 图式 1
传统镍催化、光催化及双催化体系
Scheme1.
Traditional nickel catalysis, photocatalysis and dual catalysis
图式 1
传统镍催化、光催化及双催化体系
Scheme1.
Traditional nickel catalysis, photocatalysis and dual catalysis
引用本文:
阮利衡, 董振诚, 陈春欣, 吴爽, 孙京. 过渡金属镍与可见光双催化体系的研究进展[J]. 有机化学,
2017, 37(10): 2544-2554.
doi:
10.6023/cjoc201704051

Citation: Ruan Liheng, Dong Zhencheng, Chen Chunxin, Wu Shuang, Sun Jing. Recent Progress on the Nickel/Photoredox Dual Catalysis[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2544-2554. doi: 10.6023/cjoc201704051
Citation: Ruan Liheng, Dong Zhencheng, Chen Chunxin, Wu Shuang, Sun Jing. Recent Progress on the Nickel/Photoredox Dual Catalysis[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2544-2554. doi: 10.6023/cjoc201704051
过渡金属镍与可见光双催化体系的研究进展
English
Recent Progress on the Nickel/Photoredox Dual Catalysis
Abstract:
A dual-catalysis system merging the visible light photoredox with transition metal nickel catalysis enables a new strategy to build the novel carbon-carbon and carbon-heteroatom bond, which are not generally possible via using either photoredox or nickel catalysis alone. This mild, green and promising protocol has attracted the interest of some scientific researchers. In this review, the recent progress of nickel/photoredox dual catalysis is summarized.
-
Key words:
- photocatalysis
- / nickel catalysis
- / single electron transfer
- / energy transfer
-
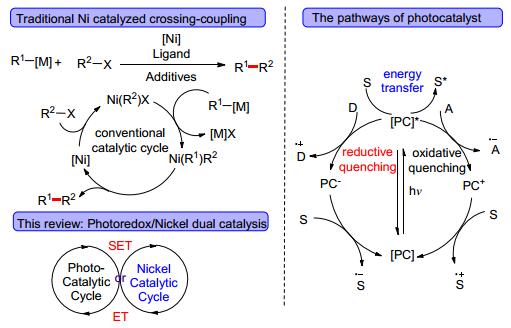
在“绿色化学”理念的推动下, 寻找更加清洁、经济、高效的能源一直是化学家努力的方向.众所周知, 太阳能是自然界中最为丰富的能源, 其中, 地面接收太阳辐射的43%为可见光[1].然而绝大多数有机分子不能直接吸收可见光, 直至科研工作者们发掘了钌、铱金属络合物以及有机染料的可见光催化性能, 许多反应在可见光条件下才可以顺利地进行.清洁绿色的可见光引发的烷基化[2]、芳基化[3]、环加成[4]及脱卤[5]等反应得到了广泛的研究和应用, 已成为有机光化学领域的研究热点[6~18], 同时也让有机合成化学迎来了新的春天.
有机合成中C(sp2)—C(sp2)键的构建目前已比较成熟, 过渡金属催化的交叉偶联反应是最有效的方法, 传统的交叉偶联反应, 经历氧化加成、金属交换与还原消除三个关键步骤(Scheme 1).在其他过程不断取得进展的情况下, 偶联反应中的金属交换部分几乎没有明显进展.金属交换通常是该类反应的决速步骤, 不同杂化形态碳的交换速率为C(sp)>C(sp2)>C(sp3)[19], 导致直接构建C(sp3)—C(sp2)或C(sp3)—C(sp3)键十分困难.相较于传统钯催化下的转金属化过程, 可见光引发的单电子转金属化使得C(sp3)容易发生交叉偶联反应.研究表明:可见光引发的单电子转金属化的速率为C(sp3)>C(sp2)>C(sp), 因此在可见光作用下, C(sp3)更容易发生交叉偶联反应[19].若将可见光催化体系引入到传统的过渡金属催化体系中便可以很好地解决C(sp3)—C(sp2)或C(sp3)—C(sp3)键不易构建的问题.
图式 1
传统镍催化、光催化及双催化体系
Scheme1.
Traditional nickel catalysis, photocatalysis and dual catalysis
过渡金属钯、镍等在交叉偶联反应中都得到了广泛的研究, 而过渡金属镍催化相较于钯催化更容易发生单电子转移, 同时过渡金属镍也更廉价.因此, 镍与可见光的双催化体系引起了广泛的关注.可见光催化体系通常经历还原淬灭循环、氧化淬灭循环或能量转移(ET)历程(Scheme 1), 其不仅具有非常良好的氧化还原性能, 还可以完成能量转移.那么在双催化体系中, 可见光催化既可以通过单电子还原或单电子氧化过程推动过渡金属镍催化, 又可以通过能量转移促进镍催化循环, 过渡金属镍和可见光起到协同催化作用.
具有清洁、绿色、高效等优势的镍与可见光双催化体系, 实现了一些在单独催化体系中难以完成的反应过程, 为有机合成中碳-碳键或碳-杂键的构建提供了一种新的思路.过渡金属与可见光双催化体系蓬勃发展, 近年来, 侧重于作者课题组的工作[20]及前期双催化体系的总结见诸报端[21].但是该领域发展异常迅猛, 最新的研究结果不断地涌现, 极大地丰富了镍与可见光的双催化体系.因此, 本文主要依据可见光催化剂参与反应的历程, 结合国内外最新的研究报道对镍与可见光双催化反应进行了分类和归纳, 以期对该领域的发展起到一定的积极作用.
1 SET交叉偶联反应
1.1 碳-碳键构建
1.1.1 脱羧偶联
羧酸衍生物经由光氧化还原过程脱去二氧化碳后可以得到烷基自由基, 是一类非常理想的自由基来源.近年来, 一些科研工作者开展了基于镍与可见光双催化体系下的羧酸及其衍生物与卤代烃的交叉偶联反应. 2014年, MacMillan课题组[19]首次报道了可见光催化剂铱络合物4和过渡金属镍催化剂共同催化的羧酸1与芳基卤化物2直接构建新型C(sp3)—C(sp2)的反应(Scheme 2).铱络合物吸收可见光能量后到达激发态, 随后单电子氧化羧酸1脱去CO2生成烷基自由基物种5, 与此同时芳基卤化物2与Ni(0)氧化加成得到Ni(Ⅱ)中间体, Ni(Ⅱ)中间体迅速捕捉体系中的烷基自由基5得到Ni(Ⅲ)加合物, 之后还原消除得到目标产物3和Ni(Ⅰ), 最后单电子还原Ni(Ⅰ)至Ni(0)完成整个催化循环.同年, 他们[22]报道了单独可见光催化体系Boc-L-脯氨酸与对苯二腈的脱羧偶联反应, 两种底物在可见光催化体系经历两次单电子转移(SET)得到两种自由基物种, 随后发生自由基之间的偶联.双催化体系同样经历两次单电子转移, 但一次形成烷基自由基, 另一次则作用于金属镍络合物.由此可见, 通过单电子转移成功地实现了可见光催化与过渡金属镍催化的有机结合, 拓展了过渡金属催化和光催化的应用范围.
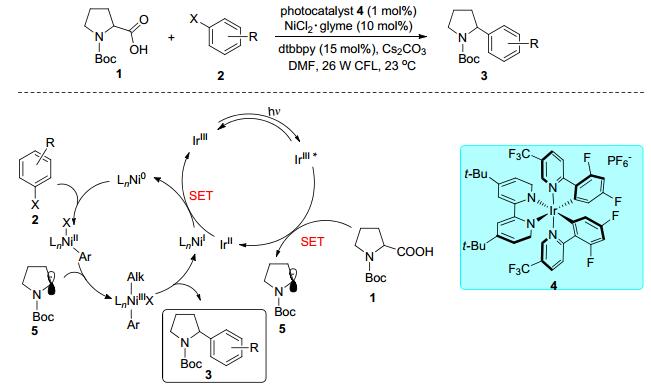 图式 2
C(sp3)—C(sp2)键的构建
Scheme2.
Formation of C(sp3)—C(sp2) bond
图式 2
C(sp3)—C(sp2)键的构建
Scheme2.
Formation of C(sp3)—C(sp2) bond
随后, MacMillan课题组[23]报道了在温和条件下简单丰富的羧酸化合物6和烷基卤化物7的偶联反应(Scheme 3).作者成功地将亲电试剂由芳基卤化物拓宽到烷基溴化物, 为构建C(sp3)—C(sp3)键提供了一种新的方法.同时, 通过镍与可见光双催化体系可以直接方便地制备医药化合物10.
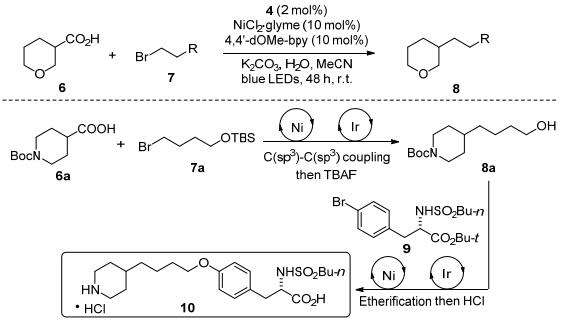 图式 3
C(sp3)—C(sp3)键的构建
Scheme3.
Formation of C(sp3)—C(sp3) bond
图式 3
C(sp3)—C(sp3)键的构建
Scheme3.
Formation of C(sp3)—C(sp3) bond
在2016年, Fu课题组与MacMillan课题组[24]开展合作成功地将镍与可见光双催化体系应用到非对称合成中, 丰富了光催化手性合成方法学(Scheme 4).从自然界普遍存在的氨基酸11和芳(杂)环卤化物12出发, 合成了一系列具有高对映选择性的苄基胺化合物13, 并进一步合成了一些具有生物活性的化合物.
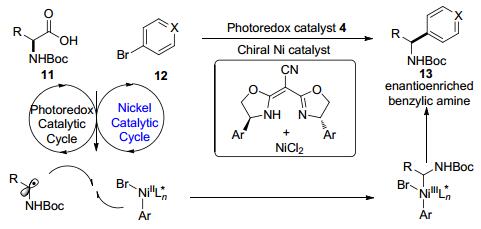 图式 4
手性苄胺的合成
Scheme4.
Preparation of chiral benzylic amines
图式 4
手性苄胺的合成
Scheme4.
Preparation of chiral benzylic amines
MacMillan课题组在双催化体系下又相继报道了原位形成的混合酸酐[25]、芳基酮羧酸[26]、烷基醇[27]与芳基卤代烃的脱羧偶联反应; 期间, Gu等[28]报道了α-羰基羧酸与吲哚的脱羧偶联反应; Oderinde等[29]详细阐述了镍与可见光双催化体系中分子氧、溶剂及光源对交叉偶联反应的影响; Tong小组[30]则报道了在镍、锌及可见光催化剂三重作用下的α-羟基酸-O-羧内酸酐脱除CO2的可控开环聚合反应.
Zhang课题组[31]合成了一系列供体-受体荧光化合物, 为非金属光催化剂的设计提供了理论支持, 其中2, 4, 5, 6-四(9-咔唑基)-间苯二腈(4CzIPN)展现出高效的光催化活性, 可以用来替代昂贵的钌、铱光催化剂(Scheme 5); Mariano等[32]研究了有机光催化剂4CzIPN与过渡金属镍催化剂组成的双催化体系催化下的新型甲酰化策略.
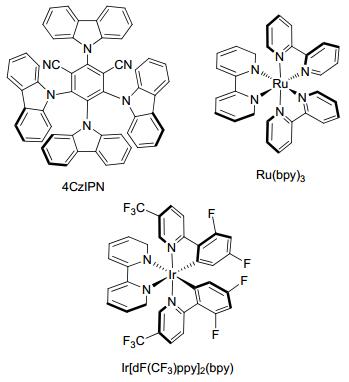 图式 5
可见光催化剂的结构
Scheme5.
Structures of photocatalyst
图式 5
可见光催化剂的结构
Scheme5.
Structures of photocatalyst
2017年, Scaiano课题组[33]在非均相催化体系下成功地实现了羧酸1与芳基碘化物14的交叉偶联反应(Eq. 1).该双催化体系由TiO2和二甲氧基乙烷氯化镍组成, TiO2在重复使用五次之后反应效率仅有稍微的下降, 然而底物的范围受到一定的限制.但是, TiO2廉价丰富及可回收性的特性足以引起更多科研工作者的兴趣, 而且非均相催化进一步拓宽了双催化体系的应用范围.
1.1.2 有机硼烷偶联
相比较于硼酸或硼酸酯, 有机三氟硼酸盐更加稳定且具有较低的氧化电势, 在交叉偶联反应中是非常良好的C(sp3)型亲核试剂. 2014年, Molander等[34]报道了过渡金属镍与可见光双催化体系下有机三氟硼酸盐15与溴苯16的交叉偶联反应(Eq. 2).与传统有机硼试剂在偶联反应中的双电子转金属化不同, 该反应体系中有机三氟硼酸盐经历了单电子转金属化.
2015年, Molander课题组[35]首次报道了α-氨基甲基三氟硼酸盐18在可见光铱催化剂及过渡金属镍催化剂协同作用下合成手性氨基酸衍生物的偶联反应(Eq. 3).手性的天然氨基酸引入了三氟硼酸盐基团后, 其在可见光条件下经历单电子转移后得到了烷基自由基物种, Ni经由氧化、还原等过程最终构建了新的C(sp3)—C(sp2)键得到了手性氨基酸衍生物20.
随后, Kozlowski与Molander等[36]展开合作通过泛函密度理论(DFT)深入研究了烷基三氟硼酸盐与芳基溴化物在双催化体系下的反应机理.各种取代的三氟硼酸盐通过可见光催化可以得到相应的烷基自由基物种21, 22, 23 (Scheme 6)[37], 而这些烷基自由基又可以与各类亲电试剂(卤代烃[38, 39]、硼化芳基卤代烃[40]、酰氯[41]及氮-酰基琥珀酰亚胺[42])发生交叉偶联反应.
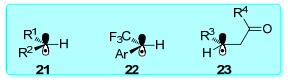 图式 6
碳自由基
Scheme6.
Carbon radical
图式 6
碳自由基
Scheme6.
Carbon radical
三氟硼酸盐的α位引入N原子使得C—B键更易于发生单电子转移而断裂.在此思路基础上, 硼酸盐的α位引入O原子, 在双催化体系下反应同样可以顺利地进行[43~45]; 仲丁基三氟硼酸盐在可见光催化体系下可以快速形成仲丁基, 仲丁基起到活化黄原酸酯的作用, 从而实现双催化体系下黄原酸酯与溴苯的交叉偶联反应[46]; Doyle等[47]则研究了双催化体系下苄基三氟硼酸盐与环状内消旋酸酐的去对称化, 高效地合成具有高立体对映选择性的酮酸化合物.
1.1.3 有机硅烷偶联
有机硅烷氧化电势很高, 通常在可见光条件下难以发生碳-硅键断裂, 高价的酚硅化合物具有高稳定性、可溶性、无害性及环境友好性, 同时其氧化还原电势较低(Eo=0.3~0.9 V), 在可见光催化剂作用下可以得到烷基自由基物种.在2015年, Fensterbank课题组[48]成功地实现了在双催化作用下高价酚硅24与对溴苯甲腈19的交叉偶联反应(Eq. 4).研究表明:在可见光催化剂作用下高价酚硅化合物可以高效地产生烷基自由基, 并且能够顺利地完成进一步镍催化循环过程.
之后不久, Molander课题组极大地拓宽了高价酚硅化合物在双催化模型下的应用范围.作者首先制备了一系列稳定的高价酚硅化合物, 在[NiCl2(dme)] (5 mol%)为催化剂, dtbbpy (5 mol%)为配体, [Ru(bpy)3](PF6)2 (2 mol%)为可见光催化剂的条件下实现了高价酚硅与芳(杂)环溴代烃的偶联反应[49](Scheme 7).接下来的研究中, 偶联试剂由芳(杂)环溴代烃扩展到了烯基卤代烃[50]、烷基溴代烃[51]、2, 1-硼氮杂萘溴化物[52]以及苯酚衍生物(三氟甲磺酸酯、甲苯磺酸酯、甲磺酸酯)[53]; 高价酚硅与硼化芳基溴化物在有机光催化剂4CzIPN与过渡金属镍双催化体系下依然可以高效地合成一系列烷基取代的芳(杂)环硼化物[54]; 高价酚硅在可见光催化体系下得到的烷基自由基, 同时是一种良好的氢原子转移试剂, 实现了芳(杂)环溴化物与硫醇的硫醚化反应[55](Scheme 7).
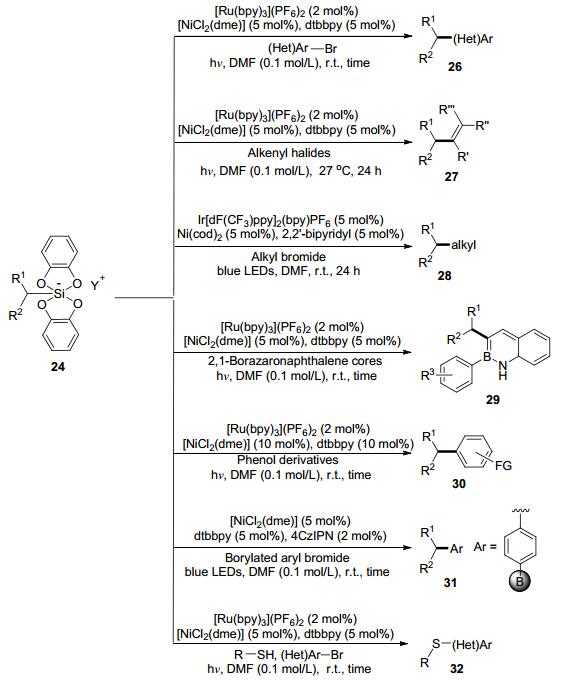 图式 7
高价酚硅作为自由基试剂
Scheme7.
Hypervalent silicates as radical agents
图式 7
高价酚硅作为自由基试剂
Scheme7.
Hypervalent silicates as radical agents
1.1.4 其它官能团化衍生物偶联
1, 4-二氢吡啶衍生物可以通过“一步法”从醛制备而来, 亦是一类非常理想的烷基自由基来源. Molander课题组[56]报道了在过渡金属镍/有机光催化剂体系下的1, 4-二氢吡啶衍生物33和芳(杂)环溴化物的偶联反应(Scheme 8), 该反应体系具有操作方便、条件温和、环境友好及良好化学选择性等特点.
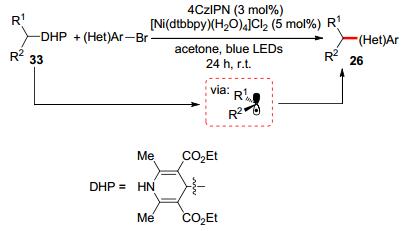 图式 8
1, 4-二氢吡啶类化合物作为自由基前体
Scheme8.
1, 4-Dihydropyridines as radical precursors
图式 8
1, 4-二氢吡啶类化合物作为自由基前体
Scheme8.
1, 4-Dihydropyridines as radical precursors
烷基溴化物同样是比较理想的碳自由基来源, 武汉大学雷爱文教授课题组[57]研究了烷基溴化物与芳(杂)环溴化物在双催化体系下直接构建C(sp3)—C(sp2)键的反应(Eq. 5).反应体系不需要化学计量的锰或者锌作还原剂, 而是使用常用的三乙胺作为最终的还原试剂. 2017年, Vannucci课题组[58]开展了相类似的烷基溴化物与芳基卤代烃的还原交叉偶联反应, 底物具有非常好的普适性, 并提出了与雷爱文教授提出的反应机理相一致的反应机理.
1.1.5 碳氢化合物偶联
碳氢化合物是自然界中最为常见的化合物, 若是在可见光作用下通过碳氢键活化得到烷基自由基, 将极大地丰富双催化模型的应用范围. 2016年, Doyle课题组[59]报道了双催化体系下芳基胺35的sp3碳-氢键酰基化反应(Eq. 6).温和条件下sp3碳-氢键的直接活化可减少预先官能团化等步骤, 是一种环境友好的合成策略.紧接着, 该课题组[60]报道了芳基氯代烃与醚的C(sp3)—H键直接交叉偶联反应, 该双催化体系烷基自由基来源不是可见光直接单电子氧化底物得到的, 而是首先芳基氯代烃与Ni(0)氧化加成, 再经单电子氧化得到Ni(Ⅲ)物种, 而Ni(Ⅲ)物种在可见光的作用下形成氯自由基, 最后氯自由基攫取醚α位的氢原子得到烷基自由基.
2016年, MacMillan课题组[61]利用溴化镍水合物作为过渡金属催化剂, 铱络合物4作为光催化剂, 乙酰克里定(3-acetoxyquinuclidine)作为氢原子转移试剂, 在温和条件下实现了C(sp3)—H亲核试剂38与芳基卤代烃16的交叉偶联反应.值得注意的是乙酰克里定在反应体系中不仅仅是氢原子转移催化剂, 还要起到有机碱的作用, 所以需要使用过量的乙酰克里定(1.1 equiv.).作者在可见光/镍双催化体系的基础上成功地引入了氢原子转移试剂, 巧妙地实现了镍催化、可见光催化及有机催化的结合(Eq. 7).
2017年, 余达刚课题组[62]报道了胺或醚40的C(sp3)—H与C(sp2)—O型亲电试剂41的直接烯基化和芳(杂)化反应(Eq. 8).该反应体系具有条件温和、官能团容忍性好、底物适用范围广及选择性优良等特点, 极大地扩展了双催化体系的应用范围.
1.2 碳-杂键构建
杂原子化合物在自然界中普遍存在, 是一类具有重要生物活性的化合物.在有机分子引入杂原子(O, S, N, P)往往能够改变整个分子的物理和化学性质, 因此构建新型碳-杂键是非常重要的研究课题.在双催化体系下构建碳-杂键的反应也引起了科研工作者的关注.
1.2.1 自由基氧化镍物种
可见光氧化还原体系通过羧酸、硼烷、硅烷等可以得到烷基自由基, 而在相似条件下硫醇能够形成相应的自由基.在2016年, Johannes与合作者[63]研究了硫醇43与芳基卤代烃14的偶联反应, 在可见光氧化还原作用下硫醇可以得到巯基自由基(Scheme 9).反应体系展现出优秀的官能团容忍性, 无论是芳基、苄基还是烷基硫醇都能高效地与芳(杂)环碘化物发生交叉偶联反应.机理研究表明该反应与之前镍催化过程不同, 该催化循环不涉及Ni(0), 而是通过瞬态的Ni(Ⅰ)物种活化整个过程.首先可见光催化体系单电子氧化硫醇43, 并在吡啶的作用下脱去质子得到巯基自由基, 单电子还原45生成46.与此同时, 46迅速捕获巯基自由基形成47, 随后47经由单电子还原得到48, 接着发生氧化加成、还原消除得到目标偶联化合物44.
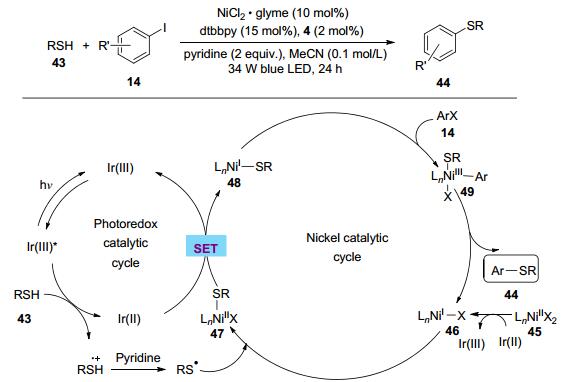 图式 9
碳-硫键的形成
Scheme9.
Bond building of C—S
图式 9
碳-硫键的形成
Scheme9.
Bond building of C—S
2015年, 肖文精课题组[64]巧妙地利用镍与可见光双催化模型在温和条件下高效地合成了一系列三芳基膦氧化合物(Eq. 9).二芳基膦氧化合物50在可见光催化循环中得到了以磷为中心的自由基物种, 继而参与镍催化循环, 最终得到目标化合物51.
1.2.2 光催化直接氧化镍物种
光氧化还原循环不仅可以通过单电子转移得到自由基, 也可以直接单电子氧化还原镍物种, 实现整个双催化循环. 2015年, MacMillan等[65]报道了醇52与卤代烃16在双催化体系下的交叉偶联反应(Scheme 10).该体系中瞬态的Ni(Ⅲ)物种是由光催化循环直接单电子氧化Ni(Ⅱ)物种得到的, 这与自由基氧化镍物种的反应历程不同.同期, Jamison课题组[66]也研究了双催化体系下具有相似催化历程的吲哚啉衍生物的高区域选择性合成.
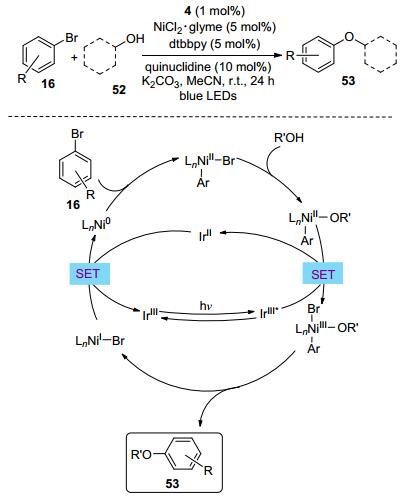 图10
碳-氧键的构建
Scheme10.
Construction of C—O bond
图10
碳-氧键的构建
Scheme10.
Construction of C—O bond
2016年, Buchwald课题组和MacMaillan课题组[67]报道了无配体参与的镍盐与可见光共同催化的芳胺化反应(Eq. 10).该反应条件温和, 底物适应性好, 为含有胺类结构单元的医药化合物、天然产物及精细化学品的合成提供了新的方法, 也是对传统过渡金属钯催化的芳胺化反应的有益补充.
2 ET交叉偶联反应
可见光催化循环不仅能够实现单电子转移, 还能发生能量转移过程.可见光催化剂吸收可见光的能量后可以传递给其它有机分子促进反应的进行.
在2016年, Molander课题组[68]报道了镍与可见光双催化体系下通过能量转移方式的碳-氢键芳基化反应, 并对反应机理过程进行了深入的研究(Scheme 11).动力学研究表明: Ni—Br键的均裂形成溴自由基, 溴自由基与底物之间发生氢原子转移得到了稳定的碳自由基.镍络合物60*到61有两种可能的途径: (A)离散的Ni(Ⅰ)和溴自由基; (B)协同形成的四中心过渡态.
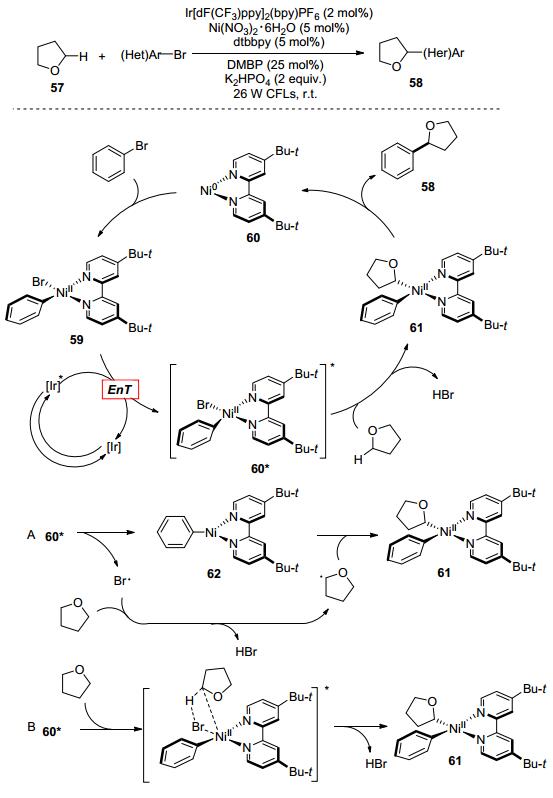 图式 11
碳-氢键芳基化
Scheme11.
Arylation of C—H bond
图式 11
碳-氢键芳基化
Scheme11.
Arylation of C—H bond
传统过渡金属催化循环一般依赖于基态有机金属配合物的氧化. 2017年, MeCusker课题组和MacMillan课题组[69]研究了镍与可见光双催化体系下羧酸63与芳基溴化物16的偶联反应, 金属镍络合物在吸收光能量后生成激发态的金属镍络合物从而促进反应的进行(Scheme 12).反应机理为:对溴苯甲酸甲酯16与Ni(0)发生氧化加成得到Ni(Ⅱ)物种65, 其经过配体的快速交换生成新的Ni(Ⅱ)物种66, 同时铱催化剂吸收能量后到达激发态, 66通过能量转移形成激发态66*, 最后66*发生还原消除得到目标偶联化合物64和Ni(0), 从而完成整个催化循环.
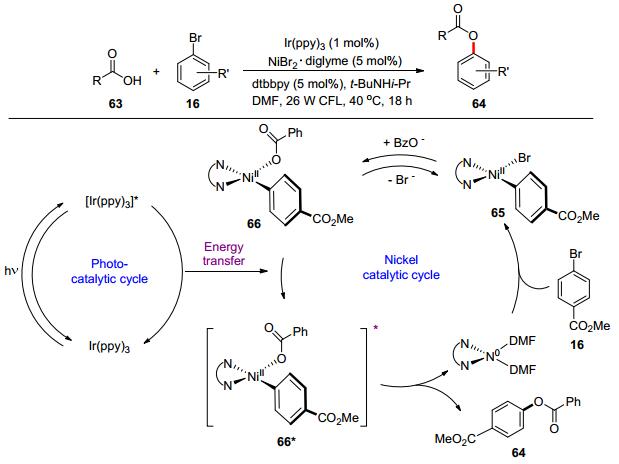 图式 12
羧酸与芳卤的偶联反应
Scheme12.
Coupling of carboxylic acids with aryl halides
图式 12
羧酸与芳卤的偶联反应
Scheme12.
Coupling of carboxylic acids with aryl halides
当然, 过渡金属镍盐在反应体系中也可以作为路易斯酸从而促进反应的发生. 2017年, 肖文精课题组[70]研究了在可见光作用下β-酮酯67的非对称氧化反应, 得到了一系列α-羟基-β-二羰基化合物68 (Eq. 10).作者将具有可见光吸收的噻吨酮与手性配体进行有机结合开发了一种新型的可见光催化剂, 该催化剂具有可见光吸收与手性配位的双重作用, 为可见光条件下的手性合成提供了一种新的思路.
3 总结与展望
综上所述, 镍与可见光协同催化体系为构建新型的碳-碳键或碳-杂键提供了一种新的方法, 拓宽了清洁的光能以及过渡金属催化在有机合成中的应用范围.而且, 双催化体系反应条件温和、清洁高效, 符合“绿色化学”发展的要求.然而, 目前协同催化体系还存在诸多问题, 如亲电试剂不够环保、底物需要预官能团化、可见光催化剂不够廉价等.此外基于能量转移方式的双催化体系研究还不够深入.随着广大科研工作者对双催化机理的深入研究及新型廉价可见光催化剂的开发, 镍与可见光双催化体系将在医药、生物、天然产物及全合成等领域得到广泛应用, 甚至有望将其推广至工业化生产中.
-
-
[1]
Zou, Z.; Ye, J.; Sayama, K.; Arakawa, H. Nature 2001, 414, 625. doi: 10.1038/414625a
-
[2]
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77. doi: 10.1126/science.1161976
-
[3]
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102. doi: 10.1039/B913880N
-
[4]
Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886. doi: 10.1021/ja805387f
-
[5]
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756. doi: 10.1021/ja9033582
-
[6]
Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239136.
-
[7]
Prier, C. K.; Rankic, D. A.; Macmillan, D. W. C. Chem. Rev. 2013, 113, 5322. doi: 10.1021/cr300503r
-
[8]
戴小军, 许孝良, 李小年, 有机化学, 2013, 33, 2046. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract342154.shtmlDai, X.; Xu, X.; Li, X. Chin. J. Org. Chem. 2013, 33, 2046(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract342154.shtml
-
[9]
关保川, 许孝良, 王红, 李小年, 有机化学, 2016, 36, 1564. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345459.shtmlGuan, B.; Xu, X.; Wang, H.; Li, X. Chin. J. Org. Chem. 2016, 36, 1564(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345459.shtml
-
[10]
刘薇, 郑昕宇, 曾建国, 程辟, 有机化学, 2017, 37, 1. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345749.shtmlLiu, W.; Zheng, X.; Zeng, J.; Cheng, P. Chin. J. Org. Chem. 2017, 37, 1(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345749.shtml
-
[11]
程骁恺, 胡新根, 陆展, 有机化学, 2017, 37, 251. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345790.shtmlCheng, X.; Hu, X.; Lu, Z. Chin. J. Org. Chem. 2017, 37, 251(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345790.shtml
-
[12]
谭芬, 肖文精, 化学学报, 2015, 73, 85. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmTan, F.; Xiao, W. Acta Chim. Sinica 2015, 73, 85(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[13]
丁奎岭, 肖文精, 吴骊珠, 化学学报, 2017, 75, 5. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmDing, K.; Xiao, W.; Wu, L. Acta Chim. Sinica 2017, 75, 5(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[14]
裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15. doi: 10.3969/j.issn.0253-2409.2017.01.003Pei, P.; Zhang, F.; Yi, H.; Lei, A. Acta Chim. Sinica 2017, 75, 15(in Chinese). doi: 10.3969/j.issn.0253-2409.2017.01.003
-
[15]
王德红, 张龙, 罗三中, 化学学报, 2017, 75, 22. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmWang, D.; Zhang, L.; Luo, S. Acta Chim. Sinica 2017, 75, 22(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[16]
钟建基, 孟庆元, 陈彬, 佟振合, 吴骊珠, 化学学报, 2017, 75, 34. doi: 10.3969/j.issn.0253-2409.2017.01.006Zhong, J.; Meng, Q.; Chen, B.; Tung, C.; Wu, L. Acta Chim. Sinica 2017, 75, 34(in Chinese). doi: 10.3969/j.issn.0253-2409.2017.01.006
-
[17]
张晶, 陈以昀, 化学学报, 2017, 75, 41. doi: 10.7503/cjcu20160643Zhang, J. Chen, Y. Acta Chim. Sinica 2017, 75, 41(in Chinese). doi: 10.7503/cjcu20160643
-
[18]
Roh, G.; Iqbal, N. Cho, E. Chin. J. Chem. 2016, 34, 459. doi: 10.1002/cjoc.201500919
-
[19]
Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C. Science 2014, 345, 437. doi: 10.1126/science.1255525
-
[20]
Tellis, J.; Kelly, C.; Primer, D.; Jouffroy, M.; Patel, N.; Molander, G. A. Acc. Chem. Res. 2016, 49, 1429. doi: 10.1021/acs.accounts.6b00214
-
[21]
Gui, Y.; Sun, L.; Lu, Z.; Yu, D. Org. Chem. Front. 2016, 3, 522. doi: 10.1039/C5QO00437C
-
[22]
Zuo, Z.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 5257. doi: 10.1021/ja501621q
-
[23]
Johnston, C. P.; Smith, R. T.; Allmendinger, S.; MacMillan, D. W. C. Nature 2016, 536, 322. doi: 10.1038/nature19056
-
[24]
Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 1832. doi: 10.1021/jacs.5b13211
-
[25]
Le, C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 11938. doi: 10.1021/jacs.5b08304
-
[26]
Chu, L.; Lipshultz, J. M.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2015, 54, 7929. doi: 10.1002/anie.201501908
-
[27]
Zhang, X.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 13862. doi: 10.1021/jacs.6b09533
-
[28]
Gu, L.; Jin, C.; Liu, J.; Zhang, H.; Yuan, M.; Li, G. Green Chem. 2015, 18, 1201.
-
[29]
Oderinde, M. S.; Varela-Alvarez, A.; Aquila, B.; Robbins, D. W.; Johannes, J. W. J. Org. Chem. 2015, 80, 7642. doi: 10.1021/acs.joc.5b01193
-
[30]
Feng, Q.; Tong, R. J. Am. Chem. Soc. 2017, 139, 6177. doi: 10.1021/jacs.7b01462
-
[31]
Luo, J.; Zhang, J. ACS Catal. 2016, 6, 873. doi: 10.1021/acscatal.5b02204
-
[32]
Huang, H.; Li, X.; Yu, C.; Zhang, Y.; Mariano, P. S.; Wang, W. Angew. Chem., Int. Ed. 2017, 56, 1500. doi: 10.1002/anie.201610108
-
[33]
McTiernan, C. D.; Leblanc, X.; Scaiano, J. C. ACS Catal. 2017, 7, 2171. doi: 10.1021/acscatal.6b03687
-
[34]
Tellis, J. C.; Primer, D. N.; Molander, G. A. Science 2014, 345, 433. doi: 10.1126/science.1253647
-
[35]
Khatib, M. E.; Serafim, R. A. M.; Molander, G. A. Angew. Chem., Int. Ed. 2016, 55, 254. doi: 10.1002/anie.201506147
-
[36]
Gutierrez, O.; Tellis, J. C.; Primer, D. N.; Molander, G. A.; Ko-zlowski, M. C. J. Am. Chem. Soc. 2015, 137, 4896. doi: 10.1021/ja513079r
-
[37]
Gutierrez-Bonet, A.; Tellis, J. C. Matsui, J. K.; Vara, B. A.; Mo-lander, G. A. ACS Catal. 2016, 6, 8004. doi: 10.1021/acscatal.6b02786
-
[38]
Tellis, J. C.; Amanu, J.; Molander, G. A. Org. Lett. 2016, 18, 2994. doi: 10.1021/acs.orglett.6b01357
-
[39]
Ryu, D.; Primer, D. N.; Tellis, J. C.; Molander, G. A. Chem. Eur. J. 2016, 22, 120. doi: 10.1002/chem.201504079
-
[40]
Yamashita, Y.; Tellis, J. C.; Molander, G. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12026. doi: 10.1073/pnas.1509715112
-
[41]
Amani, J.; Molander, G. A. J. Org. Chem. 2017, 82, 1856. doi: 10.1021/acs.joc.6b02897
-
[42]
Amani, J.; Alam, R.; Badir, S.; Molander, G. A. Org. Lett. 2017, 19, 2426. doi: 10.1021/acs.orglett.7b00989
-
[43]
Karakaya, I.; Primer, D. N.; Molander, G. A. Org. Lett. 2015, 17, 3294. doi: 10.1021/acs.orglett.5b01463
-
[44]
Karimi-Nami, R.; Tellis, J. C.; Molander, G. A. Org. Lett. 2016, 18, 2572. doi: 10.1021/acs.orglett.6b00911
-
[45]
Matsui, J. K.; Molander, G. A. Org. Lett. 2017, 19, 436. doi: 10.1021/acs.orglett.6b03448
-
[46]
Vara, B. A.; Patel, N. R.; Molander, G. A. ACS Catal. 2017, 7, 3955. doi: 10.1021/acscatal.7b00772
-
[47]
Stache, E. E.; Rovis, T.; Doyle, A. G. Angew. Chem., Int. Ed. 2017, 56, 3679. doi: 10.1002/anie.201700097
-
[48]
Corce, V.; Chamoreau, L.; Derat, E.; Coddard, J.; Ollivier, C.; Fensterbank, L. Angew. Chem., Int. Ed. 2015, 54, 11414. doi: 10.1002/anie.201504963
-
[49]
Jouffroy, M.; Primer, D. N.; Molander, G. A. J. Am. Chem. Soc. 2016, 138, 475. doi: 10.1021/jacs.5b10963
-
[50]
Patel, N. R.; Kelly, C. B.; Jouffroy, M.; Molander, G. A. Org. Lett. 2016, 18, 764. doi: 10.1021/acs.orglett.6b00024
-
[51]
Leveque, C.; Corce, V.; Chenneberg, L.; Ollivier, C.; Fensterbank. Eur. J. Org. Chem. 2017, 2017, 2118. doi: 10.1002/ejoc.201601571
-
[52]
Jouffroy, M.; Davies, G. H.; Molander, G. A. Org. Lett. 2016, 18, 1606. doi: 10.1021/acs.orglett.6b00466
-
[53]
Patel, N. R.; Molander, G. A. J. Org. Chem. 2016, 81, 7271. doi: 10.1021/acs.joc.6b00800
-
[54]
Vara, B. A.; Jouffroy, M.; Molander, G. A. Chem. Sci. 2017, 8, 530. doi: 10.1039/C6SC03236B
-
[55]
Jouffroy, M.; Kelly, C. B.; Molander, G. A. Org. Lett. 2016, 18, 876. doi: 10.1021/acs.orglett.6b00208
-
[56]
Gutiérrez-Bonet, Á.; Tellis, J. C.; Matsui, J. K.; Vara, B. A.; Molander, G. A. ACS Catal. 2016, 6, 8004. doi: 10.1021/acscatal.6b02786
-
[57]
Duan, Z.; Li, W.; Lei, A. Org. Lett. 2016, 18, 4012. doi: 10.1021/acs.orglett.6b01837
-
[58]
Paul, A.; Smith, M.; Vannucci. J. Org. Chem. 2017, 82, 1996. doi: 10.1021/acs.joc.6b02830
-
[59]
Joe, C. L.; Doyle, A. G. Angew. Chem., Int. Ed. 2016, 55, 4040. doi: 10.1002/anie.201511438
-
[60]
Shields, B. J.; Doyle, A. G. J. Am. Chem. Soc. 2016, 138, 12719. doi: 10.1021/jacs.6b08397
-
[61]
Shaw, M. H.; Shurtleff, V. W.; Terrett, J. A.; Cuthbertson, J.D.; MacMillan, D. W. C. Science 2016, 352, 1304. doi: 10.1126/science.aaf6635
-
[62]
Gui, Y. Y.; Liao, L. L.; Sun, L.; Zhang, Z.; Ye, J. H.; Shen, G.; Lu, Z. P.; Zhou, W. J.; Yu, D. G. Chem. Commun. 2017, 53, 1192. doi: 10.1039/C6CC09685A
-
[63]
Oderinde, M. S.; Frenette, M.; Robbins, D. W.; Aquila, B.; Johannes, J. W. J. Am. Chem. Soc. 2016, 138, 1760. doi: 10.1021/jacs.5b11244
-
[64]
Xuan, J.; Zeng, T. T.; Chen, J. R.; Lu, L. Q.; Xiao, W. J. Chem. Eur. J. 2015, 21, 4962. doi: 10.1002/chem.201500227
-
[65]
Terrett, J. A.; Cuthbertson, J. D.; Shurtleff, V. W.; MacMillan, D. W. C. Nature 2015, 524, 330. doi: 10.1038/nature14875
-
[66]
Tasker, S. Z.; Jamison, T. F. J. Am. Chem. Soc. 2015, 137, 9531. doi: 10.1021/jacs.5b05597
-
[67]
Corcoran, E. B.; Piront, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279. doi: 10.1126/science.aag0209
-
[68]
Heitz, D. R.; Tellis, J. C.; Molander, G. A. J. Am. Chem. Soc. 2016, 138, 12715. doi: 10.1021/jacs.6b04789
-
[69]
Welin, E. R.; Le, C.; Arias-Rotondo, D. M.; McCusker, J. K., MacMillan, D. W. C. Science 2017, 355, 380. doi: 10.1126/science.aal2490
-
[70]
Ding, W.; Lu, L.; Zhou, Q.; Wei, Y.; Chen, J.; Xiao, W. J. Am. Chem. Soc. 2017, 139, 63. doi: 10.1021/jacs.6b11418
-
[1]
-

图式 1 传统镍催化、光催化及双催化体系
Scheme 1 Traditional nickel catalysis, photocatalysis and dual catalysis
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 91
- 文章访问数: 6380
- HTML全文浏览量: 1854
 下载:
下载:
