 图1
A design for synthesis of Sorafenib analogs
Figure1.
A design for synthesis of Sorafenib analogs
图1
A design for synthesis of Sorafenib analogs
Figure1.
A design for synthesis of Sorafenib analogs
Citation:
Zhang Zhihu, Chen Yu, Chai Baoshan, Yang Xiaoman, Cai Xiaoyu, Cui Bo, You Song. Synthesis, Anticancer and Antibacterial Activities of Novel 2-Amino-4-phenylthiazole Derivatives Containing Amide Moiety[J]. Chinese Journal of Organic Chemistry,
2017, 37(9): 2377-2384.
doi:
10.6023/cjoc201704023

含酰胺结构的新型2-氨基-4-苯基噻唑类化合物的合成及抗癌和抗菌活性研究
摘要:
基于索拉非尼的结构特征设计、合成了一系列含有酰胺结构的新型2-氨基-4-苯基噻唑类化合物.所合成的化合物结构经1H NMR,13C NMR和HRMS表征,并对目标化合物的抗肿瘤和抗菌活性进行了研究.体外抗肿瘤抑制活性结果表明,部分目标化合物显示出较好的活性,特别是N-[3-(2-乙酰氨基噻唑-4-基)苯基]-3-氟苯甲酰胺(4n)对人类结肠癌细胞(HT29)和人肺上皮细胞(A549)细胞株具有显著的抗肿瘤作用,IC50值分别为6.31和7.98 μmol·L-1.进一步的研究表明化合物4n可以影响Raf/MEK/ERK信号通路.此外,体外抗菌活性筛选发现,N-[3-(2-乙酰氨基噻唑-4-基)苯基]-3,4-二氯苯甲酰胺(4h)、N-[3-(2-乙酰氨基噻唑-4-基)苯基]-3-氯苯甲酰胺(4i)和N-[3-(2-乙酰氨基噻唑-4-基)苯基]-2,4-二氯苯甲酰胺(4o)对革兰氏阳性菌、革兰氏阴性菌均具有较好抑制作用.
English
Synthesis, Anticancer and Antibacterial Activities of Novel 2-Amino-4-phenylthiazole Derivatives Containing Amide Moiety
Abstract:
A series of novel 2-amino-4-phenylthiazole derivatives containing amide moiety were designed and synthesized based on the structural features of sorafenib. The structures of synthesized compounds were characterized by 1H NMR, 13C NMR and HRMS. Both the anticancer and antibacterial activities of all the target compounds were evaluated. Most of the compounds showed potent activities, especially N-(3-(2-acetamidothiazol-4-yl)phenyl)-3-fluorobenzamide (4n) exhibited a remarkable antitumor effect against human colon cancer cell line (HT29) and human lung epithelial cells (A549) cells with IC50 values of 6.31 and 7.98 μmol·L-1, respectively. Further mechanistic study revealed that 4n can influence the Raf/MEK/ERK pathway. In addition, N-(3-(2-acetamidothiazol-4-yl)phenyl)-3, 4-dichlorobenzamide (4h), N-(3-(2-acetamidothiazol-4-yl)phenyl)-3-chlorobenzamide (4i) and N-(3-(2-acetamidothiazol-4-yl)phenyl)-2, 4-dichlorobenzamide (4o) exhibit moderate antibacterial activity against the tested bacteria.
-
Key words:
- thiazole derivative
- / amide
- / synthesis
- / anticancer
- / antibacterial
-
The mitogen-activated protein kinase (MAPK) cascade is a key intracellular signaling pathway that plays critical roles in the regulation of cell growth, cell survival and cell death. Signaling molecules in this pathway including RAS (rat sarcoma), BRAF, CRAF (rapidly accelerated fibrosarcoma) and ERK (extracellular regulated protein kinases) have been regarded as potential targets for the chemotherapy of malignant tumors.[1]
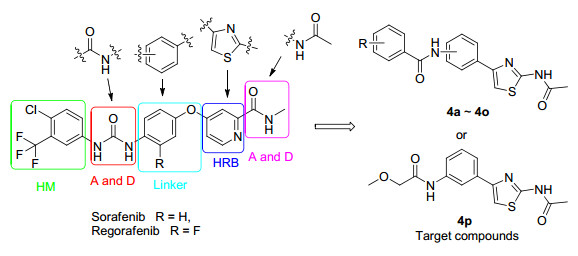
Sorafenib (Nexavar) (Figure 1), a multiple-kinase inhibitor, can inhibit multiple intracellular (Raf-1, B-Raf and mutant B-Raf) and cell surface kinases (c-Kit, Flt-3, mVEGFR2, mVEGFR3 and mPDGFRβ) and has been approved for the treatment of many human cancers including hepatocellular carcinoma (HCC), nonsmall cell lung cancer (NSCLC) and colorectal cancer (CRC) by the Food and Drug Administration (FDA). However, sorafenib has been observed the relative lack of activity in the tumors expressing mutant B-Raf[2] and unwanted side effects, [3] and more and more research focused on the optimization of sorafenib. Nowadays, the main modifications of these compounds were focused on the amide fragment, pyridine ring, phenoxy group or ureido fragments.[4] Recently, a large number of novel analogs of sorafenib have been designed in order to improve selectivity, effectiveness and safety, such as regorafenib[5] (BAY 73-4506) (Figure 1).
图1
A design for synthesis of Sorafenib analogs
Figure1.
A design for synthesis of Sorafenib analogs
Based on pharmacophore characteristics of sorafenib derivatives, we have designed and synthesized a series of amide compounds containing a thiazole ring with the aim of enhancing the antitumor activities. For this purpose, we introduced a acetamide fragment incorporated into the C-2 position of the thiazole ring to construct a bioisoster of the N-methyl-carboxamide side chain. The thiazole unit was used instead of pyridine ring of sorafenib. The ether linkage was removed in order to reduce the flexibility of the molecule. Amide-and urea-type fragments could be found in type Ⅱ inhibitors with antiproliferative properties.[6] Furthermore, urea scaffolds were replaced by amide units to construct a bioisoster by size with low electronegativity and possesses the ability to build hydrogen bonds. The design strategy and structures of the target compounds4a~4p are depicted in Figure 1. The antitumor activity of all the newly target compounds against human non-small cell lung cancer cell line A549 and human colon cancer cell line HT29 was evaluated.
Additionally, 2-amino-4-phenylthiazole is an important building block in medicinal chemistry and drug discovery research.[7] The derivatives of this scaffold are known by a wide range of antibacterial and antifungal activities.[8] Thus, target compounds were determined whether having antibacterial properties.
1 Results and discussion
1.1 Chemistry
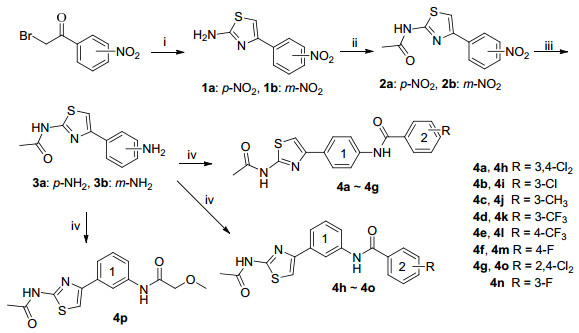
The synthesis of amide derivatives of 2-amino-4-phenylthiazole in this study is depicted in Scheme 1. The condensation-cyclization of the starting appropriate nitro substituted 2-bromo-1-phenylethanone with thiourea afforded a good yield of intermediates 1. Following acylation of intermediates 1 with acetic anhydride provided the amides 2. Aniline derivatives 3 were generated by treatment of intermediates 2 with reducing reagent Fe/NH4Cl in refluxing aqueous ethanol. Finally, the target compounds4a~4p could be prepared by condensing the intermediates 3 with the acyl chloride compounds in 74.1%~84.1% yields according to published procedures.[9]
 图Scheme 1
Synthetic route of the target compounds
Scheme1.
Synthetic route of the target compounds
图Scheme 1
Synthetic route of the target compounds
Scheme1.
Synthetic route of the target compounds
Regarding the nitrogen competition, 2-aminothiazole derivatives can exist as the amino and the corresponding imino thiazolines, the amino form is generally predominant in 2-aminothiaole.[10] Molecular orbital calculations have indicated that the ring nitrogen of 2-aminothiazole derivatives also carries a significant negative charge, which also has a certain extent nucleophilic reactivity.[11] Consequently, the exocyclic nitrogen acylation with extremely reactive acetyl chloride gaves the intermediates 2 and other byproducts (such as acetylation of the ring nitrogen[12]). Thus, acetic anhydride is used instead of the acetyl chloride to acetylate amino group of 2-aminothiazole derivatives in order to reduce side reactions. The intermediates1reacted with acetic anhydride at 130 ℃ for 24 h to give acetylation products 2 in satisfactory yield and purity. Intermediates 2 were also prepared from α-bromoketone, thiourea and acetic anhydride under microwave irradiation by a convenient one-pot reaction.[13] Compared with the stepwise reaction method, the yield and purity of the one-pot reaction are lower.
1.2 Biological activity
1.2.1 Anticancer activity
Thegrowth inhibition activities of the newly synthesized amide derivatives4a~4p against cancer cell lines A549 and HT29 were evaluated by the methylthiazolyldiphenyltetrazolium bromide (MTT) assay using sorafenib as a positive control. Results in Table 1 showed that N-(4-(2-acetamidothiazol-4-yl)phenyl)benzamide derivatives displayed better activity against A549 cell than those meta-counterparts, and a similar finding has been obtained for the target compounds against HT29 cell with the exception of 4g. The conformation of the target compounds is relatively fixed compared with sorafenib, and this result indicated that the compounds which had para-substituted aniline fragment were probably closer to the biologically active conformation of sorafenib than those with meta-substituted. To investigate the effect of substituents at the benzene ring 2 (Scheme 1) on activity, we modified the position of the substituents and introduced different substituents to the benzene ring 2. The results indicated that the anticancer activity is not obviously affected by these different substituents on the benzene ring 2, because only small differences existed in the properties of the substituents. It is worth pointing out that significant inhibition was achieved for compound 4n with an inhibition percentage of 65.20% and 91.30% for A549 and HT29, respectively. Moreover, compound 4p with shorter chain amide group demonstrated the relatively lower activities than those of the corresponding aryl amides.
 Table 1.
Inhibition of antiproliferative activity of the target compounds (inhibition/%)
Table 1.
Inhibition of antiproliferative activity of the target compounds (inhibition/%)
Compound A549 HT29 4a 53.10 65.20 4b 43.30 60.50 4c 28.80 59.90 4d 26.70 44.50 4e 55.20 57.70 4f 41.50 69.10 4g 15.50 28.60 4h 40.00 22.90 4i 25.60 61.00 4j 30.50 57.40 4k 25.10 20.60 4l 25.10 31.60 4m 8.10 60.50 4n 65.20 91.30 4o 65.30 74.90 4p 26.50 17.10 Sorafenib 98.25 99.53 Based on the evident growth inhibition activities demonstrated by most of the amide derivatives, we decided to study the concentration-response of the most potent derivatives to obtain their IC50 values in suppressing cell growth of A549 and HT29 cells. As shown in Table 2, most compounds demonstrated comparatively higher inhibitory activity against HT29 cell line and the less activity against A549 cell line. Compared to sorafenib, compounds 4a and 4n were found to have comparable antitumor activity.
Table 2.
Cytotoxic activity of targeted compounds against A549 and HT29 cells [IC50/(µmol•L-1)]
Compound A549 HT29 4a 9.33 7.67 4b 15.09 8.30 4c > 40 23.37 4e 10.39 10.51 4f > 40 9.86 4n 7.98 6.31 4o 19.24 12.61 Sorafenib 7.92 4.02 1.2.2 Analysis of targeted cellular pathways
Based on the results of in vitro anticancer activity, compounds 4n and 4c were chosen for further analysis to observe whether these derivatives shared similar targets with sorafeinib. The contrasting effects on the phosphorylation of BRAF, CRAF and ERK were analyzed by western blotting. As shown in Figure 2A, treatment of A549 cells with compounds 4n at 5 µmol•L-1 significantly inhibited the phosphorylations of BRAF, CRAF and ERK. By contrast, compound4c has little effect on the phosphorylation of BRAF, CRAF and ERK. Similiar results were obtained in HT29 cells (Figure 2B). The β-actin was used as loading control.
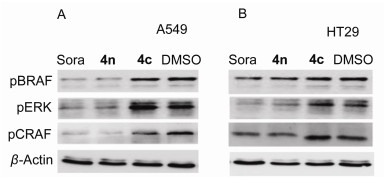 图2
The effect of representative compound on the phosphorylation of BRAF, CRAF and ERK kinases
Figure2.
The effect of representative compound on the phosphorylation of BRAF, CRAF and ERK kinases
图2
The effect of representative compound on the phosphorylation of BRAF, CRAF and ERK kinases
Figure2.
The effect of representative compound on the phosphorylation of BRAF, CRAF and ERK kinases
1.2.3 Conformational analysis
Molecular overlay studies were performed to better understand the similar activity of target compounds by superimposing the minimized conformations of sorafenib.[14] Three dimensional superimposition of sorafenib and compound 4n is shown in Figure 3. The minimized structures were obtained by calculation using the molecular mechanics force field method (MM2)[15]. They were then superimposed in virtue of the Molecular Overlay algorithm (Discovery Studio 3.5, Accelrys, Co.), which gives an automatic alignment procedure of a number of small molecules using equally weighted steric and electrostatic fields, and flexible rotatable bonds. The result showed a favorable overlap between the hydrogen bond acceptor, hydrogen bond donor and hydrophobic substituted benzyl groups (ring 2, Figure 3). It is not a good overlay between the 4-phenoxypyridine motif of sorafenib and the 4-phenylthiazole fragment of compound 4n. But considering flexibility of the ether bonds of sorafenib, this deviation may be tolerable in the target binding site.
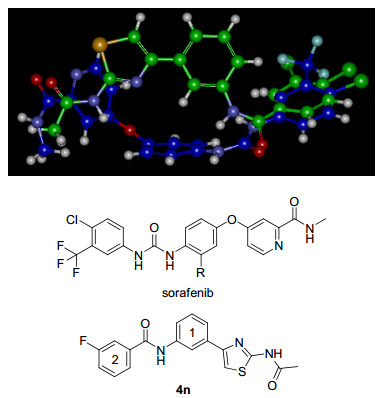 图3
A molecular overlay of sorafenib (blue) and compound 4n (green)
Figure3.
A molecular overlay of sorafenib (blue) and compound 4n (green)
图3
A molecular overlay of sorafenib (blue) and compound 4n (green)
Figure3.
A molecular overlay of sorafenib (blue) and compound 4n (green)
1.2.4 Antibacterial activity
All the target compounds were evaluated in vitro for their antibacterial activity against Staphylococcus aureus (S. aureus), Bacillus subtilis (B. subtilis) as examples of Gram-positive bacteria, and Escherichia coli (E. coli), Pseudomonas aeruginosa (P. aeruginosa) as examples of Gram-negative bacteria. The minimum inhibitory concentration values (MICs) are recorded in Table 3.
Table 3.
In vitro antibacterial activity [MIC/(µg•mL-1)]
Compound S. aureus B. subtilis E. coli P. aeruginosa 4a 100 50 200 > 200 4b 100 100 200 200 4c > 200 > 200 > 200 > 200 4d > 200 > 200 > 200 > 200 4e > 200 > 200 > 200 > 200 4f 200 100 > 200 > 200 4g 100 100 > 200 > 200 4h 12.50 25 100 200 4i 50 50 200 200 4j > 200 > 200 > 200 > 200 4k > 200 > 200 > 200 > 200 4l > 200 > 200 > 200 > 200 4m 100 50 > 200 > 200 4n 100 50 200 200 4o 25 50 100 200 4p > 200 > 200 > 200 > 200 Ciprofloxacin 6.25 6.25 12.50 25 The investigation of the antibacterial screening data revealed that all the synthesized compounds showed better activity against gram-positive bacteria. Compounds 4h, 4i, and 4o showed inhibition against the tested bacteria, especially gram-positive ones (S. aureus and B. subtilis). Overall, N-(4-(2-acetamidothiazol-4-yl)phenyl)benzamide derivatives are lower potency than meta-substituted analogs. The modification of the target compounds with various substituted benzamides and shorter chain amide group was performed to explore the structure-activity relationships. The MIC values of compounds 4a, 4b, 4f, 4g, 4h, 4i, 4m, 4n and 4o suggested that the introduction of halogen substituent on the benzene ring 2 (Scheme 1) could increase antibacterial activity. The antibacterial activity of these compounds was slightly enhanced in the following order: Cl > F. Compounds 4c, 4d, 4e, 4j, 4k and 4l were inactive for all microorganisms tested at 200 µg/mL. This suggested that synthesized compounds possessing larger groups such as methyl and trifluoromethyl may give diminished potency. In addition, the location of the substituent on the benzene ring 2 (Scheme 1) had no or little influence on the antibacterial activity. Compound 4p was found to be inactive against all tested bacterial strains, which indicated that the introduction of shorter chain amide group led to the decrease of antibacterial activity.
2 Conclusions
In conclusion, a series of new 2-amino-4-phenylthiazole derivatives possessing amide moiety were designed and synthesized. Most of these compounds exhibited significant anticancer activity against two cancer cell lines, A549 and HT29. The substituted N-(4-(2-acetamidothiazol-4-yl)phenyl)benzamides displayed higher activity than their meta-counterparts. The most active compound 4n significantly inhibited of the phosphorylations of BRAF, CRAF and ERK. In addition, compounds 4h, 4i and 4o exhibit moderate antibacterial activity against the tested bacteria. The results showed that the substituted N-(4-(2-acetamido-thiazol-4-yl)phenyl)benzamides were lower potency than meta-substituted analogs.
3 Experimental
3.1 General
Melting points were obtained in open capillaries using a WRS-1B melting point apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded on a 400/54 Premium Shielded NMR Magnet System. Mass spectral data were collected from Agilent 6200 Series TOF and 6500 Series Q-TOF LC/MS System B.05.01. (B5125) in positive ion modes.
3.2 Chemistry
3.2.1 Preparation of compounds 1a and 1b
The mixture of 2-bromo-1-(3-nitrophenyl)ethanone or 2-bromo-1-(4-nitrophenyl)ethanone (24.41 g, 0.10 mol) and thiourea (8.37 g, 0.11 mol) in anhydrous EtOH (200 mL) was heated at reflux for 5 h. After that, the solvent was concentrated, and saturated NaHCO3 (100 mL) was added to adjust the pH to about 8. The precipitates were collected by filtration, washed with H2O (100 mL×3) and dried.
4-(4-Nitrophenyl)thiazol-2-amine (1a):Yellow solid, yield 97.2%. m.p. 273.4~274.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.28~8.19 (m, 2H), 8.09~8.00 (m, 2H), 7.42 (s, 1H), 7.26 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 169.00, 148.22, 146.32, 141.28, 126.69, 124.45, 107.03, 40.53, 40.32, 40.11, 39.91, 39.70, 39.49, 39.28; HRMS calcd for C9H8N3O2S (M+H)+ 222.03372, found 222.03352.
4-(3-Nitrophenyl)thiazol-2-amine (1b): Yellow solid, yield 96.6%. m.p. 269.2~270.8 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 8.65 (dd, J=4.1, 2.2 Hz, 1H), 8.13 (d, J=10.6 Hz, 2H), 7.57 (td, J=8.1, 4.0 Hz, 1H), 6.92 (d, J=3.9 Hz, 1H), 5.07 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 167.35, 148.88, 146.96, 140.81, 131.67, 129.50, 122.23, 120.86, 105.09; HRMS calcd for C9H8N3O2S (M+H)+ 222.03372, found 222.03385.
3.2.2 Preparation of compounds 2a and 2b
Compound 1 (1.50 g, 7.0 mmol) was suspended in acetic anhydride (15 mL), and the mixture was heated at 130 ℃ with stirring for 24 h. The resulting solution was poured into ice water and stirred at room temperature until the excess anhydride was destroyed. The resulting precipitate was collected by filtering, washed with H2O (10 mL×3) and dried.
N-(4-(4-Nitrophenyl)thiazol-2-yl)acetamide (2a): Yellow solid, yield 97.0%. m.p. 312.0~312.7 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.39 (s, 1H), 8.37~8.26 (m, 2H), 8.19~8.11 (m, 2H), 7.98 (d, J=0.9 Hz, 1H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.34, 158.95, 146.95, 146.83, 140.71, 126.91, 124.67, 112.82, 40.53, 40.32, 40.12, 39.90, 39.70, 39.49, 39.28, 22.94; HRMS calcd for C11H10N3O3S (M+H)+ 264.04429, found 264.04384.
N-(4-(3-Nitrophenyl)thiazol-2-yl)acetamide (2b): Yellow solid, yield 96.8%. m.p. 296.2~298.0 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.39 (s, 1H), 8.73 (t, J=2.0 Hz, 1H), 8.35 (ddd, J=7.8, 1.7, 1.0 Hz, 1H), 8.18 (ddd, J=8.2, 2.4, 1.0 Hz, 1H), 7.93 (s, 1H), 7.74 (t, J=8.0 Hz, 1H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.27, 158.86, 148.75, 146.67, 136.22, 132.15, 130.84, 122.72, 120.46, 110.88, 40.52, 40.31, 40.10, 39.89, 39.69, 39.48, 39.27, 22.93; HRMS calcd for C11H10N3O3S (M+H)+ 264.04429, found 264.04417.
3.2.3 Preparation of compounds 3a and 3b
NH4Cl (0.19 g, 3.53 mmol) and iron powder (0.99 g, 17.65 mmol) were added to a suspension of nitro compound 2 (0.93 g, 3.53 mmol) in ethanol (60 mL) and water (30 mL). After stirring at 80 ℃ for 3 h, the mixture was filtered while hot. The filtrant was washed with hot ethanol (20 mL×3), and the filtrate was concentrated. The resulting concentrate was allowed to cool in an ice bath and the precipitate was collected, washed with H2O (10 mL) and dried.
N-(4-(4-Aminophenyl)thiazol-2-yl)acetamide (3a): Pale yellow solid, yield 83.2%. m.p. 182.0~183.7 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 10.92 (s, 1H), 7.63 (d, J=7.9 Hz, 2H), 7.28 (dd, J=3.3, 1.5 Hz, 1H), 6.95 (d, J=3.0 Hz, 1H), 6.73 (dd, J=7.9, 3.9 Hz, 2H), 1.95 (t, J=2.9 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 168.16, 158.81, 149.78, 146.52, 127.39, 124.98, 115.15, 105.02, 22.88; HRMS calcd for C11H12N3OS (M+H)+ 234.07011, found 234.07124.
N-(4-(3-Aminophenyl)thiazol-2-yl)acetamide (3b): Pale yellow solid, yield 84.4%. m.p. 189.2~189.9 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 10.66 (s, 1H), 7.30~7.14 (m, 4H), 7.11 (s, 1H), 6.69 (ddd, J=5.8, 3.9, 2.4 Hz, 1H), 2.01 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 168.18, 158.69, 149.68, 146.74, 135.24, 129.81, 116.64, 115.00, 112.72, 107.76, 22.89; HRMS calcd for C11H12N3OS (M+H)+ 234.07011, found 234.07731.
3.2.4 Preparation of compounds 4a~4p
The chloride (0.184 mmol) was added to a solution of compound 3 (50 mg, 0.123 mmol) and triethylamine (25 mg, 0.246 mmol) in CH2Cl2 (5 mL), and stirred for 8 h at room temperature. Then water (10 mL) was added and extracted with CH2Cl2 (10 mL×3). The combined organic layer was dried (Na2SO4), concentrated and purified by column chromatography on silica gel.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-3, 4-dichlorobenzamide (4a): Gray solid, yield 81.6%. m.p. 295.8~296.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.30 (s, 1H), 10.52 (s, 1H), 8.25 (d, J=1.9 Hz, 1H), 8.01~7.88 (m, 3H), 7.86 (dd, J=8.5, 2.1 Hz, 3H), 7.56 (s, 1H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.05, 163.56, 158.34, 148.82, 138.71, 135.56, 134.83, 131.73, 131.20, 130.57, 130.02, 128.48, 126.43, 120.88, 107.45, 22.94; HRMS calcd for C18H14Cl2N3O2S (M+H)+ 406.01838, found 406.01781.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-3-chlorobenzamide (4b): Gray solid, yield 81.2%. m.p.281.8~282.8 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.30 (s, 1H), 10.49 (s, 1H), 8.05 (t, J=1.9 Hz, 1H), 8.00~7.88 (m, 3H), 7.88 (d, J=8.9 Hz, 2H), 7.71 (ddd, J=8.0, 2.2, 1.1 Hz, 1H), 7.66~7.53 (m, 2H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.04, 164.48, 158.33, 148.86, 138.88, 137.28, 133.64, 131.87, 130.87, 130.46, 127.84, 126.94, 126.41, 120.85, 107.39, 22.94; HRMS calcd for C18H15ClN3O2S (M+H)+ 372.05735, found 372.05711.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-3-methylbenza-mide (4c): White solid; yield 83.8%. m.p. 154.5~156.4 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.27 (s, 1H), 10.31 (s, 1H), 7.87 (t, J=6.2 Hz, 4H), 7.77 (d, J=7.3 Hz, 2H), 7.53 (s, 1H), 7.42 (d, J=5.4 Hz, 2H), 2.41 (s, 3H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.02, 166.09, 158.31, 148.94, 139.23, 138.15, 135.36, 132.61, 130.16, 128.75, 128.56, 126.37, 125.26, 120.70, 107.23, 22.94, 21.42; HRMS calcd for C19H18N3O2S (M+H)+ 352.11197, found 352.11233.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-3-(trifluoro-methyl)benzamide (4d): White solid, yield 78.1%. m.p. 287.1~289.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.28 (s, 1H), 10.58 (s, 1H), 8.33~8.24 (m, 2H), 7.99 (d, J=7.6 Hz, 1H), 7.91 (d, J=8.8 Hz, 2H), 7.89~7.76 (m, 3H), 7.55 (s, 1H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.06, 164.47, 158.34, 148.84, 138.78, 136.16, 132.29, 130.57, 130.20, 129.76, 129.44, 128.63, 126.44, 125.76, 124.69, 124.65, 123.05, 120.97, 109.98, 107.45, 22.93; HRMS calcd for C19H15F3N3O2S (M+H)+ 406.08371, found 406.08363.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-4-(trifluoro-methyl)benzamide (4e): White solid, yield 81.3%. m.p. 298.6~299.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.31 (s, 1H), 10.61 (s, 1H), 8.19 (d, J=8.0 Hz, 2H), 8.00~7.85 (m, 6H), 7.57 (d, J=1.3 Hz, 1H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.05, 164.82, 158.34, 148.84, 139.14, 138.81, 131.95, 131.63, 130.56, 129.04, 126.44, 125.91, 125.88, 125.84, 125.80, 125.70, 122.99, 120.87, 107.45, 22.93; HRMS calcd for C19H15F3N3O2S (M+H)+ 406.08371, found 406.08363.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-4-fluorobenza-mide (4f): Gray solid, yield 83.6%. m.p. 299.8~301.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.29 (s, 1H), 10.41 (s, 1H), 8.10~8.01 (m, 2H), 7.93~7.80 (m, 4H), 7.53 (s, 1H), 7.43~7.34 (m, 2H), 2.16 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.07, 165.74, 164.85, 163.26, 158.41, 148.89, 139.09, 131.77, 131.74, 130.90, 130.81, 130.29, 126.38, 120.79, 115.90, 115.68, 107.26, 22.97; HRMS calcd for C18H15FN3O2S (M+H)+ 356.08690, found 356.08704.
N-(4-(2-Acetamidothiazol-4-yl)phenyl)-2, 4-dichloroben-zamide (4g): Gray solid; yield 77.9%. m.p. 301.5~302.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.31 (s, 1H), 10.69 (s, 1H), 7.91 (d, J=8.5 Hz, 2H), 7.84~7.76 (m, 3H), 7.69 (d, J=8.1 Hz, 1H), 7.64~7.53 (m, 2H), 2.19 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.06, 164.42, 158.39, 148.79, 138.69, 136.13, 135.31, 131.65, 130.78, 130.55, 129.64, 127.93, 126.57, 120.09, 107.44, 22.94; HRMS calcd for C18H14Cl2N3O2S (M+H)+ 406.01838, found 406.02212.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-3, 4-dichloroben-zamide (4h): White solid, yield 82.1%. m.p. 220.7~222.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.32 (s, 1H), 10.49 (s, 1H), 8.42 (t, J=2.1 Hz, 1H), 8.25 (d, J=2.2 Hz, 1H), 7.97 (ddd, J=8.4, 2.1, 0.8 Hz, 1H), 7.84 (dd, J=8.4, 0.8 Hz, 1H), 7.69~7.61 (m, 1H), 7.65~7.53 (m, 1H), 7.55 (s, 1H), 7.42 (t, J=7.9 Hz, 1H), 2.17 (d, J=0.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.13, 163.56, 158.40, 148.99, 139.54, 135.54, 135.23, 134.84, 131.73, 131.23, 130.04, 129.46, 128.52, 121.91, 120.37, 118.45, 108.55, 22.94; HRMS calcd for C18H14Cl2N3O2S (M+H)+ 406.01838, found 406.01908.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-3-chlorobenza-mide (4i): Yellow solid, yield 80.1%. m.p. 210.3~211.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.32 (s, 1H), 10.45 (s, 1H), 8.44 (t, J=1.9 Hz, 1H), 8.05 (t, J=1.9 Hz, 1H), 7.95 (ddd, J=7.7, 1.7, 1.1 Hz, 1H), 7.71~7.58 (m, 4H), 7.55 (s, 1H), 7.42 (t, J=7.9 Hz, 1H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.14, 164.47, 158.39, 149.04, 139.67, 137.24, 135.20, 133.65, 131.88, 130.89, 129.43, 127.84, 126.95, 121.81, 120.37, 118.43, 108.52, 22.94; HRMS calcd for C18H15ClN3O2S (M+H)+ 372.05735, found 372.06079.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-3-methylbenz-amide (4j): Yellow solid, yield 83.8%. m.p. 199.5~200.7 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 11.50 (s, 1H), 8.18 (s, 1H), 8.11 (s, 1H), 7.94 (s, 1H), 7.73 (s, 1H), 7.56 (dd, J=16.1, 7.8 Hz, 2H), 7.42~7.33 (m, 3H), 7.14 (d, J=3.0 Hz, 1H), 2.44 (d, J=3.2 Hz, 3H), 2.31 (d, J=2.9 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 171.49, 168.54, 165.88, 159.52, 151.98, 138.48, 132.78, 130.58, 129.58, 128.66, 128.35, 127.83, 127.23, 124.67, 124.04, 122.18, 120.20, 118.14, 108.06, 23.15, 21.40; HRMS calcd for C19H18N3O2S (M+H)+ 352.11197, found 352.11423.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-3-(trifluorome-thyl)benzamide (4k): White solid, yield 78.4%. m.p. 220.0~220.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.32 (s, 1H), 10.59 (s, 1H), 8.42 (t, J=1.9 Hz, 1H), 8.36~8.26 (m, 2H), 7.99 (dq, J=7.7, 0.9 Hz, 1H), 7.81 (t, J=7.8 Hz, 1H), 7.70~7.62 (m, 2H), 7.56 (s, 1H), 7.43 (t, J=7.9 Hz, 1H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.14, 164.42, 158.42, 148.99, 139.60, 136.12, 135.23, 132.35, 130.22, 129.74, 129.46, 129.42, 128.66, 128.63, 124.72, 124.69, 124.65, 124.61, 121.89, 120.46, 118.55, 108.54, 22.95; HRMS calcd for C19H15F3N3O2S (M+H)+ 406.08371, found 406.08875.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-4-(trifluorome-thyl)benzamide (4l): White solid, yield 84.1%. m.p. 236.7~238.7 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.35 (s, 1H), 10.60 (s, 1H), 8.47 (t, J=1.9 Hz, 1H), 8.21 (d, J=7.7 Hz, 2H), 7.96 (d, J=8.3 Hz, 2H), 7.73~7.62 (m, 2H), 7.58 (s, 1H), 7.45 (t, J=7.9 Hz, 1H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.15, 164.81, 158.40, 149.00, 139.60, 139.09, 135.23, 131.96, 131.65, 129.47, 129.05, 126.44, 125.92, 125.88, 125.84, 125.80, 125.71, 121.91, 120.87, 120.41, 118.46, 108.56, 22.93; HRMS calcd for C19H15F3N3O2S (M+H)+ 406.08371, found 406.08599.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-4-fluorobenza-mide (4m): White solid, yield 80.6%. m.p. 249.9~251.1 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.32 (s, 1H), 10.40 (s, 1H), 8.46 (t, J=1.9 Hz, 1H), 8.10 (ddd, J=8.5, 5.3, 2.6 Hz, 2H), 7.65 (tt, J=7.9, 1.3 Hz, 2H), 7.57 (s, 1H), 7.42 (td, J=8.8, 8.4, 6.6 Hz, 3H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.14, 164.84, 158.38, 149.08, 139.85, 135.17, 130.89, 130.80, 129.39, 121.64, 120.37, 118.43, 115.91, 115.69, 108.47, 22.93; HRMS calcd for C18H15FN3O2S (M+H)+ 356.08690, found 356.08801.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-3-fluorobenzamide (4n): White solid, yield 82.5%. m.p. 203.6~205.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.32 (s, 1H), 10.41 (s, 1H), 8.44 (t, J=1.9 Hz, 1H), 7.89~7.77 (m, 2H), 7.67~7.57 (m, 3H), 7.55 (d, J=0.8 Hz, 1H), 7.50~7.38 (m, 2H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.12, 166.71, 158.39, 149.04, 139.67, 137.53, 135.20, 131.10, 129.42, 124.34, 121.78, 120.38, 119.07, 118.86, 118.44, 115.05, 114.82, 108.51, 22.93; HRMS calcd. for C18H15FN3O2S (M+H)+ 356.08690, found 356.08784.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-2, 4-dichlorobenzamide (4o): Yellow solid, yield 80.9%. m.p. 207.9~209.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.36 (s, 1H), 10.67 (s, 1H), 8.44 (t, J=1.9 Hz, 1H), 7.81 (d, J=2.0 Hz, 1H), 7.71~7.65 (m, 2H), 7.60 (dd, J=8.2, 2.0 Hz, 1H), 7.58 (s, 1H), 7.52 (ddd, J=8.1, 2.2, 1.1 Hz, 1H), 7.43 (t, J=7.9 Hz, 1H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 169.16, 164.50, 158.41, 148.95, 139.51, 136.15, 135.36, 135.29, 131.66, 130.77, 129.64, 129.59, 127.92, 121.88, 119.58, 117.57, 108.66, 22.93; HRMS calcd. for C18H14Cl2N3O2S (M+H)+ 406.01838, found 406.01976.
N-(3-(2-Acetamidothiazol-4-yl)phenyl)-2-methoxyacet-amide (4p): Yellow solid, yield 74.1%. m.p. 206.0~208.1 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 10.57 (s, 1H), 8.40 (s, 1H), 8.10 (d, J=2.0 Hz, 1H), 7.61~7.50 (m, 2H), 7.39 (t, J=7.9 Hz, 1H), 7.17 (s, 1H), 4.07 (s, 2H), 3.53 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 168.32, 167.80, 158.61, 148.81, 137.56, 134.99, 129.50, 122.22, 119.45, 117.55, 108.32, 72.04, 59.29, 23.04; HRMS calcd for C14H16N3O3S (M+H)+ 306.09124, found 306.09408.
3.3 Biological assay
3.3.1 Anticancer assay
The anticancer activity of the target compounds on the A549 and HT-29 cell lines was tested using the MTT assay. The cells were seeded in 96-well plates at a density of 5×103 cells per well and incubated with 5% CO2 at 37 ℃. Next day, the target compounds were added into the culture medium. Triplicates of each concentration were used. After 48 h of incubation, 5 mg/mL fresh MTT was added to each well and incubated at 37 ℃ for 4 h. The medium was removed, and 100 μL of dimethyl sulphoxide (DMSO) was added to each well to dissolve the formazan. The absorbance at 490 nm (for absorbance of MTT formazan) and 630 nm (for the reference wavelength) were measured using the microplate reader. Cell growth inhibition rate formula is (AC-AT)/AC×100%. AC, absorbance value of the blank control group; AT, absorbance value of the experimental group. The IC50 was calculated using GraphPad Prism version 6.00 software from the non-linear curve.
3.3.2 Western blot analysis
Cells were harvested, lysed in radio immuno precipitation assay (RIPA) buffer containing 1 mmol•L-1 phenylmethanesulfonyl fluoride (PMSF), 10 μg/mL leupeptin, 1 mmol•L-1 aprotinin for 30 min. The protein concentration was determined by the bicinchoninic acid (BCA) assay. Cell lysate proteins (50 μg each lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nylon membranes. The membrane was blocked with 5% non-fat milk for 1 h, incubated with 1: 1000 diluted primary antibodies for 4 h, followed by horseradish peroxidase (HRP)-conju-gated secondary antibodies (1: 2000) for 1 h at room temperature. Bound antibodies were revealed by enhanced chemiluminescence (ECL). The levels of interested proteins were analyzed by Chemi-Genius gel imaging and analysis system (UK).
3.3.3 Antibacterial assay
The target compounds were screened for their antibacterial activity against S. aureus [CMCC (B) 26003], B. subtilis [CMCC (B) 63501], E. coli [CMCC (B) 44102] and P. aeruginosa [CMCC (B) 10104] by the broth microdilution assay. DMSO was used as a diluent to get the desired concentration. Ciprofloxacin was used as a reference standard. Serial 2-fold dilutions of each compound (200, 100, 50, 25, 12.5, 6.25 g/mL) were made to determine the MICs.
-
-
[1]
(a) Heidorn, S. J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescuduvas, I.; Dhomen, N.; Hussain, J.; Reisfilho, J. S.; Springer, C. J.; Pritchard, C. Cell 2010, 140, 209.
(b) Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B. J.; Anderson, D. J.; Alvarado, R.; Ludlam, M. J.; Stokoe, D.; Gloor, S. L.; Vigers, G. Nature 2010, 464, 431. -
[2]
Eisen, T.; Ahmad, T.; Flaherty, K. T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L. M. Br. J. Cancer 2006, 95, 581. doi: 10.1038/sj.bjc.6603291
-
[3]
(a) Wood, L. S. Community Oncol. 2006, 3, 558.
(b) Chu, E. Y. ; Wanat, K. A. ; Miller, C. J. ; Amaravadi, R. K. ; Fecher, L. A. ; Brose, M. S. ; McGettigan, S. ; Giles, L. R. ; Schuchter, L. M. ; Seykora, J. T. J. Am. Acad. Dermatol. 2012, 67, 1265.
(c) Autier, J. ; Escudier, B. ; Wechsler, J. ; Spatz, A. ; Robert, C. Arch. Dermatol. 2008, 144, 886. -
[4]
(a) Wu, C.; Wang, M.; Tang, Q.; Luo, R.; Chen, L.; Zheng, P.; Zhu, W. Molecules 2015, 20, 19361.
(b) Yao, J.; Chen, J.; He, Z.; Sun, W.; Xu, W. Bioorg. Med. Chem. 2012, 20, 2923.
(c) Jung, M. H.; El-Gamal, M. I.; Abdel-Maksoud, M. S.; Sim, T.; Yoo, K. H.; Oh, C.-H. Bioorg. Med. Chem. Lett. 2012, 22, 4362.
(d) Jiao, Y.; Xin, B. T.; Zhang, Y.; Wu, J.; Lu, X.; Zheng, Y.; Tang, W.; Zhou, X. Eur. J. Med. Chem. 2015, 90, 170.
(e) Kong, X.; Yao, Z.; He, Z.; Xu, W.; Yao, J. MedChemComm 2015, 6, 867.
(f) Daydé-Cazals, B.; Fauvel, B.; Singer, M.; Feneyrolles, C.; Bestgen, B.; Gassiot, F.; Spenlinhauer, A.; Warnault, P.; Van Hijfte, N.; Borjini, N.; Chevé, G.; Yasri, A. J. Med. Chem. 2016, 59, 3886.
(g) Yao, J.; He, Z.; Chen, J.; Chen, D.; Sun, W.; Xu, W. Chin. J. Chem. 2012, 30, 2423. -
[5]
(a) Gao, G. R.; Li, M. Y.; Lv, Y. C.; Cao, S. F.; Tong, L. J.; Wei, L. X.; Ding, J.; Xie, H.; Duan, W. H. Chin. Chem. Lett. 2016, 27, 200.
(b) Wu, Z.; Yan, M.; Hu, S. H.; Yu, Z. C.; Zhu, Y.; Cheng, Y. D.; Liu, H. C.; Zhang, Y. M.; Yao, S. H.; Tang, W. F.; Lu, T. Chin. Chem. Lett. 2014, 25, 351. -
[6]
Liu, Y.; Gray, N. S. Nat. Chem. Biol. 2006, 2, 358. doi: 10.1038/nchembio799
-
[7]
Das, D.; Sikdar, P.; Bairagi, M. Eur. J. Med. Chem. 2016, 109, 89. doi: 10.1016/j.ejmech.2015.12.022
-
[8]
(a) Bharti, S. K.; Nath, G.; Tilak, R.; Singh, S. K. Eur. J. Med. Chem. 2010, 45, 651.
(b) Qin, Y. J.; Wang, P. F.; Makawana, J. A.; Wang, Z. C.; Wang, Z. N.; Yan, G.; Jiang, A. Q.; Zhu, H. L. Bioorg. Med. Chem. Lett. 2014, 24, 5279. -
[9]
Coumar, M. S.; Chu, C. Y.; Lin, C. W.; Shiao, H. Y.; Ho, Y. L.; Reddy, R.; Lin, W. H.; Chen, C. H.; Peng, Y. H.; Leou, J. S.; Lien, T. W.; Huang, C. T.; Fang, M. Y.; Wu, S. H.; Wu, J. S.; Chittimalla, S. K.; Song, J. S.; Hsu, J. T.; Wu, S. Y.; Liao, C. C.; Chao, Y. S.; Hsieh, H. P. J. Med. Chem. 2010, 53, 4980. doi: 10.1021/jm1000198
-
[10]
(a) Tseng, C. Magn. Reson. Chem. 1987, 25, 105.
(b) Bramley, S. E.; Dupplin, V.; Goberdhan, D. G.; Meakins, G. D. J. Chem. Soc., Perkin Trans 1. 1987, 639.
(c) Forlani, L.; Tocke, A. L.; Del Vecchio, E.; Lakhdar, S.; Goumont, R.; Terrier, F. J. Org. Chem. 2006, 71, 5527. -
[11]
Metzger, J. V. In Chemistry of Heterocyclic Compounds, Vol. 34, Part 2, Ed.:Metzger, J. V., New York, 1979, p. 17.
-
[12]
Zhou, A.; Pittman, C. U. Tetrahedron Lett. 2005, 46, 3801. doi: 10.1016/j.tetlet.2005.03.205
-
[13]
袁明月, 刘敏, 张英群, 闫红飞, 李德富, 张冬暖, 刘卉闵, 有机化学, 2012, 32, 1746. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmYuan, M.; Liu, M.; Zhang, Y.; Yan, H.; Li, D.; Zhang, D.; Liu, H. Chin. J. Org. Chem. 2012, 32, 1746(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[14]
(a) Wermuth, C. G. Drug Discovery Today 2006, 11, 348.
(b) Martin, Y. C.; Kofron, J. L.; Traphagen, L. M. J. Med. Chem. 2002, 45, 4350.
(c) Vainio, M. J.; Puranen, J. S.; Johnson, M. S. J. Chem. Inf. Model. 2009, 49, 492.
-
[1]
-

Figure 1 A design for synthesis of Sorafenib analogs
A: hydrogen bond acceptor; D: hydrogen bond donor; HRB: hinge-region binding; HM: hydrophobic motif

Scheme 1 Synthetic route of the target compounds
Reagents and conditions: (ⅰ) thiourea, ethanol, reflux, 5 h; (ⅱ) acetic anhydride, 130 ℃ for 24 h; (ⅲ) Fe, NH4Cl, ethanol/water, reflux; (ⅳ) CH2Cl2, Et3N, r.t.

Figure 2 The effect of representative compound on the phosphorylation of BRAF, CRAF and ERK kinases
Table 1. Inhibition of antiproliferative activity of the target compounds (inhibition/%)
Compound A549 HT29 4a 53.10 65.20 4b 43.30 60.50 4c 28.80 59.90 4d 26.70 44.50 4e 55.20 57.70 4f 41.50 69.10 4g 15.50 28.60 4h 40.00 22.90 4i 25.60 61.00 4j 30.50 57.40 4k 25.10 20.60 4l 25.10 31.60 4m 8.10 60.50 4n 65.20 91.30 4o 65.30 74.90 4p 26.50 17.10 Sorafenib 98.25 99.53  下载: 导出CSV
下载: 导出CSV
Table 2. Cytotoxic activity of targeted compounds against A549 and HT29 cells [IC50/(µmol•L-1)]
Compound A549 HT29 4a 9.33 7.67 4b 15.09 8.30 4c > 40 23.37 4e 10.39 10.51 4f > 40 9.86 4n 7.98 6.31 4o 19.24 12.61 Sorafenib 7.92 4.02
下载: 导出CSV
Table 3. In vitro antibacterial activity [MIC/(µg•mL-1)]
Compound S. aureus B. subtilis E. coli P. aeruginosa 4a 100 50 200 > 200 4b 100 100 200 200 4c > 200 > 200 > 200 > 200 4d > 200 > 200 > 200 > 200 4e > 200 > 200 > 200 > 200 4f 200 100 > 200 > 200 4g 100 100 > 200 > 200 4h 12.50 25 100 200 4i 50 50 200 200 4j > 200 > 200 > 200 > 200 4k > 200 > 200 > 200 > 200 4l > 200 > 200 > 200 > 200 4m 100 50 > 200 > 200 4n 100 50 200 200 4o 25 50 100 200 4p > 200 > 200 > 200 > 200 Ciprofloxacin 6.25 6.25 12.50 25
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 10
- 文章访问数: 2251
- HTML全文浏览量: 205
 下载:
下载:
