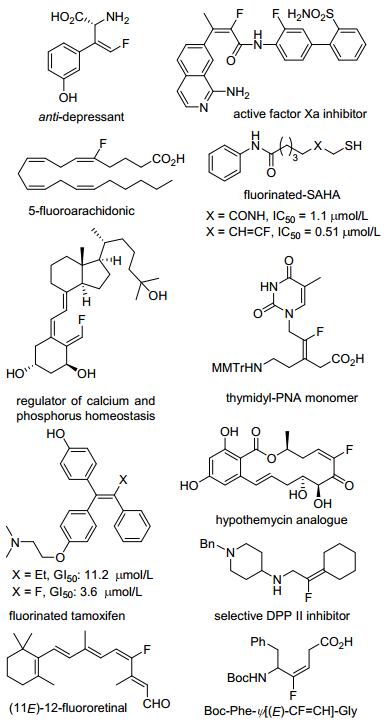
 图1
代表性的含三取代或者四取代单氟烯烃结构单元的生物活性分子
Figure1.
Representative bioactive molecules that containing tri-or tetra-substituted monofluoroalkenes structural motif.
图1
代表性的含三取代或者四取代单氟烯烃结构单元的生物活性分子
Figure1.
Representative bioactive molecules that containing tri-or tetra-substituted monofluoroalkenes structural motif.
引用本文:
廖富民, 余金生, 周剑. 三或四取代单氟烯烃的高立体选择性合成研究进展[J]. 有机化学,
2017, 37(9): 2175-2186.
doi:
10.6023/cjoc201705001

Citation: Liao Fumin, Yu Jinsheng, Zhou Jian. Recent Advances in the Highly Stereoselective Synthesis of Tri-or Tetra-substituted Monofluoroalkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2175-2186. doi: 10.6023/cjoc201705001
Citation: Liao Fumin, Yu Jinsheng, Zhou Jian. Recent Advances in the Highly Stereoselective Synthesis of Tri-or Tetra-substituted Monofluoroalkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2175-2186. doi: 10.6023/cjoc201705001
三或四取代单氟烯烃的高立体选择性合成研究进展
English
Recent Advances in the Highly Stereoselective Synthesis of Tri-or Tetra-substituted Monofluoroalkenes
Abstract:
Monofluoroalkenes have found applications in many areas of research, including the design and development new materials and drug. The highly stereoselective synthesis of this privileged structural motif has attracted great synthetic attention. This review summarizes recent progresses in highly stereoselective synthesis of monofluoroalkenes from aldehydes, ketones and diazo compounds, other substrates such as alkynes, alkenyl metallic species and alkenes are also included. The advantages and disadvantages of different methods are discussed.
-
Key words:
- monofluoroalkene
- / stereoselective synthesis
- / aldehyde and ketone
- / diazo compound
-
在有机化合物中选择性引入氟原子, 不仅可以显著改变母体化合物的物理化学性质, 还能够改善其药学活性[1].例如, 增强代谢稳定性、脂溶性、膜通透性、生物可利用性以及结合亲和力等.因此, 含氟化合物在医药、农药以及材料科学等领域中已经得到了广泛的应用[2].例如, 统计表明市面上大约30%~40%的农药分子以及20%~25%的医药分子中含有氟原子[3].
单氟烯烃是一类非常重要的含氟化合物, 不仅被广泛应用于新材料的开发, 而且还可作为生物肽键的电子等排体用于改善生物活性分子的性质.一些具有三取代或四取代含氟烯烃结构单元的生物活性分子如图 1所示.研究数据也显示, 含氟烯烃的引入, 对于改善生物活性具有明显的效果.例如, 辛二酰苯胺异羟肟酸(SA-HA)是一种组蛋白去乙酰化酶抑制剂, 当其结构中的一个酰胺基团换成其电子等排体单氟烯烃时, 其对组蛋白去乙酰化酶的抑制活性增加了一倍[4a].他莫昔芬(Tamoxifen)是一种用于治疗晚期乳腺癌的一线药物, 化学家们对其结构改造时发现当他莫昔芬中分子中的乙基被氟原子所取代时, 其对雌激素依赖性乳腺癌细胞的活性增加了三倍[4b].其次, 含氟烯烃还是一类重要的合成砌块, 能进行多种后续转化来提高结构多样性或合成其他含氟化合物[4c~4f].因此, 单氟烯烃的高选择性合成受到了合成化学家的广泛关注.经过多年研究, 目前已发展了一系列不同合成方法.根据反应底物类型, 这些方法可分为以下几种主要类型: (1) 基于醛、酮等羰基化合物的转化, 如Wittig反应、Horner-Wadsworth-Emmons (HWE)反应和Julia-Kocienski反应等; (2) 重氮类化合物的偶联反应; (3) 基于含氟化合物的转化[5], 如偕二氟烯烃和3, 3-偕二氟丙烯类化合物C—F键的断裂等反应; (4) 基于炔烃、联烯以及烯烃及其衍生物的氟化反应[6].
2011年, Taquchi和Paquin等分别综述了合成单氟代烯烃的合成方法[7b, 7c].尽管陆续还有几篇综述涉及到单氟烯烃的一些合成方法[7], 但均未包括高立体选择性地合成三取代和四取代单氟烯烃的最新进展.由于三取代或四取代非对称含氟烯烃的碳碳双键周围的空间位阻更大, 导致选择性合成的难度更高, 因此本综述拟重点讨论从醛、酮以及重氮出发来高立体选择性合成三、四取代非对称单氟烯烃, 同时也简要介绍炔烃、烯基衍生物以及烯烃氟化反应的最新研究进展.
图1
代表性的含三取代或者四取代单氟烯烃结构单元的生物活性分子
Figure1.
Representative bioactive molecules that containing tri-or tetra-substituted monofluoroalkenes structural motif.
1 基于醛或酮的合成方法
1.1 Wittig反应
氟代磷叶立德参与的Wittig反应为一步合成单氟烯烃提供了一个较好的方法.早在1969年, Schlosser和Zimmermann等[8]利用perchloryl fluoride试剂首次合成了氟亚甲基叶立德Ph3P=CHF, 并与醛酮反应, 以中等的产率合成了一系列二、三取代的单氟烯烃, 但反应的Z/E选择性很差.随后, Burton等[9]利用CFCl3为原料合成了稳定的nBu3PCFPnBu3, 并实现了其与醛的Wittig反应.产物的Z/E选择性受底物取代基的影响很大, 芳香醛主要得到(E)-单氟烯烃, 而脂肪醛在相同的条件下主要得到(Z)-单氟烯烃.
2004年, Pannecoucke等[10]发现CFBr3与PPh3原位在Et2Zn的作用下可形成氟溴亚甲基叶立德, 进而与醛或酮发生Wittig反应, 以良好到优秀的产率以及中等的Z:E选择性合成了一系列三、四取代氟溴取代的烯烃2 (Scheme 1).随后, 他们进一步发现单氟二溴乙酸酯4在相同条件下, 也能以中等的产率和Z/E选择性得到相应的(Z)-式α-氟代-α, β-不饱和酯5(Scheme 1).
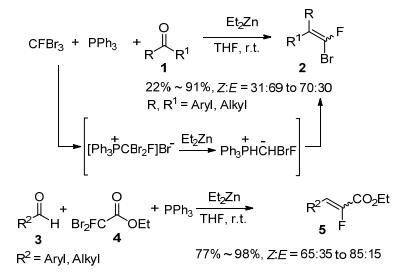 图式1
Et2Zn促进的Wittig反应
Scheme1.
Et2Zn mediated Wittig reactions
图式1
Et2Zn促进的Wittig反应
Scheme1.
Et2Zn mediated Wittig reactions
1.2 Horner-Wadsworth-Emmons (HWE)反应
尽管Wittig反应能够合成单氟烯烃, 但文献报道的例子普遍立体选择性不高, 而且氟代叶立德通常较难合成[11].因此, 自1964年Machleidt等[12]首次利用HWE反应实现了α-氟代-α, β-不饱和酯类单氟代烯烃的合成以来, 利用α-氟代膦酸酯参与的HWE反应来高效高立体选择性合成单氟烯烃也得到了研究.如1985年, Moghadam课题组[13]报道了α-氟代膦酸酯6与醛的HWE反应.作者发现以四氢呋喃作为溶剂的条件下, 该反应在低温时能高选择性地形成(E)-α-氟代-α, β-不饱和酯7 (Eq. 1).
酮类化合物与α-氟代膦酸酯的HWE反应可以合成四取代单氟烯烃, 相对于与醛的反应则研究较少. 2014年, Sano等[14]发现在正丁基锂作碱的条件下, 环状酮类化合物可以与α-氟代膦酸酯6顺利发生HWE反应, 以高达95:5的选择性合成了(E)-四取代单氟烯烃8 (Scheme 2).利用该方法为关键步骤, 作者还实现了二肽等排体9的全合成.
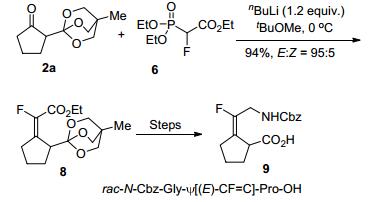 图式2
酮与6的HWE反应
Scheme2.
HWE reactions of ketone and 6
图式2
酮与6的HWE反应
Scheme2.
HWE reactions of ketone and 6
1.3 Julia-Kocienski反应
上述HWE反应虽然能高立体选择性合成三、四取代单氟烯烃, 但反应一般条件较为苛刻, 需要低温(如-78 ℃)与强碱(如nBuLi)等条件.为了开发条件更为温和的高效合成方法, 化学家们进一步研究了α-氟代砜类化合物与醛酮的Julia类型反应.
2003年, Leuqeux和Pazenok等[15]合成了苯并噻唑衍生的α-氟-α-甲基砜类化合物11a.他们尝试了在tBuOK作碱的条件下, 11a与醛的Julia类烯烃化反应(Scheme 3).虽然选择性不高, 但利用这一方法合成了除虫菊酸酯含氟类似物12a, 并发现其表现出了比非氟代菊酸酯更好的杀虫活性.
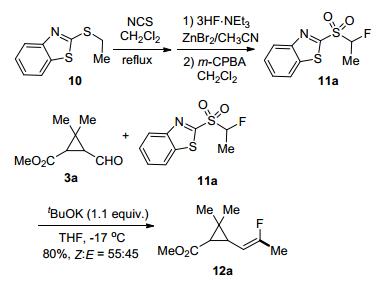 图式3
醛与11a的Julia类烯烃化反应
Scheme3.
Modified Julia olefination reaction of aldehydes and 11a
图式3
醛与11a的Julia类烯烃化反应
Scheme3.
Modified Julia olefination reaction of aldehydes and 11a
2006年, Zajc课题组[16]改进了α-氟-α-(杂)芳基砜类化合物的合成方法, 从卤代烷和钠盐13出发, 经过烷基化和氟化两步方便地合成了一系列Julia类烯烃化试剂11.其与醛酮的Julia类烯烃化反应, 能以中等到优秀的产率获得三、四取代单氟烯烃.他们还发现产物的Z/E选择性受底物以及反应条件的变化影响较大(Scheme 4).
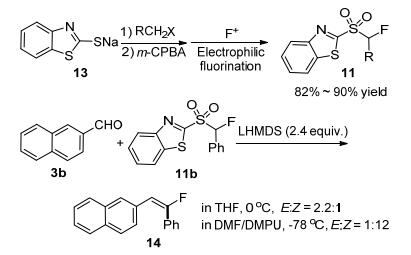 图式4
11b与醛、酮的Julia类烯烃化反应
Scheme4.
Modified Julia olefination reaction of 11b with aldehydes and ketones
图式4
11b与醛、酮的Julia类烯烃化反应
Scheme4.
Modified Julia olefination reaction of 11b with aldehydes and ketones
他们还利用DBU作碱, 实现了α-氟-α-酯基砜类化合物11c与醛的Julia类烯烃化反应, 高效地得到(E)为主的α-氟代-α, β-不饱和酯类化合物7d[17a].随后, Lequeux课题组[17b]也研究了该反应, 并发现通过添加当量的MgBr2, 可以高选择性得到(Z)为主的产物5a (Scheme 5).
 图式5
利用Julia类烯烃化反应来合成α-氟代-α, β-不饱和酯
Scheme5.
Synthesis of α-fluoro-α, β-unsaturated esters via mo-dified Julia olefination reaction
图式5
利用Julia类烯烃化反应来合成α-氟代-α, β-不饱和酯
Scheme5.
Synthesis of α-fluoro-α, β-unsaturated esters via mo-dified Julia olefination reaction
Zajc等还进一步成功实现了醛与α-氟-α-酰胺砜11e[18a]以及双砜化合物11f[18b]的Julia类烯烃化反应, 以较高选择性合成了α-氟代-α, β-不饱和酰胺15以及α-氟代-α, β-不饱和砜类化合物16 (Scheme 6).
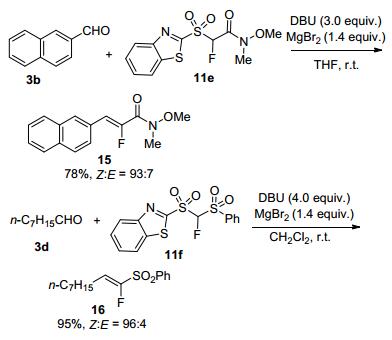 图式6
MgBr2促进的Julia类烯烃化反应
Scheme6.
MgBr2 mediated Modified Julia olefination reaction
图式6
MgBr2促进的Julia类烯烃化反应
Scheme6.
MgBr2 mediated Modified Julia olefination reaction
由于苯并噻唑衍生的α-氟代砜类化合物的α位氢的pKa值较高, 反应一般需较强的碱来攫取.因此, 设计开发活性更高的α-氟代砜类化合物来实现在更温和条件下高选择性合成单氟烯烃成为新的目标. 2008年Nájera和Alonso等[19]合成出了3, 5-二三氟甲基苯基取代的α-氟代砜类化合物17a.由于这类试剂的α位氢酸性更强, 因此在室温下, 使用K2CO3作为碱, 就能实现其与芳基
醛的高选择性Julia-Kocienski反应, 得到(Z)-式α-氟代-α, β-不饱和酯5 (Scheme 7).但其与脂肪醛反应的选择性不太理想.为解决这一问题, 作者将化合物17a的酯基换成酰胺基团, 所得化合物17b可在相同条件下, 与芳基和脂肪醛都能发生高选择性Julia-Kocienski反应, 获得单一的(Z)-α-氟代-α, β-不饱和酰胺类化合物15 (Scheme 7).作者通过改变砜基上取代基来调控α-氟代砜类化合物上α位氢的酸性的思路, 为后续发展更为温和、高选择性合成单氟烯烃的方法提供了一种有效的借鉴.
在上述Julia-Kocienski反应中, 芳香醛类底物的反应选择性通常较好, 而脂肪醛类底物则往往不太理想.
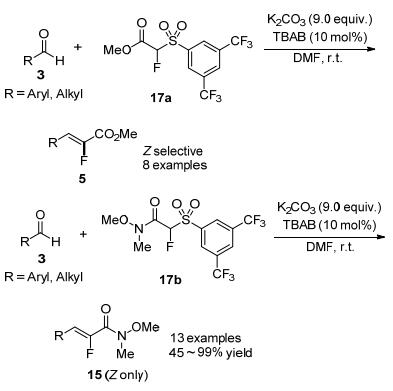 图式7
17a~17b的Julia-Kocienski类烯烃化反应
Scheme7.
Julia-Kocienski olefination reaction of 17a~17b
图式7
17a~17b的Julia-Kocienski类烯烃化反应
Scheme7.
Julia-Kocienski olefination reaction of 17a~17b
为解决这一难题, 2014年Lequeux小组[20]设计合成了嘧啶衍生的α-氟代砜类化合物18, 并研究了其与醛类化合物的反应.作者发现, 相比于苯并噻唑衍生的α-氟代砜类化合物11d, 化合物18与醛的反应在相同的条件下能取得更好的Z/E选择性(Scheme 8).
 图式8
18与醛的Julia类烯烃化反应
Scheme8.
Modified Julia olefination reaction of 18 and aldehydes
图式8
18与醛的Julia类烯烃化反应
Scheme8.
Modified Julia olefination reaction of 18 and aldehydes
2015年, 胡金波小组[21]设计合成了吡啶衍生的α-氟代砜类化合物19, 并发现其与醛的反应中间体可自发拆分, 从而通过改变反应条件巧妙地高效获得(Z)-或(E)-单氟烯烃.作者认为该反应经过以下反应历程:首先19在LiHMDS作用下形成碳负离子, 随后与醛发生加成反应, 再经过Smiles重排分别得到中间体Ⅰ和Ⅱ.中间体Ⅰ和Ⅱ均可以溶于水, 但这两个中间体在水相中的稳定性不同; 其中, (Z)中间体Ⅰ可在水相中快速形成(Z)产物, 因此可以通过乙醚萃取得到(Z)产物14-Z; 而留在水相中的中间体Ⅱ则可进一步在p-TsOH作用下形成相应的(E)产物14-E, 并最终通过萃取得到(Scheme 9).利用该方法, 作者还合成并分离了具有潜在抗癌试剂Combretastatin A-4的氟代类似物20.
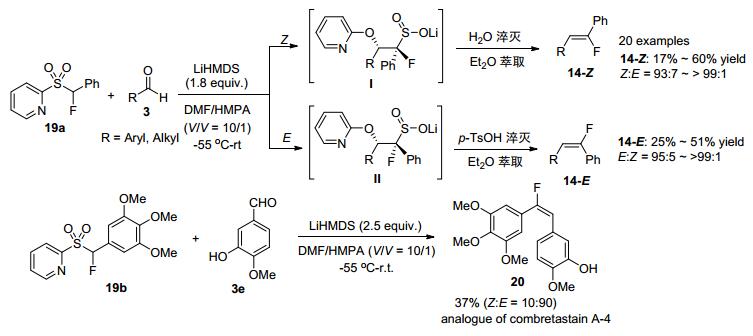 图式9
Julia-Kocienski反应中间体的自发拆分
Scheme9.
Spontaneous resolution of Julia-Kocienski intermediates
图式9
Julia-Kocienski反应中间体的自发拆分
Scheme9.
Spontaneous resolution of Julia-Kocienski intermediates
除α-氟代砜类化合物外, α-氟代亚砜、α-氟代硫化物以及α-氟代亚砜亚胺等化合物也被用于Julia反应制备单氟烯烃.例如1992年, Yamakawa等[22]合成了α-氟代亚砜类化合物21, 并利用LDA作碱, 实现了其与醛的烯烃化反应, 通过三步反应单一地合成了(Z)-α-氟代-α, β-不饱和酮类化合物23 (Scheme 9).
 图式10
α-氟代-α, β-不饱和酮的合成
Scheme10.
Synthesis of α-fluoro-α, β-Unsaturated Ketones
图式10
α-氟代-α, β-不饱和酮的合成
Scheme10.
Synthesis of α-fluoro-α, β-Unsaturated Ketones
Lequeux和Pommelet等[23]则发展了从α-氟代硫化物出发来合成单氟烯烃的方法. α-氟代硫化物24经LDA攫氢后与醛发生aldol反应形成Ⅰ, 随后利用m-CPBA将硫醚氧化成亚砜Ⅱ, 最后经过Durst反应高立体选择性地得到(Z)-式α-氟代-α, β-不饱和酯类化合物5 (Scheme 11).
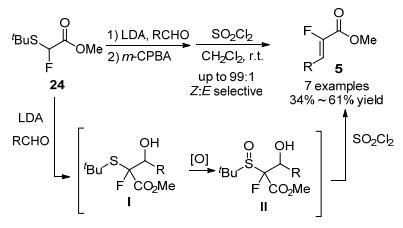 图式11
氟代硫化物与醛的烯烃化反应
Scheme11.
Olefin reactions from sulfides and aldehydes
图式11
氟代硫化物与醛的烯烃化反应
Scheme11.
Olefin reactions from sulfides and aldehydes
自从1988年Finch等[24]开发并应用α-氟代亚砜亚胺于单氟烯烃的合成以来, 利用这一新型氟代试剂来构建单氟烯烃也逐渐受到化学家们的关注.例如2009年, 胡金波小组[25a, 25b]首先改进了该类化合物的合成方法, 合成了一系列N-Ts保护的α-氟-α-芳基亚砜亚胺, 并应用于和硝酮的反应中, 高选择性地合成了一系列三取代的单氟烯烃.最近, 他们还设计开发了N-TBS保护的α-氟甲基吡啶亚砜亚胺25, 并成功应用于醛或酮的烯烃化反应, 为(E)-三取代单氟烯烃26的高选择性合成提供了一个普适性很好的方法(Scheme 12)[25c].利用这一方法, 作者还尝试了牛血浆胺氧化酶(BPAO)潜在抑制剂MDL 72161的合成以及其它一些复杂分子的后期氟化修饰中.
 图式12
氟代亚砜亚胺与酮的烯烃化反应
Scheme12.
Olefin reactions via fluorinated sulfoximine and ketones
图式12
氟代亚砜亚胺与酮的烯烃化反应
Scheme12.
Olefin reactions via fluorinated sulfoximine and ketones
1.4 其他类型的反应
除了上述Wittig, HWE以及Julia类反应外, 化学家们还发展了其它类型的基于醛或酮的反应来合成三或四取代的单氟烯烃.例如1990年, Welch课题组[26]报道了α-氟-α-硅基酯29与醛酮的Peterson类烯烃化反应来合成α-氟代-α, β-不饱和酯类化合物5.作者通过增大酯基的位阻, 同时在超低温的条件下, 实现了芳香醛底物的Peterson类烯烃化反应; 但脂肪醛和酮类底物在该条件下选择性仍然很差(Scheme 13).
2008年, Mukaiyama等[27]发现在5 mol%的nBu4N-OAc作为Lewis碱促进剂的作用下, 氟代烯酮硅基缩醛30与醛在室温下能很好地发生Peterson类烯烃化反应,以中等到优秀的产率和优秀的立体选择性得到(Z)-α-氟代-α, β-不饱和酯5 (Eq. 2).该反应的底物普适性较广, 对于芳基、烷基以及烯丙基取代的醛类化合物均适用, 且选择性很好.
 图式13
利用Peterson烯烃化反应来合成氟代烯烃
Scheme13.
Synthesis of fluoro-olefin via Peterson olefinations
图式13
利用Peterson烯烃化反应来合成氟代烯烃
Scheme13.
Synthesis of fluoro-olefin via Peterson olefinations
上述方法使用的含氟前体均需预先制备, 而且部分试剂的合成较为不易.因此, 发展从简单易得的含氟砌块出发来合成单氟烯烃的方法是一个重要的研究方向. 2003年, Falck和Mioskowski等[28a]利用单氟二溴乙酸乙酯4, 在4.5 equiv. CrCl2的作用下, 实现了其与醛的Reformatsky类反应(Scheme 14).无论脂肪醛还是芳香醛, 都能在室温下很好地反应, 以优秀的选择性获得(Z)-α-氟代-α, β-不饱和酯5.随后他们进一步使用更为环境友好的Fe粉替代CrCl2, 则实现了单氟二氯乙酸乙酯31与苯甲醛的Reformatsky类反应(Scheme 14)[28b].由于Fe粉活性较低, 因此反应需使用10 equiv.的Fe粉并在较高温度下才能顺利发生, 仍然以优秀的(Z)选择性得到α-氟代-α, β-不饱和酯5q.
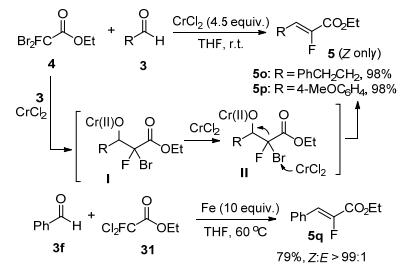 图式14
Cr(Ⅱ)和Fe(0) 促进的类Reformatsky反应
Scheme14.
Cr(Ⅱ) and Fe(0) mediated Reformatsky-type reactions
图式14
Cr(Ⅱ)和Fe(0) 促进的类Reformatsky反应
Scheme14.
Cr(Ⅱ) and Fe(0) mediated Reformatsky-type reactions
随后, Jubault等[29]发展了Et2Zn促进的单氟二溴乙酸乙酯4与醛和酮的Reformatsky类反应(Scheme 15).该方法不仅适用于各种醛类化合物, 也适用于某些酮类化合物.当反应以醛为底物时, 能以中等的产率和优秀的选择性得到(Z)-α-氟代-α, β-不饱和酯5和顺式α-溴-α-氟-β-羟基酯32; 而以酮为底物时, 反应则只得到四取代单氟烯烃, 但产物的选择性受酮上取代基的影响比较明显.
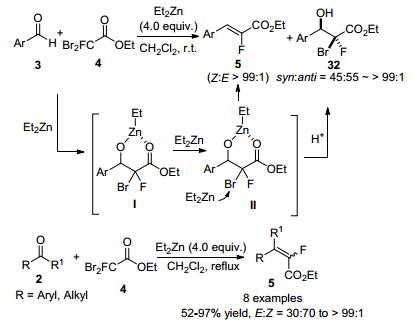 图式15
Et2Zn促进的二溴氟乙酸乙酯与醛的类Reformatsky反应
Scheme15.
Et2Zn mediated Reformatsky-type reactions of Ethyl dibromofluoroacetate and aldehydes
图式15
Et2Zn促进的二溴氟乙酸乙酯与醛的类Reformatsky反应
Scheme15.
Et2Zn mediated Reformatsky-type reactions of Ethyl dibromofluoroacetate and aldehydes
2013年, Augustine小组[30]还发现α-氟代乙酸酯33在当量的TiCl4和三乙胺的作用下, 能与醛发生烯烃化反应(Scheme 16).该反应不仅可以高选择性合成(Z)-α-氟代-α, β-不饱和酯5, 而且具有很好的底物普适性:芳香醛、烯丙基醛以及大位阻的脂肪醛均适用.
 图式16
TiCl4促进的单氟乙酸乙酯与醛的烯烃化反应
Scheme16.
TiCl4 mediated olefin reactions of ethyl monofluoroacetate and aldehydes
图式16
TiCl4促进的单氟乙酸乙酯与醛的烯烃化反应
Scheme16.
TiCl4 mediated olefin reactions of ethyl monofluoroacetate and aldehydes
α-氟代砜基化合物除了作为亲核试剂通过Julia类反应来构建单氟烯烃之外, 2014年刘金涛小组[31]还利用其作为亲电试剂来制备单氟烯烃.他们发现, 具有α氢的羰基化合物34在LDA作用下形成烯醇负离子, 再与α-三取代的α-氟代砜基化合物11g中的酮羰基发生加成反应得到中间体Ⅰ, 然后Ⅰ中的氧负离子对苯并噻唑的亚胺发生加成反应形成螺缩酮中间体Ⅱ, 最后发生消除反应即可得到目标产物.通过加入HMPA作为添加剂, 该反应能以中等到良好的产率以及单一的(E)选择性得到相应的四取代单氟烯烃12 (Scheme 17).
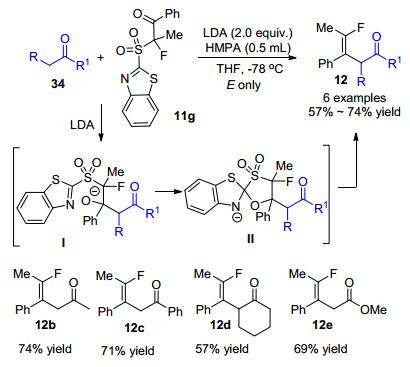 图式17
利用11g来合成四取代氟代烯烃
Scheme17.
Synthesis of tetrasubstituted fluoroalkenes via 11g
图式17
利用11g来合成四取代氟代烯烃
Scheme17.
Synthesis of tetrasubstituted fluoroalkenes via 11g
2 基于重氮的单氟烯烃化反应
重氮化合物参与的烯基化反应, 是通过汇聚式途径构建碳碳双键的重要方法[32].因此, 发展重氮与各种氟代试剂反应制备单氟烯烃的方法也受到关注, 并实现了一些成功的例子, 显示了这一方法的潜在应用价值.早在1992年, 受Fuchigami小组[33a]利用H2O2氧化α-氟-α-苯硒基羧酸酯转化成α-氟代-α, β-不饱和酯的启发, Tomoda课题组[33b]实现了从α-羰基重氮化合物出发, 通过一锅法串联反应来构建三取代单氟烯烃.作者利用现场产生的苯基氟化硒与α-羰基重氮35发生插入反应得到中间体α-氟-α-苯硒基羰基化合物, 随后在H2O2的作用下, 高选择性转化成(Z)-α-氟代-α, β-不饱和羰基化合物36 (Scheme 18).在该反应条件下, 环状和非环状的α-羰基重氮类化合物均可以很好的获得相应的单氟烯烃.
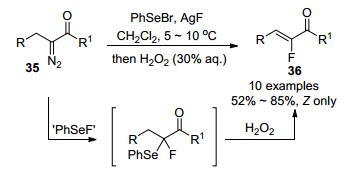 图式18
α-羰基重氮的烯烃化反应
Scheme18.
Olefin reaction of α-diazocarbonyl compounds
图式18
α-羰基重氮的烯烃化反应
Scheme18.
Olefin reaction of α-diazocarbonyl compounds
最近, Poisson小组[34a]报道了基于铜(Ⅰ)促进的α-硅基二氟甲基膦酸酯37与α-酯基重氮38的烯烃化反应来构建四取代单氟烯烃的方法.该反应在当量的CuCl的作用下, 能以中等到优秀的产率和单一的立体选择性得到(Z)-四取代氟代烯基磷酸酯类化合物39 (Scheme 19).作者认为该反应可能经历如下过程:首先氟化试剂37在铜(Ⅰ)的作用下形成活性物种Ⅰ, 随后与α-酯基重氮发生重氮插入/迁移/脱氟烯烃化反应得到相应的产物.
 图式19
α-氟代烯基膦酸酯的合成
Scheme19.
Synthesis of α-fluorovinylphosphonate
图式19
α-氟代烯基膦酸酯的合成
Scheme19.
Synthesis of α-fluorovinylphosphonate
我们小组在研究金催化的重氮参与的反应中[35], 利用金(Ⅰ)作为催化剂, 实现了α-羰基重氮和二氟烯醇硅醚[36]的烯烃化反应, 高效高立体选择性地合成了一系列(E)四取代氟代烯烃42和44 (Scheme 20)[37].该反应可能经历了如下的过程: α-羰基重氮首先在金(Ⅰ)的催化下形成活性金卡宾物种, 随后接受二氟烯醇硅醚的亲核进攻, 生成的中间体最后发生C—F键的断裂从而得到目标产物四取代单氟烯烃.当使用氧化吲哚衍生的重氮时, 不同取代基的重氮氧化吲哚以及不同芳基的二氟烯醇硅醚对反应活性影响较小.而当使用芳基重氮乙酸甲酯作为底物时, 反应选择性受二氟烯醇硅醚取代基影响较大.使用对甲氧基苯基取代的二氟烯醇硅醚时, 该反应在标准条件下能一步构建含氟四取代烯烃, 而对于苯基或者是对氯苯基取代的二氟烯醇硅醚, 则需要在第一步反应后外加0.8 equiv. DABCO来促进二氟取代的产物转化为单氟烯烃42.
3 基于其他底物的合成方法
除了利用羰基化合物和重氮来制备氟代烯烃外, 从炔烃、烯基化合物和烯烃出发的合成方法近年来也取得一些进展, 下面进行简要介绍.
3.1 炔烃的氢氟化反应
炔烃的氢氟化反应能实现一步构建含氟烯烃, 选择合适的催化剂和底物通常是实现高区域选择性的关键.自2007年Sadighi等[6b]报道了金(Ⅰ)催化的非末端炔烃的氢氟化反应以来, 这一方法受到众多关注[6].例如, Miller[6c], Gouverneur[6d]和Nevado[6e]等分别实现了金(Ⅰ)催化的其他种类炔烃的氢氟化反应.
 图式20
金(Ⅰ)催化的二氟烯醇硅醚与重氮的烯烃化反应
Scheme20.
Gold(Ⅰ) catalyzed olefin reactions of difluoroenoxysilanes and diazo reagents
图式20
金(Ⅰ)催化的二氟烯醇硅醚与重氮的烯烃化反应
Scheme20.
Gold(Ⅰ) catalyzed olefin reactions of difluoroenoxysilanes and diazo reagents
最近, 利用DMPU/HF (46)试剂作为氟源, Hammond和Xu等[38a]发展了金(Ⅰ)催化的炔烃45的氢氟化反应, 高效高区域选择性地合成了一系列多取代单氟烯烃47.该反应的底物普适性较广, 对称和非对称炔烃以及末端炔烃均适用(Scheme 21).值得一提的是, 由于DMPU亲核能力弱, 无碱性并可作为强氢键受体, DMPU/HF作为氟源与之前的Et3N/3HF相比具有明显优势.例如, DMPU/HF试剂能在不加酸或酸性添加剂的条件下与金属Lewis酸兼容.随后, Nolan等[38b]利用新合成的在空气中稳定的金(Ⅰ)二氟络合物作为催化剂, 可以很好的实现对称与非对称炔烃的氢氟化反应(Scheme 22).
 图式21
DMPU/HF与炔烃的氢氟化反应
Scheme21.
Hydrofluorination reaction of DMPU/HF and alkynes
图式21
DMPU/HF与炔烃的氢氟化反应
Scheme21.
Hydrofluorination reaction of DMPU/HF and alkynes
 图式22
金(Ⅰ)二氟络合物催化的氢氟化反应
Scheme22.
Gold(Ⅰ) bifluorides catalyzed hydrofluorination reaction
图式22
金(Ⅰ)二氟络合物催化的氢氟化反应
Scheme22.
Gold(Ⅰ) bifluorides catalyzed hydrofluorination reaction
除了金(Ⅰ)催化的炔烃的氢氟化反应外, 江焕峰等[38c]还报道了AgF促进的炔烃的氟溴化和氢氟化反应, 高区域选择性地合成了一系列含氟烯烃(Scheme 23).
 图式23
氟化银促进的氟溴烯烃的合成
Scheme23.
AgF mediated thesynthesis of bromofluoroalkenes
图式23
氟化银促进的氟溴烯烃的合成
Scheme23.
AgF mediated thesynthesis of bromofluoroalkenes
3.2 烯基化合物以及烯烃的氟化反应
烯基化合物的亲电氟化反应是一种简单、直接合成氟代烯烃的策略.自1986年Schwartz等[39a]报道了烯基碘的亲电氟化反应以来, 该领域得到了迅速的发展.在过去二十多年里, 化学家们先后发展了烯基锡[39b, 39c], 烯基三氟硼酸盐[39d], 烯基硼酸[39e], 烯基硅[39f]和烯基锂[39g]等试剂的亲电氟化反应.由于烯基金属试剂的高活性, 因此要实现高选择性的氟化较困难.
最近, Buchwald等[39h]报道了钯(Ⅱ)催化下的环状和非环状烯基三氟甲磺酸酯52的亲电氟化反应, 高区域选择性地合成了三、四取代含氟烯烃53.通过设计合成大位阻的单膦配体, 同时添加0.3 equiv.三氟甲基三乙基硅烷, 使得该反应的区域选择性得到很好的控制.该反应官能团兼容性较好, 五、六和七元环状以及部分非环状烯基三氟甲磺酸酯均适用(Scheme 24).
 图式24
钯(Ⅱ)催化的烯基三氟甲磺酸酯的氟化反应
Scheme24.
Pd(Ⅱ) catalyzed fluorination reaction of vinyl triflates
图式24
钯(Ⅱ)催化的烯基三氟甲磺酸酯的氟化反应
Scheme24.
Pd(Ⅱ) catalyzed fluorination reaction of vinyl triflates
由于烯基化合物需预先制备, 发展温和、高效的方法来实现烯烃的直接氟化构建不同的氟代烯烃无疑更为高效和实用, 但也具有更大的挑战性.
2010年, 刘国生等[40a]在发展醋酸钯催化苯乙烯的胺氟化反应时发现有少量双键C—H键直接氟化产物的生成, 显示了烯烃直接氟化来构建氟代烯烃的可行性.随后, 黄湧等[40b]利用RuCl3作为催化剂, NFSI (55)作为氟化试剂, 实现了β-取代苯乙烯以及色烯的直接C—H键氟化反应, 高效高顺式选择性地合成了系列非环状和环状三和四取代的含氟烯烃56 (Eq. 3).该反应条件温和, 官能团兼容性较好.
4 结论与展望
综上所述, 利用醛和酮来高立体选择性合成多取代单氟烯烃在最近十几年里得到了广泛研究, 特别在高立体选择性制备三取代含氟烯烃方面取得了长足进步, 并在这过程中发展出了一系列新的含氟砌块.但利用这一方法高立体选择性合成四取代烯烃的成功例子相对较少.而从重氮化合物出发来高立体选择性合成含氟烯烃近年来也得到很大发展, 并在高选择性制备四取代含氟烯烃方面具有一些独特优势.通过其他底物如炔烃、烯基化合物以及烯烃的氟化反应来高区域选择性地合成含氟烯烃的方法近年来也有一些进展.总体而言, 由于氟原子的特殊性质, 以及选择性构建三取代或者四取代含氟烯烃本身的挑战性, 发展利用简单易得的含氟砌块来高效高选择性构建复杂的多取代含氟烯烃仍将是合成化学家们需要努力的方向.
-
-
[1]
(a) Gelb, M. H. J. Am. Chem. Soc. 1986, 108, 3146.
(b) Damon, D. B. ; Hoover, D. J. J. Am. Chem. Soc. 1990, 112, 6439.
(c) Erickson, J. A. ; McLoughlin, J. I. J. Org. Chem. 1995, 60, 1626;
(d) Pesenti, C. ; Viani, F. ChemBioChem 2004, 5, 590.
(e) He, Z. ; Huang, Y, ; Francis, V. Acta Chim. Sinica 2013, 71, 700(in Chinese)
(何展荣, 黄毅勇, Francis, V. , 化学学报, 2013, 71, 700. )
(f) Ni, C. ; Zhu, L. ; Hu, J. Acta Chim. Sinica 2015, 73, 90(in Chinese).
(倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90. )
(g) Xiao, Y. ; Pan, Q. ; Zhang, X. Acta Chim. Sinica 2015, 73, 387(in Chinese).
(肖玉兰, 潘强, 张新刚, 化学学报, 2015, 73, 387. )
(h) Zhang, K. ; Xu, X. -H. ; Qing, F. -L. Chin. J. Org. Chem. 2015, 35, 556(in Chinese).
(张柯, 徐修华, 卿凤翎, 有机化学, 2015, 35, 556. )
(i) Rong, J. ; Ni, C. ; Wang, Y. ; Kuang, C. ; Gu, Y. ; Hu, J. Acta Chim. Sinica 2017, 75, 105(in Chinese).
(荣健, 倪传法, 王云泽, 匡翠文, 顾玉诚, 胡金波, 化学学报, 2017, 75, 105. ) -
[2]
(a) Hiyama, T. In Organofluorine Compounds:Chemistry and Ap-plications, Ed.:Yamamoto, H., Springer-Verlag, Berlin, 2000.
(b) Fluorine in Medicinal Chemistry and Chemical Biology, Ojima, I. Ed.; Wiley-Blackwell:United Kingdom, 2009. For reaviews:
(c) Bégué, J.-P.; Bonnet-Delphon, D. J. Fluorine Chem. 2006, 127, 992.
(d) Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013.
(e) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881.
(f) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(g) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(h) Jeschke, P. ChemBioChem 2004, 5, 570.
(i) Pagliaro, M.; Ciriminna, R. J. Mater. Chem. 2005, 15, 4981.
(j) Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441. -
[3]
Thayer, A. M. Chem. Eng. News 2006, 84, 15.
-
[4]
(a) Osada, S.; Sano, S.; Ueyama, M.; Chuman, Y.; Kodama, H.; Sakaguchi, K. Bioorg. Med. Chem. 2010, 18, 605.
(b) Malo-Forest, M.; Landelle, G.; Roy, J.-A.; Lacroix, J.; Gaudreault, R.-C.; Paquin, J.-F. Bioorg. Med. Chem. Lett. 2013, 23, 1712.
(c) Dutheuil, G.; Couve-Bonnaire, S.; Pannecoucke, X. Angew. Chem. Int. Ed. 2007, 46, 1290.
(d) Wong, O. A.; Shi, Y. A. J. Org. Chem. 2009, 74, 8377.
(e) Guérin, D.; Gaumont, A.-C.; Dez, I.; Mauduit, M.; Couve-Bonnaire, S.; Pannecoucke, X. ACS Catal. 2014, 4, 2374.
(f) Dai, W.; Xiao, J.; Jin, G.; Wu, J.; Cao, S. J. Org. Chem. 2014, 79, 10537. -
[5]
(a) Shen, Q.; Huang, Y.-G.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y. J. Fluorine Chem. 2015, 179, 14.
(b) Tian, P.; Feng, C.; Loh, T.-P. Nat. Commun. 2015, 6, 7472.
(c) Zhang, X.; Lin, Y.; Zhang, J.; Cao, S. RSC Adv. 2015, 5, 7905.
(d) Wu, J.; Xiao, J.; Dai, W.; Cao, S. RSC Adv. 2015, 5, 34498.
(e) Xiong, Y.; Huang, T.; Ji, X.; Wu, J.; Cao, S. Org. Biomol. Chem. 2015, 13, 7398.
(f) Dai, W.; Shi, H.; Zhao, X.; Cao, S. Org. Lett. 2016, 18, 4284.
(g) Kong, L.; Zhou, X.; Li, X. Org. Lett. 2016, 18, 6320. -
[6]
(a) Liu, G. Org. Biomol. Chem. 2012, 10, 6243.
(b) Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P. J. Am. Chem. Soc. 2007, 129, 7736.
(c) Gorske, B. C.; Mbofana, C. T.; Miller, S. J. Org. Lett. 2009, 11, 4318.
(d) Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem. Int. Ed. 2008, 47, 7927.
(e) de Haro, T.; Nevado, C. Chem. Commun. 2011, 47, 248;
(f) Lan, Y.; Hammond, G. B. Org. Lett. 2002, 4, 2437.
(g) Zhou, C.; Ma, Z.; Gu, Z.; Fu, C.; Ma, S. J. Org. Chem. 2008, 73, 772.
(h) Cui, H. F.; Chai, Z.; Zhao, G.; Zhu, S. Z. Chin. J. Chem. 2009, 27, 189.
(i) Lü, B.; Fu, C.; Ma, S. Org. Biomol. Chem. 2010, 8, 274. -
[7]
(a) Couve-Bonnaire, S.; Cahard, D.; Pannecoucke, X. Org. Biomol. Chem. 2007, 5, 1151.
(b) Yanai, H.; Taguchi, T. Eur. J. Org. Chem. 2011, 5939.
(c) Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Chem. Soc. Rev. 2011, 40, 2867.
(d) Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073.
(e) Drouin, M.; Hamel, J.-D.; Paquin, J.-F. Synlett 2016, 27, 821.
(f) Zhang, X.; Cao, S. Tetrahedron Lett. 2017, 58, 375. -
[8]
Schlosser, M.; Zimmermann, M. Synthesis 1969, 75.
-
[9]
Cox, D. G.; Gurusamy, N.; Burton, D. J. J. Am. Chem. Soc. 1985, 107, 2811. doi: 10.1021/ja00295a046
-
[10]
(a) Lei, X. S.; Dutheuil, G.; Pannecoucke, X.; Quirion, J. C. Org. Lett. 2004, 6, 2101.
(b) Zoute, L.; Dutheuil, G.; Quirion, J.-C.; Jubault, P.; Pannecoucke, X. Synthesis 2006, 3409. -
[11]
Burton, D. J.; Yang, Z.-Y.; Qiu, W. Chem. Rev. 1996, 96, 1641. doi: 10.1021/cr941140s
-
[12]
Machleidt, H.; Wessendorf, R. Justus Liebigs Ann. Chem. 1964, 674, 1. doi: 10.1002/(ISSN)1099-0690
-
[13]
Moghadam, G. E.; Penne, J. S. Bull. Soc. Chim. Fr. 1985, 448.
-
[14]
Sano, S.; Matsumoto, T.; Nakao, M. Tetrahedron Lett. 2014, 55, 4480. doi: 10.1016/j.tetlet.2014.06.063
-
[15]
Chevrie, D.; Lequeux, T.; Demoute, J. P.; Pazenok, S. Tetrahedron Lett. 2003, 44, 8127. doi: 10.1016/j.tetlet.2003.09.027
-
[16]
Ghosh, A. K.; Zajc, B. Org. Lett. 2006, 8, 1553. doi: 10.1021/ol060002+
-
[17]
(a) Zajc, B.; Kake, S. Org. Lett. 2006, 8, 4457.
(b) Pfund, E.; Lebargy, C.; Rouden, J.; Lequeux, T. J. Org. Chem. 2007, 72, 7871. -
[18]
(a) Ghosh, A. K.; Banerjee, S.; Sinha, S.; Kang, S. B.; Zajc, B. J. Org. Chem. 2009, 74, 3689.
(b) He, M.; Ghosh, A. K.; Zajc, B. Synlett 2008, 999. -
[19]
Alonso, D. A.; Fuensanta, M.; Gómez-Bengoa, E.; Nájera, C. Adv. Synth. Catal. 2008, 350, 1823. doi: 10.1002/adsc.v350:11/12
-
[20]
Larnaud, F.; Pfund, E.; Linclau, B.; Lequeux, T. Tetrahedron 2014, 70, 5632. doi: 10.1016/j.tet.2014.06.081
-
[21]
Zhao, Y.; Jiang, F.; Hu, J. J. Am. Chem. Soc. 2015, 137, 5199. doi: 10.1021/jacs.5b02112
-
[22]
Satoh, T.; Itoh, N.; Onda, K.; Kitoh, Y.; Yamakawa, K. Bull. Chem. Soc. Jpn. 1992, 65, 2800. doi: 10.1246/bcsj.65.2800
-
[23]
Chevrie, D.; Lequeux, T.; Pommelet, J.-C. Org. Lett. 1999, 1, 1539. doi: 10.1021/ol990178u
-
[24]
Boys, M. L.; Collington, E. W.; Finch, H.; Swanson, S.; Whitehead, J. F. Tetrahedron Lett. 1988, 29, 3365. doi: 10.1016/0040-4039(88)85163-3
-
[25]
(a) Zhang, W.; Huang, W.; Hu, J. Angew. Chem. Int. Ed. 2009, 48, 9858.
(b) Shen, X.; Hu, J. Eur. J. Org. Chem. 2014, 4437.
(c) Liu, Q.; Shen, X.; Ni, C.; Hu, J. Angew. Chem. Int. Ed. 2017, 56, 619. -
[26]
Welch, J. T.; Herbert, R. W. J. Org. Chem. 1990, 55, 4782. doi: 10.1021/jo00303a003
-
[27]
Michida, M.; Mukaiyama, T. Chem. Lett. 2008, 37, 890. doi: 10.1246/cl.2008.890
-
[28]
(a) Barma, D. K.; Kundeu, A.; Zhang, H.; Mioskowski, C.; Falck, J. R. J. Am. Chem. Soc. 2003, 125, 3218.
(b) Falck, J. R.; Bojet, R.; Barma, D. K.; Bandyopadhyay, A.; Joseph, S.; Mioskowski, C. J. Org. Chem. 2006, 71, 8178.
(c) Feng, Z.; Min, Q.-Q.; Zhao, H.-Y.; Gu, J.-W.; Zhang, X.Angew. Chem. Int. Ed. 2015, 54, 1270. -
[29]
Lemonnier, G.; Zoute, L.; Dupas, G.; Quirion, J.-C.; Jubault, P. J. Org. Chem. 2009, 74, 4124. doi: 10.1021/jo900422m
-
[30]
Augustine, J.-K.; Bombrun, A.; Venkatachaliah, S.; Jothi, A. Org. Biomol. Chem. 2013, 11, 8065. doi: 10.1039/c3ob41592a
-
[31]
Cao, C.-R.; Ou, S.; Jiang, M.; Liu, J.-T. Org. Biomol. Chem. 2014, 12, 467. doi: 10.1039/C3OB42093K
-
[32]
(a) Grundmann, C. Justus. Liebigs Ann. Chem. 1938, 536, 29.
(b) Font, J.; Serratosa, F.; Valls, J. Chem. Commun. 1970, 721.
(c) Kulkowit, S.; McKervey, M. A. J. Chem. Soc., Chem. Commun. 1978, 1069.
(d) Baratta, W.; Del Zotto, A.; Rigo, P. Chem. Commun. 1997, 2163.
(e) Del Zotto, A.; Baratta, W.; Verardo, G.; Rigo, P. Eur. J. Org. Chem. 2000, 2795.
(f) Doyle, M. P.; Hu, W.; Phillips, I. M. Org. Lett. 2000, 2, 1777.
(g) Greenman, K. L.; Carter, D. S.; Van Vranken, D. L. Tetrahedron 2001, 57, 5219.
(h) Doyle, M. P.; Yan, M. J. Org. Chem. 2002, 67, 602;
(i) Li, G.-Y.; Che, C.-M. Org. Lett. 2004, 6, 1621.
(j) Hodgson, D. M.; Angrish, D. Chem. Commun. 2005, 4902.
(k) Chen, S.; Wang, J. Chem. Commun. 2008, 4198;
(l) Xiao, Q.; Ma, J.; Yang, Y.; Zhang, Y.; Wang, J. Org. Lett. 2009, 11, 4732.
(m) Hansen, J. H.; Parr, B. T.; Pelphrey, P.; Jin, Q.; Autschbach, J.; Davies, H. M. L.Angew. Chem. Int. Ed. 2011, 50, 2544.
(n) Xia, Y.; Liu, Z.; Xiao, Q.; Qu, P.; Ge, R.; Zhang, Y.; Wang, J. Angew. Chem. Int. Ed. 2012, 51, 5714.
(o) Hu, M.; He, Z.; Gao, B.; Li, L.; Ni, C.; Hu, J. J. Am. Chem. Soc. 2013, 135, 17302.
(p) Xiao, Q.; Zhang, Y.; Wang, J. Acc. Chem. Res. 2013, 46, 236.
(q) Zhang, D.; Xu, G.; Ding, D.; Zhu, C.; Li, J.; Sun, J. Angew. Chem. Int. Ed. 2014, 53, 11070.
(r) Hu, M.; Ni, C.; Li, L.; Han, Y.; Hu, J. J. Am. Chem. Soc. 2015, 137, 14496.
(s) Zhu, C.; Xu, G.; Liu, K.; Qiu, L.; Peng, S.; Sun, J. Chem. Commun. 2015, 51, 12768.
(t) Zhu, C.; Xu, G.; Ding, D.; Qiu, L.; Sun, J. Org. Lett. 2015, 17, 4244.
(u) Zhang, Z.; Yu, W.; Wu, C.; Wang, C.; Zhang, Y.; Wang, J. Angew. Chem. Int. Ed. 2016, 55, 273.
(v) Feng, S.; Mo, F.; Xia, Y.; Liu, Z.; Liu, Z.; Zhang, Y.; Wang, J. Angew. Chem. Int. Ed. 2016, 55, 15401. -
[33]
(a) Fuchigami, T.; Hayashi, T.; Konno, A. Tetrahedron Lett. 1992, 33, 3161.
(b) Usuki, Y.; Iwaoka, M.; Tomoda, S. J. Chem. Soc., Chem. Commun. 1992, 1148. -
[34]
(a) Ivanova, M. V.; Bayle, A.; Besset, T.; Pannecoucke, X.; Poisson, T. Angew. Chem. Int. Ed. 2016, 55, 14141.
(b) Feng, Z.; Min, Q.-Q.; Xiao, Y.-L.; Zhang, B.; Zhang, X.Angew. Chem. Int. Ed. 2014, 53, 1669. -
[35]
(a) Cao, Z.-Y.; Wang, X.; Tan, C.; Zhao, X.-L.; Zhou, J.; Ding, K. J. Am. Chem. Soc. 2013, 135, 8197.
(b) Cao, Z.-Y.; Zhao, Y.-L.; Zhou, J. Chem. Commun. 2016, 52, 2537.
(c) Zhao, Y.-L.; Cao, Z.-Y.; Zeng, X.-P.; Shi, J.-M.; Yu, Y.-H.; Zhou, J. Chem. Commun. 2016, 52, 3943. -
[36]
For a review, see:
(a) Decostanzi, M.; Campagne, J.-M.; Leclerc, E. Org. Biomol. Chem. 2015, 13, 7351. For examples, see:
(b) Amii, H.; Kobayashi, T.; Hatamoto, Y.; Uneyama, K. Chem. Commun. 1999, 1323.
(c) Chu, L.; Zhang, X.; Qing, F.-L. Org. Lett. 2009, 11, 2197.
(d) Yuan, Z.; Wei, Y.; Shi, M. Chin. J. Chem. 2010, 28, 1709.
(e) Kashikura, W.; Mori, K.; Akiyama, T. Org. Lett. 2011, 13, 1860. For our continuous efforts in the functionalization of fluorinated enol silyl ethers, see:
(f) Liu, Y.-L.; Zhou, J. Chem. Commun. 2012, 48, 1919.
(g) Liu, Y.-L.; Zeng, X.-P.; Zhou, J. Acta Chim. Sinica. 2012, 70, 1451.
(h) Liu, Y.-L.; Liao, F.-M.; Niu, Y.-F.; Zhao, X.-L.; Zhou, J. Org. Chem. Front. 2014, 1, 742.
(i) Yu, J.-S.; Liu, Y.-L.; Tang, J.; Wang, X.; Zhou, J. Angew. Chem. Int. Ed. 2014, 53, 9512.
(j) Liao, F.-M.; Liu, Y.-L.; Yu, J.-S.; Zhou, F.; Zhou, J. Org. Biomol. Chem. 2015, 13, 8906.
(k) Yu, J.-S.; Zhou, J. Org. Biomol. Chem. 2015, 13, 10968.
(l) Yu, J.-S.; Liao, F.-M.; Gao, W.-M.; Liao, K.; Zuo, R.-L.; Zhou, J. Angew. Chem. Int. Ed. 2015, 54, 7381.
(m) Yu, J.-S.; Zhou, J. Org. Chem. Front. 2016, 3, 298.
(n) Zeng, X.-P.; Zhou, J. J. Am. Chem. Soc. 2016, 138, 8730. -
[37]
Liao, F.-M.; Cao, Z.-Y.; Yu, J.-S.; Zhou, J. Angew. Chem. Int. Ed. 2017, 56, 2459. doi: 10.1002/anie.201611625
-
[38]
(a) Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381.
(b) Nahra, F.; Patrick, S. R.; Bello, D.; Brill, M.; Obled, A.; Cordes, D. B.; Slawin, A. M. Z.; O'Hagan, D.; Nolan, S. P. ChemCatChem 2015, 7, 240.
(c) Li, Y.; Liu, X.; Ma, D.; Liu, B.; Jiang, H. Adv. Synth. Catal. 2012, 354, 2683. -
[39]
(a) Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445.
(b) Tius, M. A.; Kawakami, J. K. Tetrahedron 1995, 51, 3997.
(c) Sommer, H.; Fürstner A. Chem. Eur. J. 2017, 23, 558.
(d) Petasis, N. A.; Yudin, A. K.; Zavialov, I. A.; Prakash, G. K. S.; Olah, G. A. Synlett 1997, 606.
(e) Furuya, T.; Ritter, T. Org. Lett. 2009, 11, 2860.
(f) Tredwell, M.; Gourverneur, V. Org. Biomol. Chem. 2006, 4, 26.
(g) Yang, M.-H.; Matikonda, S. S.; Altman, R. A. Org. Lett. 2013, 15, 3894.
(h) Ye, Y.; Takada, T.; Buchwald, S. L. Angew. Chem. Int. Ed. 2016, 55, 15559. -
[40]
(a) Qiu, S.; Xu, T.; Zhou, J.; Guo, Y.; Liu, G. J. Am. Chem. Soc. 2010, 132, 2856;
(b) Shao, Q.; Huang, Y. Chem. Commun. 2015, 51, 6584.
-
[1]
-

图 1 代表性的含三取代或者四取代单氟烯烃结构单元的生物活性分子
Figure 1 Representative bioactive molecules that containing tri-or tetra-substituted monofluoroalkenes structural motif.

图式3 醛与11a的Julia类烯烃化反应
Scheme 3 Modified Julia olefination reaction of aldehydes and 11a

图式4 11b与醛、酮的Julia类烯烃化反应
Scheme 4 Modified Julia olefination reaction of 11b with aldehydes and ketones

图式5 利用Julia类烯烃化反应来合成α-氟代-α, β-不饱和酯
Scheme 5 Synthesis of α-fluoro-α, β-unsaturated esters via mo-dified Julia olefination reaction

图式7 17a~17b的Julia-Kocienski类烯烃化反应
Scheme 7 Julia-Kocienski olefination reaction of 17a~17b

图式9 Julia-Kocienski反应中间体的自发拆分
Scheme 9 Spontaneous resolution of Julia-Kocienski intermediates

图式12 氟代亚砜亚胺与酮的烯烃化反应
Scheme 12 Olefin reactions via fluorinated sulfoximine and ketones

图式13 利用Peterson烯烃化反应来合成氟代烯烃
Scheme 13 Synthesis of fluoro-olefin via Peterson olefinations
49%, Z:E = 29:1 40%, Z:E = 50:1 50%, Z:E = 1.4:1 51%, Z:E = 1:1

图式14 Cr(Ⅱ)和Fe(0) 促进的类Reformatsky反应
Scheme 14 Cr(Ⅱ) and Fe(0) mediated Reformatsky-type reactions

图式15 Et2Zn促进的二溴氟乙酸乙酯与醛的类Reformatsky反应
Scheme 15 Et2Zn mediated Reformatsky-type reactions of Ethyl dibromofluoroacetate and aldehydes

图式16 TiCl4促进的单氟乙酸乙酯与醛的烯烃化反应
Scheme 16 TiCl4 mediated olefin reactions of ethyl monofluoroacetate and aldehydes

图式20 金(Ⅰ)催化的二氟烯醇硅醚与重氮的烯烃化反应
Scheme 20 Gold(Ⅰ) catalyzed olefin reactions of difluoroenoxysilanes and diazo reagents

图式22 金(Ⅰ)二氟络合物催化的氢氟化反应
Scheme 22 Gold(Ⅰ) bifluorides catalyzed hydrofluorination reaction
-


 下载:
下载:
























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 42
- 文章访问数: 4344
- HTML全文浏览量: 1429
 下载:
下载:
