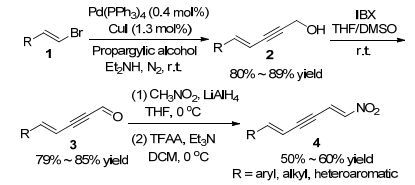
 图式 1
共轭硝基二烯炔4的合成
Scheme1.
Synthesis of conjugated nitrodienynes 4
图式 1
共轭硝基二烯炔4的合成
Scheme1.
Synthesis of conjugated nitrodienynes 4
引用本文:
刘腾, 刘建军, 贺池先, 成飞翔. 多共轭硝基二烯炔/硝基烯炔的合成以及应用研究进展[J]. 有机化学,
2017, 37(10): 2609-2618.
doi:
10.6023/cjoc201704024

Citation: Liu Teng, Liu Jianjun, He Chixian, Cheng Feixiang. Recent Progress on Polyconjugated Nitrodienynes/Nitroenynes:Synthesis and Applications[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2609-2618. doi: 10.6023/cjoc201704024
Citation: Liu Teng, Liu Jianjun, He Chixian, Cheng Feixiang. Recent Progress on Polyconjugated Nitrodienynes/Nitroenynes:Synthesis and Applications[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2609-2618. doi: 10.6023/cjoc201704024
多共轭硝基二烯炔/硝基烯炔的合成以及应用研究进展
English
Recent Progress on Polyconjugated Nitrodienynes/Nitroenynes:Synthesis and Applications
Abstract:
Polyconjugated nitrodienynes/nitroenynes as nitroolefin derivatives are good kind of electrophiles and have been widely used in organic synthesis. There are multiple reaction sites due to the conjugated system. Therefore, competitive reactions of regioselectivity in the reaction process are existence, such as 1, 4-addition reaction, 1, 6-addition reaction, and even 1, 8-addition reaction. The recent progress of the synthesis of nitrodienynes/nitroenynes and their applications in organic synthesis are summarized.
-
Key words:
- nitrodienyne
- / nitroenyne
- / organic synthesis application
- / regioselectivity
- / conjugate addition
-
近年来, 硝基烯作为良好的Michael受体在有机合成领域已被广泛地应用[1], 如用于合成具有重要用途的γ-硝基羰基化合物[2]、手性吡咯烷衍生物[3]、丁内酯衍生物[4]、环戊烷衍生物[5]以及选择性γ-氨基丁酸B型(GABAB)受体激动剂(R)-baclofen盐酸盐等[6].作为硝基烯的衍生物, 共轭硝基烯也常被用作亲电体进行共轭加成反应.而且, 由于其共轭体系的存在, 反应中存在多个亲电反应位点, 从而引入了区域选择性竞争反应可能, 如1, 4-加成反应、1, 6-加成反应甚至1, 8-加成反应. 2013年, Román课题组[7]对硝基二烯的合成与反应做了详尽的综述.基于作者前期的研究, 本文将对多共轭硝基二烯炔/硝基烯炔的合成及其在有机合成中的应用研究进行综述.
1 硝基二烯炔的合成以及应用
2014年, Shao课题组[8]设计并合成了一类新型Michael受体多共轭体系硝基二烯炔.以烯基溴化物为起始物, 二乙基胺为反应溶剂, 在0.4 mol% Pd(PPh3)4和1.3 mol% CuI催化作用下, 室温与丙炔醇发生Sonoga-shira偶联反应[9], 以80%~89%的收率得到烯炔醇产物2.随后, 化合物2经2-碘酰基苯甲酸(IBX)室温氧化, 以79%~85%的收率得到烯炔醛产物3.紧接着在LiAlH4作用下化合物3与硝基甲烷发生Henry反应, 最后在三氟乙酸酐(TFAA)和Et3N作用下脱除一分子水, 以50%~60%的收率得到一系列芳基、烷基以及杂环取代的硝基二烯炔4 (Scheme 1).该合成路线简短、反应条件温和、操作简便、底物普适性广, 且每一步反应都能以较高的收率得到目标产物.
图式 1
共轭硝基二烯炔4的合成
Scheme1.
Synthesis of conjugated nitrodienynes 4
随后, Shao课题组[8]经过大量实验条件筛选发现, 在金属镍(Ⅱ)/手性二胺配体5催化作用下硝基二烯炔4与1, 3-二羰基化合物6发生不对称1, 4-共轭加成反应.反应能够专一性以85%~95%的收率和90%~94% ee值得到一系列高功能化具有重要用途的1, 3-烯炔化合物7[10, 11], 只是当R2和R3为不同取代基时, 产物非对映选择性只有1:1 (1:1 dr) (Eq. 1).值得注意的是, 在所有反应中均未检测到1, 6-加成产物和1, 8-加成产物的生成.对于1, 4-加成产物7a, 当R2=Me时, 在甲苯为反应溶剂p-TSA催化作用下加热回流, 分子内炔键水化, 紧接着发生分子内Aldol环化反应, 以65%~76%的收率和88%~92% ee值到得具有旋光活性的环己二烯酮化合物8; 而当R2=t-BuO时, 分子内炔键水化且经历脱羧反应, 以73%~85%的收率得到不饱和羰基酸产物9.化合物9在SOCl2作用下进行酯化反应, 以80%~87%的收率和90%~93% ee值得到不饱和γ-羰基酸酯化合物10 (Scheme 2).基于多共轭体系的存在, 硝基二烯炔在有机合成中具有一定的合成实用性, 能够构建高功能化结构新颖的分子骨架.
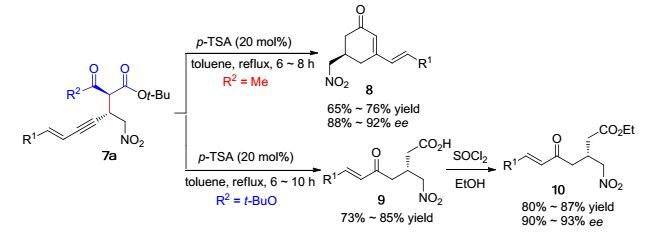 图式 2
1, 3-烯炔化合物的转化
Scheme2.
Transformation of 1, 3-enyne compounds
图式 2
1, 3-烯炔化合物的转化
Scheme2.
Transformation of 1, 3-enyne compounds
2014年, Shao课题组[12]从胡椒基取代的硝基二烯炔出发, 经3步反应以61.2%的收率、93% ee值合成得到不饱和γ-羰基酸酯10a.而且, 该课题组成功地将10a引入到石蒜碱型生物碱(+)-α-Lycorane和(+)-Lycorine不对称合成中, 利用金属催化策略实现(+)-α-Lycorane和(+)-Lycorine的催化不对称全合成.在有机碱TMG催化作用下, 化合物10a发生分子内Michael加成环化反应, 以60%的收率、93% ee值和6:1 dr值得到多取代硝基环己酮衍生物11.随后, 在BF3·OEt2作用下化合物11经乙二硫醇保护, 以77%的收率得到硫代缩酮产物12.化合物12经Zn/HCl还原硝基后在甲醇钠作用下发生分子内酰胺化反应, 以70%的收率得到二环内酰胺产物13.化合物13经LiAlH4还原后再与氯甲酸乙酯反应, 2步反应以53%的收率得到关键中间体15.随后, 中间体15在POCl3作用下发生Bischler-Napieralski反应, 以85%的收率得到多环产物16, 紧接着化合物16经Raney Ni/H2还原脱硫, 以85%的收率得到产物17.最后, 化合物17在LiAlH4还原作用下, 以12步反应1.9%的总收率立体选择性合成得到(+)-α-Lycorane.另外, 从多环产物16出发再经4步反应, 以1.1%的总收率实现(+)-Lycorine的立体选择性全合成(Scheme 3).在此之前, 有3个研究小组分别采用不对称全合成或形式合成策略完成了(-)-α-Lycorane的合成[14].反应中手性的引入采用金属镍/手性二胺配体催化体系, 该催化体系由Evans课题组2007年首次提出[15].
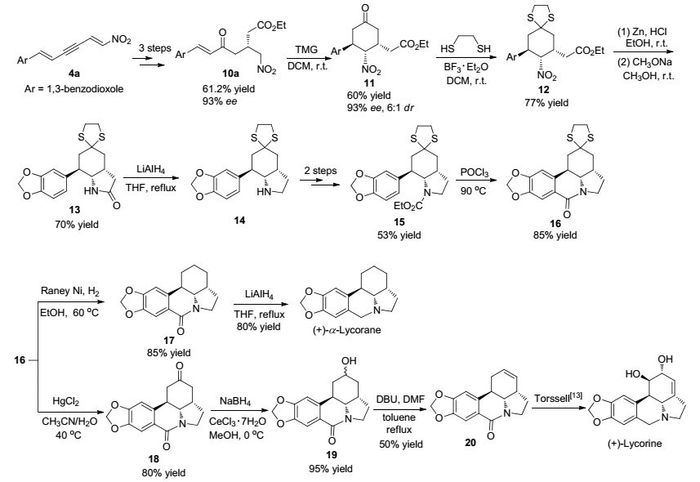 图式 3
(+)-α-Lycorane和(+)-Lycorine立体选择性合成
Scheme3.
Stereoselective synthesis (+)-α-Lycorane and (+)-Lycorine
图式 3
(+)-α-Lycorane和(+)-Lycorine立体选择性合成
Scheme3.
Stereoselective synthesis (+)-α-Lycorane and (+)-Lycorine
随后, Shao课题组[16]在此前研究基础上[12], 利用不对称有机催化策略实现了(+)-α-lycorane立体选择性二代全合成.在脯氨醇硅醚21催化作用下, 硝基二烯炔与乙醛水溶液发生不对称共轭加成反应.反应得到的1, 4-共轭加成产物不经纯化处理直接被NaBH4还原, 以51%~81%的收率和84%~98% ee值到得一系列1, 3-二烯炔化合物22 (Eq. 2).在最优催化条件下, 胡椒基取代的硝基二烯炔与乙醛水溶液发生不对称共轭加成反应, 待反应完后再经过NaClO2、KH2PO4和2-甲基异戊烯氧化作用以及p-TSA催化炔键水化3步反应, 以41%的收率得到不饱和γ-羰基酸化合物9a.根据此前报道的合成方法[12], 以化合物9a为起始物经3步反应即可得到重要中间体12.随后, 中间体12在Raney Ni/H2作用下发生还原反应和酰化反应, 以73%的收率得到内酰胺产物25.在三氟乙酸催化作用下, 化合物25与多聚甲醛发生Pictet-Spengler环化反应以83%的收率得到多环化产物26.最后, 化合物26经LiAlH4还原, 总计9步反应以4.83%总收率得到(+)-α-lycorane (Scheme 4). Shao课题组二代全合成策略以廉价易得原料出发, 合成路线简短高效, 而且实现了反应的无金属催化、环境友好.
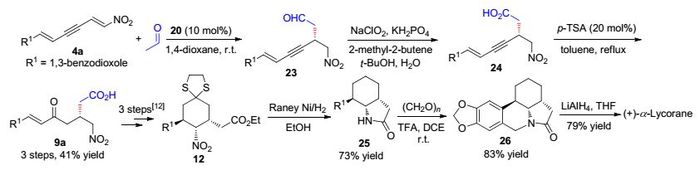 图式 4
(+)-α-lycorane不对称有机催化合成
Scheme4.
Organocatalytic asymmetric synthesis of (+)-α-lycorane
图式 4
(+)-α-lycorane不对称有机催化合成
Scheme4.
Organocatalytic asymmetric synthesis of (+)-α-lycorane
最近, 本人利用课题组设计与合成的多氢键供体伯胺硫脲类催化剂[1m], 立体选择性催化硝基二烯炔与芳酮、丙酮不对称1, 4-共轭加成反应[17].反应克服了芳酮和丙酮的弱亲核反应活性, 高收率、高对映选择性和专一区域选择性得到两类具有重要用途的高功能化1, 3-烯炔化合物.值得注意的是, 在所有反应中均未检测到1, 6-加成产物和1, 8-加成产物的生成.到目前为止, 仅有少量催化体系能够高效高立体选择性催化芳酮、丙酮与硝基烯不对称共轭加成反应[1m, 18].有趣的是, 当R2=Ar时, 在p-TSA催化作用下1, 3-烯炔化合物经过炔键水化/分子内Michael环化串联反应, 以80%~93%的收率、88%~98% ee值和大于20:1 dr值高立体选择性得到一系列具有旋光活性环1, 5-二酮化合物28; 而当R2=Me时, 在p-TSA催化作用下1, 3-烯炔化合物经过炔键水化/分子内Aldol环化串联反应, 以90%~96%的收率和85%~94% ee值高立体选择性得到一系列具有重要用途的手性环己二烯酮化合物29[19](Scheme 5).
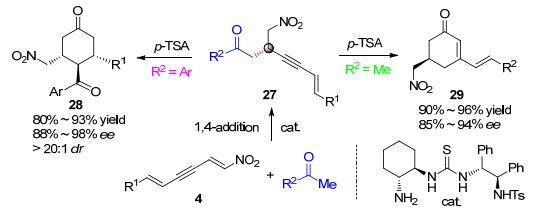 图式 5
硝基二烯炔与“困难”酮不对称共轭加成及串联环化反应
Scheme5.
Enantioselective conjugate additions of "difficult" ketones to nitrodienynes and tandem annulations
图式 5
硝基二烯炔与“困难”酮不对称共轭加成及串联环化反应
Scheme5.
Enantioselective conjugate additions of "difficult" ketones to nitrodienynes and tandem annulations
随后, 我们对高功能化1, 3-烯炔化合物分子内串联环化反应机理进行推测.首先, p-TSA催化分子内区域选择性炔键水化, 原位产生α, β-不饱和烯酮中间体30.紧接着, p-TSA促进中间体30烯醇化, 进而发生分子内Michael环化反应或分子内Aldol环化反应.反应过程中动力学控制占主导, 酸催化形成中间体33.分子内Aldol环化反应快于分子内Michael环化反应, 从而有利于Aldol环化产物29的生成(Scheme 6).
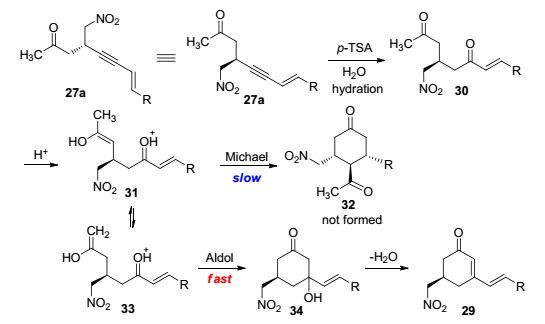 图式 6
1, 3-烯炔27a分子内串联环化反应机理
Scheme6.
Mechanism of 1, 3-enynes 27a intramolecular tandem annulations reaction
图式 6
1, 3-烯炔27a分子内串联环化反应机理
Scheme6.
Mechanism of 1, 3-enynes 27a intramolecular tandem annulations reaction
2 硝基烯炔的合成以及应用
2007年, Namboothiri课题组[20]在N-甲基吗啡啉碱性条件下, (Z)-α-溴代硝基乙烯与端炔在2.5 mol% Pd(PPh3)4和2.5 mol% CuI催化作用下发生Sonogashira偶联反应, 简便、高效以及专一选择性地得到一系列(E)-α, β-二取代硝基共轭烯炔化合物37 (Eq. 3), 该反应实现了化合物构型的反转.当R1为芳基、杂环取代, R2为芳基、烷基取代时, 反应能够顺利地进行, 以69%~86%的收率得到一系列目标产物; 而当R1为烷基取代, R2为杂环取代时, 反应均未得到目标产物, 底物的多样性还需进一步拓展.该反应中碱性N-甲基吗啡啉起了重要作用, 伯胺仲胺易与硝基烯形成稳定的氮杂环丙烷衍生物, 而N-甲基吗啡啉能够先与硝基烯发生Michael加成反应形成不稳定的氮酸酯类中间体, 随后经历Pd(PPh3)4和CuI的催化转化形成目标产物.
2009年, Alexakis课题组[21]报道了脯氨醇硅醚21立体选择性催化硝基烯炔与异戊醛的不对称1, 4-加成反应, 顺式选择性为主, 以96%~>99% ee值得到产物40.随后发现, 产物40不经纯化处理进一步在金(Ⅰ)催化作用下与醇类发生分子间串联环化反应, 能够以75%~86%的收率一锅法高效实现多取代呋喃衍生物的合成(Scheme 7). R3为芳基和杂环取代时, 反应都能顺利地进行得到相应1, 4-加成产物40.在[PPh3Au]Cl、AgBF4和p-TSA催化作用下, 中间体40进一步与各种伯醇仲醇发生串联环化反应, 高收率高立体选择性得到一系列多取代呋喃衍生物41.反应中过量的p-TSA并未对金(Ⅰ)催化剂活性起钝化作用, 而是有利于反应的进行.将反应温度降低至-10 ℃且延长反应时间, 有利于产物收率的提高.随后, Alexakis课题组[22]更加细致地对共轭体系硝基烯与醛类不对称1, 4-加成反应进行了研究, 进一步扩展了硝基烯炔与醛类底物范围, 使多取代呋喃衍生物范围得到更大范围的延伸.值得注意的是, 该反应实现了有机催化立体选择性1, 4-加成反应和金催化串联环化反应的有效结合, 一锅法接力式催化高效得到目标产物.该反应后处理简单, 利用易得的原料构建结构复杂的分子.金(Ⅰ)催化串联反应的推测机理如下(Scheme 7).
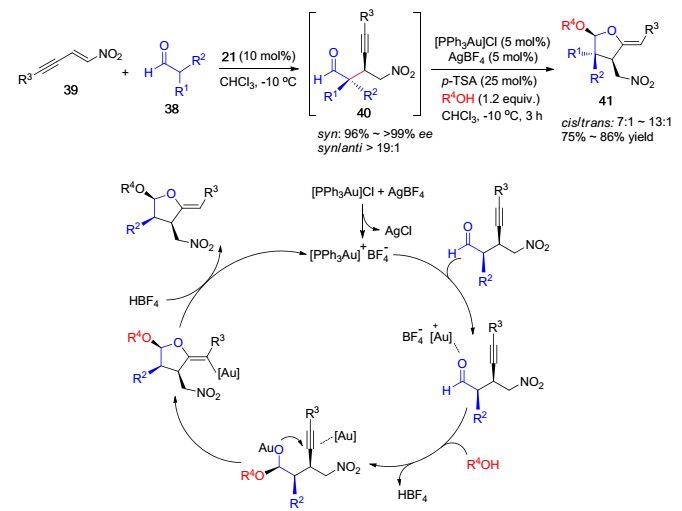 图式 7
硝基烯炔与醛类不对称共轭加成及串联反应机理
Scheme7.
Asymmetric conjugate addition reaction of nitroenynes with aldehydes and tandem cyclization mechanism
图式 7
硝基烯炔与醛类不对称共轭加成及串联反应机理
Scheme7.
Asymmetric conjugate addition reaction of nitroenynes with aldehydes and tandem cyclization mechanism
随后, Alexakis课题组[23]从炔醛出发, 经过Henry反应和TFAA脱水反应, 两步反应以42%~76%的总收率得到一系列芳基、烷基取代的硝基烯炔化合物39 (Scheme 8).紧接着, 发展了噻吩-2-甲酸铜(CuTC)/二茂铁磷配体44催化体系催化烷基铝试剂与硝基烯炔/硝基二烯不对称共轭加成反应.有意思的是, 通过控制反应条件, 可以调控硝基二烯1, 4-加成反应和1, 6-加成反应, 立体选择性得到相应产物.然而, 通过控制反应条件, 并未得到硝基烯炔的1, 6-加成产物, 而是立体专一性得到1, 4-加成产物(Eq. 4).
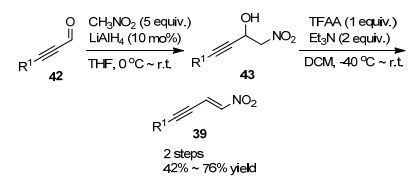 图式 8
硝基烯炔与烷基锂共轭加成反应
Scheme8.
Conjugated addition reaction of nitroenynes with alkyl lithium
图式 8
硝基烯炔与烷基锂共轭加成反应
Scheme8.
Conjugated addition reaction of nitroenynes with alkyl lithium
2010年, Ma课题组[3a]以易得的糖和手性二胺为原料设计并合成了一系列双功能伯胺硫脲有机小分子催化剂.该课题组通过大量实验条件筛选发现, 催化剂46和47能够高效高立体选择性催化硝基二烯、硝基烯炔与各种芳酮、脂肪酮共轭加成反应, 顺式选择性得到1, 4-加成产物, 且反应中均未检测到1, 6-加成产物的生成.在催化剂46和苯甲酸催化体系中, 芳酮与硝基烯炔反应能够以85%的收率和96% ee值得到相应不饱和硝基化合物; 而在催化剂47、苯甲酸和少量水催化体系中, 脂肪酮类化合物与硝基烯炔反应能够以85%~89%的收率和91%~94% ee值得到相应不饱和硝基化合物(Eq. 5).该反应底物普适性广, 反应条件温和, 稍有遗憾的是反应时间需要6 d.
2011年, Shao课题组[24]首次报道了1, 3-二羰基化合物与硝基烯炔不对称共轭加成反应.在金鸡纳生物碱衍生的硫脲催化剂51催化作用下, 以高达93%的收率和99% ee值获得1, 4-共轭加成产物.对于芳基取代和烷基取代的硝基烯炔底物, 该反应均能顺利地进行; 而当R2=t-Bu时, 只能检测到产物痕迹.当ɑ-取代-1, 3-二羰基化合物作为亲核试剂时, 反应以74%~93%收率和86%~93% ee值得到相应共轭加成产物54, 但其非对映选择性仅为1:1 (1:1 dr).此反应提供了一个概念上不同的合成药物重要前体手性炔基酸和合成上具有重要用途手性硝基炔化物的新思路, 为药物分子合成奠定了一定基础(Scheme 9).
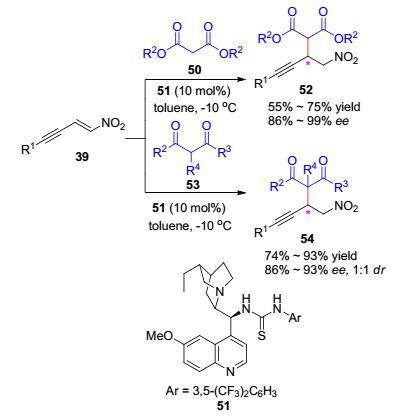 图式 9
硝基烯炔与1, 3-二羰基化合物共轭加成反应
Scheme9.
Conjugated addition reaction of nitroenynes with 1, 3-dicarbonyl compounds
图式 9
硝基烯炔与1, 3-二羰基化合物共轭加成反应
Scheme9.
Conjugated addition reaction of nitroenynes with 1, 3-dicarbonyl compounds
随后, Ooi课题组[25]报道了共轭硝基烯炔立体选择性aza-Michael加成反应.首次利用P-手性螺环芳基氨基膦阳离子56结合[3, 5-(CF3)2C6H3]4B作为Brønsted酸催化剂催化硝基烯炔与2, 4-二甲氧基芳基胺不对称共轭加成反应, 以90%~99%的收率和86%~96% ee值得到一系列β-氨基-β-炔基硝基化合物57.对于芳基取代、线性或分枝状饱和烷基取代还是卤代烷烃取代的硝基烯炔、甚至醚类取代的硝基烯炔, 反应都能够高立体选择性得到相应1, 4-加成产物.该反应扩展了低酸性芳基氨基膦阳离子在有机合成中的实用性.研究表明, 手性Brønsted酸离子在不对称催化合成中具备一定的催化潜力, 有望在不对称合成中得到更加广范的应用(Eq. 6).
2012年, Shao课题组[26]基于此前的研究[24]进一步发展了金属镍(Ⅱ)/手性二胺配体5催化体系催化硝基烯炔与丙二酸酯立体选择性共轭加成反应, 以81%~98%的收率和89%~>99% ee值得到一系列β-炔基酸衍生物52 (Eq. 7).对于此前金鸡纳生物碱硫脲有机催化剂催化研究中不反应的底物R2=t-Bu[24], 在金属镍(Ⅱ)/手性二胺催化作用下反应能够顺利地进行, 以81%的收率和92% ee值得到目标产物52a.而且, 化合物52a经p-TSA催化脱羧, 以83%的收率和90% ee值得到具有多功能用途的β-炔基酸化合物57 (Eq. 8).随后, 反应扩展到硝基烯炔与α-取代-β-酮酸酯不对称1, 4-共轭加成反应.在叔胺硫脲催化剂61催化作用下, 环状和非环状α-取代-β-酮酸酯或内酯衍生物都能作为优良的Michael供体顺利地与硝基烯炔反应, 高收率高立体选择性得到邻位双手性中心化合物62和63 (Scheme 10). Shao从不对称金属催化和不对称有机催化策略对此反应进行了详尽的研究, 两种催化策略相互补充.
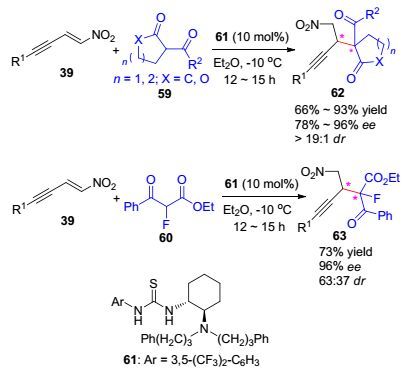 图式 10
硝基烯炔与α-取代1, 3-二羰基化合物共轭加成反应
Scheme10.
Asymmetric conjugated addition reaction of nitroenynes with α-substituted1, 3-dicarbonyl compounds
图式 10
硝基烯炔与α-取代1, 3-二羰基化合物共轭加成反应
Scheme10.
Asymmetric conjugated addition reaction of nitroenynes with α-substituted1, 3-dicarbonyl compounds
随后, Liu课题组[27]发展了氮杂环卡宾催化剂65催化硝基烯炔、硝基二烯与烯醛化合物不对称加成反应, 以51%~78%的收率、81%~97% ee值和中等非对映选择性构建高官能化5-碳-δ-硝基酯化合物66.该反应底物普适性广, 各种富电子、缺电子和无取代的α, β-不饱和醛类都能与硝基烯炔、硝基二烯顺利反应, 高收率高对映选择性得到相应1, 4-加成产物, 反应中均没有检测到1, 6-加成产物的生成(Eq. 9).该反应采用高烯醇化合成策略, 有别于之前硝基烯与醛类Stetter反应构建β-硝基酮类化合物(3-碳合成子)和硝基烯与醛/酮类Michael加成反应构建γ-硝基酮类化合物(4-碳合成子)策略, 是一种构建碳碳键的有效方法.
2013年, Punniyamurthy课题组[28]利用硝基取代的共轭1, 3-烯炔化合物与胺类化合物进行串联环化反应, 高效构筑五取代吡咯衍生物68 (Eq. 10).反应底物普适性广, R1和R2芳环上无论为供电子取代还是拉电子取代, 反应都能够顺利地进行, 以较高的收率得到相应多取代吡咯衍生物; R3为芳基和烷基取代时, 反应同样也能够顺利地进行.而且, 当用30%的氨水溶液作氮源时, 反应能够以52%~59%的收率得到一系列四取代吡咯衍生物.反应机理推测表明, 反应中缺电子硝基取代的共轭1, 3-烯炔化合物首先与胺类化合物发生aza-Michael加成反应形成中间体A.随后, 在碱作用下形成含碘中间体B, 紧接着中间体B经过分子内环化反应得到二氢吡咯衍生物C.最后, 经历碘氧化芳构化反应形成目标产物(Scheme 11).该反应条件温和, 所得高功能化产物能够进一步应用于复杂分子的合成.
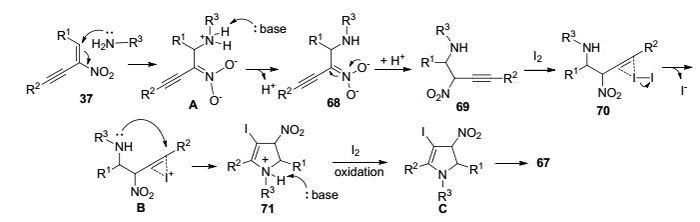 图式 11
多取代吡咯衍生物形成的可能机理
Scheme11.
Plausible mechanism for the formation of polysubstituted pyrrole derivatives
图式 11
多取代吡咯衍生物形成的可能机理
Scheme11.
Plausible mechanism for the formation of polysubstituted pyrrole derivatives
最近, Zhou课题组[29]报道了Ph3PAuOTf结合双功能叔胺硫脲催化剂74催化弱反应活性的苯甲醚或噻吩与硝基烯炔、重氮氧化吲哚三组分不对称反应(Eq. 11).研究发现, 该反应只需低催化量的催化剂Ph3PAuOTf (1 mol%)即能快速催化重氮氧化吲哚与苯甲醚或噻吩反应, 高收率形成3-芳基取代氧化吲哚中间体76a或77a (Eq. 12).值得注意的是, 这是首例金催化噻吩与重氮化合物C—H键插入型反应.随后, 在叔胺硫脲催化剂74脱质子化作用下3-芳基取代氧化吲哚与硝基烯炔发生Michael加成反应, 最终形成含C(3) 位季碳中心氧化吲哚衍生物75.该反应底物普适性广, 能够高收率高立体选择性一锅法合成得到目标产物.而且, 得到的串联反应产物能够进一步在Au(Ⅰ)催化作用下发生分子内炔键水化反应, 一锅法高收率高立体选择性得到化合物78 (Scheme 12).该类反应进一步丰富了金属/有机共催化策略构建碳碳键的反应.
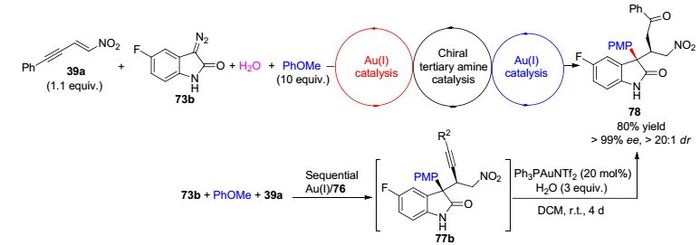 图式 12
Au(Ⅰ)催化水化反应
Scheme12.
Au(Ⅰ)-catalyzed hydration reaction
图式 12
Au(Ⅰ)催化水化反应
Scheme12.
Au(Ⅰ)-catalyzed hydration reaction
3 总结与展望
综上所述, 多共轭硝基二烯炔和硝基烯炔作为良好的亲电体在有机合成中得到了较为广泛的应用.到目前为止, 对于硝基二烯炔和硝基烯炔的研究主要集中于1, 4-共轭加成反应, 而对于其1, 6-共轭加成反应和1, 8-共轭加成反应未见报道.随着化学工作者对有机反应研究的深入, 进一步探索共轭硝基烯1, 6-共轭加成反应和1, 8-共轭加成反应构建碳-碳、碳-杂键, 得到高效、经济和绿色的合成方法仍然是今后研究的热点和具有挑战性的研究课题.
-
-
[1]
(a) Yang, Z.; Liu, J.; Liu, X.; Wang, Z.; Feng, X.; Su, Z.; Hu, C. Adv. Synth. Catal. 2008, 350, 2001.
(b) Mandal, T.; Zhao, C. Angew. Chem., Int. Ed. 2008, 47, 7714.
(c) Xue, F.; Zhang, S.; Duan, W.; Wang, W. Adv. Synth. Catal.2008, 350, 2194.
(d) Gu, Q.; Guo, X.; Wu, X. Tetrahedron 2009, 65, 5265.
(e) Morris, D. J.; Partridge, A. S.; Manville, C. V.; Racys, D. T.; Woodward, G.; Docherty, G.; Wills, M. Tetrahedron Lett. 2010, 51, 209.
(f) Peng, L.; Xu, X.; Wang, L.; Huang, J.; Bai, J.; Huang, Q.; Wang, L. Eur.J. Org. Chem. 2010, 1849.
(g) Lu, A.; Liu, T.; Wu, R.; Wang, Y.; Zhou, Z.; Wu, G.; Fang, J.; Tang, C. Eur.J. Org. Chem. 2010, 5777.
(h) Lu, A.; Liu, T.; Wu, R.; Wang, Y.; Wu, G.; Zhou, Z.; Fang, J.; Tang, C. J.Org. Chem. 2011, 76, 3872.
(i) Jiang, X.; Zhang, Y.; Chan, A. S. C.; Wang, R. Org. Lett. 2009, 11, 153.
(j) Rasappan, R.; Reiser, O. Eur. J. Org. Chem. 2009, 1305.
(k) Kokotos, C. G.; Kokotos, G. Adv. Synth. Catal. 2009, 351, 1355.
(l) Li, B.; Wang, Y.; Luo, S.; Zhong, A.; Li, Z.; Du, X.; Xu, D. Eur. J.Org. Chem. 2010, 656.
(m) Sun, Z.-W.; Peng, F.-Z.; Li, Z.-Q.; Zou, L.-W.; Zhang, S.-X.; Li, X.; Shao, Z.-H. J. Org. Chem. 2012, 77, 4103. -
[2]
For selected reviews, see: (a) Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877.
(b) Almasi, D.; Alonso, D. A.; Najera, C. Tetrahedron: Asymmetry 2007, 18, 299.
(c) Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701.
(d) Sulzer-Mosse, S.; Alexakis, A. Chem. Commun. 2007, 3123.
(e) Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065.
(f) Tsakos, M.; Kokotos, C. G. Tetrahedron 2013, 69, 10199. -
[3]
(a) Ma, H.; Liu, K.; Zhang, F.-G.; Zhu, C.-L.; Nie, J.; Ma, J.-A. J. Org. Chem. 2010, 75, 1402.
(b) Betancort, J. M.; Sakthivel, K.; Thayumanavan, R.; Tanaka, F.; Barbas Ⅲ, C. F. Synthesis 2004, 1509. -
[4]
Palomo, C.; Vera, S.; Mielgo, A.; Gomez-Bengoa, E. Angew. Chem., Int. Ed. 2006, 45, 5984. doi: 10.1002/(ISSN)1521-3773
-
[5]
Bonne, D.; Salat, L.; Dulcère, J.-P.; Rodriguez, J. Org. Lett. 2008, 10, 5409. doi: 10.1021/ol8023133
-
[6]
Corey, E. J.; Zhang, F. Org. Lett. 2000, 2, 4257. doi: 10.1021/ol0068344
-
[7]
Ballini, R.; Araújo, N.; Gil, M. V.; Román, E.; Serrano J. A. Chem. Rev. 2013, 113, 3493. doi: 10.1021/cr2002195
-
[8]
Li, X.; Peng, F.; Zhou, M.; Mo, M.; Zhao, R.; Shao, Z. Chem. Commun. 2014, 50, 1745. doi: 10.1039/C3CC48951E
-
[9]
(a) Sonogashira, K. Metal-Catalyzed Cross-Coupling Reactions, Eds.: Diederich, F.; Stang, P. J., Wiley-VCH, Weinheim, 1997, p. 203.
(b) Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 50, 4467.
(c) Stephens, R. D.; Castro, C. E. J. Org. Chem. 1963, 28, 2163. -
[10]
(a) Akiyama, T.; Takada, K.; Oikawa, T.; Matsuura, N.; Ise, Y.; Okada, S.; Matsunaga, S. Tetrahedron 2013, 69, 6560.
(b) Iverson, S. L.; Uetrecht, J. P. Chem. Res. Toxicol. 2001, 14, 175. -
[11]
(a) Zhang, W.; Zheng, S.; Liu, N.; Werness, J. B.; Guzei, I. A.; Tang, W. J. Am. Chem. Soc. 2010, 132, 3664.
(b) Werness, J. B.; Tang, W. Org. Lett. 2011, 13, 3664. -
[12]
Sun, Z.; Zhou, M.; Li, X.; Meng, X.; Peng, F.; Zhang, H.; Shao, Z. Chem. Eur. J. 2014, 20, 6112. doi: 10.1002/chem.201400178
-
[13]
Møller, O.; Steinberg, E. M.; Torssell, K. Acta Chem. Scand., Ser. B 1978, 32, 98.
-
[14]
(a) Hong, B.; Nimje, R. Y.; Wu, M.; Sadani, A. A. Eur. J.Org. Chem. 2008, 1449.
(b) Wang, Y.; Luo, Y.; Zhang, H.; Xu, P. Org. Biomol. Chem.2012, 10, 8211.
(c) Li, G.; Xie, J.; Hou, J.; Zhu, S.; Zhou, Q. Adv. Synth. Catal.2013, 355, 1597. -
[15]
Evans, D. A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583. doi: 10.1021/ja0735913
-
[16]
Meng, X.-L.; Liu, T.; Sun, Z.-W.; Wang, J.-C.; Peng, F.-Z.; Shao, Z.-H. Org. Lett. 2014, 16, 3044. doi: 10.1021/ol501158b
-
[17]
Liu, T.; Zhou, M.; Yuan, T.; Fu, B.; Wang, X.; Peng, F.; Shao, Z. Adv. Synth. Catal. 2017, 359, 89. doi: 10.1002/adsc.v359.1
-
[18]
(a) Tsakos, M.; Kokotos, C. G.; Kokotos, G. Adv. Synth.Catal. 2012, 354, 740.
(b) Flores-Ferrándiz, J.; Stiven, A.; Sotorríos, L.; Glmez-Bengoa, E.; Chinchilla, R. Tetrahedron: Asymmetry2015, 26, 970. -
[19]
(a) Hénon, H.; Mauduit, M.; Alexakis, A. Angew. Chem., Int. Ed. 2008, 47, 9122.
(b) Tian, X.; Liu, Y.-K.; Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 6439. -
[20]
Ganesh, M.; Namboothiri, I. N. N. Tetrahedron 2007, 63, 11973. doi: 10.1016/j.tet.2007.09.012
-
[21]
Belot, S.; Vogt, K. A.; Besnard, C.; Krause, N.; Alexakis, A. Angew. Chem., Int. Ed. 2009, 48, 8923. doi: 10.1002/anie.v48:47
-
[22]
Belot, S.; Quintard, A.; Krause, N.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 667. doi: 10.1002/adsc.v352:4
-
[23]
(a) Tissot, M.; Müller, D.; Belot, S.; Alexakis, A. Org. Lett. 2010, 12, 2770.
(b) Tissot, M.; Alexakis, A. Chem. Eur. J. 2013, 19, 11352. -
[24]
Li, X.-J.; Peng, F.-Z.; Li, X.; Wu, W.-T.; Sun, Z.-W.; Li, Y.-M.; Zhang, S.-X.; Shao, Z.-H. Chem.-Asian J. 2011, 6, 220. doi: 10.1002/asia.201000561
-
[25]
Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. Synlett 2011, 9, 1265.
-
[26]
Li, X.; Li, X.; P. F.; Shao, Z. Adv. Synth. Catal. 2012, 354, 2873. doi: 10.1002/adsc.v354.14/15
-
[27]
Maji, B.; Ji, L.; Wang, S.; Vedachalam, S.; Ganguly, R.; Liu, X.-W. Angew. Chem., Int. Ed. 2012, 51, 8276. doi: 10.1002/anie.v51.33
-
[28]
Bharathiraja, G.; Sakthivel, S.; Sengoden, M.; Punniyamurthy, T. Org. Lett. 2013, 15, 4996. doi: 10.1021/ol402305b
-
[29]
Cao, Z.-Y.; Zhao, Y.-L.; Zhou, J. Chem. Commun. 2016, 52, 2537. doi: 10.1039/C5CC10096H
-
[1]
-

图式 3 (+)-α-Lycorane和(+)-Lycorine立体选择性合成
Scheme 3 Stereoselective synthesis (+)-α-Lycorane and (+)-Lycorine

图式 4 (+)-α-lycorane不对称有机催化合成
Scheme 4 Organocatalytic asymmetric synthesis of (+)-α-lycorane

图式 5 硝基二烯炔与“困难”酮不对称共轭加成及串联环化反应
Scheme 5 Enantioselective conjugate additions of "difficult" ketones to nitrodienynes and tandem annulations

图式 6 1, 3-烯炔27a分子内串联环化反应机理
Scheme 6 Mechanism of 1, 3-enynes 27a intramolecular tandem annulations reaction

图式 7 硝基烯炔与醛类不对称共轭加成及串联反应机理
Scheme 7 Asymmetric conjugate addition reaction of nitroenynes with aldehydes and tandem cyclization mechanism

图式 8 硝基烯炔与烷基锂共轭加成反应
Scheme 8 Conjugated addition reaction of nitroenynes with alkyl lithium

图式 9 硝基烯炔与1, 3-二羰基化合物共轭加成反应
Scheme 9 Conjugated addition reaction of nitroenynes with 1, 3-dicarbonyl compounds

图式 10 硝基烯炔与α-取代1, 3-二羰基化合物共轭加成反应
Scheme 10 Asymmetric conjugated addition reaction of nitroenynes with α-substituted1, 3-dicarbonyl compounds

图式 11 多取代吡咯衍生物形成的可能机理
Scheme 11 Plausible mechanism for the formation of polysubstituted pyrrole derivatives
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 4636
- HTML全文浏览量: 386
 下载:
下载:
