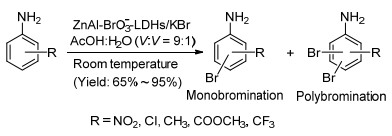
 图Scheme 1
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
Scheme1.
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
图Scheme 1
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
Scheme1.
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
Citation:
Wang Ligeng, Chen Lujun, Zhang Hualong, Yu Qin. Selective Oxidative Bromination of Anilines Using Potassium Bromide and ZnAl-BrO3--LDHs[J]. Chinese Journal of Organic Chemistry,
2017, 37(12): 3186-3190.
doi:
10.6023/cjoc201705017

ZnAl-BrO3--LDHs/KBr溴源在苯胺类化合物选择性氧化溴代的应用
English
Selective Oxidative Bromination of Anilines Using Potassium Bromide and ZnAl-BrO3--LDHs
Abstract:
The oxidative bromination of anilines was realized with ZnAl-BrO3--LDHs/KBr as the bromine source at ambient temperature in acidic medium (AcOH/H2O). High yields of monobrominated or polybrominated anilines were achieved for a wide range of anilines. Both the substrate structure and the reaction conditions will significantly influence the reaction selectivity. The use of inexpensive reagents, environmentally benign and operationally simple process, and excellent yields make it a good alternative to the synthesis of bromoanilines.
-
Key words:
- anilines
- / halogenation
- / selectivity
- / aromatic substitution
-
The bromination of anilines has received wide attention as a kind of fundamental transformation in organic synthesis.[1] Brominated anilines can serve as useful intermediates of agrochemicals and pharmaceuticals.[2, 3] Moreover, many biological reagents, [4] fine chemicals, [5] fire retardants[6] contain bromine functionality. Meanwhile, they are also important substrates in the transition-metal-catalyzed cross-coupling reactions such as Stille, Heck, and Suzuki reactions, [7] which are widely employed for C—C, C—N, and C—O bonds formation protocols.[8] Traditionally, bromoanilines are prepared by the reaction of liquid bromine in the presence of catalyst.[9] However, bromine is a hazardous chemical which is difficult to manipulate due to high vapor pressure and corroding properties.[10] To achieve high efficiency, the brominating reagent could be generated by the oxidation of bromo ion using different kinds of oxidants, such as AlBr3-H2O2-VBrPO, [11] HNO3-KBr-(CH3CO)2, [12] NaIO4-NaBr-H2SO4, [13] and H2O2-HBr[14] etc. Whereas, anilines are prone to be oxidized under these conditions and the selectivity of bromination is a major issue as these reactions often deliver the mixture of mono-, di-, and tri-brominated products. To address this issue, a number of methods have been developed, including the use of Br2-SO2Cl2-zeolite system, [15] DMSO-HX-EtAc (X=Br, I) system, [16] transition-metal-catalyzed bromination with NBS as bromide source, [17~21] and ionic tribromides.[22, 23] Conspicuously, the generation of bromide reagent is usually associated with the use of metal catalysts, and most of the above selective bromide reagents are expensive, potentially hazardous and the preparation process is complex. From a viewpoint of green chemistry, it is highly desirable to develop selective bromination with the use of non-toxic, inexpensive, and easy available brominating reagents. On account of previous reports and connection with our ongoing program, [24, 25] we applied here ZnAl-BrO3--LDHs as an oxidative brominating reagent for the selective aniline bromination reaction, in which BrO3- oxidant was released slowly in a solution of acetic acid. This bromination reaction occured smoothly at ambient temperature to selectively afford mono-, di-, and tri-bromo anilines in a moderate to excellent yields without the need of any additional catalyst (Scheme 1).
图Scheme 1
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
Scheme1.
Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
1 Results and discussion
1.1 Optimization of reaction conditions
Initially, we chose o-nitroaniline as the substrate, and the results are shown in Scheme 2 and Table 1. Under the lower reaction temperature, releasing rate of bromate in ZnAl-BrO3--LDHs was slow and the active molecules were weak. The reaction was completed within 3 h (Table 1, Entry 2). With further elevating the reaction temperature, the yield didn't increase, resulting in a mixture of mono-and di-brominated products (Table 1, Entry 4).
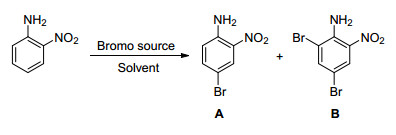 图Scheme 2
Selective bromination of 2-nitroaniline
Scheme2.
Selective bromination of 2-nitroaniline
图Scheme 2
Selective bromination of 2-nitroaniline
Scheme2.
Selective bromination of 2-nitroaniline
 Table 1.
Bromination of 2-nitroanilinea
Table 1.
Bromination of 2-nitroanilinea
Entry Solvent (V:V) Oxidant Molar ratiob Time/h Product (yieldc/%) 1 AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:1:1 1.1 A:B (70:16) 2d AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 3 A (95) 3 AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 1 A (95) 4e AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 0.8 A:B (10:40) 5e AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:1.2:1.2 0.6 B (88.7) 6 CH2Cl2/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 24 — 7 CH3CN/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 10 A (10) 8 EA/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 7 A (15) 9 AcOH/H2O (9:1) NaIO4 1:1.2:1.2 6 A (91.6) 10 AcOH/H2O (9:1) H2O2 1:1.2:1.2 0.8 A:B (29.6:25.7) 11 AcOH/H2O (9:1) KBrO3 1:0.6:0.6 0.5 A:B (9:43) aReaction condition: room temperature unless otherwise stated; bmolar ratio of substrate:oxidant:potassium bromide; c isolated yield by column chromatography; d temperature around 15 ℃; e temperature around 40 ℃. To our delight, with 0.6 equiv. of ZnAl-BrO3--LDHs (1.0×10-3 mol BrO3-/g ZnAl-BrO3--LDHs)[24] and KBr in AcOH/H2O at 25 ℃ for 1 h, monobromination product was obtained as the sole product in 95% yield (Table 1, Entry 3). The bromination of 2-nitroaniline with 1.2 equiv. of brominating reagents in AcOH/H2O as solvent at 40 ℃ afforded B in 88.7% yield (Table 1, Entry 5).
The substrate did not undergo bromination even prolonged the reaction time up to 24 h when CH2Cl2-H2O was used as solvent (Table 1, Entry 6). The advantages of acetic acid as the main solvent was obvious, not only completely miscible with water, but also can be used as a Brnsted-Lowry acid to provide the proton, which improved the oxidizability of BrO3- to activate the reaction. Free hydrogen ion affected the oxidation of bromate directly, giving monobromination compounds with good control over dibromination. Pure acetic acid was used as solvent. However, without water, the release of BrO3- was too slow and not sufficient. Finally, several common oxidizing agents were selected (Table 1, Entries 9, 10, 11). Because of the strong oxidation of hydrogen peroxide and bromate, a mixture of monobromination and dibromination were produced during the reaction. Moreover, due to the higher redox potential under acidic conditions, BrO3- was exposed directly, leading to a mixture of monosubstituted and disubstituted products for the strong oxidized characteristic. Consequently, the optimal conditions of monobromination were identified as: mixed solvent of AcOH/H2O (V:V=9:1), molar ratio for substrate/oxi-dent/bromine source (1:0.6:0.6). The optimal conditions of dibromination were identified as: mixed solvent of AcOH/H2O (V:V=9:1), molar ratio for substrate/oxi-dent/bromine source (1:1.2:1.2).
1.2 Substrate scope of the reaction
To explore the scope of the methodology, we examined a variety of activated aniline derivatives to electrophilic bromination. And the results are shown in Table 2. As can be seen, the activated substrates were converted to mono or poly-bromination in good yields within a short period of time at 25 ℃. We selected the ortho-, meta-, para-position of nitroaniline and chloroaniline to brominate. Contrast with 2-nitroaniline in Table 1, 4-nitroaniline (Table 2, Entries 1, 2) afforded the desired product of monobromination and dibromination under the same reaction conditions. But 3-nitroaniline (Table 2, Entry 3) was formed the corresponding tribromination compounds as the sole products. For the deactivated group halogen substituents (Cl), in addition to the substitution reaction of 3-chloroaniline, which formed the tribromination product directly. We attempted to control bromination by reducing the reaction at 5 ℃, however, the conversion decrease. At the same time, the substrate with COOCH3 was subjected to the bromination (Table 2, Entries 4, 5). Although the yield of products was reduced, it was well converted substrate into the monobrominated or dibrominated products in the similar conditions. The result showed good selectivity to the corresponding parp-substituted product. Moreover, regioselective bromination was observed in the case of aromatics substituted bearing two electron withdrawing groups like 2-chloro-4-nitrophenylamine (Table 2, Entry 15) to give 2-chloro-6-bromo-4-nitroaniline in 95% yield within 0.6 h. The aniline derivatives in the case of electron-donating groups such as OCH3, OH did not work in this method. We hypothesized that Al3+ and Zn2+, which dissociated from ZnAl-BrO3--LDHs, played a catalytic role as Lewis acid.
Table 2.
Bromination of aniline derivativesa
Entry Substrate Time/ h Molar ratiob Product Yieldc/ % 1 
1 1:0.6:0.6 
92 2 
1.2 1:1.2:1.2 
87 3 
1.2 1:1.2:1.2 
66 4 
0.5 1:0.6:0.6 
78 5 
1 1:1.2:1.2 
90 6 
1 1:1.2:1.2 
83 7 
0.6 1:0.6:0.6 
77 8 
1 1:0.6:0.6 
73 9 
2 1:1.2:1.2 
65 10 
1 1:0.6:0.6 
78 11 
1.5 1:1.2:1.2 
88 12 
0.8 1:1.2:1.2 
65 13 
1 1:0.6:0.6 
70 14 
5 1:1.2:1.2 
72 15 
0.6 1:0.6:0.6 
95 16 
2 1:0.6:0.6 
93 aReaction condition: room temperature; bmolar ratio of substrate:oxidant:potassium bromide; cisolated yield by column chromatography. 2 Conclusions
In summary, this work provides a new method for the production of monobrominated or polybrominated anilines in good yield and within short reaction times. The synergistic effects from the ZnAl-BrO3--LDHs can release gradually and AcOH/H2O solvent has enhanced the activity and chemoselectivity for the selective bromination of anilines. This method offers remarkable improvements according to the simplicity reaction condition, easy to operate, shorten the reaction time and satisfactory yield. On top of that, the reagents are environmentally friendly. The complex catalysts and hazardous organic solvents can thus be avoided. Therefore, the process can be considered as 'green' compared to other procedures.
3 Experimental section
3.1 Reagents and instruments
X-4 melting point detector (Beijing Tech Instrument Co., Ltd), thermometer is not corrected; BRUKER 500 MHz, NMR (CDCl3 or DMSO-d6 as solvent, TMS as internal standard); Agilent LC-QTOF-MS high resolution mass spectrometer. All of the substrates were obtained from commercial suppliers and without further purification.
3.2 Experiment methods
3.2.1 Synthesis of ZnAl-BrO3--LDHs
Bromate intercalated ZnAl-layered double hydroxides were synthesized by the coprecipitation method according to the procedures described in literature[24].
3.2.2 Synthesis of target products A and B
The synthesis of compound A was chose as an example. A 50 mL of three-necks-flask equipped with a reflux condenser, a magnetic and a thermoneter. To a stirred solution of AcOH/H2O (V:V=9:1) with substrate (2 mmol) and KBr (1.2 mmol, 0.15 g). The reaction mixture was stirred vigorously when ZnAl-BrO3--LDHs (1.2 g) was added in batches slowly over a period of 20 min, so that the temperature of the mixture did not exceed 40 ℃. After complete addition of the LDHs, it was left at room temperature and continued for another 40 min whilst monitoring its progress by TLC. The undissolved ZnAl-BrO3--LDHs was removed by centrifuge under 5000 r/min. The reaction mixture was extracted with dichloromethane (10 mL×3), the organic layers was separated and washed with sodium sulphite solution and water individually, dried over sodium sulphate anhydrous. The crude product was purified by careful column chromatography on silica gel (200~300 mech) under a mixture of ethyl acetate in petroleum ether.
4-Bromo-2-nitrobenzenanime (A): Yellow solid, 95% yield, m.p. 111~113 ℃ (lit.[26a]: 113~114 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.29 (d, J=2 Hz, 1H), 7.45 (d, J=9, 2 Hz, 1H), 6.73~6.76 (m, 1H), 6.11 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ: 145.29, 138.03, 130.58, 126.96, 121.39, 105.06; HRMS calcd for C6H5BrN2O2 [M+H]+ 216.9613, found 216.9611.
2, 4-Dibromo-6-nitrobenzenamine (B): Orange solid, 89% yield, m.p. 126~128 ℃ (lit.[26b]: 126 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.31 (d, J=2 Hz, 1H), 7.83 (d, J=2 Hz, 1H), 6.65 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ: 142.00, 140.58, 132.15, 127.53, 112.57, 105.08; HRMS calcd for C6H4Br2N2O2[M+H]+ 294.8718, found 294.8715.
3.2.3 Synthesise of target products in Table 2
2-Bromo-4-nitrobenzenamine (1a): Yellow solid, 92% yield, m.p. 104~105 ℃ (lit.[26b]: 104 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.40 (d, J=3 Hz, 1H), 8.05 (d, J=9 Hz, 1H), 6.76 (d, J=9 Hz, 1H), 4.83 (s, 2H).
2, 6-Dibromo-4-nitrobenzenamine (2b): Yellow solid, 87% yield, m.p. 200~202 ℃ (lit.[26b]: 197 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.36 (s, 2H), 5.30 (s, 2H).
2, 4, 6-Tribromo-3-nitrobenzenamine (3c): Yellow solid, 66% yield, m.p. 101~103 ℃ (lit.[26b]: 102 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.71 (s, 1H), 4.93 (s, 2H).
2-Amino-5-bromobenzoic acid methyl ester (4d): Gray solid, 78% yield, m.p. 73~74 ℃ (lit.[26c]: 74 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.98 (d, J=2 Hz, 1H), 7.34 (d, J=9 Hz, 1H), 6.58 (d, J=9 Hz, 1H), 5.77 (s, 2H), 3.89 (s, 3H).
Methyl 2-amino-3, 5-dibromobenzoate (5e): Gray solid, 90% yield, m.p. 88~90 ℃ (lit.[26d]: 90 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.99 (d, J=2 Hz, 1H), 7.70 (d, J=2 Hz, 1H), 6.38 (s, 2H), 3.90 (s, 3H).
2, 6-Dibromo-4-chlorobenzenamine (6f): Gray solid, 83% yield, m.p. 94~95 ℃ (lit.[26b]: 93 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.40 (s, 2H), 4.56 (s, 2H).
2-Bromo-4-chlorobenzenamine (7g): Gray solid, 77% yield, m.p. 65~67 ℃ (lit.[26b]: 69 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.42 (d, J=2 Hz, 1H), 7.09 (d, J=9 Hz, 1H), 6.70 (d, J=9 Hz, 1H), 4.14 (s, 2H).
4-Bromo-3-chlorobenzenamine (8h): Gray solid, 73% yield, m.p. 66~67 ℃ (lit.[26e]: 63~64 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.32 (d, J=9 Hz, 1H), 6.76 (d, J=2 Hz, 1H), 6.61 (d, J=8.5 Hz, 1H), 4.16 (s, 2H).
2, 4, 6-Tribromo-3-chlorobenzenamine (9i): Gray solid, 65% yield, m.p. 123~124 ℃ (lit.[26b]: 123 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.69 (s, 1H), 4.78 (s, 2H).
4-Bromo-2-chlorobenzenamine (10j): Gray solid, 78% yield, m.p. 70~72 ℃ (lit.[26b]: 70 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.39 (d, J=2 Hz, 1H), 7.17 (d, J=9 Hz, 1H), 6.66 (d, J=9 Hz, 1H), 4.05 (s, 2H).
2, 4-Dibromo-6-chlorobenzenamine (11k): Gray solid, 88% yield, m.p. 94~95 ℃ (lit.[26b]: 95 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.48 (d, J=2 Hz, 1H), 7.37 (d, J=2 Hz, 1H), 4.53 (s, 2H).
2, 6-Dibromo-4-methylbenzenamine (12l): Gray solid, 65% yield, m.p. 78~80 ℃ (lit.[26b]: 73~75 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.22 (d, J=1 Hz, 2H), 4.37 (s, 2H), 2.22 (s, 3H).
4-Bromo-3-methylbenzenamine (13m): Gray solid, 70% yield, m.p. 80~81 ℃ (lit.[26f]: 81~82 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.28 (d, J=2 Hz, 1H), 6.59 (d, J=3 Hz, 1H), 6.41 (d, J=8 Hz, 1H), 3.61 (s, 2H), 2.31 (s, 3H).
2, 4-Dibromo-1-naphthalenamine (14n): Rufous solid, 72% yield, m.p. 114~115 ℃ (lit.[26g]: 121~123 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.15~8.22 (m, 1H), 7.81~7.83 (m, 2H), 7.60~7.63 (m, 1H), 7.54~7.57 (m, 1H), 4.66 (s, 2H).
2-Bromo-6-chloro-4-nitrobenzenamine (15o): Yellow solid, 95% yield, m.p. 175~177 ℃ (lit.[26h]: 177~179 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.33 (d, J=3 Hz, 1H), 8.21 (d, J=2 Hz, 1H), 5.25 (s, 2H).
4-Bromo-2-chloro-5-(trifluoromethyl)benzenamine (16p): Brown liquid, 93% yield; 1H NMR (500 MHz, CDCl3) δ: 7.57 (s, 1H), 7.08 (d, J=5 Hz, 1H), 4.27 (s, 2H).
Supporting Information The products of the NMR spectrum. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
-
-
[1]
Wischang, D.; Br€ucher, O.; Hartung, J. Chem. Rev. 2011, 255, 2204.
-
[2]
Albadi, J.; Tajik, H.; Keshavarz, M. Monatsh. Chem. 2012, 144, 179.
-
[3]
Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937. doi: 10.1021/cr00021a014
-
[4]
Roy, S. C.; Guin, C.; Rana, K. K.; Maiti, G. Tetrahedron Lett. 2001, 42, 6941. doi: 10.1016/S0040-4039(01)01412-5
-
[5]
Stavber, S.; Jereb, M.; Zupan, M. Synthesis 2008, 1487. doi: 10.1055/s-2003-38689
-
[6]
Venkateswarlu, K.; Suneel, K.; Das, B.; Reddy, K. N.; Reddy, T. S. Synth. Commun. 2009, 40, 215.
-
[7]
Sammakia, T.; Stangeland, E. L.; Whitcomb, M. C. J. Org. Lett. 2002, 4, 2385. doi: 10.1021/ol026135m
-
[8]
Kalyani, D.; Dick, A. R.; Anani, W. Q. Org. Lett. 2006, 8, 2523. doi: 10.1021/ol060747f
-
[9]
Khan, A. T.; Goswami, P.; Choudhary, L. H. Tetrahedron Lett. 2006, 47, 2751. doi: 10.1016/j.tetlet.2006.02.075
-
[10]
张国富, 王涌, 丁成荣, 刘仁华, 梁鑫淼, 有机化学, 2011, 31, 804.Zhang, G. F.; Wang, Y.; Ding, C. R.; Liu, R. H.; Liang, X. M. Chin. J. Org. Chem. 2011, 31, 804(in Chinese).
-
[11]
Kikushima, K.; Moriuchi, T.; Hirao, T. Tetrahedron Lett. 2010, 41, 340. https://www.ajol.info/index.php/sajc/article/view/123725
-
[12]
Tsoukala, A.; Liguori, L.; Occhipinti, G. Tetrahedron Lett. 2009, 50, 831. doi: 10.1016/j.tetlet.2008.12.016
-
[13]
Kumar, L.; Mahajan, T.; Agarwal, D. D. Ind. Eng. Chem. Res. 2012, 51, 11593. doi: 10.1021/ie202851k
-
[14]
Podgorsek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009. 65, 4429. doi: 10.1002/chin.200938040
-
[15]
Gnaim, J. M.; Sheldon, R. A. Tetrahedron Lett. 2005, 46, 4465. doi: 10.1016/j.tetlet.2005.04.116
-
[16]
Song, S.; Sun, X.; Li, X. W.; Yuan, Y. Z.; Jiao, N. Org. Lett. 2015, 17, 2886. doi: 10.1021/acs.orglett.5b00932
-
[17]
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e
-
[18]
Neufeldt, S. R.; Sanford, M. S. Chem. Res. 2012, 45, 936. doi: 10.1021/ar300014f
-
[19]
Schroder, N.; Wencel-Delord, J.; Glorius, F. J. Am. Chem. Soc. 2012, 134, 8298. doi: 10.1021/ja302631j
-
[20]
Du, Z. J.; Gao, L. X.; Lin, Y. J.; Han, F. S. ChemCatChem 2014, 6, 123. doi: 10.1002/cctc.v6.1
-
[21]
Mo, F.; Yan, J. M.; Qiu, D.; Li, F.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 2028. doi: 10.1002/anie.200906699
-
[22]
Bliman, D.; Pettersson, M.; Bood, M.; Grøtli, M. Tetrahedron Lett. 2014, 55, 2929. doi: 10.1016/j.tetlet.2014.03.084
-
[23]
Brimmer, A. F.; Dahl, F. R. J. Cheminf. 2015, 71, 6112. https://e-archivo.uc3m.es/handle/10016/5804
-
[24]
王力耕, 郑飞翔, 蒋晨星, 倪哲明, 硅酸盐学报, 2015, 43, 672.Wang, L. G.; Zheng, F. X.; Jiang, C. X.; Ni, Z. M. J. Chin. Ceram. Soc. 2015, 43, 672(in Chinese).
-
[25]
Ghiaci, M.; Sedaghat, M. E.; Ranjbari, S. Appl. Catal. A 2010, 384, 18. doi: 10.1016/j.apcata.2010.05.053
-
[26]
(a) Chrétien, J. M. ; Zammattio, F. ; Le Grognec, E. ; Paris, M. ; Cahingt, B. ; Montavon, G. ; Quintard, J. P. J. Org. Chem. 2005, 70, 2870.
(b) Bagmanov, B. T. Russ. J. Appl. Chem. 2009, 82, 1570.
(c) Parthasarathy, M. ; Gopalakrishnan, R. Spectrochim. Acta, Part A 2012, 97, 1152.
(d) Gopal, N. ; Saravanan, V. S. ; Jagadeeswaran, M. Asian J. Chem. 2006, 18, 2611.
(e) Choy, J. ; Jaime-Figueroa, S. ; Jiang, L. ; Wagner, P. Synth. Commun. 2007, 40, 3840.
(f) Trunz, B. B. ; Jedrysiak, R. ; Tweats, D. ; Brun, R. ; Kaiser, M. ; Suwinski, J. ; Torreele, E. Eur. J. Med. Chem. 2011, 46, 1524.
(g) Mandadapu, A. K. ; Saifuddin, M. ; Agarwal, P. K. ; Kundu, B. Org. Biomol. Chem. 2009, 7, 2796.
(h) Glidewel, C. ; Low, J. N. ; Skakle, J. M. ; Wardell, J. L. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2005, C61, 336.
-
[1]
-

Scheme 1 Aniline bromination using ZnAl-BrO3--LDHs/KBr as oxidative brominating reagents
Table 1. Bromination of 2-nitroanilinea
Entry Solvent (V:V) Oxidant Molar ratiob Time/h Product (yieldc/%) 1 AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:1:1 1.1 A:B (70:16) 2d AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 3 A (95) 3 AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 1 A (95) 4e AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 0.8 A:B (10:40) 5e AcOH/H2O (9:1) ZnAl-BrO3--LDHs 1:1.2:1.2 0.6 B (88.7) 6 CH2Cl2/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 24 — 7 CH3CN/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 10 A (10) 8 EA/H2O (9:1) ZnAl-BrO3--LDHs 1:0.6:0.6 7 A (15) 9 AcOH/H2O (9:1) NaIO4 1:1.2:1.2 6 A (91.6) 10 AcOH/H2O (9:1) H2O2 1:1.2:1.2 0.8 A:B (29.6:25.7) 11 AcOH/H2O (9:1) KBrO3 1:0.6:0.6 0.5 A:B (9:43) aReaction condition: room temperature unless otherwise stated; bmolar ratio of substrate:oxidant:potassium bromide; c isolated yield by column chromatography; d temperature around 15 ℃; e temperature around 40 ℃.  下载: 导出CSV
下载: 导出CSV
Table 2. Bromination of aniline derivativesa
Entry Substrate Time/ h Molar ratiob Product Yieldc/ % 1 1 1:0.6:0.6 92 2 1.2 1:1.2:1.2 87 3 1.2 1:1.2:1.2 66 4 0.5 1:0.6:0.6 78 5 1 1:1.2:1.2 90 6 1 1:1.2:1.2 83 7 0.6 1:0.6:0.6 77 8 1 1:0.6:0.6 73 9 2 1:1.2:1.2 65 10 1 1:0.6:0.6 78 11 1.5 1:1.2:1.2 88 12 0.8 1:1.2:1.2 65 13 1 1:0.6:0.6 70 14 5 1:1.2:1.2 72 15 0.6 1:0.6:0.6 95 16 2 1:0.6:0.6 93 aReaction condition: room temperature; bmolar ratio of substrate:oxidant:potassium bromide; cisolated yield by column chromatography.
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 10
- 文章访问数: 2924
- HTML全文浏览量: 317
 下载:
下载:
