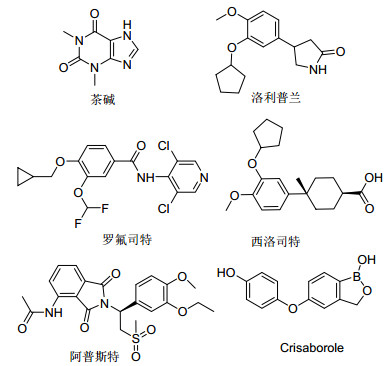
 图1
已上市PDE-4抑制剂的结构类型
Figure1.
PDE-4 inhibitors on the market
图1
已上市PDE-4抑制剂的结构类型
Figure1.
PDE-4 inhibitors on the market
引用本文:
高粟繁, 许勤龙, 李家明, 储昭兴, 何广卫, 林高峰, 朱正伟, 崔勇, 莫佳佳, 郭敬, 赵炎. PDE-4抑制剂的设计、合成及生物活性研究[J]. 有机化学,
2018, 38(2): 478-485.
doi:
10.6023/cjoc201705042

Citation: Gao Sufan, Xu Qinlong, Li Jiaming, Chu Zhaoxing, He Guangwei, Lin Gaofeng, Zhu Zhenwei, Cui Yong, Mo Jiajia, Guo Jing, Zhao Yan. Design, Synthesis, and Biological Evaluation of Novel PDE-4 Inhibitors[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 478-485. doi: 10.6023/cjoc201705042
Citation: Gao Sufan, Xu Qinlong, Li Jiaming, Chu Zhaoxing, He Guangwei, Lin Gaofeng, Zhu Zhenwei, Cui Yong, Mo Jiajia, Guo Jing, Zhao Yan. Design, Synthesis, and Biological Evaluation of Novel PDE-4 Inhibitors[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 478-485. doi: 10.6023/cjoc201705042
PDE-4抑制剂的设计、合成及生物活性研究
摘要:
依据药效团原理,对已报道的磷酸二酯酶(PDE-4)抑制剂Crisaborole进行结构修饰和改造,设计并合成了7个全新的小分子化合物,其结构经1H NMR、13C NMR和HRMS确证.研究其对磷酸二酯酶-4A(PDE-4A)的抑制活性、抑制炎症因子TNF-α释放效果以及抗炎活性.结果表明,所设计的7个化合物均表现出良好生物活性,其中一个化合物活性明显优于阳性对照药.
-
关键词:
- PDE-4抑制剂
- / Crisaborole
- / 合成
- / 抗炎
English
Design, Synthesis, and Biological Evaluation of Novel PDE-4 Inhibitors
Abstract:
Based on the reported phosphodiesterase-4(PDE-4) inhibitor of crisaborole, seven compounds with structural novelty were designed and synthesized according to the pharmacophore-combination strategy. The structures of them were identified by NMR and HRMS. Their inhibitory activities against phosphodiesterase-4A (PDE-4A) have been investigated. The inhibitory activities of inflammatory factor induced by lipopolysaccharide (LPS) or phorbol ester have been measured by mouse model. The results showed that all compounds exhibited high anti-inflammatory activities. In particular, one compound activity was significantly better than that of positive control drug.
-
Key words:
- PDE-4 inhibitor
- / crisaborole
- / synthesis
- / anti-inflammatory
-
炎症是一种十分常见而又重要的基本病理过程, 具有血管系统的活体组织对损伤因子的防御性反应都称为炎症.炎症反应与多种疾病的关系密切, 如皮炎、银屑病、系统性红斑狼疮[1]等.例如系统性红斑狼疮是由于基因的改变[2], 引起表皮增生, 从而引起一系列炎症反应.所以, 治疗炎症对于上述疾病的治疗具有重要作用.研究表明, 体内各类炎症反应和细胞内cAMP的摩尔浓度有关[3, 4]. cAMP和cGMP对于细胞活动起着重要的调节作用, 其浓度的调节主要是由核苷酸环化酶的合成和磷酸二酯酶(PDEs)水解作用之间的平衡来决定.
磷酸二酯酶(PDEs)具有水解细胞内第二信使环磷酸腺苷(cAMP)和环磷酸鸟苷(cGMP)的功能[5].细胞内cAMP或cGMP含量的下降会造成这些第二信使所传导的生化作用失常. PDEs有11个亚型, 它们在人体内分布广泛, 其生理作用涉及多个研究领域[6~10].尤其是亚型之一的PDE-4在炎症细胞中的作用明显, 它能选择性水解cAMP. cAMP浓度下降从而诱发诸如哮喘、抑郁症、银屑病、炎症等疾病[11~14]. PDE-4又可以分为4个亚型(PDE-4A、B、C、D), 其中PDE-4A在炎症细胞中表达强烈.因此, 抑制PDE-4A的活性对炎症疾病的治疗有很大帮助.
目前已有多种PDE-4抑制剂进入临床使用, 包括非选择性抑制剂茶碱和选择性抑制剂如洛利普兰、罗氟司特、西洛司特、阿普斯特等(图 1), 用于治疗炎症相关性疾病, 上述药物全部为口服药物, 不能进行局部使用. Crisaborole(图 1)是2016年上市的第一个外用非固醇类治疗特应性皮炎的药物, 市场前景广阔.因此, 以Crisaborole为先导化合物, 设计一系列新的PDE-4外用药物, 具有重要的临床意义.
图1
已上市PDE-4抑制剂的结构类型
Figure1.
PDE-4 inhibitors on the market
研究表明[15], PDE-4有3个活性区域, M区(含有金属离子)、S区(溶剂填充口袋)和Q区(由谷氨酰胺酸、苯丙氨酸和异亮氨酸组成)(图 2), PDE-4抑制剂通过和这3个活性区域结合, 从而增加cAMP的浓度水平, 治疗炎症、哮喘以及其他病症[16, 17]. Liu等[18]以黄酮类为模板研究设计了一系列化合物, 其中化合物1对PDE-4抑制效果良好. Kato等[19]报道合成了化合物2, 位于M区的R基团用吡啶替换氰基合成的化合物活性更强.推测吡啶环和M区有很好的结合能力. Gewald等[20]合成化合物3, 并指出和M区结合的R1基团用带氰基环取代活性能进一步增强. Elansary等[21]设计并合成了化合物4, 由于化合物中2位芳基无法和M区结合, 所以抑制活性弱, 化合物5改成了三氮唑后芳基延伸进入M区并与金属离子作用, 活性得到大大增强.表明连接基团能否将活性片段延伸至酶活性区域对活性影响很大.
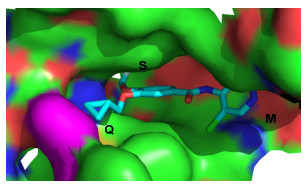 图2
罗氟司特与PDE-4的蛋白晶体图
Figure2.
Crystalline crystals of Roflumilast and PDE-4
图2
罗氟司特与PDE-4的蛋白晶体图
Figure2.
Crystalline crystals of Roflumilast and PDE-4
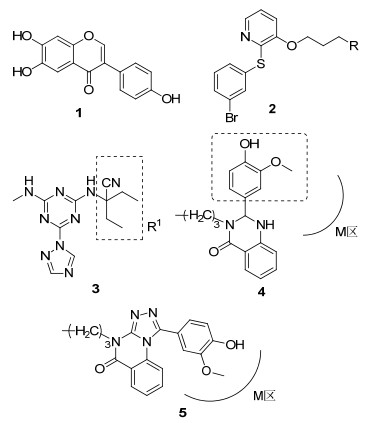 图3
PDE-4相关衍生物的结构类型
Figure3.
Structural types of PDE-4 related derivatives
图3
PDE-4相关衍生物的结构类型
Figure3.
Structural types of PDE-4 related derivatives
基于以上研究, 我们以Crisaborole为先导化合物进行结构修饰和改造.保留C部分的母核结构, 增加B部分的链长, 同时考虑将A部分苯环改成吡啶环并将替换环上氰基为三氟甲基, 以增加药物代谢稳定性.或者用具有抗炎活性的中药小分子丹皮酚[22]、川芎嗪[23]替换活性片段A, 考察它们和M区的结合能力.另外, 以文献[18]化合物为模板, 设计了具有苯并呋喃或苯并吡喃结构的化合物(图 4).共设计合成了7个未见报道的化合物, 具体合成路线见Schemes 1~3.分子对接结果显示, 所合成的化合物均能结合到PDE-4蛋白中的重要活性位点和口袋.药效试验表明这些化合物表现出优异的抗炎作用和效果, 优于已上市药物.
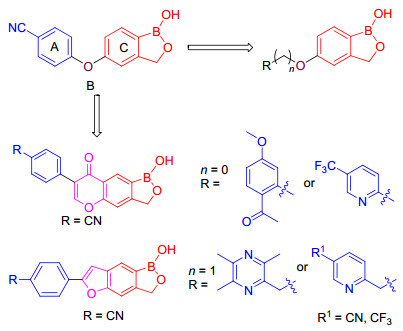 图4
PDE-4抑制剂的设计思路
Figure4.
Design ideas of PDE-4 inhibitors
图4
PDE-4抑制剂的设计思路
Figure4.
Design ideas of PDE-4 inhibitors
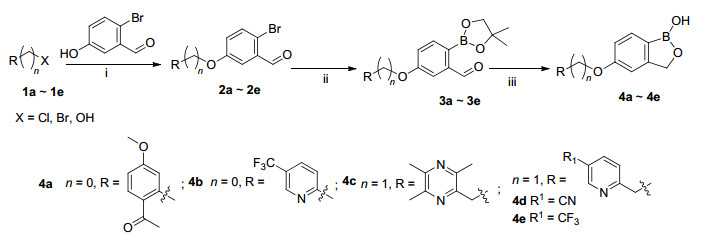 图式 1
化合物4a~4e的合成路线
Scheme1.
Synthetic route of compounds 4a~4e
图式 1
化合物4a~4e的合成路线
Scheme1.
Synthetic route of compounds 4a~4e
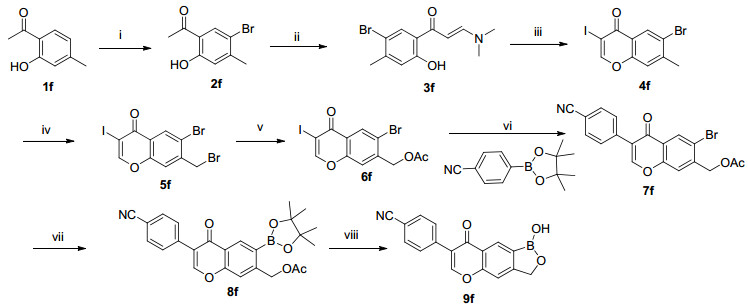 图式 2
化合物9f合成路线
Scheme2.
Synthetic route of compounds 9f
图式 2
化合物9f合成路线
Scheme2.
Synthetic route of compounds 9f
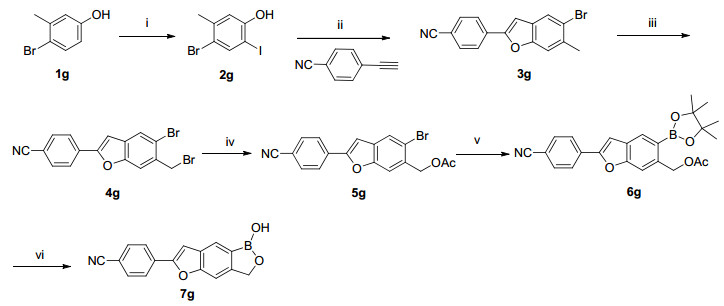 图式 3
化合物7g合成路线
Scheme3.
Synthetic route of compound 7g
图式 3
化合物7g合成路线
Scheme3.
Synthetic route of compound 7g
1 结果与讨论
1.1 目标化合物的合成
目标化合物4a~4e合成方法见Scheme 1.首先在碱催化剂和氮气保护存在下, 缩合不同的起始原料和2-溴-5-羟基苯甲醛, 然后将中间体2a~2e用二氧六环溶解, 加入频哪醇硼酸酯、乙酸钾、钯试剂, 氮气氛围下100 ℃搅拌2 h得中间体3a~3e.将中间体3a~3e.用甲醇溶解, 缓慢加入硼氢化钠, 再加入盐酸, 得到目标化合物4a~4e.目标化合物9f的合成方法见Scheme 2, 7g的合成方法见Scheme 3.目标化合物的结构经过1H NMR、13C NMR和HRMS分析确证.
1.2 目标化合物对PDE-4A的抑制活性
目标化合物对PDE-4A的抑制活性结果见表 1.从表 1可以发现, 所合成的7个化合物在三个浓度下酶活性均小于50%, 表明具有显著的酶抑制活性.其中化合物4a、4b、4d对PDE-4A的抑制活性在0.08 μmol/L浓度下优于Crisaborole.为了验证所设计的化合物与PDE-4A的作用位点和结合模式, 通过Autodock4.2D分子模拟软件将所合成的化合物与PDE-4A进行计算机分子对接, 对接结果如表 2及图 5(采用Pymol1.8进行处理)所示.结果发现分子对接与酶测试的结果基本一致.
 表 1
化合物对PDE-4A的抑制活性结果
Table 1.
Inhibitory activity of the compounds on PDE-4A
表 1
化合物对PDE-4A的抑制活性结果
Table 1.
Inhibitory activity of the compounds on PDE-4A
化合物 酶活性/% 8 μmol/L 0.8 μmol/L 0.08 μmol/L Crisaborole 4.76 13.63 47.24 4a 2.83 6.07 20.58 4b 2.56 5.32 15.31 4c 4.55 12.97 39.94 4d 3.55 11.56 29.77 4e 4.02 12.85 35.62 9f 4.70 13.15 45.53 7g 5.01 15.27 49.34 DMSO 82.69
表 2
合成化合物的分子对接最低能量打分
Table 2.
Molecular docking lowest energy score for synthetic compounds
化合物 ΔG/(kcal·mol-1) 4a -22.4084 4b -21.4423 4c -20.6015 4d -20.9113 4e -20.8225 7g -19.9887 9f -17.8381 Crisaborole -18.0403 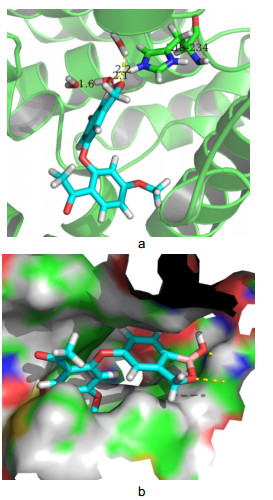 图5
化合物4a与PDE-4A作用位点
Figure5.
Binding site of the compound 4a interact with PDE-4A
图5
化合物4a与PDE-4A作用位点
Figure5.
Binding site of the compound 4a interact with PDE-4A
1.3 抗炎活性研究
为了考察所合成的化合物抗炎活性, 测试了化合物对佛波醇脂诱导的小鼠耳水肿模型的影响, 结果见表 3.
表 3
化合物对佛波醇脂致小鼠耳廓肿胀度及肿胀抑制率的影响(Mean±SD, n=8)a
Table 3.
Effects of compounds on the inhibitory rate of mouse auricular swelling induced by phorbol ester
组别 剂量/(mg·耳-1) 左耳/(10-2 mm) 右耳/(10-2 mm) 肿胀度/(10-2 mm) 抑制率/% 空白 — 32.46±1.17 32.71±1.36 — — 模型 — 32.86±1.79 55.41±7.65 22.55±4.71 — 地塞米松 1 32.81±2.06 42.34±6.65 9.53±3.19△△ 57.74 Crisaborole 1 33.95±2.06 46.68±2.62 12.73±2.42△△ 43.55 4a 1 35.15±1.45 44.53±2.44 9.38±2.46△△* 58.41 4b 1 33.79±1.86 42.88±3.21 9.09±2.16△△* 59.69 4c 1 35.35±1.76 44.98±2.45 9.63±2.98△△* 57.32 4d 1 33.09±1.99 42.55±4.02 9.46±2.79△△* 58.04 4e 1 33.59±1.91 42.99±3.39 9.40±2.68△△* 58.31 9f 1 33.70±1.27 45.20±2.02 11.50±2.73△△ 49.00 7g 1 32.51±2.28 44.64±5.02 12.13±3.19△△ 46.23 a ΔP<0.05, △△P<0.01 vs.模型; *P<0.05, **P<0.01 vs. crisaborole. 从表 3数据可以看出, 与空白组相比, 模型组涂抹佛波醇脂造模后, 肿胀度达到22.55×10-2 mm, 表明造模效果明显.与模型组相比, 阳性药地塞米松组、Crisa-borole组及各化合物组均能极显著降低耳肿胀度(P<0.01).与Crisaborole组比较, 化合物4a~4e均能显著降低耳肿胀度(P<0.05), 作用强于Crisaborole.上述结果与酶测试结果基本一致.
1.4 抑制炎症因子释放
为了考察所合成的化合物抗炎活性, 测试了化合物对LPS诱导BALB/C小鼠炎症因子释放的影响, 结果见表 4.从表 4数据可以看出, 与模型组比较, 各化合物及阳性药地塞米松、Crisaborole能极显著性降低血清中TNF-α的含量(P<0.01).与Crisaborole比较, 化合物4b能极显著降低血清中TNF-α的含量(P<0.01), 4a、4c、4d、4e能显著降低TNF-α的含量(P<0.05), 表明4a~4e作用均强于Crisaborole.
表 4
化合物对LPS诱导BALB/C小鼠炎症因子释放的影响(Mean±SD, n=6)a
Table 4.
Effects of compounds on the release of inflammatory factors in BALB/C mice induced by LPS
组别 剂量(mg·kg-1) TNF-α/(ng·mL-1) 空白组 — 0.07±0.04 模型组 — 3.22±0.40△△ 地塞米松 10 1.22±0.33▲▲ Crisaborole 2 1.67±0.34▲▲ 4a 2 1.22±0.14▲▲* 4b 2 1.18±0.27▲▲** 4c 2 1.32±0.27▲▲* 4d 2 1.28±0.26▲▲* 4e 2 1.25±0.24▲▲* 9f 2 1.47±0.28▲▲ 7g 2 1.53±0.25▲▲ a △P<0.05, △△P<0.01 vs.空白组; ▲P<0.05, ▲▲P<0.01 vs.模型组; *P<0.05, **P<0.01 vs crisaborole. 2 结论
本研究对已上市药物Crisaborole进行结构修饰和改造, 设计并合成了7个未见报道的新型化合物.化合物结构均通过1H NMR、13C NMR和HRMS确证.药效试验显示阳性对照Crisaborole和7个目标化合物对PDE-4A的抑制、炎症因子TNF-α释放含量的降低和小鼠耳水肿胀的治疗有良好的作用, 其中化合物4b显示出了比阳性对照药Crisaborole更为出色的抗炎作用.表明在保留Crisaborole母核结构的基础上进行结构修饰和改造得到的化合物可以取得很好的抗炎活性.为寻找到选择性更高, 活性更强, 副作用更少的治疗炎症药物做铺垫, 为后续研究提供参考.
3 实验部分
3.1 仪器与试剂
DF-101S集热式恒温加热磁力搅拌器、DFY-5/20低温恒温反应浴、DHG-9140恒温干燥箱、CL-A平板搅拌器均为河南巩义市予华仪器有限责任公司产品; CKX31型倒置显微镜(奥林巴斯公司). NBS、BPO、Pd/C、Pd(dppf)Cl2、各反应起始原料、Cs2CO3、DMF-DMA、Br2、I2均购自安耐吉化学有限公司(纯度≥98%); 所用的其他试剂购自国药集团化学试剂有限公司(分析纯).
3.2 实验方法
3.2.1 目标化合物4a~4e的合成
以1-(2-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)-4-甲氧基苯基)乙酮(4a)的合成为例.在100 mL单口瓶中加入(2-溴-5-碘苯基)甲醇(5 g, 0.016 mol)、樟脑磺酸(350 mg, 1.5 mmol), 加入50 mL二氯甲烷溶解.缓慢加入THP-OH (3 mL, 0.032 mol), 搅拌40 min, 反应完全后直接浓缩, 柱层析得2-(2-溴-5-碘苄氧基)四氢吡喃(3 g, 收率47%).
在50 mL单口瓶加入丹皮酚(1 g, 6 mmol), 原料1a (2 g, 5 mmol), 加入20 mL吡啶溶解.再加入碳酸铯(3 g, 9 mmol)、氧化亚铜(572 mg, 4 mmol), 氮气氛围下130 ℃搅拌过夜, 反应完全后用乙酸乙酯萃取, 饱和食盐水洗涤三次, 有机层干燥, 过滤, 浓缩, 柱层析得中间体2a (1.3 g, 收率60%).
在50 mL单口瓶加入中间体2a (1 g, 2.28 mmol)、频哪醇硼酸酯(0.69 g, 2.74 mmol), 加入10 mL二氧六环溶解.再加入乙酸钾(782 mg, 8 mmol)、钯试剂(160 mg, 0.23 mmol), 氮气氛围下100 ℃搅拌2 h, 反应完全后用乙酸乙酯萃取, 饱和食盐水洗涤三次, 有机层干燥, 过滤, 浓缩, 柱层析得中间体3a (1.1 g, 收率98%).
在50 mL单口瓶中加入中间体3a (1.1 g, 2.28 mmol), 加入10 mL乙醇溶解.再加入3 mol/L盐酸, 0 ℃搅拌0.5 h, 缓慢升至室温, 反应3 h.薄层色谱(TLC)检测反应完全, 加入饱和碳酸氢钠调pH至7, 析出白色固体, 抽滤, 水洗三遍烘干得4a.
1-(2-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)-4-甲氧基苯基)乙酮(4a):白色固体, 收率67.0%. m.p. 117.3~119.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.17 (s, 1H), 7.84~7.86 (d, J=4.01 Hz, 1H), 7.76~7.78 (d, J=8.03 Hz, 1H), 7.03 (s, 2H), 6.88~6.90 (s, 1H), 6.53 (s, 1H), 4.96 (s, 2H), 3.77 (s, 3H), 2.48 (s, 3H); 13C NMR (150 Hz, DMSO-d6) δ: 198.67, 167.01, 161.99, 159.86, 159.70, 135.50, 135.34, 126.15, 120.30, 113.57, 113.53, 108.96, 72.78, 58.90, 17.18. HRMS calcd for C16H15BO5 [M+H]+ 299.1085, found 299.1095.
2-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)-5-三氟甲基-吡啶(4b):白色固体, 收率44.8%. m.p. 142.4~144.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.21 (s, 1H), 8.58 (s, 1H), 8.25 (dd, J=8.66, 2.38 Hz, 1H), 7.79 (d, J=8.03 Hz, 1H), 7.24~7.32 (m, 2H), 7.16 (dd, J=7.78, 1.76 Hz, 1H), 4.99 (s, 2H); 13C NMR (150 Hz, DMSO-d6) δ: 168.55, 159.19, 158.41, 148.45, 140.79, 135.12, 127.87, 126.07, 123.67, 117.59, 115.10, 72.77. HRMS calcd for C13H9BF3NO3 [M+H]+ 296.0700, found 296.0702.
2-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)甲基-3, 5, 6-三甲基吡嗪(4c):白色固体, 收率74.5%. m.p. 103.3~104.8 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 7.63 (d, J=8.28 Hz, 1H), 7.08 (s, 1H), 6.99 (dd, J=8.03, 1.76 Hz, 1H), 5.18 (s, 2H), 4.93 (s, 2H), 2.49 (br. s, 3H), 2.46 (d, J=2.51 Hz, 6H); 13C NMR (150 Hz, DMSO-d6) δ: 163.83, 159.41, 153.50, 152.26, 151.61, 148.95, 134.94, 72.75, 72.13, 23.99, 23.73, 22.62. HRMS calcd for C15H17BN2O3 [M+H]+ 285.1405, found 285.1403.
6-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)甲基-烟腈(4d):白色固体, 收率45.6%. m.p. 112.6~115.1 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.04 (d, J=1.00 Hz, 1H), 9.01 (s, 1H), 8.36 (dd, J=8.16, 1.88 Hz, 1H), 7.72 (d, J=8.03 Hz, 1H), 7.64 (d, J=8.03 Hz, 1H), 7.06 (s, 1H), 7.02 (dd, J=8.16, 1.88 Hz, 1H), 5.33 (s, 2 H), 4.92 (s, 2H); 13C NMR (150 Hz, DMSO-d6) δ: 164.21, 163.29, 159.49, 155.23, 143.98, 134.96, 124.44, 120.08, 117.95, 111.13, 109.99, 72.77, 72.68. HRMS calcd for C14H11BN2O3 [M+H]+ 267.0935, found 267.0932.
2-(1-羟基-1, 3-二氢苯并[c][1, 2]氧硼杂环戊熳-5-基氧基)甲基-5-三氟甲基-吡啶(4e):白色固体, 收率28.6%. m.p. 121.4~123.0 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.96~9.04 (m, 2H), 8.27 (dd, J=8.41, 2.13 Hz, 1H), 7.75 (d, J=8.28 Hz, 1H), 7.65 (d, J=8.03 Hz, 1H), 7.08 (d, J=1.76 Hz, 1H), 7.02 (dd, J=8.16, 2.13 Hz, 1H), 5.35 (s, 2H), 4.92 (s, 2H); 13C NMR (150 Hz, DMSO-d6) δ: 164.44, 163.38, 159.51, 149.04, 137.65, 134.97, 127.24, 125.90, 124.57, 117.94, 109.98, 72.77, 72.71. HRMS calcd for C14H11BF3NO3 [M+H]+ 310.0857, found 310.0861.
3.2.2 目标化合物9f的合成
化合物4-羟基苯硼酸频那醇酯(500.0 mg, 2.27 mmol)溶于四氢呋喃(THF) (5 mL), 0 ℃下加入NaH (113.6 mg, 2.84 mmoL), 逐滴加入MOMCl (274.4 mg, 3.41 mmol).缓慢升至室温, 在此温度下反应1 h.反应完成后在0 ℃下, 加水(15 mL)淬灭该反应, 乙酸乙酯萃取(10 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=30:1]得到MOM保护的苯硼酸频那醇酯(360 mg, 60.8%).取2-羟基-4-甲基苯乙酮(8.0 g, 53.3 mmol)溶于CHCl3 (80 mL), -10 ℃下逐滴加入Br2 (2.73 mL, 53.3 mmol)的CHCl3 (20 mL)溶液.在此温度下反应3 h.反应液倒入200 mL水中, 分离有机层.有机相分别用水洗(100 mL×2), 饱和的亚硫酸钠溶液洗(100 mL×3), 无水硫酸钠干燥, 过滤、浓缩并用热的正己烷重结晶(100 mL)得到中间体2f (9.3 g, 76.2%).
取中间体2f (6.0 g, 26.2 mmol)溶于N, N-二甲基甲酰胺二甲基缩醛(DMFDMA) (60 mL), 80 ℃反应1 h.反应液浓缩得到中间体3f (7.4 g, 99.4%).
取中间体3f (7.4 g, 26.0 mmol)溶于CHCl3 (74 mL), 搅拌下加入吡啶(2.27 g, 28.6 mmol)和碘(13.2 g, 52.1 mmol).室温下反应过夜.反应完成后加入饱和的硫代硫酸钠溶液(120 mL), 二氯甲烷(DCM)萃取(80 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=30:1]得到目标化合物中间体4f (5.3 g, 55.8%).
取中间体4f (3.45 g, 9.45 mmol)溶于CCl4 (80 mL), 搅拌下加入N-溴代琥珀酰亚胺(NBS) (1.68 g, 9.45 mmol)和过氧化二苯甲酰(BPO) (343.5 mg, 1.42 mmol). 80 ℃反应过夜. TLC显示反应完成.冷却至室温, 加入H2O (50 mL), DCM萃取(50 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=30:1]得到中间体5f (2.9 g, 69.1%).
取中间体5f (2.9 g, 6.53 mmol)溶于DMF (35 mL), 搅拌下加入AcONa (2.68 g, 32.7 mmol). 80 ℃反应30 min. TLC显示反应完成.冷却至室温, 加入H2O (50 mL), 乙酸乙酯萃取(30 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=10:1]得到中间体6f (2.1 g, 75.9%).
化合物6f (1.0 g, 2.36 mmol)溶于DME (20 mL)和H2O (8 mL), 搅拌下加入4-氰基苯硼酸(347.4 mg, 2.36 mmol), Na2CO3 (751.7 mg, 7.09 mmol)和Pd/C (100 mg, 10% Pd).氮气保护下, 45 ℃反应4 h. TLC显示反应完成.冷却至室温, 过滤, 加入H2O (50 mL), 乙酸乙酯萃取(30 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=8:1]得到中间体7f (520.0 mg, 55.2%).
中间体7f (185.0 mg, 0.46 mmol)溶于dioxane (20 mL), 搅拌下加入B2pin2 (153.4 mg, 0.60 mmol), AcOK (136.8 mg, 1.39 mmol)和Pd(dppf)Cl2 (34.1 mg, 0.046 mmol).氮气保护下, 90 ℃反应过夜. TLC显示反应完成.冷却至室温, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=10:1]得到中间体8f (96.0 mg, 46.4%).
中间体8f (48.0 mg, 0.11 mmol)溶于MeOH (6 mL)和6 mol/L HCl溶液(2 mL). 45 ℃反应1 h.冷却至室温, 过滤, 滤饼水洗(2 mL×3), 烘干得到目标化合物9f (24.0 mg, 73.5%).
4-(1-羟基-8-氧代-3, 8-二氢-1H-[1, 2]氧硼杂环戊熳[3, 4-g]苯并吡喃-7-基)苄腈(9f):白色固体, 收率73.5%. m.p. 153.4~155.1 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.71 (s, 1H), 8.63 (s, 1H), 7.92~7.96 (m, 2H), 7.84~7.88 (m, 2H), 7.75 (s, 1H), 5.17 (s, 2H); 13C NMR (150 Hz, DMSO-d6) δ: 178.24, 162.21, 160.28, 159.05, 140.00, 135.17, 132.80, 132.14, 126.13, 125.36, 121.94, 114.20, 113.53, 72.94. HRMS calcd for C17H10BNO4 [M+H]+304.0776, found 304.0781.
3.2.3 目标化合物7g的合成
取1g (3.5 g, 18.7 mmol)溶于NH3·H2O (175 mL), 0 ℃下加入KI (9.32 g, 56.1 mmoL)和I2 (4.75 g, 18.7 mmol)的水溶液(88 mL).缓慢升至室温, 在此温度下反应20 min.反应完成后0 ℃下用浓盐酸调节pH值至5~6.乙酸乙酯萃取(35 mL×3), 合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=30:1]得到中间体2g (2.6 g, 44.4%).
取2g (4.5 g, 14.4 mmol)和4-乙炔基苯氰(2.01 g, 15.8 mmol)溶于toluene (100 mL), 加入Cs2CO3 (9.37 g, 28.8 mmoL)和(1, 10-菲啰啉)双(三苯基磷)硝酸铜二氯甲烷络合物(1.79 g, 2.16 mmol).氮气保护下, 100 ℃反应过夜.反应完成后过滤、旋干, 粗品用THF (100 mL)和乙酸乙酯(100 mL)打浆得到中间体3g (2.5 g, 55.7%).
取中间体3g (2.32 g, 7.43 mmol)溶于CCl4 (40 mL), 搅拌下加入NBS (1.32 g, 7.43 mmol)和BPO (360.1 mg, 1.49 mmol).氮气保护下80 ℃反应过夜. TLC显示反应完成.冷却至室温, 加入H2O (50 mL), DCM萃取(30 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=50:1]得到4g (1.92 g, 66.1%).
取中间体4g (1.3 g, 3.32 mmol)溶于DMF (40 mL), 搅拌下加入AcONa (1.36 g, 16.6 mmol). 80 ℃反应1.5 h. TLC显示反应完成.冷却至室温, 加入H2O (50 mL), 乙酸乙酯萃取(25 mL×3).合并有机相, 无水硫酸钠干燥, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=50:1)]得到5g (670 mg, 85.0% purity, 46.3% yield).
取中间体5g (300.0 mg, 0.81 mmol)溶于dioxane (10 mL), 搅拌下加入B2pin2 (267.5 mg, 1.05 mmol), AcOK (238.6 mg, 2.43 mmol)和Pd(dppf)Cl2 (59.3 mg, 0.08 mmol).氮气保护下, 100 ℃反应2.5 h.反应完成后冷却至室温, 过滤、浓缩并纯化[SiO2, V(石油醚):V(乙酸乙酯)=15:1]得到6g (280.0 mg, 48.8%).
取中间体6g (280.0 mg, 0.67 mmol)溶于MeOH (6 mL), 搅拌下加入NaOH溶液(2.68 mL, 2.68 mmol), 室温搅拌30 min.逐滴加入3 mol/L HCl溶液, 调节pH值到2~3, 继续室温反应10 min.过滤, 滤饼水洗(3 mL×3), 烘干得到目标化合物7g.
4-(1-羟基-1, 3-二氢-[1, 2]氧硼杂环戊熳[3, 4-f]苯并呋喃-6-基)苄腈(7g):白色固体, 收率81.3%. m.p. 167.5~168.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.25 (s, 1H), 8.11 (d, J=8.28 Hz, 2H), 8.06 (s, 1H), 7.98 (d, J=8.53 Hz, 2H), 7.79 (s, 1H), 7.69 (s, 1H), 5.11 (s, 2H); 13C NMR (150 Hz, DMSO-d6) δ: 159.89, 156.48, 154.66, 136.83, 136.09, 131.52, 128.14, 127.08, 121.79, 113.76, 108.62, 107.48, 72.84. HRMS calcd for C16H10BNO3 [M+H]+276.0827, found 276.0834.
3.3 目标化合物对PDE-4A的抑制作用
使用从Sf9昆虫细胞中使用N-末端GST标记由杆状病毒表达的PDE-4A.在新制备的pH 7.5 Tris缓冲液中加入指定的PDE-4A和1 mmol/L cAMP, 将酶溶液输送到反应孔中, 将化合物在DMSO中超声溶解然后加入至酶溶液中, 在室温下孵育10 min, 将底物溶液倒入反应孔中以引发反应.在室温下孵育1 h, 加入检测示踪剂(AMP2/GMP2AlexaFluor 633 Tracer)和抗体(Transcreener® AMP2/GMP2 Antibody)以终止反应, 并温和混合孵育90 min.在Ex/Em 620/688处测量荧光偏振, 8、0.8、0.08 μmol/L, 测试实验物.酶活性小于50%, 视作具有显著抑制活性.
3.4 分子对接
PDE-4A晶体结构(PDB code: 1XMY)由Protein Data Bank下载, 通过ChemBio3D ultra 14.0和Autodock4.2D中的Ligand模块进行小分子结构预处理.使用Autodock4.2D对接软件进行对接, 口袋盒子以晶体结构中的配体定义, 其对接盒子边长设置为30 Å, 并使用半经验自由能进行评价, 拉马克遗传算法循环为80次, 其他参数保持默认.
3.5 本发明化合物对佛波醇脂诱导的小鼠耳水肿模型的抗炎作用
取雄性ICR小鼠, 体重18~22 g, 适应性饲养7 d, 随机分为12组, 每组8只, 即空白组、模型组、阳性对照地塞米松组、阳性对照Crisaborole组、7个化合物组, 于佛波醇脂(5 μg/20 μL/耳朵)涂抹前30 min及涂抹后15 min, 各组大鼠右耳分别均匀涂抹相应化合物或阳性对照药物1 mg/20 μL/耳朵, 左耳均涂抹溶媒20 μL(丙酮+乙醇), 空白组及模型组两耳均涂抹等剂量的溶媒20 μL(丙酮+乙醇).在造模后6 h, 通过测厚仪测量耳朵肿胀作为炎症指标, 公式计算肿胀度及抑制率[肿胀度=(右耳平均厚度-左耳平均厚度)/右耳平均厚度×100%, 抑制率(%)=(模型组平均肿胀度-给药组平均肿胀度)/模型组平均肿胀度×100%].
3.6 目标化合物对LPS诱导BALB/C小鼠炎症因子释放的影响
取雌性BALB/C小鼠, 体重18~22 g, 适应性饲养7 d, 随机分为空白组、模型组、阳性对照地塞米松10 mg/kg组、阳性对照Crisaborole 2 mg/kg组、7个目标化合物各2 mg/kg组, 每组6只.各组小鼠口服给予相应化合物及对照药物, 空白组、模型组给予相应溶媒.给药0.5 h后除空白组外, 其余各组LPS腹腔注射0.2 mL造模, 2 h采血, 3500 r/min 10 min离心后分装保存, ELISA试剂盒检测血清TNF-α含量.
辅助材料(Supporting Information) 7个目标化合物的1H NMR, 13C NMR和HRMS图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
Du, Y.; Su, Y.; He, J. Ann. Rheum. Dis. 2015, 74, 2070. doi: 10.1136/annrheumdis-2013-204441
-
[2]
Zuo, X. -B.; Sheng, Y. -J.; Hu, S. -J. Rheumatol. Int. 2014, 34, 459. doi: 10.1007/s00296-013-2864-3
-
[3]
Weishaar, R. E.; Cain, M. H.; Bristol, J. A. J. Med. Chem. 1985, 28, 537. doi: 10.1021/jm50001a001
-
[4]
Houslay, M. D.; Sullivan, M.; Bolger, G. B. Adv. Pharmacol. 1998, 44, 225. doi: 10.1016/S1054-3589(08)60128-3
-
[5]
Murthy, V. S.; Mangot, A. G. Indian J. Pharmacol. 2015, 47, 594. doi: 10.4103/0253-7613.169593
-
[6]
Omori, K.; Kotera, J. Circ. Res. 2007, 100, 309. doi: 10.1161/01.RES.0000256354.95791.f1
-
[7]
Chen, S. K.; Zhao, P.; Shao, Y. X. Bioorg. Med. Chem. Lett. 2012, 22, 3261. doi: 10.1016/j.bmcl.2012.03.026
-
[8]
Li, Z.; Cai, Y. -H.; Cheng, Y. -K. J. Chem. Inf. Model. 2013, 53, 972. doi: 10.1021/ci400063s
-
[9]
Zhao, P.; Chen, S. -K.; Cai, Y. -H. Biochim. Biophys. Acta, Biomembr. 2013, 1834, 2089. doi: 10.1016/j.bbapap.2013.07.004
-
[10]
Zhong. J. -S.; Huang, Y. -Y.; Ding, W. -J. Fitoterapia 2013, 91, 159. doi: 10.1016/j.fitote.2013.08.027
-
[11]
Conti, M.; Jin, S. L.; Monaco, L. Endocr. Rev. 1991, 12, 218. doi: 10.1210/edrv-12-3-218
-
[12]
Lugnier, C. Pharmacol. Ther. 2006, 109, 366. doi: 10.1016/j.pharmthera.2005.07.003
-
[13]
Nicholson, C. D.; Challiss, R. A.; Shahid, M. Trends Pharmacol. Sci. 1991, 12, 19. doi: 10.1016/0165-6147(91)90484-A
-
[14]
Nicholson, C. D.; Shahid, M. Pulm. Pharmacol. 1994, 7, 1. http://www.ncbi.nlm.nih.gov/pubmed/8003849
-
[15]
Card, G. L.; England, B. P.; Suzuki, Y. Structure 2004, 12, 2233. doi: 10.1016/j.str.2004.10.004
-
[16]
Cooper, N. Trends Pharmacol. Sci. 1997, 18, 164. doi: 10.1016/S0165-6147(97)90613-1
-
[17]
Parikh, N.; Chakraborti, A. K. Curr. Med. Chem. 2015, 23. http://www.ncbi.nlm.nih.gov/pubmed/26572614
-
[18]
Liu, Y. -N.; Huang, Y. -Y.; Bao, J. -M. Fitoterapia 2014, 94, 177. doi: 10.1016/j.fitote.2014.02.010
-
[19]
Kato, Y.; Kawasaki, M.; Nigo, T. Bioorg. Med. Chem. 2013, 21, 5851. doi: 10.1016/j.bmc.2013.07.007
-
[20]
Gewald, R.; Grunwald, C.; Egerland, U. Bioorg. Med. Chem. Lett. 2013, 44, 4308. http://www.ncbi.nlm.nih.gov/pubmed/23806553/
-
[21]
Elansary, A. K.; Kadry, H. H.; Ahmed, E. M. Med. Chem. Res. 2011, 21, 3327. doi: 10.1007%2Fs00044-011-9846-3
-
[22]
Liao, W.; Tsai, T.; Ho, T. Evidence-Based Complementary Altern. Med. 2016, 2016, 3704647. http://www.ncbi.nlm.nih.gov/pubmed/28101118
-
[23]
Ying, W.; Liu, J.; Zhang, H. Environ. Toxicol. Pharmacol. 2016, 46, 55. doi: 10.1016/j.etap.2016.07.005
-
[1]
-

图 2 罗氟司特与PDE-4的蛋白晶体图
Figure 2 Crystalline crystals of Roflumilast and PDE-4
M: (metal binding pocket), Q: (hydrophobic clamp pocket), S:(solvent filled side pocket)

图式 1 化合物4a~4e的合成路线
Scheme 1 Synthetic route of compounds 4a~4e
Reagents and conditions: (ⅰ) K2CO3, DMF, 80~120 ℃; (ⅱ) B2pin2, Pd(dppf)Cl2, AcOK, 100~105 ℃, 1 h; (ⅲ) NaBH4, HCl, THF/CH3OH, 0 ℃

图式 2 化合物9f合成路线
Scheme 2 Synthetic route of compounds 9f
Reagents and conditions:(ⅰ) Br2, CHCl3, -10 ℃, 3 h; (ⅱ) DMF-DMA, 80 ℃, 1h; (ⅲ) pyridine, I2, CHCl3, r.t.; (ⅳ) NBS, CCl4, 80 ℃; (ⅴ) AcONa, DMF, 80 ℃; (ⅵ) Na2CO3, Pd/C, DMF, H2O, 45~50 ℃, 5 h; (ⅶ) B2pin2, AcOK, Pd(dppf)Cl2, dioxane, 100 ℃; (ⅷ) NaOH, MeOH, 3 mol/L HCl, r.t.

图式 3 化合物7g合成路线
Scheme 3 Synthetic route of compound 7g
Reagents and conditions: (ⅰ) KI, I2, NH3·H2O, r.t.; (ⅱ) Cs2CO3, toluene, 100 ℃; (ⅲ) NBS, BPO, CCl4, 80 ℃; (ⅳ) AcONa, DMF, 80 ℃; (ⅴ) B2pin2, AcOK, Pd(dppf)Cl2, dioxane, 100 ℃; (ⅵ) NaOH, MeOH, 3 mol/L HCl, r.t.

图 5 化合物4a与PDE-4A作用位点
Figure 5 Binding site of the compound 4a interact with PDE-4A
(a) the binding mode of compound 4a with PDE-4; (b) the binding site of compound 4a in PED-4
表 1 化合物对PDE-4A的抑制活性结果
Table 1. Inhibitory activity of the compounds on PDE-4A
化合物 酶活性/% 8 μmol/L 0.8 μmol/L 0.08 μmol/L Crisaborole 4.76 13.63 47.24 4a 2.83 6.07 20.58 4b 2.56 5.32 15.31 4c 4.55 12.97 39.94 4d 3.55 11.56 29.77 4e 4.02 12.85 35.62 9f 4.70 13.15 45.53 7g 5.01 15.27 49.34 DMSO 82.69  下载: 导出CSV
下载: 导出CSV
表 2 合成化合物的分子对接最低能量打分
Table 2. Molecular docking lowest energy score for synthetic compounds
化合物 ΔG/(kcal·mol-1) 4a -22.4084 4b -21.4423 4c -20.6015 4d -20.9113 4e -20.8225 7g -19.9887 9f -17.8381 Crisaborole -18.0403
下载: 导出CSV
表 3 化合物对佛波醇脂致小鼠耳廓肿胀度及肿胀抑制率的影响(Mean±SD, n=8)a
Table 3. Effects of compounds on the inhibitory rate of mouse auricular swelling induced by phorbol ester
组别 剂量/(mg·耳-1) 左耳/(10-2 mm) 右耳/(10-2 mm) 肿胀度/(10-2 mm) 抑制率/% 空白 — 32.46±1.17 32.71±1.36 — — 模型 — 32.86±1.79 55.41±7.65 22.55±4.71 — 地塞米松 1 32.81±2.06 42.34±6.65 9.53±3.19△△ 57.74 Crisaborole 1 33.95±2.06 46.68±2.62 12.73±2.42△△ 43.55 4a 1 35.15±1.45 44.53±2.44 9.38±2.46△△* 58.41 4b 1 33.79±1.86 42.88±3.21 9.09±2.16△△* 59.69 4c 1 35.35±1.76 44.98±2.45 9.63±2.98△△* 57.32 4d 1 33.09±1.99 42.55±4.02 9.46±2.79△△* 58.04 4e 1 33.59±1.91 42.99±3.39 9.40±2.68△△* 58.31 9f 1 33.70±1.27 45.20±2.02 11.50±2.73△△ 49.00 7g 1 32.51±2.28 44.64±5.02 12.13±3.19△△ 46.23 a ΔP<0.05, △△P<0.01 vs.模型; *P<0.05, **P<0.01 vs. crisaborole.
下载: 导出CSV
表 4 化合物对LPS诱导BALB/C小鼠炎症因子释放的影响(Mean±SD, n=6)a
Table 4. Effects of compounds on the release of inflammatory factors in BALB/C mice induced by LPS
组别 剂量(mg·kg-1) TNF-α/(ng·mL-1) 空白组 — 0.07±0.04 模型组 — 3.22±0.40△△ 地塞米松 10 1.22±0.33▲▲ Crisaborole 2 1.67±0.34▲▲ 4a 2 1.22±0.14▲▲* 4b 2 1.18±0.27▲▲** 4c 2 1.32±0.27▲▲* 4d 2 1.28±0.26▲▲* 4e 2 1.25±0.24▲▲* 9f 2 1.47±0.28▲▲ 7g 2 1.53±0.25▲▲ a △P<0.05, △△P<0.01 vs.空白组; ▲P<0.05, ▲▲P<0.01 vs.模型组; *P<0.05, **P<0.01 vs crisaborole.
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 3051
- HTML全文浏览量: 616
 下载:
下载:
