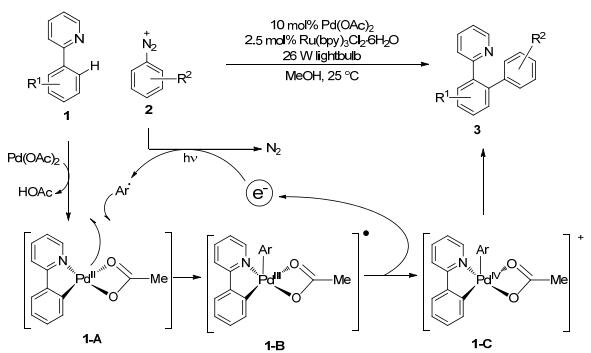
 图图1
经由光催化产生钯(Ⅳ)中间体的钯催化的C—H键官能团化反应
Scheme1.
Palladium-catalyzed C—H functionalization via Pd(Ⅳ) intermediates generated by photoredox catalysis
图图1
经由光催化产生钯(Ⅳ)中间体的钯催化的C—H键官能团化反应
Scheme1.
Palladium-catalyzed C—H functionalization via Pd(Ⅳ) intermediates generated by photoredox catalysis
引用本文:
吴江, 李嘉雯, 李昊, 朱纯银. 可见光催化促进的金属有机基元反应[J]. 有机化学,
2017, 37(9): 2203-2210.
doi:
10.6023/cjoc201704030

Citation: Wu Jiang, Li Jiawen, Li Hao, Zhu Chunyin. Visible Light Photoredox Catalysis Mediated Elementary Steps in Organometallic Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2203-2210. doi: 10.6023/cjoc201704030
Citation: Wu Jiang, Li Jiawen, Li Hao, Zhu Chunyin. Visible Light Photoredox Catalysis Mediated Elementary Steps in Organometallic Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2203-2210. doi: 10.6023/cjoc201704030
可见光催化促进的金属有机基元反应
English
Visible Light Photoredox Catalysis Mediated Elementary Steps in Organometallic Reactions
Abstract:
Many organometallic reactions involve three elementary steps including oxidative addition, transamination, and reductive elimination. But sometimes very high barrier exists for this approach, leading to the failure of the reactions. The key development in recent reports is the implementation of visible-light photocatalysts as a means to induce the desired redox processes in a mild and selective manner. In this context, organometallic reactions mediated by photoredox catalysts could give rise to novel reactivity, thus this area has drawn much attention in organic chemistry community. In this review, prominent examples from the recent literatures are organized on the basis of the elementary transformation enabled by photoredox catalysis.
-
Key words:
- organometallic reactions
- / elementary steps
- / visible light photoredox
-
金属有机反应是现代合成化学的重要基石之一, 可高效地构建各种碳碳键和碳杂键, 已被广泛地应用于药物、天然产物等复杂化合物的合成中[1].大部分金属有机反应通常包含氧化加成和还原消除等基元反应, 而这些基元反应一般会涉及到金属价态的改变.一直以来, 人们较多地依赖于金属和配体自身的特性来调控金属变价, 实现氧化加成和还原消除等基元反应.可见光催化方法学是近年来发展起来的一种强大的合成手段, 以其低能耗、条件温和、无污染等特点而广受关注, 给有机合成化学带来了新的机遇[2].这一催化模式具有高活性和氧化还原特性, 可以通过单电子转移的机制实现金属的氧化还原, 从而促进金属有机基元反应, 特别是那些仅依赖金属和配体自身能力而无法实现的基元反应.近年来, 很多文献报道了可见光催化促进的氧化加成、还原消除以及转金属化反应, 因此, 本文综述了这些反应的最新进展及其反应机理的探讨.
1 可见光催化促进的氧化加成
氧化加成是亲电组分与金属中心的成键作用, 同时伴随着金属中心形式上的双电子氧化, 大多数反应表现为碳卤键或类碳卤键对金属的一步协同的氧化加成.但是, 很多金属和亲电试剂经常无法通过这一协同的反应机理进行反应.而很多研究表明烷基自由基可以方便地加成到金属中心[3], 例如:光催化产生的异丙基自由基可以加速异丙基碘对Pt(Ⅱ)配合物的氧化加成[4]; 类似地, 氟代烷基碘也是通过链式的自由基机理对Au(Ⅰ)配合物进行氧化加成[5].基于此, 人们开始考虑利用光催化产生自由基促进氧化加成, 从而实现催化的有机合成.这方面最早的例子是2011年Sanford等[6]报道的钯催化的导向C—H键活化.他们利用芳基重氮盐在可见光催化下, 室温即可产生高价钯中间体, 无需高温加热的剧烈条件.所得Pd(Ⅳ)中间体1-C发生还原消除可以得到各种取代的芳基化产物.此法适用于不同的定位基团和芳基重氮盐, 以及二芳基碘盐(Scheme 1).
图图1
经由光催化产生钯(Ⅳ)中间体的钯催化的C—H键官能团化反应
Scheme1.
Palladium-catalyzed C—H functionalization via Pd(Ⅳ) intermediates generated by photoredox catalysis
随后这一策略被利用到了金催化的反应中, 人们通过芳基重氮盐或者二芳基碘盐可获得高活性的三价金中间体, 从而利用该中间体在温和的条件下实现了一系列的重要有机转化.例如, Glorius等[7]在2013年报道了利用金和光催化的组合催化策略实现的烯烃的双功能化反应.该反应适用于不同取代的含氧/氮烯烃和芳基重氮盐, 在室温和家用荧光灯的照射下, 可以合成各类芳基杂环化合物, 产率中等到良好(Eq. 1).
作者认为该反应的第一步是被金配位的双键4被分子内的氧或者氮亲核试剂进攻, 从而生成一价烷基金中间体4-A.接着, 该一价金中间体首先与芳基自由基加成为二价金中间体4-B, 随后被光催化剂单电子氧化生成三价金中间体4-C.最后通过还原消除得到目标产物, 同时再生一价金催化剂(Scheme 2).
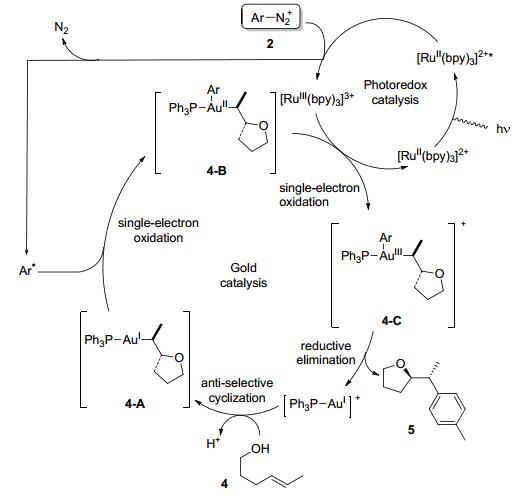 图图2
金和光催化剂共同催化的含氧/氮烯烃与芳基重氮盐的反应机理
Scheme2.
Proposed mechanism of the dual visible light photoredox and gold-catalyzed oxy-and aminoarylation of alkenes with aryldiazonium salts
图图2
金和光催化剂共同催化的含氧/氮烯烃与芳基重氮盐的反应机理
Scheme2.
Proposed mechanism of the dual visible light photoredox and gold-catalyzed oxy-and aminoarylation of alkenes with aryldiazonium salts
利用类似策略, 该课题组[8]还报道了分子间烯烃的醚化芳基化反应.该反应适用于不同取代的末端烯烃和芳基重氮盐或者二芳基碘盐, 在室温和家用荧光灯的照射下, 可以合成各类芳基杂环化合物, 个别底物产率可达优秀(Scheme 3).
 图图3
金和光催化剂共同催化的烯烃的醚化芳基化反应
Scheme3.
Dual visible light photoredox and gold-catalyzed oxyarylation of alkenes
图图3
金和光催化剂共同催化的烯烃的醚化芳基化反应
Scheme3.
Dual visible light photoredox and gold-catalyzed oxyarylation of alkenes
类似地, 最近Ollivier和Fensterbank等[9]报道了利用炔烃的醚化烷基化合成苯并呋喃的反应.该反应以各种取代的炔基苯酚和芳基重氮盐为底物, 条件温和, 产率良好到优秀.作者认为该反应也经历了光催化氧化产生三价金中间体的历程(Eq. 2).
可见光促进的氧化加成反应还被应用在一系列金催化的芳基化重排反应.例如, 2014年Toste小组[10]报道了利用金和光催化的组合策略实现的氧化芳基化扩环反应.该反应以不同的烯基或联烯基环醇, 和重氮盐为底物, 在金和光催化作用下, 可以制得各种官能团化的环酮(Eq. 3).
通过包括时间分辨FT-IR在内的各种机理实验的考察, 作者认为该反应首先通过光催化将芳基重氮盐还原为芳基自由基, 而该自由基可与一价金催化剂反应生成二价金中间体.接着, 二价金中间体又经光催化氧化为三价金中间体, 同时再生光催化剂.最后, 三价金中间体与π键配位, 导致开环和随后的还原消除而生成最终产物(Scheme 4).
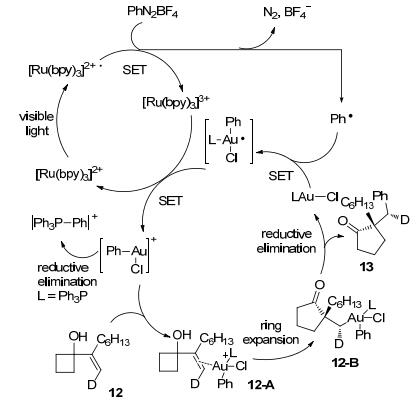 图图4
光催化促进的、金催化氧化芳基化扩环反应机理
Scheme4.
Proposed mechanism of the photoredox-promoted, gold-catalyzed ring expansion-arylation reaction
图图4
光催化促进的、金催化氧化芳基化扩环反应机理
Scheme4.
Proposed mechanism of the photoredox-promoted, gold-catalyzed ring expansion-arylation reaction
炔丙醇也可在此策略下发生芳基化重排反应. 2016年, Shin等[11]报道了利用炔丙醇和芳基重氮盐合成α-芳基酮的反应.以Ph3PAuCl和[Ru(bpy)3](PF6)2为催化剂, 在蓝光LED灯的照射下, 该反应产率可达95% (Eq. 4).
作者认为该反应首先通过光催化利用芳基重氮盐将一价金氧化为三价金配合物.然后, 在氢键的介导下, 甲醇进攻被三价金配位的炔烃而生成烯基金中间体14-B.该烯基金中间体再发生消除反应得到联烯基金中间体14-C, 进而发生异构和水解生成最终产物(Scheme 5).
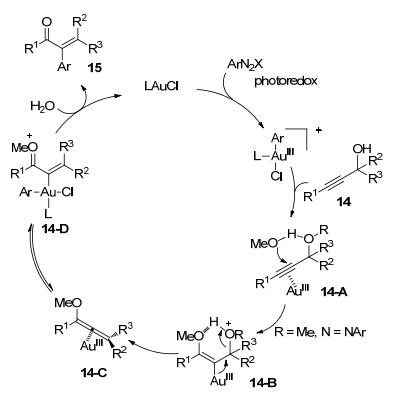 图图5
利用炔丙醇和芳基重氮盐合成α-芳基酮的反应机理
Scheme5.
Proposed mechanism of the reaction of propargyl alcohols with aryl diazonium salts
图图5
利用炔丙醇和芳基重氮盐合成α-芳基酮的反应机理
Scheme5.
Proposed mechanism of the reaction of propargyl alcohols with aryl diazonium salts
在Shin等的工作报道后不久, Glorius[12]和Alcaide等[13]也报道了他们利用光和金催化剂共催化炔丙醇和芳基重氮盐的串联重排/偶联反应.他们利用易于制备的炔丙醇作为底物通过Meyer-Schuster重排反应合成α-芳基烯酮; 而炔的水合反应也可以与交叉偶联反应串联选择性地合成α-芳基酮.两个反应均是在室温、可见光照射的条件下进行, 可见光来源于易于获得的光源、甚至是日光(Scheme 6).
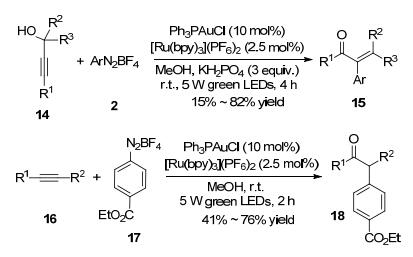 图Scheme6
金/光共催化的炔双功能化
Scheme6.
Alkyne difunctionalization by dual gold/photoredox catalysis
图Scheme6
金/光共催化的炔双功能化
Scheme6.
Alkyne difunctionalization by dual gold/photoredox catalysis
利用类似的光和金催化剂共同催化的策略, Alcaide等[13]研究了含有脂肪族、芳香族取代基的伯/仲炔丙醇与芳基重氮盐的Meyer-Schuster/芳基化反应, 产率中等到良好(Eq. 5).
另外, 可见光促进的氧化加成反应还被应用在一系列金催化的交叉偶联反应中. 2015年, Toste小组[14]报道了金和光催化剂共催化的C—P交叉偶联反应.在Ph3PAuCl (10 mol%)和[Ru(bpy)3](PF6)2 (2 mol%)的存在下, 芳基重氮盐可以与不同类型的P(O)H化合物反应得到偶联的芳基膦酸酯/亚膦酸酯, 收率可达到90%.值得一提的是, 如果将金催化剂替换为钯、银或铜盐, 产物的收率很低、甚至没有产物产生, 这些结果表明, 金催化在这类反应中的独特性(Eq. 6).
包括C(sp2)—C(sp2)和C(sp)—C(sp2)在内的C—C的交叉偶联反应也有报道.这些反应以芳基重氮盐作为自由基源在光催化的条件下将Au(Ⅰ)氧化为活性的Au(Ⅲ), 另一偶联的组分可以是炔或芳基硼酸. 2016年Glorius小组[15]报道了一个光和金催化剂共催化的端基炔芳基化反应.在10 mol% ((p-MeO)C6H4)3PAuCl和0.5 mol% [Ru(bpy)3](PF6)2的条件下, 不同的烃基/芳基端炔与不同的芳基重氮盐反应, 得到偶联产物, 产率可达79% (Scheme 7).
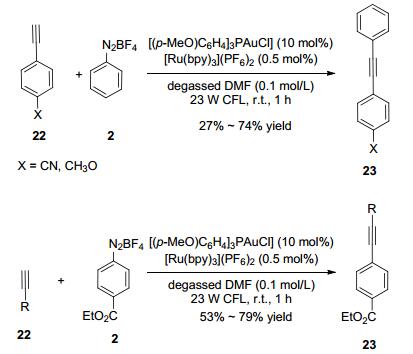 图Scheme7
金/光共催化的端基炔和芳基重氮盐的C(sp)—H芳基化反应
Scheme7.
Dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts
图Scheme7
金/光共催化的端基炔和芳基重氮盐的C(sp)—H芳基化反应
Scheme7.
Dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts
该反应的可能机理是, 首先, 光激发[Ru(bpy)3]2+为激发态的*[Ru(bpy)3]2+, 然后与芳基重氮盐经历单电子转移(SET)产生芳基自由基和氧化态的[Ru(bpy)3]3+, 芳基自由基与Au(Ⅰ)催化剂反应产生Au(Ⅱ).其次, Au(Ⅱ)再和另芳基重氮盐发生单电子转移反应产生亲电阳离子Au(Ⅲ)中间体, 然后Au(Ⅲ)中间体与炔配位, 得到炔-Au(Ⅲ)配合物.最后, 炔-Au(Ⅲ)配合物经还原消除反应再生Au(Ⅰ)催化剂和交叉偶联的产物(Scheme 8).
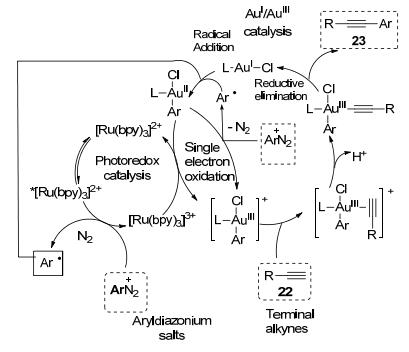 图Scheme8
金和光催化剂共催化的端基炔和重氮盐的C(sp)—H芳基化的可能的反应机理
Scheme8.
Proposed mechanism for the dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts
图Scheme8
金和光催化剂共催化的端基炔和重氮盐的C(sp)—H芳基化的可能的反应机理
Scheme8.
Proposed mechanism for the dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts
除了端基炔, 炔基三甲基硅烷也可以用作C(sp)— C(sp2)交叉偶联反应的底物. 2016年, Toste等[16]报道了Ar3PAuCl和Ru(bpy)3(PF6)2共催化的炔基三甲基硅烷和芳基重氮盐的偶联反应.该反应条件温和, 产率可以达到82%, 并具有好的官能团耐受性(Eq. 7).
金/光催化剂共同催化的C(sp2)—C(sp2)的交叉偶联反应通常是以芳基重氮盐和芳基硼酸作为底物的. 2016年, Hermange/Fouquet和Lee两个研究小组分别独立报道了他们在该领域的研究工作. Hermange和Fouque研究小组[17]发展了一种合成二芳基化合物的方法.该反应是在蓝光LED灯照射下, 以芳基重氮盐和芳基硼酸为反应物, PPh3AuCl作催化剂, CsF作碱, 乙腈作溶剂, 9-mesityl-10-acridinium tetrafluoroborate或者Ru(bpy)3-(PF6)2作为光敏剂.该反应也适用于带有多种官能团的底物, 如溴代芳基、碘代芳基、醛和醇(Eq. 8).通过化学计量的PPh3AuPh的31P和1H NMR谱图, 作者研究了该反应的机理.研究表明, 在9-mesityl-10-acridinium tetrafluoroborate存在下, 蓝光LED灯照射, PPh3AuPh和芳基重氮盐反应得到目标的偶联产物.
根据这些研究结果和以前的关于金/光催化剂共催化的文献报道, 该反应的机理可能是:首先, PPh3AuCl和芳基硼酸发生转金属化产生PPh3AuIAr′, 从而触发了反应.其次, 与芳基自由基发生氧化加成反应, 生成氧化态的Au(Ⅱ)中间体26-B, Au(Ⅱ)中间体再发生单电子氧化反应生成Au(Ⅲ)中间体26-C.最后, Au(Ⅲ)配合物经过还原消除反应生成二芳基化合物, 并再生Au(Ⅰ)催化剂(Scheme 9).
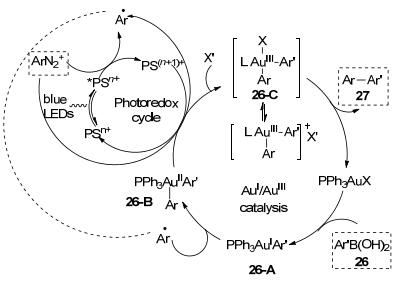 图Scheme9
芳基硼酸与芳基重氮盐偶联反应的可能机理
Scheme9.
Proposed mechanism for the couplings of arylboronic acids with aryldiazonium salts
图Scheme9
芳基硼酸与芳基重氮盐偶联反应的可能机理
Scheme9.
Proposed mechanism for the couplings of arylboronic acids with aryldiazonium salts
几乎与此同时, Lee等[18]也报道了芳基硼酸和芳基重氮盐的温和的、氧化的、非碱性的交叉偶联反应.在5 mol% PPh3AuNTf2和2.5 mol% [Ru(bpy)3](PF6)2存在下, 各种芳基硼酸可以顺利地与不同的芳基重氮盐反应生成目标的偶联产物, 收率可达到92% (Eq. 9).
通过对31P NMR的谱图分析, 对该反应的机理进行了详细的研究, 并给出了两个可能的反应机理.一种可能的机理是:芳基硼酸和Au(Ⅰ)的转金属化发生在Au(Ⅰ)氧化为Au(Ⅲ)之前; 另外一个可能的机理是: Au(Ⅰ)氧化为Au(Ⅲ)先于Au(Ⅰ)的转移金属化(Scheme 19).
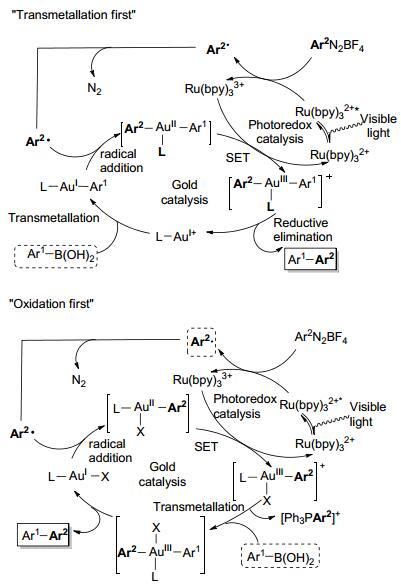 图Scheme10
金和光催化剂共催化的C(sp2)—C(sp2)交叉偶联反应的两个可能机理
Scheme10.
Two distinct pathways for the dual gold photoredox C(sp2)—C(sp2) cross coupling
图Scheme10
金和光催化剂共催化的C(sp2)—C(sp2)交叉偶联反应的两个可能机理
Scheme10.
Two distinct pathways for the dual gold photoredox C(sp2)—C(sp2) cross coupling
2 可见光催化促进的还原消除
还原消除一般是金属配体之间的成键, 同时伴随着金属中心形式上的双电子还原.作为偶联反应的关键步骤, 还原消除被广泛研究, 大量的金属和配体被应用于不同的反应, 但是仍然有很多还原消除很慢, 甚至很难发生的情况.光催化的介入也可以促进还原消除反应. 2014年Doyle & MacMillan等[19]报道了镍和光催化剂协同催化的C(sp3)—C(sp2)偶联反应, 成功实现了一系列烷基羧酸与芳基卤化物的脱羧偶联(Eq. 10).
对于反应的机理, 作者认为首先芳基卤化物与Ni(0) 发生氧化加成产生Ni(Ⅱ)中间体29-A, 同时, 羧酸在光催化条件下脱羧产生烷基自由基, 然后该自由基加成到Ni(Ⅱ)中间体上生成Ni(Ⅲ)中间体29-B.接着, Ni(Ⅲ)中间体发生还原消除生成Ni(Ⅰ)中间体, 最后Ni(Ⅰ)被处于低价态的光敏剂还原为Ni(0).整个过程通过单电子转移实现了Ni(0)→Ni(Ⅱ)→Ni(Ⅲ)→Ni(Ⅰ)的循环, 克服了传统Ni(0)→Ni(Ⅱ)→Ni(0) 机制中还原消除较困难的问题(Scheme 11).
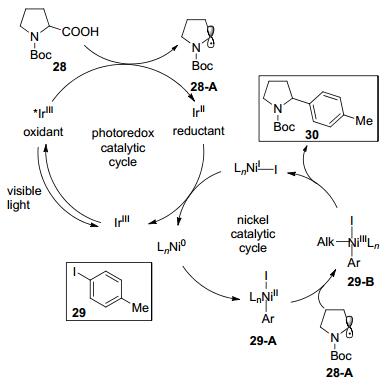 图Scheme11
镍和光催化剂协同催化的C(sp3)—C(sp2)烷基羧酸与芳基卤化物的脱羧偶联机理
Scheme11.
Proposed mechanism for the dual nickel photoredox C(sp3)—C(sp2) cross coupling between aryl halides and carboxylic acids
图Scheme11
镍和光催化剂协同催化的C(sp3)—C(sp2)烷基羧酸与芳基卤化物的脱羧偶联机理
Scheme11.
Proposed mechanism for the dual nickel photoredox C(sp3)—C(sp2) cross coupling between aryl halides and carboxylic acids
利用光催化下的这一Ni(0)→Ni(Ⅱ)→Ni(Ⅲ)→Ni(Ⅰ)机制, MacMillan小组[20]于2015年报道了利用醇和芳基卤化物在镍和光催化剂的共同作用下偶联为芳基醚的反应.该反应直接利用处于氧化态的光敏剂将不易发生还原消除的Ni(Ⅱ)物种氧化为Ni(Ⅲ)物种, 从而促进还原消除, 实现了C(sp2)—O偶联反应(Scheme 12).
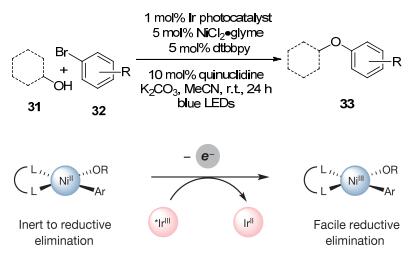 图Scheme12
镍和光催化剂协同催化的C(sp2)—O醇与芳基卤化物的偶联反应
Scheme12.
Dual nickel photoredox C(sp2)—O cross coupling between aryl halides and alcohols
图Scheme12
镍和光催化剂协同催化的C(sp2)—O醇与芳基卤化物的偶联反应
Scheme12.
Dual nickel photoredox C(sp2)—O cross coupling between aryl halides and alcohols
除了C(sp3)—C(sp2)偶联反应, 这一机制还被应用于C—P键的构建中. 2015年, 肖文精课题组[21]报道了镍和光催化剂协同催化的二芳基氧磷与芳基碘化物的偶联反应.该反应具有广泛的底物范围, 可以兼容各种官能团, 产率最高可达91% (Eq. 11).
3 可见光催化促进的转金属化反应
转金属化反应是亲核底物与金属中心的成键作用, 形式上金属价态没有变化, 但是, 光催化的介入也可以促进转金属化反应.其一般的机理可能是:首先, 亲核底物通过单电子转移的机制在光催化的条件下被氧化为自由基, 然后加成到金属上形成配合物, 这一配合物中间体再被光催化还原为低价态.基于这一原理, 2014年Molander课题组[22]实现了苄基三氟硼酸钾与芳基溴化物在光催化剂和镍催化剂协同作用下的偶联反应, 该反应经由光促进的单电子转移机制完成转金属化反应, 从而克服了传统转金属化的高能垒(Eq. 12).
对于具体的反应机理, 作者认为首先芳基卤化物与Ni(0) 发生氧化加成生产Ni(Ⅱ)中间体, 同时, 苄基三氟硼酸钾在光催化条件下脱三氟硼酸基产生烷基自由基, 然后该自由基加成到Ni(Ⅱ)中间体上生成Ni(Ⅲ)中间体.接着, Ni(Ⅲ)中间体再发生还原消除生成Ni(Ⅰ)中间体, 最后Ni(Ⅰ)再被处于低价态的光敏剂还原为Ni(0) (Scheme 13).
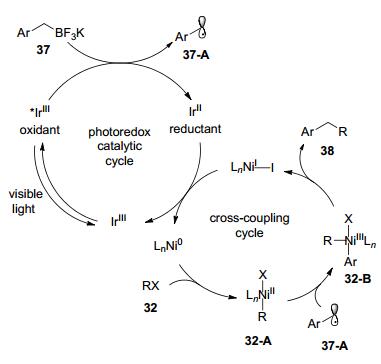 图Scheme13
镍和光催化剂协同催化的苄基三氟硼酸钾与芳基溴化物的偶联反应机理
Scheme13.
Proposed mechanism for dual nickel photo redox cross coupling between diarylphosphine oxides and aryl iodides
图Scheme13
镍和光催化剂协同催化的苄基三氟硼酸钾与芳基溴化物的偶联反应机理
Scheme13.
Proposed mechanism for dual nickel photo redox cross coupling between diarylphosphine oxides and aryl iodides
4 结论
本综述阐述了可见光催化促进的金属有机基元反应, 包括氧化加成、还原消除以及转金属化反应等.这类反应的特点是利用单电子转移的方式实现金属的氧化还原, 从而实现传统金属有机中较为困难的反应.目前这一策略的研究主要集中在钯、金、镍等金属催化的反应上, 未来的研究有望进一步拓展到其它金属, 特别是廉价金属催化的反应.另外, 这些反应的其中一个偶联组分——自由基源的类型太少, 大多数采用的是芳基重氮盐和二芳基高碘盐.我们相信, 在不久的将来, 更多的偶联组分可被应用于该反应.
-
-
[1]
(a) Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062.
(b) Hartwig, J. F. Organotransition Metal Chemistry:From Bonding to Catalysis, University Science Books, Herndon, VA, 2010. -
[2]
For selected reviews on photoredox catalysis:
(a) Xuan, J.; Xiao, W. J. Angew. Chem., Int. Ed. 2012, 51, 6828.
(b) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. Chem. Rev. 2013, 113, 5322.
(c) Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176.
(d) Angnes, R. A.; Li, Z.; Correia, C. R. D.; Hammond, G. B. Org. Biomol. Chem. 2015, 13, 9152.
(e) Xuan, J.; Zhang, Z. G.; Xiao, W. J. Angew. Chem., Int. Ed. 2015, 54, 15632.
(f) Luo, J.; Zhang, J. ACS Catal. 2016, 6, 873.
(g) Chen, J. R.; Hu, X. Q.; Lu, L. Q.; Xiao, W. J. Chem. Soc. Rev. 2016, 45, 2044.
(h) Lang, X.; Zhao, J.; Chen X., Chem. Soc. Rev. 2016, 45, 3026.
(i) Levin, M. D.; Kim, S.; Toste, F. D. ACS Cent. Sci. 2016, 2, 293.
(j) Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035.
(d) Hopkinson, M. N.; Tlahuext-Aca, A.; Glorius, F. Acc. Chem. Res. 2016, 49, 2261.
(k) Tellis, J. C.; Kelly, C. B.; Primer, D. N.; Jouffroy, M.; Patel, N. R.; Molander, G. A. Acc. Chem. Res. 2016, 49, 1429.
(l) Gui, Y.-Y.; Sun, L.; Lu, Z.-P.; Yu, D.-G. Org. Chem. Front. 2016, 3, 522.
(m) Fabry, D.-C.; Rueping, M. Acc. Chem. Res. 2016, 49, 1969.
(n) Zhang, M.; Zhu, C.; Ye, L.-W. Synthesis 2017, 1150.
(o) Zuo, X.; Wu, W.-L.; Su, W.-P. Acta Chim. Sinica 2015, 73, 1298(in Chinese).
(左璇, 吴文亮, 苏伟平, 化学学报, 2015, 73, 1298.)
(p) Guan, B.-C.; Xu, X.-L.; Wang, H.; Li, X.-N. Chin. J. Org. Chem. 2016, 36, 1564(in Chinese).
(关保川, 许孝良, 王红, 李小年, 有机化学, 2016, 36, 1564.)
(q) Tan, F.; Xiao, W.-J. Acta Chim. Sinica 2015, 73, 85(in Chinese).
(谭芬, 肖文精, 化学学报, 2015, 73, 85.)
(r) Roh, G.-B.; Iqbal N.; Cho. E. J. Chin. J. Chem. 2016, 34, 459. -
[3]
Halpern, J. Acc. Chem. Res. 1970, 3, 386. doi: 10.1021/ar50035a004
-
[4]
Hill, R. H.; Puddephatt, R. J. J. Am. Chem. Soc. 1985, 107, 1218. doi: 10.1021/ja00291a022
-
[5]
(a) Johnson, A.; Puddephatt, R. J. J. Chem. Soc., Dalton Trans. 1976, 1360.
(b) Winston, M. S.; Wolf, W. J.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 7777. -
[6]
(a) Kalyani, D.; McMurtrey, K. B.; Neufeldt, S. R.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 18566.
(b) Neufeldt, S. R.; Sanford, M. S. Adv. Synth. Catal. 2012, 354, 3517. -
[7]
Sahoo, B.; Hopkinson, M. N.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 5505. doi: 10.1021/ja400311h
-
[8]
Hopkinson, M. N.; Sahoo, B.; Glorius, F. Adv. Synth. Catal. 2014, 356, 2794. doi: 10.1002/adsc.201400580
-
[9]
Xia, Z.; Khaled, O.; Mouriès-Mansuy, V.; Ollivier, C.; Fensterbank, L. J. Org. Chem. 2016, 81, 7182. doi: 10.1021/acs.joc.6b01060
-
[10]
Shu, X.-Z.; Zhang, M.; He, Y.; Frei, H.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 5844. doi: 10.1021/ja500716j
-
[11]
Um, J.; Yun, H.; Shin, S. Org. Lett. 2016, 18, 484. doi: 10.1021/acs.orglett.5b03531
-
[12]
Tlahuext-Aca, A.; Hopkinson, M. N.; Garza-Sanchez, R. A.; Glorius, F. Chem.-Eur. J. 2016, 22, 5909. doi: 10.1002/chem.201600710
-
[13]
Alcaide, B.; Almendros, P.; Busto, E.; Luna, A. Adv. Synth. Catal. 2016, 358, 1526. doi: 10.1002/adsc.201600158
-
[14]
He, Y.; Wu, H.; Toste, F. D. Chem. Sci. 2015, 6, 1194. doi: 10.1039/C4SC03092C
-
[15]
Tlahuext-Aca, A.; Hopkinson, M. N.; Sahoo, B.; Glorius, F. Chem. Sci. 2016, 7, 89. doi: 10.1039/C5SC02583D
-
[16]
Kim, S.; Rojas-Martin, J.; Toste, F. D. Chem. Sci. 2016, 7, 85. doi: 10.1039/C5SC03025K
-
[17]
Cornilleau, T.; Hermange, P.; Fouquet, E. Chem. Commun. 2016, 52, 10040. doi: 10.1039/C6CC04239B
-
[18]
Gauchot, V.; Lee, A.-L. Chem. Commun. 2016, 52, 10163. doi: 10.1039/C6CC05078F
-
[19]
(a) Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C. Science 2014, 345, 437.
(b) Le, C. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 11938.
(c) Noble, A.; McCarver, S. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 624. -
[20]
(a) Terrett, J. A.; Cuthbertson, J. D.; Shurtleff, V. W.; MacMillan, D. W. C. Nature 2015, 524, 330.
(b) Chu, L.; Lipshultz, J. M.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2015, 54, 7929. -
[21]
Xuan, J.; Zeng, T.; Chen, J.; Lu, L.; Xiao, W. Chem.-Eur. J. 2015, 21, 4962. doi: 10.1002/chem.201500227
-
[22]
(a) Tellis, J. C.; Primer, D. N.; Molander, G. A. Science 2014, 345, 433.
(b) El Khatib, M.; Serafim, R. A. M.; Molander, G. A. Angew. Chem., Int. Ed. 2016, 55, 254.
(c) Primer, D. N.; Karakaya, I.; Tellis, J. C.; Molander, G. A. J. Am. Chem. Soc. 2015, 137, 2195.
-
[1]
-

图 1 经由光催化产生钯(Ⅳ)中间体的钯催化的C—H键官能团化反应
Figure 1 Palladium-catalyzed C—H functionalization via Pd(Ⅳ) intermediates generated by photoredox catalysis

图 2 金和光催化剂共同催化的含氧/氮烯烃与芳基重氮盐的反应机理
Figure 2 Proposed mechanism of the dual visible light photoredox and gold-catalyzed oxy-and aminoarylation of alkenes with aryldiazonium salts

图 3 金和光催化剂共同催化的烯烃的醚化芳基化反应
Figure 3 Dual visible light photoredox and gold-catalyzed oxyarylation of alkenes

图 4 光催化促进的、金催化氧化芳基化扩环反应机理
Figure 4 Proposed mechanism of the photoredox-promoted, gold-catalyzed ring expansion-arylation reaction

图 5 利用炔丙醇和芳基重氮盐合成α-芳基酮的反应机理
Figure 5 Proposed mechanism of the reaction of propargyl alcohols with aryl diazonium salts

Scheme6 金/光共催化的炔双功能化
Scheme6 Alkyne difunctionalization by dual gold/photoredox catalysis

Scheme7 金/光共催化的端基炔和芳基重氮盐的C(sp)—H芳基化反应
Scheme7 Dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts

Scheme8 金和光催化剂共催化的端基炔和重氮盐的C(sp)—H芳基化的可能的反应机理
Scheme8 Proposed mechanism for the dual gold/photoredox-catalyzed C(sp)—H arylation of terminal alkynes with diazonium salts

Scheme9 芳基硼酸与芳基重氮盐偶联反应的可能机理
Scheme9 Proposed mechanism for the couplings of arylboronic acids with aryldiazonium salts

Scheme10 金和光催化剂共催化的C(sp2)—C(sp2)交叉偶联反应的两个可能机理
Scheme10 Two distinct pathways for the dual gold photoredox C(sp2)—C(sp2) cross coupling

Scheme11 镍和光催化剂协同催化的C(sp3)—C(sp2)烷基羧酸与芳基卤化物的脱羧偶联机理
Scheme11 Proposed mechanism for the dual nickel photoredox C(sp3)—C(sp2) cross coupling between aryl halides and carboxylic acids

Scheme12 镍和光催化剂协同催化的C(sp2)—O醇与芳基卤化物的偶联反应
Scheme12 Dual nickel photoredox C(sp2)—O cross coupling between aryl halides and alcohols
-


 下载:
下载:












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 23
- 文章访问数: 3915
- HTML全文浏览量: 452
 下载:
下载:
