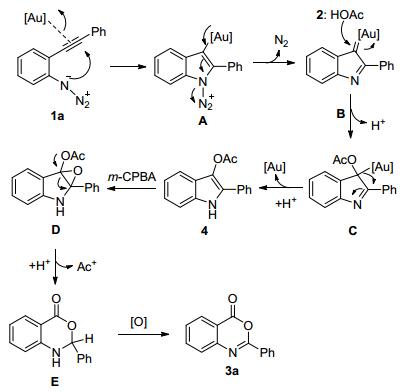 Scheme2.
Proposed mechanism for the formation of 3a
Scheme2.
Proposed mechanism for the formation of 3a
2 Conclusions
In summary, a mild and efficient synthetic route to 4H-3, 1-benzoxazin-4-ones via one-pot one-step gold-cata-lyzed oxidative rearrangement of 2-alkynyl arylazides has been reported. These results show that the reaction tolerates a structurally diverse set of 2-alkynyl arylazides. This method can also provided the anthranilic acids in excellent yields. Our studies show that the reaction can be performed well in very short time by using acetic acid as solvent. Efforts are currently underway to apply the method for the synthesis of biologically active compounds.
3 Experimental section
3.1 General considerations
Analytical thin layer chromatography (TLC) was performed using Merck 60 F254 pre-coated silica gel plate. Visualization was achieved by UV light (254 nm). Flash chromatography was performed using a Merck silica gel 60 with freshly distilled solvents. Unless otherwise stated, 1H and 13C NMR spectra were measured on a Bruker AVANCE Ⅲ 400 MHz spectrometer. Unless otherwise stated, chemical shifts were recorded with respect to TMS in CDCl3. Infrared spectra were recorded on a NICOLET 6700 FTIR Spectrometer. High Resolution Mass (HRMS) spectra were obtained using an LTQ Orbitrap.
3.2 General procedure for gold-catalyzed formation of 4H-3, 1-benzoxazin-4-ones (3a~3q)
A pressure tube equipped with a magnetic stirrer bar was charged with tBuXPhosAuNTf2(2 mol%), 2-alkynyl arylazides 1 (0.1 mmol), m-CPBA (0.2 mmol, 2 equiv.), and acetic acid 2 (1 mL). The reaction solution was stirred for 30 min at room temperature. Then the mixture was washed several times with saturated sodium bicarbonate solution and extracted with DCM (10 mL×2). The combined organic layer was washed with brine (10 mL) and dried over anhydrous MgSO4. After removing the solvent under reduced pressure, the residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, V:V=1:10), providing the desired compound 3.
2-Phenyl-4H-benzo[d][1, 3]oxazin-4-one (3a)[7h]: 90% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.31~8.33 (m, 2H), 8.24~8.26 (m, 1H), 7.81~7.86 (m, 1H), 7.70 (d, J=8.1 Hz, 1H), 7.56~7.59 (m, 1H), 7.50~7.54 (m, 3H).
2-(p-Tolyl)-4H-benzo[d][1, 3]oxazin-4-one (3b)[7h]: 74% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.19~8.25 (m, 3H), 7.77~7.83 (m, 1H), 7.68 (d, J=7.6 Hz, 1H), 7.50 (t, J=7.6 Hz, 1H), 7.31 (d, J=8.0 Hz, 2H), 2.45 (s, 3H).
2-(4-Methoxyphenyl)-4H-benzo[d][1, 3]oxazin-4-one (3c)[7a]: 81% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.20~8.26 (m, 3H), 7.80 (t, J=7.1 Hz, 1H), 7.64 (d, J=8.0 Hz, 1H), 7.47 (t, J=7.2 Hz, 1H), 6.99 (d, J=8.9 Hz, 2H), 3.89 (s, 3H).
2-(4-Chlorophenyl)-4H-benzo[d][1, 3]oxazin-4-one (3d)[7h]: 75% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.23~8.27 (m, 3H), 7.82~7.86 (m, 1H), 7.68 (d, J=7.6 Hz, 1H), 7.54 (t, J=7.6 Hz, 1H), 7.49 (d, J=8.8 Hz, 2H).
Methyl 4-(4-oxo-4H-benzo[d][1, 3]oxazin-2-yl)benzoate (3e)[7a]: 80% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.38 (d, J=8.6 Hz, 2H), 8.26 (d, J=7.8 Hz, 1H), 8.17 (d, J=8.6 Hz, 2H), 7.86 (t, J=8.2 Hz, 1H), 7.72 (d, J=7.7 Hz, 1H), 7.56 (t, J=6.9 Hz, 1H), 3.97 (s, 3H).
2-(3-Fluorophenyl)-4H-benzo[d][1, 3]oxazin-4-one (3f)[7a]: 82% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.24 (d, J=7.8 Hz, 1H), 8.10 (d, J=7.8 Hz, 1H), 7.99~8.02 (m, 1H), 7.82~7.86 (m, 1H), 7.70 (d, J=8.0 Hz, 1H), 7.46~7.56 (m, 2H), 7.25~7.29 (m, 1H).
6-Chloro-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3g)[7h]: 93% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.29~8.31 (m, 2H), 8.20 (d, J=2.4 Hz, 1H), 7.75~7.78 (m, 1H), 7.57~7.66 (m, 2H), 7.50~7.54 (m, 2H).
7-Chloro-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3h)[11]: 85% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.30 (d, J=7.2 Hz, 2H), 8.17 (d, J=8.4 Hz, 1H), 7.70 (d, J=2.0 Hz, 1H), 7.60 (t, J=7.4 Hz, 1H), 7.52 (t, J=7.2 Hz, 2H), 7.46~7.49 (m, 1H), 3.97 (s, 3H).
2-Phenyl-6-(trifluoromethyl)-4H-benzo[d][1,3]oxazin-4-one (3i)[7h]: 89% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.52 (s, 1H), 8.33 (d, J=7.3 Hz, 2H), 8.02~8.05 (m, 1H), 7.81 (d, J=8.5 Hz, 1H), 7.52~7.64 (m, 3H).
2-Phenyl-7-(trifluoromethyl)-4H-benzo[d][1, 3]oxazin-4-one (3j)[11]: 83% yield. 1H NMR (CDCl3, 400 MHz): 8.39 (d, J=8.2 Hz, 1H), 8.35 (d, J=7.2 Hz, 2H), 8.00 (s, 1H), 7.75 (d, J=8.2 Hz, 1H), 7.64 (t, J=7.4 Hz, 1H), 7.56 (t, J=7.2 Hz, 2H).
6-Fluoro-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3k)[7a]: 92% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.28~8.30 (m, 2H), 7.87~7.90 (m, 1H), 7.69~7.73 (m, 1H), 7.50~7.60 (m, 4H).
7-Chloro-2-(4-chlorophenyl)-4H-benzo[d][1, 3]oxazin-4-one (3l)[7a]: 86% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.24 (d, J=8.7 Hz, 2H), 8.17 (d, J=8.4 Hz, 1H), 7.69 (s, 1H), 7.48~7.51 (m, 3H).
Methyl 4-oxo-2-phenyl-4H-benzo[d][1, 3]oxazine-6-car-boxylate (3m)[7a]: 56% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.90 (d, J=1.92 Hz, 1H), 8.44~8.46 (m, 1H), 8.32~8.34 (m, 2H), 7.74 (d, J=8.4 Hz, 1H), 7.59~7.63 (m, 1H), 7.51~7.55 (m, 2H), 3.98 (s, 3H).
6, 8-Dichloro-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3n)[12]: 77% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.35 (d, J=7.2 Hz, 2H), 8.12 (d, J=2.4 Hz, 1H), 7.87 (d, J=2.4 Hz, 1H), 7.60~7.64 (m, 1H), 7.52~7.55 (m, 2H).
6-Methyl-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3o)[7h]: 82% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.30 (d, J=7.1 Hz, 2H), 8.04 (s, 1H), 7.49~7.65 (m, 5H), 2.49 (s, 3H).
6, 8-Dimethyl-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3p)[11]: 52% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.32 (d, J=6.9 Hz, 2H), 7.88 (s, 1H), 7.48~7.54 (m, 4H), 2.62 (s, 3H), 2.44 (s, 3H).
7-Methoxy-2-phenyl-4H-benzo[d][1, 3]oxazin-4-one (3q)[6b]: 54% yield. 1H NMR (CDCl3, 400 MHz) δ: 8.31 (d, J=7.1 Hz, 2H), 8.14 (d, J=8.7 Hz, 1H), 7.49~7.59 (m, 3H), 7.11 (d, J=2.5 Hz, 1H), 7.04~7.07 (m, 1H), 3.96 (s, 3H).
3.3 General procedure for the synthesis of 2-phenyl-1H-indol-3-yl acetate (4)
To a solution of 1-azido-2-(phenylethynyl)benzene (1a) (0.1 mmol) and acetic acid 2 (1 mL) was added gold catalyst (2 mol %). The mixture was stirred at 60 ℃ and monitored by TLC analysis. On completion, the reaction mixture was directly subjected to purification by flash column chromatography on silica gel (petrol ether/ethyl acetate, V:V=20:1 to 10:1) to give the desired product 2-phenyl-1H-indol-3-yl acetate (4)[10d], 80% yield. 1H NMR (CDCl3, 400 MHz) δ: 2.41 (s, 3H), 7.12~7.16 (m, 1H), 7.18~7.22 (m, 1H), 7.28~7.35 (m, 2H), 7.41~7.45 (m, 3H), 7.61 (d, J=7.5 Hz, 2H), 8.10 (br, 1H).
3.4 General procedure for the synthesis of anthranilic acids (5a~5d)
A pressure tube equipped with a magnetic stirrer bar was charged with tBuXPhosAuNTf2 (2 mol%), 2-alkynyl arylazides 1 (0.1 mmol), m-CPBA (0.2 mmol, 2 equiv), and acetic acid 2 (1 mL). The reaction solution was stirred for 30 min at room temperature. Removing the solvent 2 under reduced pressure with proper heating, the residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, V:V=1:4), providing the desired compound 5.
2-Pentanamidobenzoic acid (5a): 91% yield, white solid. m.p. 73.2 ℃; 1H NMR (CDCl3, 400 MHz) δ: 10.95 (s, 1H), 8.76~8.78 (d, J=8.5 Hz, 1H), 8.13~8.15 (dd, J=8.0, 1.6 Hz, 1H), 7.59~7.63 (m, 1H), 7.11~7.15 (m, 1H), 2.47~2.50 (m, 2H), 1.72~1.80 (m, 2H), 1.40~1.49 (m, 2H), 0.97 (t, J=7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 13.8, 22.3, 27.6, 38.4, 114.0, 120.6, 122.7, 131.8, 135.7, 142.1, 172.5, 172.7; IR (neat) v: 3324, 2958, 2927, 2860, 1686, 1649, 1584, 1242, 956, 758, 654 cm-1; HRMS (ESI) calcd for C12H16NO3 [M+H]+ 222.1130, found 222.1128.
2-Hexanamidobenzoic acid (5b): 97% yield, yellow solid. m.p. 77.4 ℃; 1H NMR (CDCl3, 400 MHz) δ: 10.97 (s, 1H), 8.76~8.78 (d, J=8.5 Hz, 1H), 8.13~8.15 (dd, J=8.0, 1.3 Hz, 1H), 7.59~7.63 (m, 1H), 7.11~7.15 (m, 1H), 2.46~2.50 (m, 2H), 1.74~1.81 (m, 2H), 1.36~1.44 (m, 4H), 0.92 (t, J=7.1 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ:13.9, 22.4, 25.2, 31.3, 38.7, 113.9, 120.6, 122.7, 131.8, 135.7, 142.1, 172.5, 172.7; IR (neat) v: 3330, 2954, 2929, 2856, 1691, 1672, 1586, 1264, 900, 755, 653 cm-1; HRMS (ESI) calcd for C13H18NO3 [M+H]+ 236.1287, found 236.1285.
2-(Cyclopropanecarboxamido)benzoic acid (5c): 88% yield, brown solid. m.p. 98.3 ℃; 1H NMR (CDCl3, 400 MHz) δ: 11.20 (s, 1H), 8.72~8.74 (d, J=8.5 Hz, 1H), 8.11~8.13 (dd, J=8.0, 1.6 Hz, 1H), 7.55~7.59 (m, 1H), 7.08~7.12 (m, 1H), 1.62~1.68 (m, 1H), 1.11~1.15 (m, 2H), 0.88~0.92 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ: 8.3, 16.8, 114.3, 120.5, 122.4, 131.7, 135.3, 142.1, 173.0, 173.0; IR (neat) v: 3349, 2925, 2854, 1689, 1662, 1582, 1271, 957, 757, 660 cm-1; HRMS (ESI) calcd for C11H12NO3 [M+H]+ 206.0817, found 206.0815.
2-Pivalamidobenzoic acid (5d): 82% NMR yield (mixed with 3-chlorobenzoic acid). 1H NMR (CDCl3, 400 MHz) δ: 11.20 (s, 1H), 8.82~8.79 (d, J=8.5 Hz, 1H), 8.17~8.14 (dd, J=8.5, 1.6 Hz, 1H), 7.61~7.58 (m, 1H), 7.14~7.10 (m, 1H), 1.36 (s, 9H).
-
-
[1]
(a) Padwal, R. Curr. Opin. Invest. Drugs 2008, 9, 414.
(b) Eissa, A. M. F. ; El-Sayed, R. J. Heterocycl. Chem. 2006, 43, 1.
(c) Neumann, U. ; Schechter, N. M. ; Gutschow, M. Bioorg. Med. Chem. 2001, 9, 947.
(d) Gutschow, M. ; Neumann, U. Bioorg. Med. Chem. 1997, 5, 1935.
(e) Krantz, A. ; Spencer, R. W. ; Tam, T. F. ; Liak, T. J. ; Copp, L. J. ; Thomas, E. M. ; Rafferty, S. P. J. Med. Chem. 1990, 33, 464. -
[2]
(a) Krantz, A. ; Spencer, R. W. ; Tam, T. F. ; Liak, T. J. ; Copp, L. J. ; Thomas, E. M. ; Rafferty, S. P. J. Med. Chem. 1990, 33, 464.
(b) Stein, R. L. ; Strimpler, A. M. ; Viscarello, B. R. ; Wildonger, R. A. ; Mauger, R. C. ; Trainor, D. A. Biochemistry 1987, 26, 4126. -
[3]
For some examples, see: (a) Yamada, Y. ; Kato, T. ; Ogino, H. ; Ashina, S. ; Kato, K. Horm. Metab. Res. 2008, 40, 539.
(b) Padwal, R. Curr. Opin. Invest. Drugs 2008, 9, 414. -
[4]
Hedstrom, L.; Moorman, A. R.; Dobbs, J.; Abeles, R. H. Biochemistry 1984, 23, 1753. doi: 10.1021/bi00303a026
-
[5]
Jarvest, R. L.; Parratt, M. J.; Debouck, C. M.; Gorniak, J. G.; Jennings, L. J.; Serafinowska, H. T.; Strickler, J. E. Bioorg. Med. Chem. Lett. 1996, 6, 2463. doi: 10.1016/0960-894X(96)00455-6
-
[6]
(a) Li, W. F. ; Wu, X. F. J. Org. Chem. 2014, 79, 10410.
(b) Yamashita, M. ; Iida, A. Tetrahedron Lett. 2014, 55, 2991.
(c) Lian, X. L. ; Lei, H. ; Quan, X. J. ; Ren, Z. H. ; Wang, Y. Y. ; . Guan, Z. H. Chem. Commun. 2013, 49, 8196.
(d) Ge, Z. Y. ; Xu, Q. M. ; Fei, X. D. ; Tang, T. ; Zhu, Y. M. ; Ji, S. J. J. Org. Chem. 2013, 78, 4524.
(e) Xie, Y. Y. ; Wang, S. P. J. Chem. Res. 2012, 36, 123.
(f) Wu X. F. ; Neumann, H. ; Beller, M. Chem. Eur. J. 2012, 18, 12599.
(g) Kumar, R. A. ; Maheswari, C. U. ; Ghantasala, S. ; Jyothi, C. ; Reddy, K. R. Adv. Synth. Catal. 2011, 353, 401.
(h) Xue, S. ; McKenna, J. ; Shieh, W. C. ; Repic, O. J. Org. Chem. 2004, 69, 6474.
(i) Chenard, B. L. ; Welch, W. M. ; Blake, J. F. ; Butler, T. W. ; Reinhold, A. ; Ewing, F. E. ; Menniti, F. S. ; Pagnozzi, M. J. J. Med. Chem. 2001, 44, 1710.
(j) Coppola, G. M. J. Heterocycl. Chem. 1999, 36, 563.
(k) Larksarp, C. ; Alper, H. Org. Lett. 1999, 1, 1619.
(l) Kotsuki, H. ; Sakai, H. ; Morimoto, H. ; Suenaga, H. Synlett 1999, 1993.
(m) Cacchi, S. ; Fabrizi, G. ; Marinelli, F. Synlett 1996, 997.
(n) Larock, R. C; . Fellows, C. A. J. Org. Chem. 1980, 45, 363.
(o) Errede, L. A. J. Org. Chem. 1976, 41, 1763.
(p) Jackson, T. G. ; Morris, S. R. ; Turner, R. H. J. Chem. Soc. 1968, 13, 1592.
(q) Taylor, E. C. ; Knopf, R. I. ; Borror, A. L. J. Am. Chem. Soc. 1960, 82, 3152. -
[7]
For recent examples on the synthesis of 4H-3, 1-benzoxazin-4-ones 3, see: (a) Wang, L. ; Xie, Y. -B; Huang, N. -Y. ; Yan, J. -Y. ; Hu, W. -M. ; Liu, M. -G. ; Ding, M. -W. ACS Catal. 2016, 6, 4010.
(b) Verma, A. ; Kumar, S. Org. Lett. 2016, 18, 4388.
(c) Arcadi, A. ; Chiarini, M. ; Vecchio, L. D. ; Marinelli, F. ; Michelet, V. Chem. Commun. 2016, 52, 1458.
(d) Laha, J. K. ; Patel, K. V. ; Tummalapalli, K. S. S. ; Dayal, N. Chem. Commun. 2016, 52, 10245.
(e) Prakash, R. ; Gogoi, S. Adv. Synth. Catal. 2016, 19, 3046.
(f) Yu, J. ; Zhang-Negrerie, D. ; Du, Y. Eur. J. Org. Chem. 2016, 2016, 562.
(g) Wu, X. F. ; Schranck, J. ; Neumann, H. ; Beller, M. Chem. -Eur. J. 2011, 17, 12246.
(h) Liu, Q. ; Chen, P. ; Liu, G. ACS Catal. 2013, 3, 178.
(i) Li, W. ; Wu, X. F. J. Org. Chem. 2014, 79, 10410.
(j) Chavan, S. P. ; Bhanage, B. M. Eur. J. Org. Chem. 2015, 2015, 2405.
(k) Munusamy, S. ; Venkatesan, S. ; Sathiyanarayanan, K. I. Tetrahedron Lett. 2015, 56, 203.
(l) Larksarp, C. ; Alper, H. Org. Lett. 1999, 1, 1619.
(m) Salvadori, J. ; Balducci, E. ; Zaza, S. ; Petricci, E. ; Taddei, M. J. Org. Chem. 2010, 75, 1841.
(n) Nayak, M. K. ; Kim, B. H. ; Kwon, J. E. ; Park, S. ; Seo, J. ; Chung, J. W. ; Park, S. Y. Chem. -Eur. J. 2010, 16, 7437. -
[8]
For recent selected reviews on gold-catalyzed reactions, see: (a) Harris, R. J. ; Widenhoefer, R. A. Chem. Soc. Rev. 2016, 45, 4533.
(b) Asiri, A. M. ; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 4471.
(c) Zheng, Z. ; Wang, Z. ; Wang, Y. ; Zhang, L. Chem. Soc. Rev. 2016, 45, 4448.
(d) Ziand, W. ; Toste, F. D. Chem. Soc. Rev. 2016, 45, 4567.
(e) Huple, D. B. ; Ghorpade, S. ; Liu, R. -S. Adv. Synth. Catal. 2016, 358, 1348.
(f) Dorel, R. ; Echavarren, A. M. Chem. Rev. 2015, 115, 9028.
(g) Jia, M. ; Bandini, M. ACS Catal. 2015, 5, 1638.
(h) Wang, Y. ; Muratore, M. E. ; Echavarren, A. M. Chem. Eur. J. 2015, 21, 7332.
(i) Zhang, L. Acc. Chem. Res. 2014, 47, 877.
(j) Yeom, H. -S. ; Shin, S. Acc. Chem. Res. 2014, 47, 966.
(k) Fensterbank, L. ; Malacria, M. Acc. Chem. Res. 2014, 47, 953.
(l) Benitez, D. ; Shapiro, N. D. ; Tkatchouk, E. ; Wang, Y. ; God-dard Ⅲ, W. A. ; Toste, F. D. Nat. Chem. 2009, 1, 482.
(m) Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180.
(n) Wu, W. -T. ; Zhang, L. ; You, S. -L. Acta Chim. Sinica 2017, 75, 419(in Chinese). (吴文挺, 张立明, 化学学报, 2017, 75, 419. )
for recent selected examples, see: (o) Zhao, J. -R. ; Yuan, X. ; Wang, Z. ; Chen, S. ; Zhang, Z. -X. ; Xue, W. Org. Chem. Front. 2015, 2, 34.
(p) Li, L. ; Zhou, B. ; Ye, L. Chin. J. Org. Chem. 2015, 35, 655(in Chinese). (李龙, 周波, 叶龙武, 有机化学, 2015, 35, 655. )
(q) Zhang, P. -C. ; Wang, Y. ; Qian, D. ; Li, W. ; Zhang, J. Chin. J. Chem. 2017, 35, 849. -
[9]
For recent selected a review on the generation of α-imino gold carbenes, see: Davies, P. W. ; Garz n, M. Asian J. Org. Chem. 2015, 4, 694.
-
[10]
For recent selected examples, see: (a) Lonca, G. H. ; Tejo, C. ; Chan, H. L. ; Chiba, S. ; Gagosz, F. Chem. Commun. 2017, 53, 736.
(b) Jin, H. ; Tian, B. ; Song, X. ; Xie, J. ; Rudolph, M. ; Rominger, F. ; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2016, 55, 12688.
(c) Pan, Y. ; Chen, G. -W. ; Shen, C. -H. ; He, W. ; Ye, L. -W. Org. Chem. Front. 2016, 3, 391.
(d) Li, N. ; Lian, X. -L. ; Li, Y. -H. ; Wang, T. -Y. ; Han, Z. -Y. ; Zhang, L. ; Gong, L. -Z. Org. Lett. 2016, 18, 4178.
(e) Jin, H. ; Huang, L. ; Xie, J. ; Rudolph, M. ; Rominger, F. ; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2016, 55, 794.
(f) Zhou, A. -H. ; He, Q. ; Shu, C. ; Yu, Y. -F. ; Liu, S. ; Zhao, T. ; Zhang, W. ; Lu, X. ; Ye, L. -W. Chem. Sci. 2015, 6, 1265.
(g) Xiao, X. -Y. ; Zhou, A. -H. ; Shu, C. ; Pan, F. ; Li, T. ; Ye, L-W. Chem. -Asian J. 2015, 10, 1854.
(h) Shen, C. -H. ; Pan, Y. ; Yu, Y. -F. ; Wang, Z. -S. ; He, W. ; Li, T. ; Ye, L. -W. J. Organomet. Chem. 2015, 795, 63.
(i) Li, N. ; Wang, T. -Y. ; Gong, L. -Z; Zhang, L. Chem. -Eur. J. 2015, 21, 3585.
(j) Shu, C. ; Wang, Y. -H. ; Zhou, B. ; Li, X. -L. ; Ping, Y. -F. ; Lu, X. ; Ye, L. -W. J. Am. Chem. Soc. 2015, 137, 9567.
(k) Zhu, L. ; Yu, Y. ; Mao, Z. ; Huang, X. Org. Lett. 2015, 17, 30.
(l) Wu, Y. ; Zhu, L. ; Yu, Y. ; Luo, X. ; Huang, X. J. Org. Chem. 2015, 80, 11407.
(m) Pawar, S. K. ; Sahani, R. L. ; Liu, R. -S. Chem. -Eur. J. 2015, 21, 10843.
(n) Prechter, A. ; Henrion, G. ; dit Bel, P. F. ; Gagosz, F. Angew. Chem., Int. Ed. 2014, 53, 4959.
(o) Garzón M. ; Davies, P. W. Org. Lett. 2014, 16, 4850.
(p) Tokimizu, Y. ; Oishi, S. ; Fujii, N. ; Ohno, H. Org. Lett. 2014, 16, 3138.
(q) Chatzopoulou, E. ; Davies, P. W. Chem. Commun. 2013, 49, 8617.
(r) Yan, Z. -Y. ; Xiao, Y. ; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 8624.
(s) Xiao, Y. ; Zhang, L. Org. Lett. 2012, 14, 4662.
(t) Lu, B. ; Luo, Y. ; Liu, L. ; Ye, L. ; Wang, Y. ; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 8358.
(u) Wetzel, A. ; Gagosz, F. Angew. Chem., Int. Ed. 2011, 50, 7354.
(v) Li, C. ; Zhang, L. Org. Lett. 2011, 13, 1738.
(w) Davies, P. W. ; Cremones, A. ; Dumitrescu, L. Angew. Chem., Int. Ed. 2011, 50, 8931.
(x) Gorin, D. J. ; Davis, N. R. ; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 11260. -
[11]
For our recent studies on gold-catalyzed tandem reactions, see: (a) Chen, C. ; Zou, Y. ; Chen, X. ; Zhang, X. ; Rao, W. ; Chan, P. W. H. Org. Lett. 2016, 18, 4730.
(b) Zhang, X. ; Sun, X. ; Fan, H. ; Lyu, C. ; Li, P. ; Zhang, H. ; Rao, W. RSC Adv. 2016, 6, 56319.
(c) Zhang, X. ; Sun, X. ; Fan, H. ; Li, P. ; Lyu, C. ; Rao, W. Eur. J. Org. Chem. 2016, 25, 4265.
(d) Zhang, X. ; Sun, X. ; Fan, H. ; Cui, X. ; Ma, M. Chin. J. Org. Chem. 2015, 35, 1469(in Chinese). (张小祥, 孙小萍, 张海飞, 崔杏丽, 马猛涛, 有机化学, 2015, 35, 1469. ) -
[12]
Wang, J.; Jiang, X.; Zhang, Y.; Zhu, Y.; Shen, J. Tetrahedron Lett. 2015, 56, 2349. doi: 10.1016/j.tetlet.2015.03.121
-
[13]
Eid, A. I. Eur. J. Med. Chem. 1979, 14, 463.
-
[1]
-
Table 1. Optimization of reaction conditionsa
Entry Oxidant Solvent Time/h Yieldb/% 1 m-CPBA HOAc 0.5 86 2c m-CPBA HOAc 0.5 85 3c, d Air HOAc 24 — 4e oxone HOAc 0.5 67 5 TBHP HOAc 0.5 40 6f m-CPBA MeCN 0.5 49 7f m-CPBA PhCl 0.5 39 8f m-CPBA (CH2Cl)2 0.5 54 9g m-CPBA (CH2Cl)2 0.5 34 10h m-CPBA HOAc 0.5 69 11i m-CPBA HOAc 0.5 NR aUnless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with tBuXPhosAuNTf2:1a:oxidant ratio = 0.02:1:2. bIsolated yield. c At 60 ℃.d 85% of 4a obtained.e12% of 4a was obtained. f0.4 mL of HOAc was used.g Without HOAc.h1.5 equiv of m-CPBA was used.iWithout catalyst. NR=no reaction  下载: 导出CSV
下载: 导出CSV
Table 2. Synthesis of 4H-3, 1-benzoxazin-4-ones 3a, b
aUnless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with t-BuXPhosAuNTf2:1a:m-CPBA ratio=0.02:1:2. bIsolated yield.
下载: 导出CSV
Table 3. Synthesis of anthranilic acids 5a, b
a Unless stated otherwise, all reactions were performed at room temperature in 1 mL of HOAc with t-BuXPhosAuNTf2:1a:m-CPBA ratio=0.02:1:2. bNMR yield (mixed with 3-chlorobenzoic acid).
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 2956
- HTML全文浏览量: 304
 下载:
下载:
