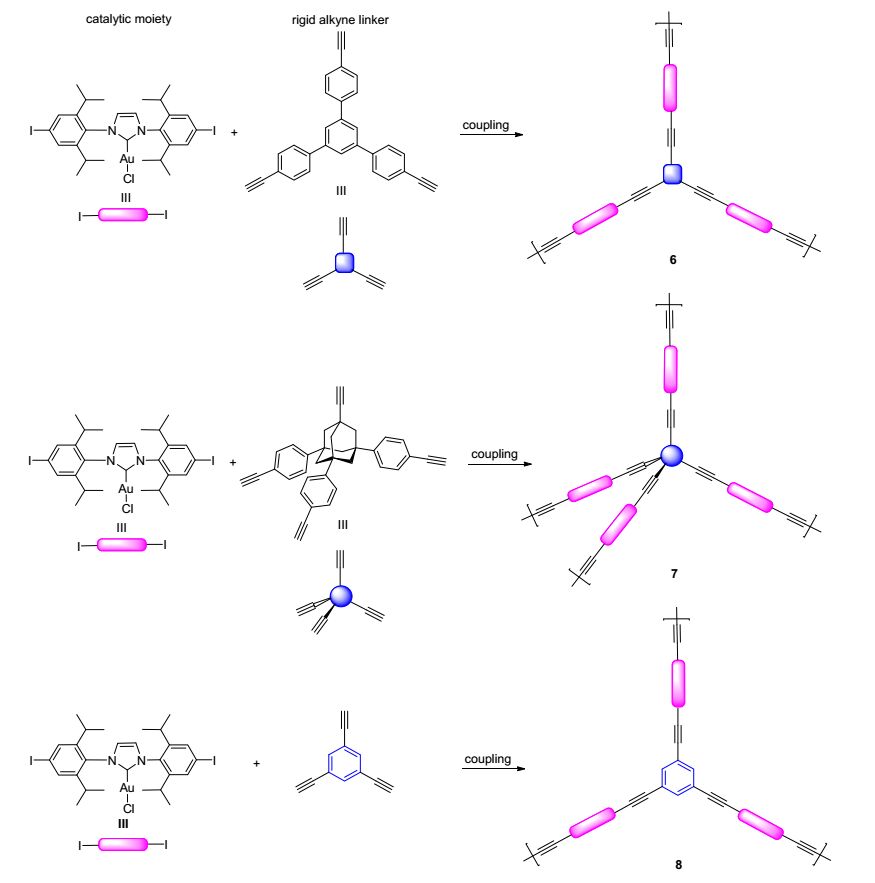
 图式 1
Pd-催化的Sonogashira偶联合成(NHC)Au@POPs
Scheme1.
Synthesis of (NHC)Au@POPs using the Pd-catalyzed Sonogashira coupling method
图式 1
Pd-催化的Sonogashira偶联合成(NHC)Au@POPs
Scheme1.
Synthesis of (NHC)Au@POPs using the Pd-catalyzed Sonogashira coupling method
引用本文:
杨璐, 徐彩霞. 催化炔烃马氏水合反应研究进展[J]. 有机化学,
2017, 37(12): 3130-3145.
doi:
10.6023/cjoc201704050

Citation: Yang Lu, Xu Caixia. Advances in the Catalytic Markovnikov Hydration of Alkynes toward Ketones[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3130-3145. doi: 10.6023/cjoc201704050
Citation: Yang Lu, Xu Caixia. Advances in the Catalytic Markovnikov Hydration of Alkynes toward Ketones[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3130-3145. doi: 10.6023/cjoc201704050
催化炔烃马氏水合反应研究进展
English
Advances in the Catalytic Markovnikov Hydration of Alkynes toward Ketones
Abstract:
Carbonyl group is one of the important building blocks in organic synthesis. The catalytic Markovnikov hydration of alkynes is an eco-friendly reaction with 100% atom economy to construct carbonyl compounds, and is one of hot spots of organic chemistry over recent years. Research progress concerning catalytic hydration of alkynes to ketones on the view of the types of catalysts, and catalytic mechanisms are concluded. Finally, the further development and challenge of catalytic hydration of alkynes are dis-cussed.
-
Key words:
- alkyne
- / Markovnikov hydration
- / catalytic
- / ketone
- / mechanism
-
羰基是现代有机合成的重要骨架结构单元.通过催化炔烃水合为酮来构筑羰基是最直接、环境友好、原子经济的方法之一.传统的炔烃水合反应采用的是汞-酸催化体系, 该催化体系具有很好的活性和选择性.但自汞的毒性被认识之后, 对非汞催化体系的研究便大量涌现.到目前为止, 已经发展了包括Au、Ag、Pt、Pd、Fe等金属催化剂和Brønsted酸(B酸)等多种催化剂.
近年来对炔烃水合反应的研究报道很多, 但关于这方面的研究进展报道并不多见.仅在2015年, Aponick等[1]就金催化的炔烃水合和氢化烷氧基化反应的区域选择性方面的研究工作进行了总结评述.本文将对炔烃水合反应中的各类催化体系做一详细的综述, 并对炔烃水合反应的催化体系加以展望.
1 金催化炔烃水合反应
由于金催化剂所具有的独特活性, 所以对金催化剂的研究吸引了越来越多化学研究工作者的参与, 并且活性金物种对炔烃水合反应所具有的特殊亲和力使得炔烃水合反应已经成为衡量金催化剂活性的基准反应之一, 因此利用各种金络合物催化炔烃水合反应的研究不断被报道.研究发现金以Au(0)、Au(Ⅰ)和Au(Ⅲ)形式存在时, 都可以催化炔烃水合反应, 其中尤以Au(Ⅰ)的研究报道最多.
1.1 Au(Ⅰ)催化炔烃水合反应
在炔烃水合反应中, 利用Au(Ⅰ)为活性成分的催化剂基本都含有有机配体.早在2002年, Tanaka等[2]用(Ph3P)AuCH3催化炔烃水合反应.发现在甲醇水溶液中, 炔烃高选择性地水合生成马氏产物酮.但在无溶剂条件下反应并不发生, 说明甲醇是水合反应高效进行的关键.而且配体(如(PhO)3P或CO)的加入改善了催化剂的性能, 从而降低催化剂用量.共催化剂(如CH3SO3H、CF3SO3H、H3PW12O40和H2SO4)存在时, 无须加入配体且仅需少量催化剂就能取得很好的催化效果(Eq. 1). (Ph3P)AuCH3/H2SO4/MeOH催化体系对脂肪炔、芳香炔都有很好的催化性能.但该催化体系对双烷基取代的不对称内炔区域选择性不高, 并且需要较多酸助剂.
Hammond等[3]用含高位阻膦配体的金络合物为催化剂, 催化剂用量为ppm级的条件实现了炔烃的水合(Eq. 2). L1AuCl/AgOTf/MeOH催化体系对端炔和对称内炔都具有很好的催化活性.并且当增加酸助剂(TfOH)时, 反应温度可由80 ℃降为室温, 且金催化剂用量可减半, 说明酸助剂能够明显促进催化剂性能.但该催化体系仍需少量银盐作助剂.
Fujita等[4]利用偶联反应合成了一系列树形水溶性膦配体, 然后制备成相应的树形膦金络合物催化剂.发现在AgOTf和TfOH助剂存在下, 该催化体系在溶剂甲醇中对端炔和对称内炔具有很好的催化活性(Eq. 3), 羟基和羧基等官能团具有很好的兼容性.该催化剂循环使用4次活性没有降低, 但该催化剂需要酸助剂和银盐助剂.
Li等[5]在表面活性剂Brij76自组装作用下, 有机金(Ⅰ)硅烷与烷氧基硅烷苯共聚制备有序介孔硅基催化剂Au-PPh2-PMO(Ph).此法成功地将Au(Ⅰ)引入有序介孔硅基材料骨架中, 所制备的催化剂具有高的表面积、有序的介孔通道和好的水热稳定性, 对炔烃水合反应的催化活性明显高于未固载的Au(PPh3)Cl.研究发现Au(Ⅰ)与PPh2的特殊配位方式是Au-PPh2-PMO(Ph)高效催化炔烃水合反应的重要原因.而且催化剂的高比表面和大孔径有利于反应物的扩散和吸附, 可使催化剂具有更好的性能.在该催化体系中电子效应对反应有明显的影响, 其中脂肪族和富电子芳基端炔都能取得很高的收率, 但贫电子芳基端炔收率很低(Eq. 4).该催化剂很容易实现重复使用, 但该催化体系需要1 equiv.的酸助剂.
Laguna等[6]以水溶性的三苯基膦三间磺酸钠盐(TPPTS)为膦配体, 制得各种膦配位的炔基金络合物.发现AuC≡CR(TPPTS) (R为叔丁基或3-噻吩基)活性最佳.其对芳基端炔具有较好的催化活性, 但反应的电子效应明显, 只有富电子芳基端炔为底物时转化率很高(Eq. 5).该催化体系需要酸助剂, 并且内炔在该催化体系中完全不反应.
Leyva等[7]制备了一系列膦配位的AuPR3NTf2(PR3=SPhos, PPh3, Pt-Bu3), 并应用于炔烃水合反应.发现在不加入酸且以甲醇为溶剂的条件下, 该方法将各种炔烃接近定量转化为相应的酮(Eq. 6), 包括端炔、烯炔、内炔、炔丙醇.而各种取代基包括羟基、卤素、酯基、醚类以及对酸敏感的基团, 都具有很好的兼容性, 而TMS在反应中会完全脱去.并认为该催化剂的催化活性极大地取决于与金属离子配位的膦配体的性质以及金属络合物的平衡阴离子的软度.该催化体系对不对称内炔的区域选择性不好.
Schmidbaur等[8]制备了膦配位的Au(Ⅰ)的磺酸盐和羧酸盐催化剂, 在BF3•Et2O为共催化剂的条件下进行炔烃的水合反应.发现在所有的催化剂中(Ph3P)Au-OC(O)C2F5的催化活性最高, 可在室温下实现3-己炔水合.并且Au(Ⅰ)的催化活性远大于Ag(Ⅰ)的原因可能是因为Ag(Ⅰ)比Au(Ⅰ)具有更强的配位能力, Ag(Ⅰ)不仅与配体氧牢固地键合, 而且会与溶剂中的氧结合, 所以很难再与炔烃进行配位, 导致对炔烃水合反应没有活性.同时发现共催化剂在反应中除了与金属络合物反应生成更具催化活性的金属阳离子, 而且BF3•Et2O在反应条件下可能生成氢氟酸, 而氢氟酸不仅能够促进反应中间体中C—Au键的断裂, 而且B酸本身就有利于水合反应的进行, 因此BF3•Et2O的存在有利于水合反应的进行.研究了3-己炔、苯乙炔、环戊基乙炔的水合反应.在相同实验条件下, 反应4 h时只有3-己炔水合的产率较高(85%), 另两种炔烃水合的产率都不到10%.
刘盛广等[9]合成了一系列新型茚基膦金络合物, 并研究了对炔烃水合反应的催化性能.该茚基膦金催化剂对炔烃水合反应具有很好的催化性能(Eq. 7).该催化体系不仅对内炔有很好的活性, 而且不对称内炔反应的区域选择性也很高, 但不对称内炔只研究了苯基甲基乙炔.该催化体系也需要银盐助剂.
Nolan等[10]发现(NHC)AuCl/AgSbF6/二氧六环催化体系能高收率地将炔烃水合为酮(Eq. 8).并且发现当(NHC)AuCl为(IPr)AuCl时, 反应活性非常好.通常催化剂用量仅需50~100 (0.005~0.01 mol%)即可实现炔烃的高效水合.而使用其他的NHC配体时或者其他银源时, 反应活性都会变差.当单独使用银盐作催化剂时, 反应活性很低.此催化体系对芳香炔(端炔、对称内炔)、脂肪炔(端炔和内炔)和单芳基取代的不对称内炔都具有非常好的适用性.但不对称内炔的区域选择性较差.后来Nolan等[10]以(IPr)AuOH为催化剂前体, 将(IPr)AuOH在B酸(HSbF6)存在的条件下原位转变为(IPr)AuCl后催化炔烃水合反应(Eq. 9).发现该催化体系在无需银盐的条件下就能够将各种炔烃包括端炔和内炔有效地转化为相应的酮.
Joó等[12, 13]用制备的带有磺酸钠官能团化的水溶性(NHC)Au络合物(3和4)催化炔烃水合反应.发现此类络合物在没有酸的情况下对端炔具有很高催化活性, 能够将端炔接近定量地转化为酮, 并且酸的存在能更好地促进水合反应, 但研究的反应底物比较有限, 主要研究了苯乙炔和对甲氧基苯乙炔. Silbestri等[14]也用类似的(NHC)Au络合物开展了炔烃水合反应的研究.发现位阻更大的(NHC)Au活性更高, 性能最好的(NHC)Au络合物为5, 其结构与3类似, 但位阻更大.该催化剂在纯水溶剂中活性很高, 炔烃能接近定量地转化为酮(Eq. 10).由于催化剂的水溶性和反应物、产物的水不溶性, 所以催化剂可以方便地实现回收再利用.他们都进行了纯水溶剂和混合溶剂(水与甲醇)的对照实验, 发现溶剂水中加入甲醇后, 可加速反应进行.但该反应体系仅对端炔适用, 内炔的区域选择性很低.
Li等[15]通过Sonogashira偶联的方法设计合成了一系列(NHC)Au@POPs (Scheme 1), 此方法将(NHC)Au构筑到有机聚合物的结构中.通过改变有机单体及其浓度, 可以控制合成具有不同孔径结构和表面积的(NHC)Au@POPs.研究发现(NHC)Au@POPs/AgSbF6/ MeOH催化体系对芳香族端炔和对称内炔的水合反应都具有优异的催化性能(Eq. 11), 而且催化剂孔径结构会影响其催化性能.该催化剂循环使用5次活性几乎没有明显降低, 但此催化体系需要用AgSbF6作助剂, 并且反应需要较高温度.
图式 1
Pd-催化的Sonogashira偶联合成(NHC)Au@POPs
Scheme1.
Synthesis of (NHC)Au@POPs using the Pd-catalyzed Sonogashira coupling method
2016年, Dobereiner等[16]利用含硼的阴离子型氮杂环卡宾(BNHC)为配体的Au(Ⅰ)络合物为催化剂研究炔烃的水合及内炔水合的区域选择性.发现(BNHC)Au-(SMe2)/二氧六环催化体系在无酸、无银盐助剂的条件下, 能够很好地实现炔烃水合(Eq. 12), 并且内炔比端炔反应的产率更高.但不对称内炔水合的区域选择性不高, 当不对称内炔的芳环上有弱吸电子的取代基时, 反应根本不能进行.
Scarso等[17]将杯芳烃10 (Scheme 2)在溶剂中自组装得到笼状的六聚体106•8H2O, 然后将(NHC)Au 11封装在六聚体空腔内得到11@106•8H2O.研究了未封装的11和封装后的11@106•8H2O对炔烃水合反应的速率和底物选择性方面的影响.发现封装后, 由于底物不能快速进入通过氢键结合的六聚体空腔, 导致反应的速率明显降低.用具有不同电子云密度和尺寸大小的炔烃进行竞争反应, 发现两种催化剂在底物选择性上具有相反的结果. 11催化时有利于富电子的芳香炔(尺寸更大), 而封装后的11@106•8H2O催化时, 由于空腔的限制更有利于尺寸更小的芳香炔(电子云密度相对最小).虽然该方法所得的底物选择性差异较小, 但却为设计底物选择性催化剂提供了研究思路.之后, Scarso等[18]又发现在B酸(HBF4)作助剂时, 在没有金属的条件下六聚体106•8H2O就可以直接催化炔烃的水合反应(Eq. 13), 并且底物分子尺寸越大, 产率越低, 显示出笼状结构六聚体催化反应的尺寸选择性.内炔的反应活性相对较低, 当苯环上连有弱吸电子基团(如溴)时, 并没有相应的产物生成.
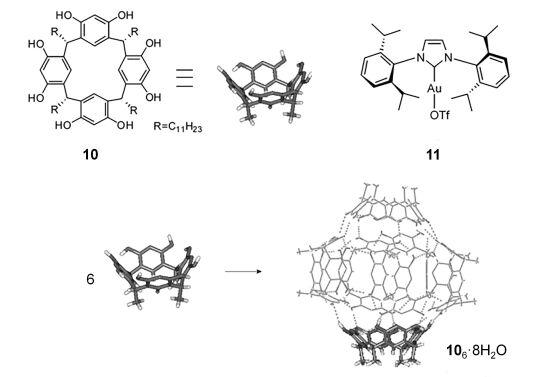 图式 2
杯芳烃10, 106•8H2O和(IPr)AuOTf络合物11的结构[17]
Scheme2.
Chemical structures of resorcin[4]arene 10,
106•8H2O and (IPr)AuOTf complex 11
图式 2
杯芳烃10, 106•8H2O和(IPr)AuOTf络合物11的结构[17]
Scheme2.
Chemical structures of resorcin[4]arene 10,
106•8H2O and (IPr)AuOTf complex 11
Hu等[19, 20]以甲醇为溶剂, 开展了(RNC)AuCl(异腈基金)和(NHC)AuCl催化炔烃水合反应的研究.发现(NHC)AuCl对端炔水合反应具有很好的活性(Eq. 14), 包括脂肪族和芳香族端炔, 但研究底物较少.作为合成(NHC)AuCl的前体, (RNC)AuCl 12对炔烃水合反应也具有较好的活性.并且它们在室温下即可实现脂肪族和富电子芳香族端炔的高效水合(Eq. 15), 而苯环上连有强吸电子基团时, 产率很低(仅8%), 说明在此催化体系中电子效应对反应影响明显.其中(NHC)AuCl在室温下非常稳定, 反应后含有催化剂的水相无需处理可以直接再使用, 重复使用6次催化活性没有明显变化.但是这两种催化剂都需要有KB(C6H5)4存在下才能反应.此研究为合成(NHC)Au的前体(RNC)AuCl在有机反应中的应用提供了新的方向.
2015年, Cisnetti等[21]制备了水溶性的(NHC)Au, 并且将其用于亲水性的端炔(含羟基或乙酰氨基)的水合反应, 发现(NHC)AuCln(n=1或3)/MeOH催化体系在无需酸或者银盐助剂的条件下, 无论Au(Ⅲ)还是Au(Ⅰ), 都能很好地催化水合反应(Eq. 16), 且官能团的兼容性很好.该反应体系无需酸和银盐助剂.结合动力学和DFT研究, 提出了金催化炔烃水合反应的机理(Scheme 3).
 图式 3
(NHC)Au(Ⅰ)和(NHC)Au(Ⅲ)催化炔烃水合反应机理
Scheme3.
Proposed alkyne hydration pathway for (NHC)Au(Ⅰ) and (NHC)Au(Ⅲ) complexes
图式 3
(NHC)Au(Ⅰ)和(NHC)Au(Ⅲ)催化炔烃水合反应机理
Scheme3.
Proposed alkyne hydration pathway for (NHC)Au(Ⅰ) and (NHC)Au(Ⅲ) complexes
Hashmi等[22]用异腈基金与一级或二级胺反应制备了一系列非环状的含氮卡宾金络合物, 此类催化剂在有共催化剂AgSbF6、助剂三氟醋酸(TFA)存在下, 在室温就能很好地将苯乙炔转化为苯乙酮.催化性能好于很多文献报道的Au(Ⅰ)催化剂.但是该催化体系不仅需要银盐助剂, 还需要酸助剂.
最近Zuccaccia等[23]研究了(NHC)Au催化的炔烃水合反应.该反应体系无需酸助剂、银助剂和溶剂, 在室温或50 ℃的温和条件下即可实现炔烃的水合.其催化性能要远好于其他无酸、无银盐助剂的金催化体系.研究还发现该催化体系的关键在于金催化剂的阴离子, 只有当阴离子为配位能力和碱性较强的OTf -和NTf2-时, 催化剂性能很好(Eq. 17).反之如阴离子的配位能力和碱性较弱时, 如OTs-和TFA-等时, 反应几乎不进行.但该催化体系必须在助剂NBu4OTf存在下, 反应才能快速进行.
除了具有有机配体的Au(Ⅰ)催化剂, 利用简单一价金盐催化炔烃水合反应的报道很少. Hong等[24]利用AuCl/MeOH催化体系, 在没有其他配体和酸等助剂的条件下, 很好地实现了炔烃水合反应.该催化体系对端炔具有较好的催化性能(Eq. 18).该方法既无需配体和酸等助剂, 也没有复杂的催化剂制备过程.
1.2 Au(Ⅲ)催化炔烃水合反应
早在2002年, Raubenheimer等[25]制备了噻唑基Au(Ⅲ)离子液体(IL), 并利用该离子液体得到了首个噻唑基卡宾金化合物.发现该离子液体可在苯乙炔水合反应中具有催化剂和溶剂的双重作用, 并且离子液体具有容易回收再利用的特点.但是以该离子液体为溶剂和催化剂时不如用咪唑型离子液体为溶剂、氯金酸钠为催化剂的催化效果.在30 ℃时, 苯乙炔水合反应产率最高仅75%, 由于该离子液体稳定性差, 所以升高温度水合反应产率降低.之后很少有关于金离子液体催化炔烃水合反应的报道, 但是利用各种金络合物和卡宾金化合物催化炔烃水合反应的报道却很多.
Nolan等[26]利用(NHC)Au(Ⅰ)合成了一系列的(NHC)Au(Ⅲ), 并首次用(NHC)Au(Ⅲ)Br3催化炔烃水合反应. (IPr)AuBr3/MeOH体系对富电子芳香端炔具有很好的催化性能(Eq. 19), 对脂肪族端炔活性较低, 对内炔完全没有活性.当加入1 equiv.的AgPF6后, 将(NHC)Au的用量由10 mol%降至2 mol%, 苯乙炔水合反应时间由24 h缩短为1 h, 苯乙酮的产率却由95%提高到99%, 说明AgPF6能极大地提高催化剂的性能.
Laguna等[27]研究无机和有机Au(Ⅰ)、Au(Ⅲ)络合物催化苯乙炔水合反应.发现有机Au(Ⅲ)络合物在中性条件下能有效地催化炔烃水合反应, 并且在络合物中至少有一个氯离子与中心金属配位才能有效地催化炔烃水合反应; 反应中加入酸能够提高催化剂的催化性能, 其原因除了酸本身能催化炔烃水合反应之外, 酸还能有效地抑制络合物分解而转化为没有催化活性的单质金.通过光谱研究指出该反应的催化活性中心是Au(Ⅲ), 而不是Au(Ⅰ).并提出了Au(Ⅲ)催化的炔烃水合反应机理(Scheme 4).包括: (1)炔烃碳碳叁键与Au(Ⅲ)配位, (2)水分子的氢氧根亲核进攻活化的碳碳叁键, (3)烯醇式结构互变异构, (4)烷基金络合物的质子解.
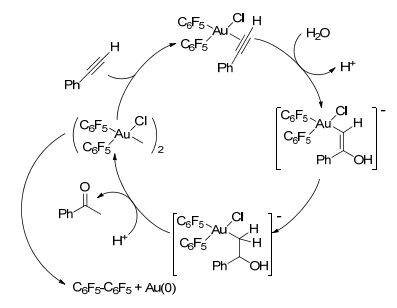 图式 4
[Au(C6F5)2Cl]2催化苯乙炔水合反应机理
Scheme4.
Mechanism of [Au(C6F5)2Cl]2-catalyzed hydration of phenylacetylene
图式 4
[Au(C6F5)2Cl]2催化苯乙炔水合反应机理
Scheme4.
Mechanism of [Au(C6F5)2Cl]2-catalyzed hydration of phenylacetylene
金催化炔烃水合反应通常都是利用结构复杂的金络合物, 此类催化剂活性较高, 但是制备麻烦, 价格高.而利用结构简单且容易获得的Au(Ⅲ)盐催化炔烃水合反应的研究很少. Vasudevan等[28]发现微波辐射下炔烃在过热水中即可实现水合反应.但该方法有明显的电子效应, 仅对富电子的芳基端炔有很好的效果.当在此体系中加AuBr3催化剂时, 反应会取得更高的产率, 但仍然仅对富电子芳炔有效.而Pu等[29]在最近直接利用NaAuCl4•2H2O为催化剂, 在混和溶剂中水合得到了各种β-胺基酮(Eq. 20).该方法虽然产率不是很高, 但反应条件温和, 催化剂易得, 且反应物的手性不受影响.
1.3 Au(0)催化炔烃水合反应
Au(0)曾被认为对炔烃水合反应没有催化活性[27].但Li课题组[30]在前期Au(Ⅰ)-PPh2-PMO(Ph)研究的基础上, 利用含巯基硅烷与乙基桥联硅烷共聚得到有序介孔有机硅材料SH-PMO(Et), 然后通过有机硅材料中的巯基与氯金酸发生氧化还原反应得到有序介孔催化剂Au-
HS@SO3H-PMO(Et) (Scheme 5).有序介孔硅材料的巯基能将Au(Ⅲ)原位还原为单质金, 而巯基则被氧化为磺酸基, 还原后的单质金以纳米颗粒的形式均匀分散在介孔材料孔壁的内表面.金颗粒分布在内表面能有效地防止金在催化反应过程中从介孔材料上脱落, 而且生成的磺酸基本身对炔烃的水合反应具有一定的催化作用.研究发现Au-HS@SO3H-PMO(Et)双功能催化剂对炔烃水合反应具有优秀的催化性能, 其催化性能要明显高于只用金催化剂或磺酸类催化剂.并且由于Au-HS@SO3H-PMO(Et)介孔材料的两亲性, 所以反应在水相介质或者有机相介质中都能顺利进行.在底物普适性方面, 对各类炔烃, 包括芳香族、脂肪族端炔和对称内炔以及单芳基取代的不对称内炔(苯基甲基乙炔)都有非常优秀的催化性能(Eq. 21).该催化剂重复使用10次之后活性才略有下降. Li课题组[31]又在此基础上开展了Au-SH@SO3H-SBA-15催化炔烃水合反应的研究, 也取得了很好的结果.
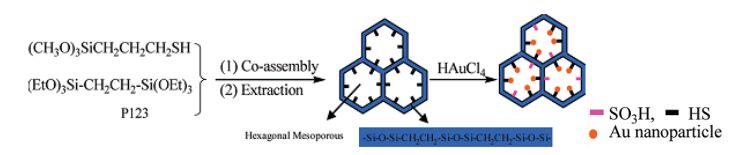 图式 5
Au-SH@SO3H-PMO(Et)有序介孔催化剂的制备[30]
Scheme5.
Illustration of the preparation of the Au-SH@SO3H-PMO(Et)
图式 5
Au-SH@SO3H-PMO(Et)有序介孔催化剂的制备[30]
Scheme5.
Illustration of the preparation of the Au-SH@SO3H-PMO(Et)
2 银催化炔烃水合反应
在炔烃水合反应中, 银盐经常以金催化剂的助剂形式出现, 而直接利用银盐为催化剂的研究报道相对较少. 2012年, Mann等[32]开展了AgSbF6催化的端基炔烃水合反应研究, 发现该催化体系在无需配体和酸等助剂的条件下, 就能高效地将炔烃水合为酮(Eq. 22).研究发现银盐的阴离子对催化性能的影响很大.该催化体系中水合反应的底物普适性广, 而且电子效应对反应几乎没有影响, 羧基、羟基、氯、氰基、烯基、酰氨基等官能团都有好的兼容性.由于2-乙炔基吡啶的氮和催化剂发生配位, 会使催化剂失去活性, 所以没有水合产物生成.
Bera等[33]用含氟的芳基硼酸银催化炔烃的水合反应.发现该催化剂对芳香族端炔具有很好的催化活性, 对脂肪族端炔活性较低(Eq. 23), 而对称内炔完全没有活性.但该催化体系无需任何助剂和有机溶剂.
Lingaiah等[34]在无溶剂条件下, 利用银离子部分交换的硅钨酸(AgxSTA)催化炔烃的水合反应.发现硅钨酸(AgxSTA)中, 当x=3时具有最优的催化性能. AgxSTA(0<x<4)很好地保持了STA的Keggin结构, Ag交换量的增多, AgxSTA的B酸位减少, L酸位增多, 由于银离子含量的增多增加了质子的流动性, 因此增大了B酸的酸强度, 当x=3时, 酸强度达到最大.发现催化剂的催化性能与其酸性有关, 并且B酸和L酸位对催化性能都有贡献.由于银离子的掺入, Ag3STA具有很好的热稳定性, 而且很容易回收利用. Ag3STA催化体系对端炔具有很好的催化性能(Eq. 24), 其中脂肪族端炔活性要低一些.对称的芳基内炔完全不反应, 单芳基取代的不对称内炔仅有很低的活性.并提出了Ag3STA催化炔烃水合反应的机理(Scheme 6).其催化机理与Au(Ⅲ)催化的机理基本一致.
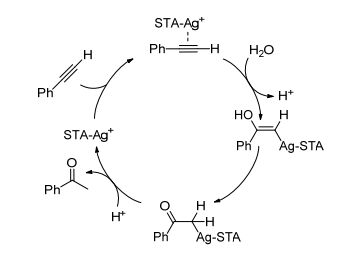 图式 6
Ag3STA催化的炔烃水合反应机理
Scheme6.
Mechanism of Ag3STA-catalyzed hydration of alkynes
图式 6
Ag3STA催化的炔烃水合反应机理
Scheme6.
Mechanism of Ag3STA-catalyzed hydration of alkynes
Srivastava等[35]利用水热法合成的锆硅纳米晶沸石为载体, 负载银纳米颗粒后催化端炔水合反应.该催化体系是首例利用单质银为活性组分的报道.此催化体系无需其他助剂, 其活性高的原因既有活性组分纳米银的催化作用, 还有载体表面B酸和L酸的共同作用.纳米银在反应中的作用是活化碳碳叁键, 以利于水分子的亲核进攻.此催化体系对端炔均有活性(Eq. 25), 但脂肪族端炔活性较芳香族端炔活性低.催化剂重复利用5次活性几乎没有下降.负载银后催化水合反应的收率比只用载体时仅增加了20%左右, 说明纳米银的催化活性不是很高, 但是此研究打开了单质银催化炔烃水合的研究路线.
3 钯催化炔烃水合反应
钯是有机反应中常用的催化剂, 但是钯催化炔烃水合反应的研究相对较少[36].早在1983年, Utimoto等[37, 38]就用PdCl2为催化剂, 在乙腈的水溶液中将含有羟基或羰基的脂肪族内炔转化为酮.在回流条件下反应30~60 min, 炔烃能高效地水合为酮, 产率大于80%, 对羟基兼容性也很好.在催化剂为PdCl2(MeCN)2[39], 且用量为5 mo%的条件下, 室温下进行超声辐射, 各种炔基酮都能很好地进行水合反应, 且反应的区域选择性很高, 产率在27%~99%, 个别底物有较多的异构体生成, 造成产率较低(27%).
Xu等[40]以PdCl2/二氧六环催化体系将取代的炔基磷酸酯水合为相应的酮酸酯.该催化体系对脂肪族、芳香族炔基磷酸酯都具有很好的活性(Eq. 26).对羟基、酯基、卤素等官能团也都有很好的兼容性.
我们课题组[41]研究了零价钯催化芳基端炔水合反应.发现采用Pd(PPh3)4/HCl催化体系时, 反应不仅有明显的电子效应, 而且取代基的位置效应也很明显(Eq. 27).并提出了零价钯催化的炔烃水合反应机理(Scheme 7).包括: (1) Pd(PPh3)4发生配体的解离, 生成Pd(PPh3)2; (2) Pd(PPh3)2与盐酸发生氧化加成, 生成钯氢中间体; (3)炔烃碳碳叁键插入钯氢键, 生成烯基钯中间体; (4)烯基钯中间体发生配体交换; (5)发生还原消除, 生成烯醇式中间体, 并实现Pd(PPh3)2的再生.而烯醇式中间体发生互变异构, 生成水合产物酮.
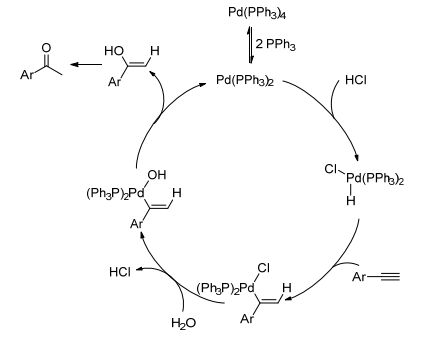 图式 7
Pd(PPh3)4催化炔烃水合反应的机理
Scheme7.
Proposed mechanism of Pd(PPh3)4-catalyzed hydration reaction of alkynes
图式 7
Pd(PPh3)4催化炔烃水合反应的机理
Scheme7.
Proposed mechanism of Pd(PPh3)4-catalyzed hydration reaction of alkynes
4 铂催化炔烃水合反应
早在1990年Jennings等[42, 43]就以Pt(Ⅱ)为中心金属的Zeise二聚体为催化剂研究, 炔烃水合反应.发现此催化体系对脂肪族端炔、内炔都具有很好的催化性能(产率30%~100%), 但对内炔的区域选择性不高.并用PtCl2、PtBr2、PtI2为催化剂研究了非活化内炔的水合反应, 发现它们对脂肪族内炔均有催化活性.由于PtBr2在四氢呋喃(THF)中溶解性较差, 所以催化炔烃水合反应的速率最低, 但反应区域选择性最高, 说明低的反应速率有助于提高反应的区域选择性.
Scarso等[44]用含双膦胺配体(PNP)的Pt(Ⅱ)高效地催化炔烃的水合反应.发现PNP配体上的取代基会对炔烃水合反应产生影响.当连有双邻甲氧基苯基的PNP为配体时催化剂的性能最佳.在反应介质的研究中发现, 当在水中添加表面活性剂, 特别是十二烷基苯磺酸钠(SDS)加入时, 反应效果有了明显的提升.其原因是表面活性剂加入后形成的胶束对催化剂和炔烃具有较大的溶解度, 因此可提供给炔烃与催化剂更多的接触机会, 从而实现高效催化反应(Eq. 28).而固体因在胶束中的溶解度太小, 所以固态的芳香炔烃较液态芳香炔烃水合反应的结果差很多.
Blum等[45]将PtCl4用CO进行预处理, 用处理后的铂羰基络合物催化炔烃水合反应.发现PtCl4水溶液在CO气氛下处理时, 会生成H2[Pt3(CO)6]n(n=5, 6), 然后H2[Pt3(CO)6]n会与Pt盐分解生成的HCl反应生成HPtCl(CO)2, 而HPtCl(CO)2是催化炔烃水合反应的活性组分.该催化剂体系对端炔、内炔具有很好的催化活性(产率61%~100%), 但部分不对称内炔的区域选择性不高.他们[46]又将PtCl4在不做预处理的条件下直接在CO气氛中催化炔烃的水合反应.发现羰基化合物能够促进炔烃的水合反应.但该方法对电子效应和位阻效应敏感, 而少量膦配体的加入, 例如手性膦配体(DIOP)、聚苯乙烯基二苯基膦, 有利于水合反应的进行.
5 钴催化炔烃水合反应
Lei等[47]利用Salen-Co(Ⅲ)络合物催化炔烃水合反应, 发现这三种Salen-Co络合物均可催化炔烃水合反应, 但与17、18相比, 19为水溶性的, 因此反应后19可直接通过萃取的方法与产品分离, 实现催化剂的循环使用. Salen-Co/H2SO4/MeOH催化体系对富电子芳基端炔具有很好的活性(Eq. 29), 脂肪族端炔效果较差, 内炔不反应.但该催化体系需要H2SO4作共催化剂.
2012年, Naka等[48]用水溶性钴卟啉络合物催化炔烃水合反应(Eq. 30).发现钴卟啉络合物/HNTf2/MeOH催化体系仅适用于端炔, 但催化剂用量很低, 反应收率高, 在该催化体系中, 电子效应对反应几乎无影响, 且烷硅醚、烯丙醚、羧酸酯、苄醚、硼酸酯、酰胺、碘、氰基、缩醛基等对酸/碱或氧化还原敏感的官能团都具有很好的兼容性.但该催化体系仍需要HNTf2作为助剂, 并且催化剂所用配体卟啉价格昂贵, 这会极大地影响该催化剂的广泛应用.
最近, Li等[49]以钴肟[Co(dmgBF2)2•2H2O]为催化剂, 以端炔为原料高收率地得到了各种甲基酮(Eq. 31).该方法的优点是无需酸和银盐助剂, 催化剂价廉且可大量制备, 反应条件温和, 对烯丙醚、苄醚、酰胺、羧酸酯、卤素、硝基等官能团都具有很好的兼容性.并根据氘代甲醇溶剂中进行的实验, 利用GC-MS和XPS表征结果提出了钴催化炔烃水合反应的机理(Scheme 8).
Lin等[50]发现In-Co(Ⅲ)(TBP)-MOFs催化端炔水合反应的效果要明显优于均相的Na3[Co(TPPS)][48]和Co(Ⅲ)(TBP)催化剂, 也明显优于与In-Co(Ⅲ)(TBP)-MOFs结构类似的In-In(TBP)-MOF和Zr-Co(TBP)-MOF催化剂.其原因是在In-Co-MOFs催化剂结构中, 平行的两个网状结构中的Co(TBP)之间的距离为8.8 Å, 而此距离恰好是同时活化反应底物炔烃和甲醇的理想距离, 并给出了催化反应的可能机理(Scheme 9).首先Co(TBP)作为Lewis酸与炔烃的碳碳叁键配位使叁键活化, 而相邻的Co(TBP)又与MeOH配位, 然后配位后的甲醇亲核进攻碳碳叁键得到烯基醚, 最后烯基醚水解为甲基酮.该催化体系的优点是催化剂用量低, 仅0.1~0.2 mol%, 无需其他添加剂, 反应条件温和.但是反应速率较慢, 反应时间需要一天或更长, 仅适用于端炔, 并且芳香族端炔取代基位置效应明显, 取代基为对位时, 能高产率的得到产物(Eq. 32), 而为邻位和间位时, 几乎没有产物生成, 这应该是由于邻位、间位取代的炔烃较对位取代炔烃的分子尺寸更大, 所以受两个相邻的Co(TBP)之间的距离为8.8 Å所限制.此工作的重要性更多地在于展示了MOFs的协同催化作用, 并为高效的多相催化剂设计提供了一种新的策略.
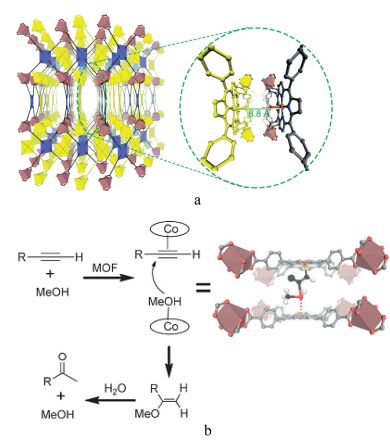 图式 9
(a) In-Co(Ⅲ)(TBP)-MOFs结构及(b) In-Co(Ⅲ)(TBP)-MOFs协同催化的炔烃水合水合反应的可能机理[50]
Scheme9.
(a) Structure of In-Co(Ⅲ)(TBP)-MOFs and (b) proposed mechanism for cooperative activation in In-Co(Ⅲ)-(TBP)-MOFs)-catalyzed hydration of alkynes
图式 9
(a) In-Co(Ⅲ)(TBP)-MOFs结构及(b) In-Co(Ⅲ)(TBP)-MOFs协同催化的炔烃水合水合反应的可能机理[50]
Scheme9.
(a) Structure of In-Co(Ⅲ)(TBP)-MOFs and (b) proposed mechanism for cooperative activation in In-Co(Ⅲ)-(TBP)-MOFs)-catalyzed hydration of alkynes
6 铁催化炔烃水合反应
铁用于催化炔烃水合反应研究较晚, 2009年Darcel等[51]报道了FeCl3催化的炔烃水合反应.发现FeCl3催化时炔烃能够高选择性地水合为酮, 且不需要酸等各种助剂.此催化体系仅对端炔有效(Eq. 33), 包括富电子和贫电子端炔, 贫电子端炔活性略低.而内炔和连有羟基、乙酰基的脂肪族端炔在该条件下完全不反应, 此结果与Pastel等[52]的实验结果基本一致.炔烃中含有羟基、乙酰基时水合反应不能进行, 其原因可能是因为这些基团和FeCl3发生配位, 从而失去催化活性.
Lee等[53]用FeCl2/CH3SO3H/DCE催化体系进行炔烃水合反应的研究.该催化剂对端炔、对称内炔、单芳基取代的不对称内炔都具有很好的活性(Eq. 34), 且不对称内炔的区域选择性很好.而且催化剂比较廉价易得, 但CH3SO3H的用量较大.
Bassetti等[54]用Fe2(SO4)3/AcOH催化体系在95 ℃的条件下实现了芳香族端炔的水合(Eq. 35).但在该催化体系中, 电子效应影响很大, 贫电子芳基端炔产率很低, 而且反应采用B酸作溶剂.
Leyva-Pérez等[55]在AgNTf2为助剂的条件下, 用FeCl3催化炔烃的水合反应.该催化体系使各种端炔和对称内炔高效地转化为水合产物酮(Eq. 36).反应中催化剂和助剂反应生成[Fe(NTf2)3] (Scheme 10), 弱配位的三氟酰亚氨阴离子提高了Fe3+的亲碳性, 这是其具有高催化活性的关键.并且[Fe(NTf2)3]在反应条件下会发生水解反应, 生成具有催化活性的B酸, 因此FeCl3/AgNTf2催化体系是一个L酸和B酸共同催化.
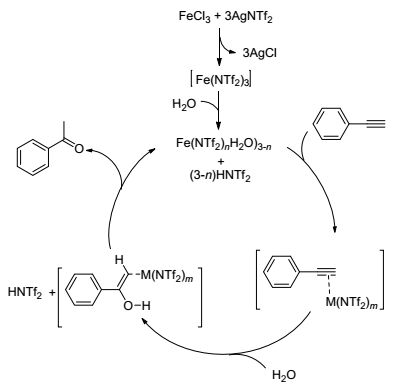 图式 10
[Fe(NTf2)3]催化的苯乙炔水合反应机理
Scheme10.
Tentative mechanism for the Markovnikov hydration of phenylacetylene catalyzed by [Fe(NTf2)3]
图式 10
[Fe(NTf2)3]催化的苯乙炔水合反应机理
Scheme10.
Tentative mechanism for the Markovnikov hydration of phenylacetylene catalyzed by [Fe(NTf2)3]
7 铑催化炔烃水合反应
20世纪60年代, 就有关于Rh(Ⅲ)络合物催化炔烃水合反应的报道[56].之后关于铑催化炔烃水合反应的报道就很少见了.直到2015年, Xu等[57]报道了首例铑催化下可见光促进的炔烃水合反应, 该催化体系对脂肪族和芳香族端炔均具有很好的催化活性(Eq. 37).通过ESI-MS观察到了反应的铑-酮中间体, 并利用合成的铑-酮中间体进行了动力学实验.提出了催化可见光促进的炔烃水合反应机理(Scheme 11), 包括(1)炔烃与催化剂铑(Ⅲ)卟啉络合物配位, 之后甲醇亲核进攻, 生成β-缩醛烷基铑(Ⅲ)卟啉; (2)发生缩醛和酮的异构化, 生成铑(Ⅲ)-酮中间体; (3)铑(Ⅲ)-酮中间体发生光解, 生成PhCOCH2自由基和铑(Ⅱ)络合物; (4)铑(Ⅱ)络合物与甲醇反应生成铑(Ⅲ)卟啉络合物和CH3O-Rh络合物, 实现催化循环.该研究为将可见光应用在炔烃水合反应的研究提供了很好的研究思路.
8 Brønsted酸催化炔烃水合反应
2004年, Alami等[58]以TsOH为催化剂进行不对称炔烃的水合反应(Eq. 38).该体系能够很好地催化单芳基取代的不对称内炔水合, 并且总是生成羰基区域选择性的靠近芳环的水合产物.当用水作溶剂时, 羟基官能团具有很好的兼容性.
Cai等[59]用TsOH/AcOH催化体系实现了炔烃的水合反应(Eq. 39).该催化体系对端炔和部分单芳基取代的不对称内炔都能实现高效、高区域选择性的水合, 包括富电子和贫电子炔烃.但TsOH和AcOH用量较大.
2012年, Srivastava等[60]制备了一系列咪唑基B酸离子液体(BAIL), 并研究了对炔烃水合反应的催化性能(Eq. 40).发现离子液体的酸性与催化活性基本一致, 酸性越强, 活性越高, 而且磺酸基官能团化的离子液体比没有磺酸基的离子液体具有更高的催化活性.该催化体系对端炔有较好的活性, 对对称内炔活性较差.通过理论计算提出BAIL催化炔烃水合反应的机理.首先碳碳叁键质子化, 然后水分子亲核进攻生成烯醇式结构, 之后烯醇式互变异构, 最后酮式结构发生质子解, 生成产物酮, 同时催化剂再生.
Blum等[61]在水中加入表面活性剂形成微乳液, 然后用盐酸作催化剂, 将炔烃高选择性地水合为酮(Eq.41).该催化体系对芳香炔非常适用, 包括端炔和内炔.脂肪族端炔和单芳基取代的不对称内炔活性很低.但不对称内炔反应的区域选择性很高.
Wong等[62]在离子液体为溶剂的条件下直接用H2SO4催化炔烃水合反应(Eq. 42).反应在40 ℃的温和条件下仅需0.5 h就能高效、高产率地得到水合产物酮.此催化体系高效的原因可能是在离子液体中酸的质子具有更高的化学活性.此方法适用于端炔和对称内炔, 但催化剂用量很大.
Hammond等[63]报道了组合酸(LBA和BBA)/AcOH催化体系, 其中AcOH为溶剂兼共催化剂、少量L酸[0.2 mol% Ga(OTf)3]或B酸(3%含三氟磺酸基的Nafion)为共催化剂的条件下, 实现了端炔和对称内炔的高效水合(Eq. 43).固载化的B酸催化剂重复利用3次活性没有下降.但此催化体系采用B酸(AcOH)作溶剂.
Li等[64]在室温下, 利用TfOH/CF3CH2OH催化体系研究了炔烃水合反应(Eq. 44).该反应体系对于脂肪族端炔、内炔均有很好的催化活性, 且单芳基取代的不对称内炔(苯基乙基乙炔)反应的区域选择性很高.取代基的电子效应对反应几乎没有影响, 酯基、羧基、羟基、卤素、氰基和氨基等官能团在反应中都具有很好的兼容性, 但反应时间较长.并通过DFT研究发现, 炔烃水合反应的质子化过程在溶剂CF3CH2OH中具有比乙醇中更低的能垒.所以CF3CH2OH的使用也是该催化体系性能很好的关键所在.
Wang等[65]以没食子酸为催化剂实现了炔烃在温和条件下的高效水合(Eq. 45).该催化体系无需有机溶剂、无需金属催化剂和其他强酸为助剂, 对脂肪族和芳香族端炔均适用, 氯、羟基和羧基等官能团在反应中具有很好的兼容性, 该催化剂循环使用5次活性没有明显降低.而且发现天然产物鞣酸对此反应也有很好的催化性能, 这打开了炔烃的水合反应体系绿色化通道.
Tanemura等[66]利用磺酸化的缩合多核芳烃树脂(S-COPNA)催化炔烃水合反应(Eq. 46).该B酸催化剂用量较少, 而且作为固体酸催化剂容易回收利用, 循环使用5次活性几乎没有下降.使用相同量H+的条件下, 其催化活性明显高于H2SO4、TfOH、TsOH和C8F17SO3H.但该催化体系主要适用于芳基端炔, 对脂肪族端炔和对称内炔完全没有活性.
Nama等[67]以Hβ分子筛为催化剂, 在无需有机溶剂条件下实现了炔烃水合(Eq. 47).此催化剂不仅适用于端炔, 对单芳基取代的不对称内炔也能以优秀的区域选择性获得酮, 但吡啶基乙炔却完全没有水合产物生成.该方法无需银盐和酸助剂, 催化剂重复使用3次活性没有明显降低.
Yun等[68]合成了同时含有微孔、介孔和大孔, 并具有超强酸性的Y型纳米沸石.发现该沸石无需有机溶剂的条件下对芳香族端炔水合反应具有很好的催化活性(Eq. 48).其活性除了源于表面的大量酸性位点外, 还源于其具有的介孔和大孔结构有利于反应物质的传质.该催化剂重复使用6次活性几乎没有下降.
9 其他催化剂催化炔烃水合反应
除了金、银、钯、铂、钴、铁、铑和B酸催化炔烃水合反应的报道外, 还有钌、铱等金属络合物催化剂和氧化物型催化剂的报道.在2004年, Gimeno等[69]合成了一系列有机膦配位的Ru(Ⅱ)络合物.此类络合物催化脂肪族和芳香族端炔(1-辛炔和苯乙炔)水合为酮的产率最高为84%和89%.其中脂肪族端炔反应时生成较多的反马氏产物醛, 但产物仍以马氏产物为主.
Ishii等[70]在Ir(Ⅰ)为催化剂、有机膦为配体、L酸为助剂和正丁醇为溶剂的条件下, 发现非活化的端炔均能顺利地水合为酮(Eq. 49), 而内炔完全没有活性.
Jha等[71]在微波作用下利用Cu(OTf)2/AcOH催化体系将各种芳香族端炔水合得到了目标产物酮(Eq. 50).该催化体系的特点是催化剂非贵金属、用量低, 反应快速, 但反应温度较高, AcOH在作溶剂的同时, 还起到了助剂的作用, 该催化体系虽然对酯基和醛基具有兼容性, 但是产率很低.
Wu等[72]用SnCl4•5H2O/MeOH催化体系实现了端炔的高效水合(Eq. 51), 该催化体系无需酸和银盐作助剂, 但是催化剂用量较多, 反应温度较高.
李等[73]利用Ce(SO4)2/H2SO4/Benzene催化体系进行炔烃的水合反应研究(Eq. 52).此催化体系对所研究的端炔都具有活性, 但电子效应明显, 贫电子芳炔产率很低, 而含有酯基的端炔和内炔均不反应. Xiang等[74]用此催化体系催化卤代炔烃水合反应制备了各种卤代酮(Eq. 53).该催化体系对含有强吸电子和含有给电子的的芳基炔烃都具有很好的适用性.并且催化剂廉价易得, 但反应仍需要较多的酸作为助剂.
Bielawski等[75]首次用氧化石墨烯催化炔烃水合反应(Eq. 54).在无需助剂的条件下, 氧化石墨烯能够很好地将芳香族端炔转化为酮, 而对脂肪炔、二炔和内炔的催化性能一般.该催化剂不含金属, 并且通过简单的过滤即可实现回收再重复利用.
Mizuno等[76]在高温下焙烧得到Sn-W混合氧化物催化剂.发现在焙烧温度为800 ℃, Sn/W(物质的量比)为2时, 催化剂B/L酸比值达最大, 此催化剂在炔烃水合反应中性能也达到最佳(Eq. 55).其性能明显高于之前报道的多相催化剂.该混合氧化物的催化性能主要源于催化剂表面的B酸位.该催化体系对端炔和内炔都适用, 并且取代基的电子效应对反应没有明显影响.
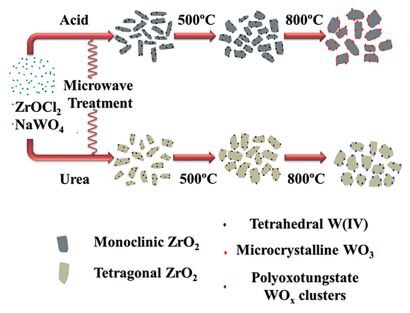
Corma等[77]利用微波一步法制备了WOx-ZrO2纳米固体酸催化剂(Scheme 12).发现加入尿素合成的WOx-ZrO2对苯乙炔水合反应具有更好的催化活性.催化活性相是钨酸盐族、WO3微晶和四方相的ZrO2.四方相ZrO2提高了钨物种的酸性特征, 从而更有利于催化水合反应.
10 总结及展望
催化炔烃水合反应已有大量的研究和报道, 所用催化剂主要包括金属络合物催化剂、B酸、负载型氧化物催化剂等.其中尤以金属络合物催化剂研究最多, 包括对活性组分金属离子的研究及配体和助剂等的研究.实现了各种脂肪族端炔、芳香族端炔以及对称内炔为底物的水合反应, 但是炔烃水合反应的研究仍存在诸多不足, 比如多种金属络合物为催化剂时, 必须由银盐或者B酸等作助剂的问题, 廉价、高效的金属催化剂还较少, 在室温下能高效催化反应的催化剂较少, 很多高效的均相催化剂不能循环利用, 底物普适性广的催化体系较少, 最具挑战性的是不对称内炔(特别是双烷基取代的不对称内炔)的区域选择性控制问题.因此, 今后炔烃水合反应催化体系还有广阔的发展空间.比如高效、廉价金属催化剂的开发, 贵金属催化剂的固载化、具有高度区域选择性催化剂的开发以及环境友好B酸催化剂的开发等.相信随着科研工作者不断地努力, 将会有区域选择性更高、官能团兼容性更广、底物普适性更好、制备更简单、更廉价的炔烃水合反应催化体系不断出现.
-
-
[1]
Goodwin, J. A.; Aponick, A. Chem. Commun. 2015, 51, 8730. doi: 10.1039/C5CC00120J
-
[2]
Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Angew. Chem., Int. Ed. 2002, 41, 4563. doi: 10.1002/1521-3773(20021202)41:23<4563::AID-ANIE4563>3.0.CO;2-U
-
[3]
Ebule, R. E.; Malhotra, D.; Hammond, G. B.; Xu, B. Adv. Synth. Catal. 2016, 358, 1478. doi: 10.1002/adsc.201501079
-
[4]
Fujita, K.-I.; Kujime, M.; Muraki, T. Bull. Chem. Soc. Jpn. 2009, 82, 261. doi: 10.1246/bcsj.82.261
-
[5]
Zhu, F.; Zhang, F.; Yang, X.; Huang, J.; Li, H. J. Mol. Catal. A:Chem. 2011, 336, 1. doi: 10.1016/j.molcata.2010.11.020
-
[6]
Sanz, S.; Jones, L. A.; Mohr, F.; Laguna, M. Organometallics 2007, 26, 952. doi: 10.1021/om060821y
-
[7]
Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067. doi: 10.1021/jo802558e
-
[8]
Roembke, P.; Schmidbaur, H.; Cronje, S.; Raubenheimer, H. J. Mol. Catal. A:Chem. 2004, 212, 35. doi: 10.1016/j.molcata.2003.11.011
-
[9]
韩治军, 毛树兰, 彭晖, 皮云宵, 陈友, 刘盛华, 余广鳌, 有机化学, 2014, 34, 893. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201405007&dbname=CJFD&dbcode=CJFQHan, Z.; Mao, S.; Peng, H.; Pi, Y.; Chen, Y.; Liu, S.; Yu, G. Chin. J. Org. Chem. 2014, 34, 893(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201405007&dbname=CJFD&dbcode=CJFQ
-
[10]
Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448. doi: 10.1021/ja809403e
-
[11]
Nun, P.; Ramón, R. S.; Gaillard, S.; Nolan, S. P. J. Organomet. Chem. 2011, 696, 7. doi: 10.1016/j.jorganchem.2010.08.052
-
[12]
Czégéni, C. E.; Papp, G.; Kathó, Á.; Joó, F. J. Mol. Catal. A:Chem. 2011, 340, 1. doi: 10.1016/j.molcata.2011.03.009
-
[13]
Almássy, A.; Nagy, C. E.; Bényei, A. C.; Joó, F. Organometallics 2010, 29, 2484. doi: 10.1021/om1001292
-
[14]
Fernández, G. A.; Chopa, A. B.; Silbestri, G. F. Catal. Sci. Technol. 2016, 6, 1921. doi: 10.1039/C5CY01278C
-
[15]
Wang, W.; Zheng, A.; Zhao, P.; Xia, C.; Li, F. ACS Catal. 2014, 4, 321. doi: 10.1021/cs400983y
-
[16]
Weerasiri, K. C.; Chen, D.; Wozniak, D. I.; Dobereiner, G. E. Adv. Synth. Catal. 2016, 358, 4106. doi: 10.1002/adsc.201601013
-
[17]
Cavarzan, A.; Reek, J. N. H.; Trentin, F.; Scarso, A.; Strukul, G. Catal. Sci. Technol. 2013, 3, 2898. doi: 10.1039/c3cy00300k
-
[18]
Sorella, G. L.; Sperni, L.; Ballester, P.; Strukul, G.; Scarso, A. Catal. Sci. Technol. 2016, 6, 6031. doi: 10.1039/C6CY00307A
-
[19]
Xu, Y.; Hu, X.; Zhang, S.; Xi, X.; Wu, Y. ChemCatChem 2016, 8, 262. doi: 10.1002/cctc.201501065
-
[20]
Xu, Y.; Hu, X.; Shao, J.; Yang, G.; Wu, Y.; Zhang, Z. Green Chem. 2015, 17, 532. doi: 10.1039/C4GC01322K
-
[21]
Ibrahim, H.; Frémont, D. P.; Braunstein, P.; Théry, V.; Nauton, L.; Cisnetti, F.; Gautier, A. Adv. Synth. Catal. 2015, 357, 3893. doi: 10.1002/adsc.201500729
-
[22]
Hashmi, A. S. K.; Hengst, T.; Lothschütz, C.; Rominger, F. Adv. Synth. Catal. 2010, 352, 1315. doi: 10.1002/adsc.v352:8
-
[23]
Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. ACS Catal. 2016, 6, 7363. doi: 10.1021/acscatal.6b01626
-
[24]
Hong, S.; Das, A.; Park, S.; Muthaiah, S. Synlett 2015, 26, 2517. doi: 10.1055/s-00000083
-
[25]
Deetlefs, M.; Raubenheimer, H. G.; Esterhuysen, M. W. Catal. Today 2002, 72, 29. doi: 10.1016/S0920-5861(01)00475-8
-
[26]
Frémont, P.; Singh, R.; Stevens, E. D.; Petersen, J. L.; Nolan, S. P. Organometallics 2007, 26, 1376. doi: 10.1021/om060887t
-
[27]
Casado, R.; Contel, M.; Laguna, M.; Romero, P.; Sanz, S. J. Am. Chem. Soc. 2003, 125, 11925. doi: 10.1021/ja036049x
-
[28]
Vasudevan, A.; Verzal, M. K. Synlett 2004, 631.
-
[29]
Ying, J.; Pu, L. J. Org. Chem. 2016, 81, 8135. doi: 10.1021/acs.joc.6b01111
-
[30]
Zhu, F.-X.; Wang, W.; Li, H.-X. J. Am. Chem. Soc. 2011, 133, 11632. doi: 10.1021/ja203450g
-
[31]
Zhu, F.; Li, H. Chin. J. Chem. 2014, 32, 1072. doi: 10.1002/cjoc.201400408
-
[32]
Thuong, M. B. T.; Mann, A.; Wagner, A. Chem. Commun. 2012, 48, 434. doi: 10.1039/C1CC12928G
-
[33]
Saha, S.; Sarbajna, A.; Bera, J. K. Tetrahedron Lett. 2014, 55, 1444. doi: 10.1016/j.tetlet.2014.01.045
-
[34]
Rao, K. T. V.; Prasad, P. S. S.; Lingaiah, N. Green Chem. 2012, 14, 1507. doi: 10.1039/c2gc35130g
-
[35]
Sarmah, B.; Srivastava, R.; Satpati, B. ChemistrySelect 2016, 1, 1047. doi: 10.1002/slct.201600132
-
[36]
Meier, I. K.; Marsella, J. A. J. Mol. Catal. 1993, 78, 31. doi: 10.1016/0304-5102(93)87031-3
-
[37]
Utimoto, K. Pure Appl. Chem. 1983, 55, 1845.
-
[38]
Fukuda, Y.; Shiragami, H.; Utimoto, K.; Nozaki, H. J. Org. Chem. 1991, 56, 5816. doi: 10.1021/jo00020a024
-
[39]
Imi, K.; Imai, K.; Utimoto, K. Tetrahedron Lett. 1987, 28, 3127. doi: 10.1016/S0040-4039(00)96302-0
-
[40]
Li, X.; Hu, G.; Luo, P.; Tang, G.; Gao, Y.; Xu, P.; Zhao, Y. Adv. Synth. Catal. 2012, 354, 2427. doi: 10.1002/adsc.v354.13
-
[41]
Xu, C.; Du, W.; Zeng, Y.; Dai, B.; Guo, H. Org. Lett. 2014, 16, 948. doi: 10.1021/ol403684a
-
[42]
Hiscox, W.; Jennings, P. W. Organometallics 1990, 9, 1997. doi: 10.1021/om00157a005
-
[43]
Hartman, J. W.; Hiscox, W. C.; Jennings, P. W. J. Org. Chem. 1993, 58, 7613. doi: 10.1021/jo00078a056
-
[44]
Trentin, F.; Chapman, A. M.; Scarso, A.; Sgarbossa, P.; Michelin, R. A.; Strukul, G.; Wass, D. F. Adv. Synth. Catal. 2012, 354, 1095. doi: 10.1002/adsc.201100326
-
[45]
Baidossi, W.; Lahav, M.; Blum, J. J. Org. Chem. 1997, 62, 669. doi: 10.1021/jo961740m
-
[46]
Israelsohn, O.; Vollhardt, K. P. C.; Blum, J. J. Mol. Catal. A:Chem. 2002, 184, 1. doi: 10.1016/S1381-1169(01)00425-3
-
[47]
Wang, S.; Miao, C.; Wang, W.; Lei, Z.; Sun, W. Chin. J. Catal. 2014, 35, 1695. doi: 10.1016/S1872-2067(14)60105-4
-
[48]
Tachinami, T.; Nishimura, T.; Ushimaru, R.; Noyori, R.; Naka, H. J. Am. Chem. Soc. 2012, 135, 50.
-
[49]
Hou, S.; Yang, H.; Cheng, B.; Zhai, H.; Li, Y. Chem. Commun. 2017, 53, 6926. doi: 10.1039/C7CC03919K
-
[50]
Lin, Z.; Zhang, Z.-M.; Chen, Y.-S.; Lin, W. Angew. Chem. 2016 128, 13943.
-
[51]
Wu, X.-F.; Bezier, D.; Darcel, C. Adv. Synth. Catal. 2009, 351, 367. doi: 10.1002/adsc.200800666
-
[52]
Damiano, J. P.; Pastel, M. J. Organomet. Chem. 1996, 522, 303. doi: 10.1016/0022-328X(96)06294-8
-
[53]
Park, J.; Yeon, J.; Lee, P. H.; Lee, K. Tetrahedron Lett. 2013, 54, 4414. doi: 10.1016/j.tetlet.2013.06.015
-
[54]
Bassetti, M.; Ciceri, S.; Lancia, F.; Pasquini, C. Tetrahedron Lett. 2014, 55, 1608. doi: 10.1016/j.tetlet.2014.01.083
-
[55]
Cabrero-Antonino, J. R.; Leyva-Pérez, A.; Corma, A. Chem. Eur. J. 2012, 18, 11107. doi: 10.1002/chem.201200580
-
[56]
James, B. R.; Rempel, G. L. J. Am. Chem. Soc. 1969, 91, 863. doi: 10.1021/ja01032a012
-
[57]
Liu, X.; Liu, L.; Wang, Z.; Fu, X. Chem. Commun. 2015, 51, 11896. doi: 10.1039/C5CC04015A
-
[58]
Olivi, N.; Thomas, E.; Peyrat, J.-F.; Alami, M.; Brion, J.-D. Synlett 2004, 2175.
-
[59]
Cai, C.; Liu, H.; Wei, Y. Synlett 2016, 27, 2378. doi: 10.1055/s-0035-1562779
-
[60]
Kore, R.; Kumar, T. J. D.; Srivastava, R. J. Mol. Catal. A:Chem. 2012, 360, 61. doi: 10.1016/j.molcata.2012.04.010
-
[61]
Nairoukh, Z.; Avnir, D.; Blum, J. ChemSusChem 2013, 6, 430. doi: 10.1002/cssc.201200838
-
[62]
Wong, W.-L.; Ho, K.-P.; Lee, L. Y. S.; Lam, K.-M.; Zhou, Z.-Y.; Chan, T. H.; Wong, K.-Y. ACS Catal. 2011, 1, 116. doi: 10.1021/cs100016h
-
[63]
Liang, S.; Hammond, G. B.; Xu, B. Chem. Commun. 2015, 51, 903. doi: 10.1039/C4CC08938C
-
[64]
Liu, W.; Wang, H.; Li, C.-J. Org. Lett. 2016, 18, 2184. doi: 10.1021/acs.orglett.6b00801
-
[65]
Deng, T.; Wang, C.-Z. Catal. Sci. Technol. 2016, 6, 7029. doi: 10.1039/C6CY01629D
-
[66]
Tanemura, K.; Suzuki, T. Tetrahedron Lett. 2017, 58, 955. doi: 10.1016/j.tetlet.2017.01.072
-
[67]
Mameda, N.; Peraka, S.; Marri, M. R.; Kodumuri, S.; Chevella, D.; Gutta, N.; Nama, N. Appl. Catal. A:Gen. 2015, 505, 213. doi: 10.1016/j.apcata.2015.07.038
-
[68]
Xu, S.; Yun, Z.; Feng, Y.; Tang, T.; Fang, Z.; Tang, T. RSC Adv. 2016, 6, 69822. doi: 10.1039/C6RA11489J
-
[69]
Cadierno, V.; Crochet, P.; García-Garrido, S. E.; Gimeno, J. Dalton Trans. 2004, 3635.
-
[70]
Hirabayashi, T.; Okimoto, Y.; Saito, A.; Morita, M.; Sakaguchi, S.; Ishii, Y. Tetrahedron 2006, 62, 2231. doi: 10.1016/j.tet.2005.12.010
-
[71]
Jha, M.; Shelke, G. M.; Pericherla, K.; Kumar, A. Tetrahedron Lett. 2014, 55, 4814. doi: 10.1016/j.tetlet.2014.06.116
-
[72]
Chen, D.; Wang, D.; Wu, W.; Xiao, L. Appl. Sci. 2015, 5, 114. doi: 10.3390/app5020114
-
[73]
刘文杰, 李金恒, 有机化学, 2006, 26, 1073. doi: 10.3321/j.issn:0253-2786.2006.08.007Liu, W.; Li, J. Chin. Org. Chem. 2006, 26, 1073(in Chinese). doi: 10.3321/j.issn:0253-2786.2006.08.007
-
[74]
Zou, H.; Jiang, J.; Yi, N.; Fu, W.; Deng, W.; Xiang, J. Chin. J. Chem. 2016, 34, 1251. doi: 10.1002/cjoc.v34.12
-
[75]
Dreyer, D. R.; Jia, H.-P.; Bielawski, C. W. Angew. Chem., Int. Ed. 2010, 49, 6813.
-
[76]
Jin, X.; Oishi, T.; Yamaguchi, K.; Mizuno, N. Chem. Eur. J. 2011, 17, 1261. doi: 10.1002/chem.v17.4
-
[77]
Gonell, F.; Portehault, D.; Julián-López, B.; Vallé, K.; Sanchez, C.; Corma, A. Catal. Sci. Technol. 2016, 6, 8257. doi: 10.1039/C6CY01082B
-
[1]
-

图式 1 Pd-催化的Sonogashira偶联合成(NHC)Au@POPs
Scheme 1 Synthesis of (NHC)Au@POPs using the Pd-catalyzed Sonogashira coupling method

图式 2 杯芳烃10, 106•8H2O和(IPr)AuOTf络合物11的结构[17]
Scheme 2 Chemical structures of resorcin[4]arene 10, 106•8H2O and (IPr)AuOTf complex 11

图式 3 (NHC)Au(Ⅰ)和(NHC)Au(Ⅲ)催化炔烃水合反应机理
Scheme 3 Proposed alkyne hydration pathway for (NHC)Au(Ⅰ) and (NHC)Au(Ⅲ) complexes

图式 4 [Au(C6F5)2Cl]2催化苯乙炔水合反应机理
Scheme 4 Mechanism of [Au(C6F5)2Cl]2-catalyzed hydration of phenylacetylene

图式 5 Au-SH@SO3H-PMO(Et)有序介孔催化剂的制备[30]
Scheme 5 Illustration of the preparation of the Au-SH@SO3H-PMO(Et)

图式 7 Pd(PPh3)4催化炔烃水合反应的机理
Scheme 7 Proposed mechanism of Pd(PPh3)4-catalyzed hydration reaction of alkynes

图式 8 钴肟催化炔烃水合反应的可能机理[49]
Scheme 8 Proposed mechanism of cobaloxime-catalyzed hydration of alkynes

图式 9 (a) In-Co(Ⅲ)(TBP)-MOFs结构及(b) In-Co(Ⅲ)(TBP)-MOFs协同催化的炔烃水合水合反应的可能机理[50]
Scheme 9 (a) Structure of In-Co(Ⅲ)(TBP)-MOFs and (b) proposed mechanism for cooperative activation in In-Co(Ⅲ)-(TBP)-MOFs)-catalyzed hydration of alkynes

图式 10 [Fe(NTf2)3]催化的苯乙炔水合反应机理
Scheme 10 Tentative mechanism for the Markovnikov hydration of phenylacetylene catalyzed by [Fe(NTf2)3]

-




 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 43
- 文章访问数: 5230
- HTML全文浏览量: 1628
 下载:
下载:
