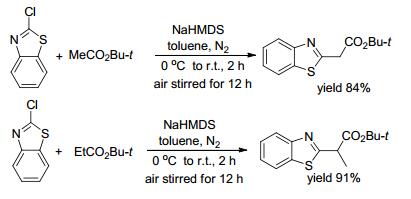
 图式1
NaHMDS催化苯并噻唑2-位烷基化
图式1.
NaHMDS catalyzed 2-position alkylation of benzothiazole
图式1
NaHMDS催化苯并噻唑2-位烷基化
图式1.
NaHMDS catalyzed 2-position alkylation of benzothiazole
引用本文:
戴小强, 朱亚波, 汪洲洋, 翁建全. 2-位官能团化合成2-取代苯并噻唑的研究进展[J]. 有机化学,
2017, 37(8): 1924-1938.
doi:
10.6023/cjoc201703034

Citation: Dai Xiaoqiang, Zhu Yabo, Wang Zhouyang, Weng Jianquan. Progress in 2-Position Functionalized Synthesis of 2-Substitutedbenzothiazoles[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 1924-1938. doi: 10.6023/cjoc201703034
Citation: Dai Xiaoqiang, Zhu Yabo, Wang Zhouyang, Weng Jianquan. Progress in 2-Position Functionalized Synthesis of 2-Substitutedbenzothiazoles[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 1924-1938. doi: 10.6023/cjoc201703034
2-位官能团化合成2-取代苯并噻唑的研究进展
English
Progress in 2-Position Functionalized Synthesis of 2-Substitutedbenzothiazoles
Abstract:
2-Substitutedbenzothiazole derivatives were widely used in chemical, pharmaceutical and agricultural fields. 2-Position functionalized method was a convenient and effective way to synthesize 2-substitutedbenzothiazoles. In this paper, the methods for the synthesis of 2-substitutedbenzothiazoles by the alkylation, acylation, arylation, amination and phosphorylation reactions at the 2-position are reviewed, and their future outlook is also discussed.
-
Key words:
- benzothiazole
- / 2-position functionalization
- / alkylation
- / acylation
- / arylation
- / amination
- / phosphorylation
-
苯并噻唑及其衍生物作为一类含氮、硫的稠杂环化合物, 具有广泛的用途.由于具有良好的生物活性, 其作为重要的母体和先导骨架被广泛应用于农药[1, 2]、医药[3~5]等药物创制中.在工业上, 其还可用作橡胶硫化促进剂[6]、塑料染色剂[7]等.鉴于此, 自1879年Hofmann[8]首次合成了2-氯与2-苯基苯并噻唑之后, 对其的结构修饰和合成便得到了广泛的关注和发展.
苯并噻唑衍生物的结构修饰主要是在苯环上或是噻唑环2-位上.由于2-位取代基对苯并噻唑衍生物活性影响较大, 因此其2-位取代衍生物的合成最受关注[9]. 2-取代苯并噻唑的合成主要有两种方法: (1) 通过缩合成环得到2-位取代的苯并噻唑; (2) 采用2-位官能团化方法在苯并噻唑的2-位引入取代基合成2-取代苯并噻唑.近年来2-取代苯并噻唑的合成方法层出不穷, 关于缩合成环构建2-取代苯并噻唑, 王玉炉[9]及竺宁[10]等已分别按合成方法和反应底物进行分类对其合成方法进行了详细总结.但对于采用2-位官能团化方法合成2-取代苯并噻唑的合成方法, 尚无文献对其进行系统总结与讨论.基于此, 本文主要根据反应类型的不同, 对近10年在金属催化、非金属催化以及光催化等催化体系下, 采用2-位官能团化法合成2-取代苯并噻唑的研究进行综述和总结.
1 2-烷基化苯并噻唑
苯并噻唑作为一种常见的原料, 其在药物合成、天然产物以及化工中间体中具有广泛的用途, 因此合成2-烷基化苯并噻唑的方法也得到了广泛的研究.早在1994年, Nishio课题组[11]利用光催化将苯并噻唑-2-硫酮和烯烃进行[2+2]环加成, 再开环制备了2-取代烷基苯并噻唑.这是首次利用光催化方法合成2-取代苯并噻唑衍生物.
随后, 2003年Kondo课题组[12]报道了一种t-Bu-P4碱在反应体系中催化芳香烃去质子功能化反应, 实现了苯并噻唑和二苯甲酮的直接偶联反应(Eq. 1).该反应对杂环芳烃和甲酮衍生物具有广泛的适用性, 且产率高. 2012年该课题组[13]再次报道了利用胺基取代硅烷和鎓的氟化物结合实现了脱质子功能烷基化反应.反应以P5F作碱, 胺基取代硅烷(Me3SiNR2)作添加剂, 使苯并噻唑和二苯甲酮直接偶联(Eq. 2).该体系反应条件温和, 分离产率高, 且对各种芳杂环具有广泛的适用性, 但也存在反应时间长、碱不易得等缺点.
2009年, Daugulis等[14]在有机碱t-BuOLi作用下, 促使苯并噻唑和二芳酮发生偶联反应(Eq. 3).反应体系简便, 无任何金属催化剂作用, 仅在逐步升温的条件下就发生酮羰基的还原加成, 并获得77%的分离产率.该碱廉价易得, 反应体系简便.
2014年, Inamoto等[15]以四甲基氟化胺(TMAF)/碱金属醇盐作活化剂催化了杂芳环去质子功能化反应(Eq. 4).反应以TMAF作氟化物源, MeOK作催化剂, N, N-二甲基甲酰胺(DMF)作溶剂, 室温下反应24 h, 实现了苯并噻唑和二芳甲酮的烷基化.该反应具有很高的实用性, 是一种有效的去质子化方法.
2006年, Shen等[16]以碱催化了卤代芳杂环α-烷基化(Scheme 1).反应以二-(三甲基硅)-氨基钠(NaHMDS)作碱, 甲苯作溶剂, 氮气保护下, 0 ℃加碱搅拌至室温反应2 h, 再通空气搅拌12 h得目标产物.该反应体系简便、无任何金属催化剂, 反应条件温和, 且对各种酯、内酯、酰胺、内酰胺具有广泛的适用性.但这类反应也存在操作复杂、不易控制、需氮保等不足.
图式1
NaHMDS催化苯并噻唑2-位烷基化
图式1.
NaHMDS catalyzed 2-position alkylation of benzothiazole
2010年, Miura课题组[17]报道了钯催化的直接苄基化反应, 反应以2.5 mol% Pd2(dba)3作催化剂, 5 mol% dppp作添加剂, 醋酸钠作碱, 120 ℃反应条件下实现了目标产物的合成(Eq. 5).反应得到良好的产率, 且直接实现了C—H键的活化.
2008年, Doucet等[18]首次报道了Pd催化的芳杂环和溴代烯烃的烯基化, 反应以PdCl(C3H5)(dppb)作催化剂, DMF作溶剂, Cs2CO3作碱, 实现了芳杂环和溴代烷烯的直接偶联反应(Eq. 6).该实验证明, 芳杂环功能化反应具有广泛的适用性, 各种卤代烯、卤代芳烃和三氟甲磺酸类化合物均适用, 且都具有良好的产率.但反应体系需高温和氩气保护, 反应时间长.
2010年, Olsson等[19]合成了含苯并噻唑结构的药物, 在基于功能细胞的测定中被鉴定为钙传感受体(CaST)的正变构调节剂(PAM).该反应通过苯并噻唑和芳香酮的直接偶联得到目标产物(Scheme 2).反应采用常规的丁基锂/-78 ℃反应体系, 合成出具有药物活性的苯并噻唑衍生物, 该类物质的合成进一步说明了苯并噻唑基团在药物分子创制中的重要作用.
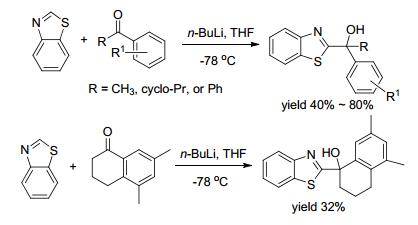 图式2
苯并噻唑和芳酮的直接偶联
图式2.
Direct coupling of benzothiazole and arone
图式2
苯并噻唑和芳酮的直接偶联
图式2.
Direct coupling of benzothiazole and arone
2011年, 王剑波等[20]报道了一种Cu催化的交叉偶联反应, 在强碱t-BuOLi作用下, 实现了苯并噻(噁)唑的C—H键活化(Eq. 7).反应体系先利用Cu催化剂和苯并噻(噁)唑2-位络合, 再利用Cu的孤对电子和另一底物络合, 再发生迁移, 酸化得到目标产物.该反应路径独特、官能团容忍性好, 且得到良好的分离产率, 对这一类反应的探究具有重要意义.
随后2012年, Prabhu等[21]也采用Pd催化的烷基化反应合成取代烯, 反应以2.5 mol% Pd(PPh3)2Cl2作催化剂, t-BuOLi作碱, 100 ℃反应条件下实现了2-卤代苯并噻唑和腙的烷基化反应(Scheme 3).相比同类反应, 该体系不需加入其他任何添加剂, 催化剂用量少, 分离产率高.同年, Miura课题组[22]也以N-对甲苯磺酰腙为烷基化试剂, 采用相似的催化条件, 额外加入苯酚作配体, 采用Ni/Co-催化实现苯并噁唑(苯并噻唑)的2-位烷基化反应(Eq. 8).
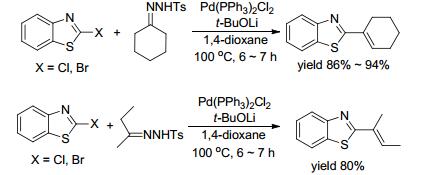 图式3
Pd催化2-卤代苯并噻唑和腙的烷基化反应
图式3.
Palladium catalyzed alkylation reactions of 2-halo-benzothiazole with hydrazones
图式3
Pd催化2-卤代苯并噻唑和腙的烷基化反应
图式3.
Palladium catalyzed alkylation reactions of 2-halo-benzothiazole with hydrazones
2012年, 王锐等[23]以强碱作金属试剂, 在-78 ℃下首先合成苯并噻唑-锂(钾)试剂, 再在添加剂的作用下, 和N-叔丁基亚硫酰基亚胺反应生成2-位的不对称选择性偶联产物(Eq. 9).反应体系中苯并噻唑-锂(钾)试剂起到非常好的亲核性, 反应10 min得到很好的产率. R为取代基是吸电子的芳环能使目标产物的d.r.>99:1, 且给电子芳环有助于提高产率.
2013年, Weaver等[24]报道了可见光诱导的C—H键功能化偶联反应(Eq. 10).反应在0.75 mol% Ir(ppy)3作光催化剂, 咪唑作配体, 乙腈作溶剂, Blue LED灯照射下, 实现了2-氯苯并噻唑和脂肪胺的直接偶联反应.此反应体系温和、反应条件简便, 且具有很好的区域选择性.
2014年, O'Shea等[25]以Me3SiO-/Bu4N+作活化剂, 在室温条件下促使了三甲基有机硅烷对羰基化合物和亚胺衍生物的加成反应(Eq. 11).该反应对各种三甲基硅烷衍生物具有很广泛的适用性, 且反应条件温和、产率良好.
同年, 韩建林等[26]报道了Mannich-Type加成反应(Eq. 12).反应通过苯并噻唑和有机碱(丁基锂)反应形成2-锂化的苯并噻唑, 然后以其作为亲核试剂进攻(S)-N-叔丁基亚磺酰基-酮亚胺衍生物, 合成了含有两个苯并噻唑取代基的亚磺酰胺衍生物.该反应对各种苯并噻唑衍生物进行了实验, 都得到良好的分离产率.
2015年, 韩建林等[27]再次报道了一种相似的不对称合成氨基-苯并噻唑衍生物的方法, 反应体系中以二异丙基氨基锂(LDA)作碱, 以2-锂化苯并噻唑作亲核性试剂去进攻(S)-N-叔丁基亚磺酰基-酮亚胺衍生物合成目标产物(Eq. 13).该反应体系拓展了含亚磺酰胺的苯并噻唑衍生物的合成路径, 这一方法对合成潜在的药物具有重要的价值, 而且反应具有很好的不对映选择性和高的分离产率.
2005年, Klapars和Waldman等[28]以碱催化腈的α-烷基化反应(Eq. 14).反应体系以NaHMDS或KHMDS作碱, 甲苯或THF作溶剂, 实现卤代芳杂环和α-烷基腈的直接偶联.反应成功的关键在于腈阴离子的形成, 促进了卤代芳杂环的进攻, 实现烷基化的偶联, 反应对各种烷基腈、卤代芳杂环具有广泛的适用性, 反应简便.
随后, 2012年黄汉民和夏春谷等[29]报道了类似于Miura课题组的Pd-催化体系Pd2(dba)3/dppp/NaOAc, 以5 mol% Pd(MeCN)2Cl2作催化剂, 6 mol% dppp作配体, 碱的作用下选择性催化杂芳苄基化偶联(Scheme 4).作者发现, 通过控制使用不同的碱, 苯并噁唑和苄氯可以得到一苄基、二苄基、三苄基偶联产物, 苯并噻唑未发现此现象, 仅提供了三苄基偶联产物.该烷基化体系中, Pd-催化剂在反应中起到至关重要的作用, 碱促进芳杂环形成2-位的金属键, 也促进了多苄基的形成.
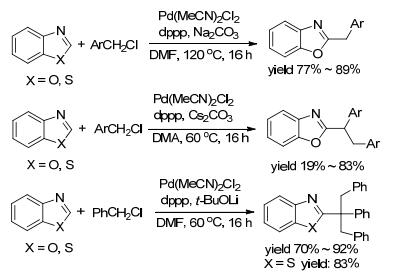 图式4
Pd催化芳杂环的亲核加成
图式4.
Palladium catalyzed nucleophilic addition of aromatic heterocycles
图式4
Pd催化芳杂环的亲核加成
图式4.
Palladium catalyzed nucleophilic addition of aromatic heterocycles
2013年, 杨尚东等[30]报道了Pd催化的C—H加成烷基化反应(Eq. 15).作者以10 mol% Pd(OAc)2作催化剂, 双齿氮2, 2-二吡啶作配体, 二氧六环作溶剂, 100 ℃条件下实现了N-取代吲哚醌和苯并杂芳的偶联.该反应体系无需其他的碱和添加剂, 对官能团具有很好的适应性, 生物测试结果表明该目标产物对肾癌细胞和肝癌细胞具有很好的抑制作用, 对苯并噻唑类化合物应用到药物合成中具有重要的参考价值.
2014年, 陈晓岚和赵玉芬等[31]报道了Ag催化脱羧促进的苯并噻唑直接烷基化反应(Eq. 16).反应以20 mol% AgNO3作催化剂, K2S2O8作氧化剂, 二氯甲烷/水作溶剂, 室温下反应8 h合成了30个目标产物.该反应直接采用烷基羧酸作为烷基化试剂, 原料廉价易得, 反应条件温和, 反应时间短, 但是反应催化剂和氧化剂用量较大.
2015年, Guin等[32]报道了一种氧气介导的脱羧反应, 实现了芳杂环和醛的C—C键偶联(Eq. 17).该反应首先在氧气作用下将醛氧化成酸, 再进行脱羧和苯并噻唑反应合成目标产物.该方法操作简便、环境友好、产率良好, 且对各种杂环芳烃的C—H键活化具有广泛的适用性.
2016年, 黄金波和朱强等[33]报道了无过渡金属催化的酰胺烷基化反应(Eq. 18).反应以廉价的K2S2O8作氧化剂, 70 ℃下反应23 h, 促使苯并噻唑和酰胺衍生物偶联.该反应是自由基介导的交叉脱氢偶联反应(CDC)反应, 合成路线简便、绿色环保, 对替换过渡金属催化具有重要的参考意义.但相比同类型反应而言, 反应时间偏长.
同年, Shenvi等[34]报道了一种Mn催化氢电子转移(HAT)的自由基芳基化8-芳基薄荷醇的合成.该反应采用两步进行, 首先通过HAT芳基磺酰氯和异蒲勒醇磺酰化, 再进行芳基迁移、脱磺酰基合成目标产物(Scheme 5).由于醇官能团的选择性电离导致直接采用H2SO4/PhH介导的Friedel-Crafts芳基化产物复杂, 故作者研究了自由基催化的HAT烷基化偶联反应.该反应设计巧妙, 免了Friedel-Crafts芳基化的复杂产物、具有良好的产率, 自由基氢电子转移机理对后续的研究具有十分重要的参考意义.
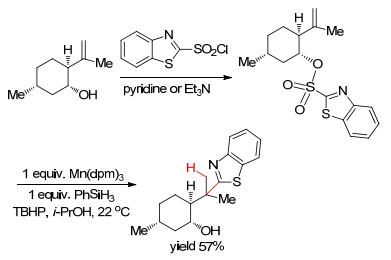 图式5
Mn催化8-芳基薄荷醇的合成
图式5.
Mn-catalyzed synthesis of 8-arylmenthol
图式5
Mn催化8-芳基薄荷醇的合成
图式5.
Mn-catalyzed synthesis of 8-arylmenthol
2013年, 强琚莉和陈兆旭等[35]报道了Cu催化交叉脱氢偶联反应(Eq. 23).该反应以Cu(OTf)2作催化剂, K2S2O8作氧化剂, 60 ℃条件下反应14 h实现了苯并噻唑和醚的直接偶联.该反应具有广泛的适用性, 对各种噻唑衍生物、苯并噻唑衍生物和四氢呋喃、1, 4-二氧六烷均能进行偶联, 且反应条件温和、分离产率良好.
2015年, Shah等[36]报道了可见光催化的C—H键功能化反应(Scheme 6).该反应在K2S2O8作催化剂, 水作溶剂, 27 W CFL灯照射下实现了醚和缺电子芳烃的直接偶联.反应通过光引发使K2S2O8和缺电子芳烃形成复合物, 激发醚自由基的形成, 再和缺电子芳烃发生偶联.该反应对各种醚具有广泛的适用性, 反应条件温和, 溶剂绿色环保等优点.
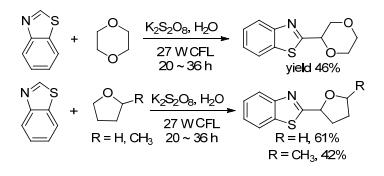 图式6
可见光诱导的苯并噻唑和醚的反应
图式6.
Visible light-induced reactions of benzothiazole with ethers
图式6
可见光诱导的苯并噻唑和醚的反应
图式6.
Visible light-induced reactions of benzothiazole with ethers
2 2-酰基化苯并噻唑
1971年, Thames等[37]对Beel课题组的方法进行改进, 将低温反应体系逐渐升温至室温, 使产率达到75%~80%.作者发现该C—Si断裂能和氯甲酸乙酯、醛、邻苯二甲酸酐进行偶联, 形成2-位的羰基类取代产物, 且都得到很好的产率(Scheme 7).该反应体系也存在着反应敏感、反应温度高等缺点. 2007年Koskinen等[38]报道了一种CuI/ZnCl2催化合成二芳酮的方法(Eq. 20).此路线是采用很常见的合成方法, n-BuLi作碱夺取苯并噻唑2-位的氢, 再和苯甲酰氯偶联.反应中二芳酮主要是以中间体的形式存在, 该杂二芳酮最终是被合成新型的脂肪酸酰胺水解酶抑制剂, 从而说明苯并噻唑在新药物合成中具有重要的价值.
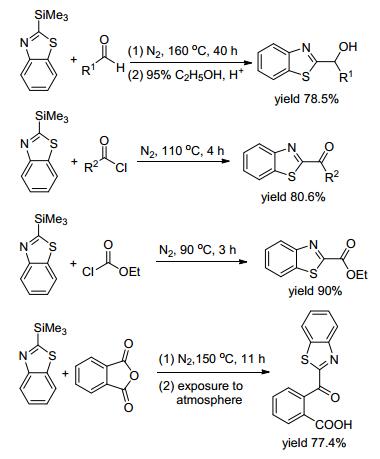 图式7
2-酰基化苯并噻唑的合成
图式7.
Synthesis of 2-acylated benzothiazole
图式7
2-酰基化苯并噻唑的合成
图式7.
Synthesis of 2-acylated benzothiazole
2011年, 王磊等[39]报道了一种新型、简洁、有效的酰基化反应, 仅以过氧化苯甲酸叔丁酯(TBPB)作氧化剂, 甲苯作溶剂, 在100 ℃下反应12 h就实现了苯并噻唑和甲酰胺的直接脱氢偶联反应(Eq. 21).该方法的优点在于不使用金属催化剂、无碱、环境友好、底物适用性好, 但相比同类反应而言, 所用溶剂污染大, 反应温度高.
2004年, 王涛等[40]报道了一种以乙腈衍生物作羰基合成子, 采用一锅法实现了卤代芳烃(杂芳烃)和乙腈衍生物发生SNAr取代和氧化合成二芳酮(Eq. 22).反应体系中采取分步投料方式, NaHMDS促使SNAr取代反应的进行, Na2O2起氧化作用.该反应具有很广泛的适用性, 对卤代芳烃、杂环芳烃、稠杂环芳烃和各种乙腈衍生物具有很好的反应效果.
2011年, 王志刚等[41]以最简单的氧化剂(NaH/air)来氧化各种醇成酮(Eq. 23).该方法对各种杂芳烯丙基醇、环丙基醇的氧化具有很好的适用性.反应中对杂芳烯丙基醇的氧化仅通过NaH/N2就实现了该反应, 但产率不高, 氢化钠使用及处理也需比较谨慎.
2015年, 雷爱文等[42]报道了一种铜催化的选择性氧化杂芳亚甲基成酮(Eq. 24).通过控制实验发现, 反应中氯乙酸乙酯充当启动子, 自由基参与整个反应过程, 实现了这一有效的C—H键氧化和功能化.该反应对各种N-杂环化合物具有很好的适用性, 为合成杂环芳酮提供了新的路径.
2013年, 吴安心等[43]报道了一种I2/KOH催化的2-烷基酮苯并噻唑的合成方法(Eq. 25).该反应通过碘氧化芳酮, 苯并噻唑在碱作用下开环, 再进行环缩合得到目标产物.该反应体系对各种芳酮具有很广泛的适用性, 且具有很高的分离产率, 对合成二芳基酮具有重要的参考价值.同年, 邓国军等[44]报道了Fe催化的2-芳酮苯并噻唑的合成, 反应以FeCl3·6H2O作催化剂, P(Cy)3HBF4作配体, 实现了苯并噻唑2-位的羰基化反应(Eq. 26).作者对这一机理进行阐述, 为先氧化、再开环、然后环缩合得目标产物.反应对各种铁盐进行了探究, 均具有不错的催化效果, 通过控制反应条件, 使得2-芳基酮为主产物.但该反应时间长、温度高.
2014年, 宋秋玲等[45]以CuI (0.005~2 mol%)作催化剂, HBF4作酸, 经过苯并噻唑的开环和再缩合环化, 实现了苯并噻唑和苯乙酮的偶联(Eq. 27).该反应具有催化剂用量少、产率高、对官能团具有很好的容忍性.
2013年, 张艳和葛海波等[46]报道了Ni催化的脱羧酰基化, 反应以Ni(ClO4)2·6H2O作催化剂, Ag2CO3作氧化剂, 苯作溶剂, 实现了α-羰基羧酸脱羧和芳杂环的酰基化偶联(Eq. 28).该反应体系对各种芳杂环和羰基羧酸衍生物具有很好的适用性.随后, Saxena等[47]也报道了一种Pd催化铁络合物作氧化剂催化的芳基酮脱羧偶联(Eq. 29).反应以Pd(OAc)2作催化剂, [Fe(Ⅲ)EDTA-(η2-O2)]3-作氧化剂, 水作溶剂, 室温下实现了芳杂环的交叉脱羧偶联反应.相比其他酰基化反应, 该催化体系具有反应条件温和、溶剂绿色环保、分离产率高等优点, 且反应时间短, 克服了高温长时间反应的缺点.
2015年, 陆红健和张艳等[48]以[Co]/Ag2CO3作共催化剂, 3-FC6H4CF3作溶剂, 催化α-羰基羧酸脱羧和苯并噻(噁)唑发生交叉偶联反应(Eq. 30).该体系中Ag2CO3促使脱羧反应, [Co]催化剂和芳杂环2-位络合, 再进行偶联生成2-羰基芳杂环衍生物.该反应得到很好的分离产率, 对芳杂环衍生物具有很好的适用性, 该类催化体系对芳杂环衍生物2-位C—H键活化的研究具有重要意义.
2013年, Hu等[49]报道合成了一种含苯并噻唑-2-芳酮的新药(Eq. 31).该反应在LDA, -70 ℃低温条件下, 实现了4-取代苯甲酸甲酯和苯并噻唑的二芳酮合成.该产物可作为合成目标药物的中间体, 对苯并噻唑衍生物应用于药物合成中具有重要的借鉴价值.
随后, 2014年, 陈晓岚和屈凌波等[50]报道了以无金属催化体系过氧化物催化的苯并噻唑酰基化(Eq. 32).反应以TBHP作氧化剂, 乙腈作溶剂, 80 ℃反应24 h就实现了磷酸酯作为酰基化试剂的偶联.该反应体系通过TBHP氧化磷酸酯中的α-位亚甲基形成自由基, 然后进攻苯并噻唑实现氧化偶联反应.反应对各种取代基具有广泛的适用性, 且具有不错的分离产率.
2015年, Eycken等[51]报道了Pd-催化的杂芳C(sp2)—H功能化(Eq. 33).反应以Pd(OAc)2作催化剂, Xantphos作配体, Cs2CO3作碱, 异腈作插入体, 分两步实现杂芳的酰基化反应.该反应体系打破固定的直接氧化、脱羧偶联等方法, 采用异腈作插入体先和卤代苯偶联, 再和杂芳酰化得到目标产物, 合成方法路径新颖, 但反应体系太复杂, 后处理困难, 反应条件也不温和.
2016年, Miura等[52]报道了一种无金属的催化体系, 以空气作氧化剂, 在可见光下, 实现了2-甲基芳杂环的吸氧催化氧化成醛反应(Eq. 34).反应中I2先插入, 再发生C—I键均裂氧化成目标产物, 该反应避免了过渡金属催化的不足, 反应条件温和、高效.同年, 季海涛等[53]报道了苯甲醛介导的光氧化还原交叉脱氢偶联反应(CDC) (Eqs. 35, 36).该反应在以苯甲醛作光敏剂, (NH)4S2O8作催化剂, CFL灯的照射下实现了苯并噻唑和N, N-二取代乙酰胺(或甲酰胺)脱氢偶联反应.相比于过渡金属催化的反应, 此反应具有条件温和、环境友好、绿色节能等优点.
3 2-芳基化苯并噻唑
由于杂环-芳环骨架结构在许多天然产物、医药、功能材料中随处可见, 因此在有机合成中芳基化的杂环化合物的合成方法得到广泛的关注[54].过渡金属催化的交叉偶联是最可靠的方法之一, 其中包括了卤代芳烃的偶联、芳醛作芳基化试剂促进的开环再环合偶联以及直接催化C—H键活化的交叉偶联反应.
2009年, Miura等[55]报道了Ni催化的直接芳基化, 反应以NiBr2作催化剂, 1, 10-邻菲罗啉作配体, t-BuOLi作碱, 二甘醇二甲醚作溶剂, 150 ℃实现了苯并噻唑(苯并噁唑)和溴代苯衍生物的直接偶联(Eq. 37).该体系对各种溴代芳烃均具有很好的适用性, 且具有良好的产率.但反应催化体系用量大、反应温度高.
同年, Itami课题组[56]以Ni催化实现了卤代芳烃和芳杂环化合物的直接偶联(Eq. 38).反应以Ni(OAc)2作催化剂, bipy作配体, t-BuOLi作碱, 实现了目标产物的合成.该方法被初步证实了可用于快速合成一种用于痛风和高尿酸血症的药物非布索坦.且该催化体系能实现苯并噻唑和各种卤代芳烃、卤代芳杂环的偶联, 具有良好的催化效果.
2010年, Gosmini等[57]报道了钴-催化的C—SMe键活化, 实现了杂芳环的交叉偶联反应(Eq. 39).反应在CoBr2/Zn的催化下, 先使溴代苯生成格氏试剂, 再断杂芳硫醚中的C—S键实现目标产物的偶联.同年, Ranu等[58]以Pd纳米颗粒催化了芳基化反应, 反应以Pd(OAc)2/TBAB作催化剂, K2CO3作碱, AgOAc作添加剂, 分子筛干燥的条件下实现了苯并噻唑和碘代芳烃的直接偶联(Eq. 40).反应中通过Pd和碘代芳烃偶联成ArPdI, 再在添加剂的作用下产生Pd阳离子亲电子中间体, 碱最后促进偶联的形成.该反应对各种碘代芳烃衍生物均适用, 且具有良好的分离产率.但该反应体系复杂、不易处理、催化剂添加剂昂贵, 且反应温度高、反应时间长.
2011年, 黄志真等[59]报道了一种实用的Pd-Cu共催化体系, 采用廉价的PPh3作配体, 实现了苯并噻唑(苯并噁唑)和溴代芳烃的直接偶联反应(Eq. 41).该反应体系条件温和、操作简便, 且得到很好的产率.随后, Itami课题组[60]采用Ni催化了芳基化偶联反应(Eq. 42).反应在Ni(OAc)2/bipy/t-BuOLi催化体系中进行, 120 ℃条件下实现芳基化反应, 相比Pd-Cu体系, 该体系更廉价环保.并且该方法被成功应用于合成黄嘌呤氧化酶抑制剂(有效治疗痛风和高尿酸血的药物)、氯苯唑酸[有效治疗TTR-FAP(转甲状腺素蛋白相关性家族性淀粉样多发性神经病)药物]以及具有抗结核活性的天然产物.
2014年, Hong等[61]报道了一种Cu介导的C—H键功能化反应, 实现苯并噻唑的2-位官能团化(Eq. 43).在反应体系中强碱夺取苯并噻唑2-位的氢使其和Cu离子形成2-位络合, 3-碘代喹诺酮再和苯并噻唑铜络合物发生氧化加成、还原消除得到目标产物.该反应体系具有高效、反应方便、适用性广等特点, 但也存在反应条件不够温和、需高温和氮气保护且反应时间长等缺点.
Punji课题组[62]合成了单核和双核钯的螯合物, 并将其应用于催化区域选择性C—H键活化, 实现了苯并噻唑和对甲基碘苯的芳基化反应, 得到95%的分离产率(Eq. 44).随后在2016年, 该课题组[63]再次报道了在一种Pd的螯合物和CuI共催化的作用下, 实现了苯并噻唑和对甲基碘苯的芳基化(Eq. 45).作者研究发现, 反应路径是以PdⅡ-PdⅣ-PdⅡ, 反应先是苯并噻唑和CuI, Pd螯合剂进行偶联, 对甲基碘苯进行氧化加成, 再进行还原消除得目标产物.
2015年, 武杰和侯红卫等[64]报道了一种铜金属有机框架(Cu-MOFs)结构的催化剂催化的杂芳环C—H键活化(Eq. 46).作者以这种Cu-MOFs结构作催化剂, 200 mol% K2CO3作碱, DMF作溶剂实现了芳杂环和卤代芳环的直接偶联.该MOFs结构类似多孔沸石, 通过可逆变换捕捉O2实现这一氧化/还原变化, 从而实现Ullmann偶联反应.相比过渡金属盐催化有着独特的反应优势, 但此反应添加剂用量大、反应温度高.
2011年, 游劲松等[65]报道了Pd催化氧化的芳杂环与吲哚(或吡咯类)的交叉偶联(Eqs. 47, 48).该体系采用Pd(dppf)Cl2, X-Phos, CuCl, Cu(OAc)2·H2O、吡啶结合共催化, 于1, 4-二氧六环/DMSO中催化芳杂环的偶联.该反应对各种杂芳结构、吡咯类、吲哚具有广泛的适用性, 且直接实现了C—H键的活化.但催化体系太过复杂, 后处理不容易, 且采用各种贵金属盐、过渡金属盐, 成本昂贵, 污染大.
2014年, 王中夏等[66]报道Pd催化的芳基化, 反应采用[Pd(π-allyl)Cl]2作催化剂, PCy3或IPr·HCl作配体, 有机碱作用下促使芳基季铵盐和芳杂环的偶联(Eq. 49).该反应通过钯催化C—H/C—N键的断裂, 对各种芳香季铵盐、萘环季铵盐具有广泛的适用性, 且得到良好的产率.
2015年, 崔秀玲和吴养洁等[67]报道了一种有效的交叉脱氢偶联反应, 在无金属的状态下, 以空气为氧化剂, 实现了N-氧喹诺啉和苯并噻(噁)唑的偶联(Eq. 50).反应体系中仅以叔丁醇锂就实现了该偶联反应, 作者通过控制实验发现, 喹诺啉中的N—O键对反应起到至关重要的作用.该路线简便、高效、环境友好, 但反应时间长、反应温度高.
2011年, 刘小刚等[68]以[Pd]/[Cu]催化了芳杂环的芳基化反应(Eq. 51).反应在以Pd(OAc)2/Cu(OAc)2作共催化剂, 1, 10-菲咯啉作配体, K3PO4作碱的条件下, 100 ℃实现了苯并噻唑(苯并噁唑)和芳基硼酸的直接偶联反应.该反应体系提供了一条新的合成杂芳和芳烃的偶联反应路线, 且对各种官能团具有很好的容忍性.同年, 王锐等[69]报道了Cu催化氧化的C—C键偶联反应(Eq. 52).该反应以10 mol% CuCl作催化剂, t-BuOLi作碱, 在氧气作用下实现了芳杂环和芳基硼酸酯的交叉偶联反应.相比而言, 此合成路线反应条件温和、反应时间短、催化剂廉价且具有很高的分离产率.
2015年, Muthusubramanian等[70]也报道了一种有效的Cu介导Pd催化的脱硫C—C键交叉偶联(Eq. 53).反应中Pd(OAc)2作催化剂, Cu(I)噻吩-2-羧酸盐(CuTC)作脱硫试剂, PPh3作配体, 实现了噻唑烷-2-硫酮和芳基硼酸的偶联反应.反应体系简便、产率高, 且芳基硼酸适用性广, 对合成各种取代苯并噻唑提供了一条新思路.
2012年, 谭泽等[71]以K2S2O8介导催化氧化合成了2-芳基化苯并噻唑(Eq. 54).仅以K2S2O8作氧化剂, 100 ℃反应3 h成功合成了26个2-芳基化偶联产物, 产率24%~74%.该反应避免了贵金属/过渡金属的催化体系, 无需任何配体, 氧化剂廉价易得, 且首次采用芳醛(苯基乙醛酸)作为芳基化试剂.但反应温度高、产率一般.
2013年, 宋秋玲等[72]报道了以廉价铜盐作催化剂, 促使苯乙酸脱羧偶联(Eq. 55).该反应在铜盐的作用下脱羧然后被氧化成醛, 苯并噻唑在高温下开环, 再和醛环合生成目标产物.该体系对各种苯乙酸衍生物具有广泛的适用性, 且具有很好的分离产率.但路线反应时间长、反应温度高.
2014年, Bhanage等[73]以Fe催化有效合成了2-芳基苯并噻唑(Eq. 56).反应体系以Fe(NO3)3·9H2O作催化剂, P(t-Bu)3·HBF4作配体, 在氧气条件下通过先氧化苯乙烯成苯甲醛, 苯并噻唑开环再和苯甲醛环合生成目标产物.该反应催化剂廉价易得, 氧化剂绿色丰富, 且得到良好的产率, 但存在配体用量大、反应时间长、反应温度高等不足.
同年, 陈善勇等[74]以Fe催化了苯并噻唑芳基化和芳酰化反应(Eq. 57).反应以FeCl3·6H2O作催化剂, K2S2O8作氧化剂, 在100 ℃条件下实现了苯并噻唑和苯甲醇的芳基化反应.反应也是通过先氧化醇、苯并噻唑氧化开环、再进行二者环化得目标产物.相比于同类型反应, 该反应原料廉价易得、反应时间短、无需任何配体.随后张慧君等[75]以过渡金属Cu催化了芳基化反应(Eq. 58).反应以廉价的CuCl2作催化剂, 70% TBHP作氧化剂, 无溶剂条件下实现苯并噻唑和苄醇(芳醛)的偶联.此反应机理包括苯并噻唑开环、醛胺偶联、再分子内环合.该反应优点在于无溶剂、反应体系温和、方法简便.
2014年, 崔秀玲等[76]也报道了KI催化的芳基化反应(Eq. 59).反应在无金属条件下, 以KI作催化剂, TBHP作氧化剂, 水作溶剂, 100 ℃下反应8 h合成了28个芳基化产物, 产率36%~79%.该反应体系的优点在于不使用任何配体、无金属污染、溶剂绿色、反应时间短.相比同类型反应, 该反应产率不高.
2015年, 王锐等[77]报道了Cu(OTf)2/K2S2O8共催化体系, 实现了2-芳基苯并噻唑的氧化偶联(Eq. 60).反应体系通过K2S2O8氧化α-氨基苯乙酸, 再脱羧, 苯并噻唑在K2S2O8/高温条件下开环, 和苯甲醛再环合得目标产物.该反应首次探究了α-氨基苯乙酸的氧化偶联, 对后续研究这类反应具有重要意义.
2016年, Weaver课题组[78]报道了光氧化还原介导的C—H键功能化偶联反应(Eq. 61).该反应以0.3 mol% fac-Ir(ppy)3作光催化剂, K2CO3作碱, Blue LED灯照射下实现了2-溴苯并噻唑和富电子芳烃的交叉偶联反应.此反应条件温和、操作简单、官能团容忍性好, 且具有很好的分离产率.
苯并噻唑的直接芳基化反应主要以过渡金属催化为主, 添加配体、氧化剂促进反应的实现, 从整个反应体系看, 主要以卤代芳烃、芳醛作芳基化试剂, 过渡金属盐/氧化剂形成的催化体系实现苯并噻唑的芳基化反应是一个主要的途径.虽然这一反应类型能得到很好的催化效果, 但也存在着金属催化剂带来的污染大、反应温度高、反应时间长等不足.
4 胺基化的苯并噻唑(C—N键的形成)
C—N键的形成主要通过交叉偶联反应实现, 其在合成具有药物活性的产物具有潜在的研究价值.含芳杂环的C—N键偶联产物广泛应用于天然产物合成、农用化学品、医药以及材料科学领域[79].苯并噻唑的胺基化反应种类很多, 本文简单介绍了近几年合成胺基化的方法.
2013年, Hu等[80]报道了一种含苯并噻唑的具有抑制磷酸二酯酶10A (PDE10A)的新化合物(Eq. 62).反应在Pd2(dba)3催化下, 叔丁醇钾作碱促使2-溴苯并噻唑和4-取代苯胺的C—N键偶联.该新化合物对PDE10A酶具有良好的抑制作用(IC50=4.3±0.42 nmol·L-1), 且对人和鼠具有P-糖蛋白流失率低的优点, 此新药也说明了苯并噻唑在药物中具有重要的潜在价值.
2014年, Nageswar课题组[81]以Ru/C作非均相催化剂, t-BuOLi作碱, 80 ℃反应8 h获得了54种目标产物, 产率58%~90% (Eq. 63).该反应的优点在于操作简单、不使用任何配体、对水和空气不敏感、底物适用性广且催化剂可重复利用多次仍有活性.同年, 该课题组[82]报道磁性纳米材料CuFe2O4催化的C—N键偶联反应(Eq. 64).反应体系以CuFe2O4作催化剂, Cs2CO3作碱, 110 ℃反应15 h实现苯并噻唑和胺的直接偶联.相比Ru/C催化体系, 该纳米催化剂催化效果一般、反应条件也不温和.但该方法说明, 磁性纳米材料能够催化促进C—N键的偶联, 且该磁性材料可回收重复利用, 对探究纳米材料催化这类反应具有重要意义.
2016年, Nageswar课题组[83]再次报道了类似的2-卤代苯并噻唑和胺的偶联反应(Eq. 65).该反应体系既不用金属催化剂, 也不用任何配体, 更不使用任何碱, 仅在水作溶剂、室温下进行偶联反应.这种理想化的合成方法类似于酸碱中和, 为合成2-胺基化苯并噻唑提供了一条绿色途径.
5 磷酸化的苯并噻唑(C—P键的形成)
在过去几十年, 有机磷化合物在生物化学、材料化学、有机合成和催化领域得到广泛的应用[84].通常, 含磷取代基在生物、药物和功能材料方面具有重要作用, 也作为磷配体或直接作官能团应用在过渡金属催化中[85].基于此, 2-位磷酸化的苯并噻唑化合物在药物研发和有机合成领域也受到广泛关注.因此, 本文简单介绍近年来2-位磷酸化苯并噻唑化合物的合成及应用.
2012年, 李福伟等[86]报道了首个Pd-催化直接磷酸化作用(Eq. 66).反应以Pd(OAc)2作催化剂, 脯氨酸(L1)或2, 2-二吡啶(L2)作配体, K2S2O8作氧化剂, 100 ℃反应24 h实现了芳杂环和磷酸酯的直接偶联.该方法通过氧化裂解C—H键断裂, 实现了芳杂环的C—H键活化.但反应体系复杂、催化剂昂贵、反应温度高、反应时间长.同年, 杨尚东等[87]报道了Ni催化C—P键的交叉偶联(Eq. 67).反应以NiCl2(PPh3)2作催化剂, K2CO3作碱, DMF作溶剂, 50 ℃下反应实现了卤代芳杂环和芳基磷酸的交叉偶联反应.该反应操作简单、反应条件温和, 对各种卤代芳、卤代杂芳具有广泛的适用性, 且具有良好的分离产率.
2014年, Chamarthi等[88]以LaCl3·7H2O催化的Michaelis-Arbuzov反应, 实现了C—P键的偶联(Eq. 68).作者以LaCl3·7H2O作催化剂, 无溶剂状态下, 促进卤代芳杂环和二甲氧基苯基膦的直接交叉偶联, 该反应避免毒性催化剂和溶剂的危害, 反应条件温和、且产率高, 并且作者对各种偶联产物进行生物活性测试, 具有很好的抗菌活性.同年, 陈晓岚和曲凌波等[89]报道了过氧化物催化的C—P键的偶联反应(Eq. 69).反应以过氧化二叔丁基(DTBP)作氧化剂, 乙腈作溶剂, 80 ℃反应30 h实现了苯并噻唑和磷酸酯的交叉脱氢偶联.此催化体系克服了过渡金属、贵金属催化的不足, 但反应时间较长.
2015年, 张慧君等[90]报道了Ag介导的直接磷酸化作用, 反应以AgNO3作催化氧化剂, 乙腈作溶剂实现了苯并噻唑和二芳基氧磷的氧化偶联(Eq. 70).该体系比Pd催化磷酸化反应[91]简便, 不需额外添加氧化剂, 但相比于DTBP作氧化剂, 硝酸银比较昂贵.
2016年, 吴磊等[92]在无金属、无溶剂状态下, 直接自氧化形成C—P键.该反应仅以氧气作为氧化源, 65 ℃反应18 h就得到杂芳C—P键的偶联(Eq. 71).该反应通过氧气使二苯基氧膦变成磷酸过氧化物, 产生过氧自由基和二苯基膦自由基, ·OOH自由基氧化芳杂环形成自由基, 再自由基偶联形成目标产物.该反应体系简便、绿色环保、产率高.同年, 该课题组[93]再次报道了一种以染料作光敏剂催化的芳杂环C—H键功能化偶联反应(Eq. 72).该反应体系采用无金属催化, 以5 mol% Eosin B作光敏剂, 氯仿作溶剂, 11 W LED灯照射下, 实现了苯并噻唑和芳基磷酸的交叉脱氢偶联.此反应合成路线简便, 无任何氧化剂和添加剂, 仅有的副产物是H2, 且反应对各种官能团具有很好的适应性, 原子经济利用率高, 是一条绿色环保的合成路线.
随后2016年, 王官武等[94]也报道了Mn-促进的交叉偶联磷酸化反应(Eq. 73).反应以Mn(OAc)3·2H2O作催化剂, 球磨条件下, 实现了苯并噻唑的磷酸化.相比同类反应, 该体系克服了有机溶剂的污染, 不添加任何氧化剂, 也无配体, 是一条绿色环保的路径; 能室温反应, 反应时间短, 以空气作氧化剂, 是一类反应温和的催化体系; 且对官能团具有很好的适应性.
6 结论与展望
综上所述, 2-位官能团化合成2-取代苯并噻唑的合成方法很多, 主要有过渡金属催化、金属氧化物催化、强碱催化以及逐渐被研究的可见光介导催化.合成的目标产物主要有烷基化、酰基化、芳基化、胺基化和磷酸化等偶联产物.相对于缩合环化方法, 2-位官能团化法合成2-取代苯并噻唑具有设计连接方便、反应体系简便、原料易得、原子利用率高等优点.
随着苯并噻唑衍生物的抗菌、杀虫、抗癌等各种生物活性的发现, 2-取代苯并噻唑在医药和农药等领域的应用日益广泛, 对其合成方法的研究也越来越多.纵观全文, 2-位官能团化合成2-取代苯并噻唑主要有两种反应机理:一种是直接交叉脱氢偶联, 另一种是噻唑先开环再环合.总的来说, 大多采用过渡金属盐、贵金属盐在高温条件下实现反应, 而且反应时间较长, 相比而言污染大、催化剂贵、能耗也高.从文中少量的可见光介导催化的反应中可以看出, 以自由基反应的可见光催化室温条件下就能进行, 克服了反应中的高温能耗; 环境友好的光敏剂种类也繁多, 可以替代过渡金属.但可见光催化目前仍处在研究的初期, 有待我们不断深入研究, 因此利用可见光催化的自由基反应合成2-取代苯并噻唑的方法研究尤其值得关注.
-
-
[1]
王海林, 阮铃莉, 陈勇, 刘幸海, 翁建全, 有机化学, 2014, 34, 419. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343601.shtmlWang, H. L.; Ruan, L. L.; Chen, Y.; Liu, X. H.; Weng, J. Q. Chin. J. Org. Chem. 2014, 34, 419(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343601.shtml
-
[2]
翁建全, 黄华, 谭成侠, 刘幸海, 储为盛, 陈杰, 有机化学, 2012, 32, 957. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340917.shtmlWeng, J. Q.; Huang, H.; Tan, C. X.; Liu, X. H.; Cu, W. S.; Chen, J. Chin. J. Org. Chem. 2012, 32, 957(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340917.shtml
-
[3]
Facchinetti, V.; Reis, R. R.; Gomes, C. R. B.; Vasconcelos, T. R. A. Mini-Rev. Org. Chem. 2012, 9, 44. doi: 10.2174/157019312799079929
-
[4]
Weekes, A.; Westwell, A. Curr. Med. Chem. 2009, 16, 2430. doi: 10.2174/092986709788682137
-
[5]
Mouri, T.; Tokumura, J.; Kochi, S.; Fukui, H.; Nakano, J.; Ando, T. Nippon Noyaku Gakkaishi 2002, 27, 353. http://ci.nii.ac.jp/naid/110001713342
-
[6]
Debnatch, S. C.; Basu, D. K. J. Appl. Polym. Sci. 1994, 52, 597. doi: 10.1002/app.1994.070520503
-
[7]
Chevrie, D.; Lequeux, T.; Demoute, J. P.; Pazenok, S. Tetrahedron Lett. 2003, 44, 8127. doi: 10.1016/j.tetlet.2003.09.027
-
[8]
Hofmann, A. W. Ber. Dtsch. Chem. Ges. 1879, 12, 1126. doi: 10.1002/(ISSN)1099-0682
-
[9]
李焱, 王玉炉, 有机化学, 2006, 26, 878. doi: 10.3321/j.issn:0253-2786.2006.06.023Li, Y.; Wang, Y. L. Chin. J. Org. Chem. 2006, 26, 878(in Chinese). doi: 10.3321/j.issn:0253-2786.2006.06.023
-
[10]
竺宁, 张志伟, 高敏, 韩利民, 索全伶, 洪海龙, 有机化学, 2013, 33, 1423. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341871.shtmlZu, N.; Zhang, Z. W.; Gao, M.; Han, L. M.; Suo, Q. L.; Hong, H. L. Chin. J. Org. Chem. 3013, 33, 1423(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341871.shtml
-
[11]
Nishio, T.; Mori, Y.; Iida, I.; Hosomi, A. HeIv. Chim. Acta 1994, 77, 981. doi: 10.1002/(ISSN)1522-2675
-
[12]
Imahori, T.; Kondo, Y. J. Am. Chem. Soc. 2003, 125, 8082. doi: 10.1021/ja0342300
-
[13]
Inamoto, K.; Okawa, H.; Taneda, H.; Sato, M.; Hirono, Y.; Yonemoto, M.; Kikkawa, S.; Kondo, Y. Chem. Commun. 2012, 48, 9771. doi: 10.1039/c2cc35701a
-
[14]
Popov, I.; Do, H. Q.; Daugulis, O. J. Org. Chem. 2009, 74, 8309. doi: 10.1021/jo9015369
-
[15]
Inamoto, K.; Okawa, H.; Kikkawa, S.; Kondo, Y. Tetrahedron 2014, 70, 7917. doi: 10.1016/j.tet.2014.08.054
-
[16]
Shen, H. C.; Ding, F. X.; Colletti, S. L. Org. Lett. 2006, 8, 1447. doi: 10.1021/ol060246u
-
[17]
Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2010, 12, 1360. doi: 10.1021/ol1002576
-
[18]
Gottumukkala, A. L.; Derridj, F.; Djebbar, S.; Doucet, H. Tetrahedron Lett. 2008, 49, 2926. doi: 10.1016/j.tetlet.2008.03.020
-
[19]
Gustafsson, M.; Jensen, J.; Bertozzi, S. M.; Currier, E. A.; Ma, J. N.; Burstein, E. S.; Olsson, R. Bioorg. Med. Chem. Lett. 2010, 20, 5918. doi: 10.1016/j.bmcl.2010.07.077
-
[20]
Zhao, X.; Wu, G. J.; Zhang, Y.; Wang, J. B. J. Am. Chem. Soc. 2011, 133, 3296. doi: 10.1021/ja111249p
-
[21]
Ojha, D. P.; Prabhu, K. R. J. Org. Chem. 2012, 77, 11027. doi: 10.1021/jo301987c
-
[22]
Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2012, 51, 775. doi: 10.1002/anie.201106825
-
[23]
Zhang, J. L.; Yang, Y. H.; Wang, M.; Lin, L.; Wang, R. Tetrahedron Lett. 2012, 53, 6893. doi: 10.1016/j.tetlet.2012.09.131
-
[24]
Singh, A.; Arora, A.; Weaver, J. D. Org. Lett. 2013, 15, 5390. doi: 10.1021/ol402751j
-
[25]
Das, M.; O'Shea, D. F. J. Org. Chem. 2014, 79, 5595. doi: 10.1021/jo5007637
-
[26]
Mei, H. B.; Dai, Y. L.; Wu, L. M.; Soloshonok, V. A.; Han, J. L.; Pan, Y. Eur. J. Org. Chem. 2014, 12, 2429.
-
[27]
Dai, Y. L.; Xie, C.; Wu, L. M.; Mei, H. B.; Soloshonok, V. A.; Han, J. L.; Pan, Y. RSC Adv. 2015, 5, 3491. doi: 10.1039/C4RA15405C
-
[28]
Klapars, A.; Waldman, J. H.; Campos, K. R.; Jensen, M. S.; McLaughlin, M.; Chung, J. Y. L.; Cvetovich, R. J.; Chen, C. Y. J. Org. Chem. 2005, 70, 10186. doi: 10.1021/jo051737f
-
[29]
Xie, P.; Huang, H. M.; Xie, Y. J.; Guo, S. M.; Xia, C. G. Adv. Synth. Catal. 2012, 354, 1692. doi: 10.1002/adsc.201200025
-
[30]
Wang, G. W.; Zhou, A. X.; Wang, J. J.; Hu, R. B.; Yang, S. D. Org. Lett. 2013, 15, 5270. doi: 10.1021/ol402494e
-
[31]
Zhao, W. M.; Chen, X. L.; Yuan, J. W.; Qu, L. B.; Duan, L. K.; Zhao, Y. F. Chem. Commun. 2014, 50, 2018. doi: 10.1039/c3cc48069k
-
[32]
Paul, S.; Guin, J. Chem. Eur. J. 2015, 21, 17618. doi: 10.1002/chem.201503809
-
[33]
Wang, J.; Li, J.; Huang, J. B.; Zhu, Q. J. Org. Chem. 2016, 81, 3017. doi: 10.1021/acs.joc.6b00096
-
[34]
Crossley, S. W. M.; Martinez, R. M.; Zuluaga, S. G.; Shenvi, R. A. Org. Lett. 2016, 18, 2620. doi: 10.1021/acs.orglett.6b01047
-
[35]
Xie, Z. Y.; Cai, Y. P.; Hu, H. W.; Lin, C.; Jiang, J. L.; Chen, Z. X.; Wang, L. Y.; Pan, Y. Org. Lett. 2013, 15, 4600. doi: 10.1021/ol4022113
-
[36]
Devari, S.; Shah, B. A. Chem. Commun. 2016, 52, 1490. doi: 10.1039/C5CC08817H
-
[37]
Pinkeron, F. H.; Thames, S. F. J. Heterocycl. Chem. 1971, 8, 257. doi: 10.1002/(ISSN)1943-5193
-
[38]
Myllymaki, M. J.; Saario, S. M.; Kataja, A. O.; Castillo-Melendez, J. A.; Nevalainen, J. R. O.; Jarvinen, T.; Koskinen, A. M. P. J. Med. Chem. 2007, 50, 4236. doi: 10.1021/jm070501w
-
[39]
He, T.; Li, H. J.; Li, P. H.; Wang, L. Chem. Commun. 2011, 47, 8946. doi: 10.1039/c1cc13086b
-
[40]
Yin, Z. W.; Zhang, Z. X.; Kadow, J. F.; Meanwell, N. A.; Wang, T. J. Org. Chem. 2004, 69, 1364. doi: 10.1021/jo030234b
-
[41]
Wang, X. B.; Wang, Z. G. Tetrahedron 2011, 67, 3406. doi: 10.1016/j.tet.2011.03.052
-
[42]
Liu, J. M.; Zhang, X.; Yi, H.; Liu, C.; Liu, R.; Zhang, H.; Zhuo, K. L.; Lei, A. W. Angew. Chem., Int. Ed. 2015, 54, 1261. doi: 10.1002/anie.201409580
-
[43]
Gao, Q. H.; Wu, X.; Jia, F. C.; Liu, M. C.; Zhu, Y. P.; Cai, Q.; Wu, A. X. J. Org. Chem. 2013, 78, 2792. doi: 10.1021/jo302754c
-
[44]
Liu, S. W.; Chen, R.; Chen, H.; Deng, G. J. Tetrahedron Lett. 2013, 54, 3838. doi: 10.1016/j.tetlet.2013.05.050
-
[45]
Feng, Q.; Song, Q. L. Adv. Synth. Catal. 2014, 356, 2445. doi: 10.1002/adsc.v356.11/12
-
[46]
Yang, K.; Zhang, C.; Wang, P.; Zhang, Y.; Ge, H. B. Chem. Eur. J. 2014, 20, 7241. doi: 10.1002/chem.201402516
-
[47]
Sharma, S.; Khan, I. A.; Saxena, A. K. Adv. Synth. Catal. 2013, 355, 673. doi: 10.1002/adsc.201201085
-
[48]
Yang, K.; Chen, X. Y.; Wang, Y. Q.; Li, W. Q.; Kadi, A. A.; Fun, H. K.; Sun, H.; Zhang, Y.; Li, G. G.; Lu, H. J. J. Org. Chem. 2015, 80, 11065. doi: 10.1021/acs.joc.5b01450
-
[49]
Hu, E.; Kunz, R. K.; Chen, N.; Rumfelt, S. N.; Siegmund, A.; Andrews, K.; Chmait, S.; Zhao, S. R.; Davis, C.; Chen, H.; Zeiner, D. L.; Ma, J.; Biorn, C.; Shi, J. X.; Porter, A.; Treanor, J.; Allen, J. R. J. Med. Chem. 2013, 56, 8781. doi: 10.1021/jm401234w
-
[50]
Chen, X. L.; Li, X.; Qu, L. B.; Tang, Y. C.; Mai, W. P.; Wei, D. H.; Bi, W. Z.; Duan, L. K.; Sun, K.; Chen, J. Y.; Ke, D. D.; Zhao, Y. F. J. Org. Chem. 2014, 79, 8407. doi: 10.1021/jo501791n
-
[51]
Sharma, U. K.; Sharma, N.; Xu, J.; Song, G. H.; Eycken, E. V. V. Chem. Eur. J. 2015, 21, 4908. doi: 10.1002/chem.201406562
-
[52]
Nagasawa, Y.; Tachikawa, Y.; Yamaguchi, E.; Tada, N.; Miura, T.; Itoh, A. Adv. Synth. Catal. 2016, 358, 178. doi: 10.1002/adsc.201500811
-
[53]
Zhang, Y. Q.; Teuscher, K. B.; Ji, H. T. Chem. Sci. 2016, 7, 2111. doi: 10.1039/C5SC03640B
-
[54]
Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359. doi: 10.1021/cr000664r
-
[55]
Hachiya, H.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2009, 11, 1737. doi: 10.1021/ol900159a
-
[56]
Canivet, J.; Yamaguchi, J.; Ban, I.; Itami, K. Org. Lett. 2009, 11, 1733. doi: 10.1021/ol9001587
-
[57]
Begouin, J. M.; Rivard, M.; Gosmini, C. Chem. Commun. 2010, 46, 5972. doi: 10.1039/c0cc01055c
-
[58]
Saha, D.; Adak, L.; Ranu, B. C. Tetrahedron Lett. 2010, 51, 5624. doi: 10.1016/j.tetlet.2010.08.063
-
[59]
Yan, X. M.; Mao, X. R.; Huang, Z. Z. Heterocycles 2011, 83, 1371. doi: 10.3987/COM-11-12176
-
[60]
Yamamoto, T.; Muto, K.; Komiyama, M.; Canivet, J.; Yamaguchi, J.; Itami, K. Chem. Eur. J. 2011, 17, 10113. doi: 10.1002/chem.v17.36
-
[61]
Shin, S.; Kim, Y.; Kim, K.; Hong, S. Org. Biomol. Chem. 2014, 12, 5719. doi: 10.1039/C4OB00939H
-
[62]
Pandey, D. K.; Khake, S. M.; Gonnade, R. G.; Punji, B. RSC Adv. 2015, 5, 81502. doi: 10.1039/C5RA18289A
-
[63]
Khake, S. M.; Jagtap, R. A.; Dangat, Y. B.; Gonnade, R. G.; Vanka, K.; Punji, B. Organometallics 2016, 35, 875. doi: 10.1021/acs.organomet.6b00003
-
[64]
Huang, C.; Wu, J.; Song, C. J.; Ding, R.; Qiao, Y.; Hou, H. W.; Chang, J. B.; Fan, Y. T. Chem. Commun. 2015, 51, 10353. doi: 10.1039/C5CC02432C
-
[65]
Wang, Z.; Li, K. Z.; Zhao, D. B.; Lan, J. B.; You, J. S. Angew. Chem., Int. Ed. 2011, 50, 5365. doi: 10.1002/anie.201101416
-
[66]
Zhu, F.; Tao, J. L.; Wang, Z. X. Org. Lett. 2015, 17, 4926. doi: 10.1021/acs.orglett.5b02458
-
[67]
Chen, X. P.; Cui, X. L.; Yang, F. F.; Wu, Y. J. Org. Lett. 2015, 17, 1445. doi: 10.1021/acs.orglett.5b00330
-
[68]
Ranjit, S.; Liu, X. G. Chem. Eur. J. 2011, 17, 1105. doi: 10.1002/chem.v17.4
-
[69]
Yang, F. Z.; Xu, Z. Q.; Wang, Z.; Yu, Z. K.; Wang, R. Chem. Eur. J. 2011, 17, 6321. doi: 10.1002/chem.201100136
-
[70]
Rajaguru, K.; Mariappan, A.; Manjusri, R.; Muthusubramanian, S.; Bhuvanesh, N. RSC Adv. 2015, 5, 86832. doi: 10.1039/C5RA17827D
-
[71]
Yang, Z. Y.; Chen, X.; Wang, S. Z.; Liu, J. D.; Xie, K.; Wang, A. W.; Tan, Z. J. Org. Chem. 2012, 77, 7086. doi: 10.1021/jo300740j
-
[72]
Song, Q. L.; Feng, Q.; Zhou, M. X. Org. Lett. 2013, 15, 5990. doi: 10.1021/ol402871f
-
[73]
Khemnar, A. B.; Bhanage, B. M. RSC Adv. 2014, 4, 8939. doi: 10.1039/C3RA46955G
-
[74]
Wang, J.; Zhang, X. Z.; Chen, S. Y.; Yu, X. Q. Tetrahedron 2014, 70, 245. doi: 10.1016/j.tet.2013.11.078
-
[75]
Zhang, M. L.; Lu, W. T.; Ruan, W. Q.; Zhang, H. J.; Wen, T. B. Tetrahedron Lett. 2014, 55, 1806. doi: 10.1016/j.tetlet.2014.01.120
-
[76]
Gao, Y. Y.; Song, Q. L.; Cheng, G. L.; Cui, X. L. Org. Biomol. Chem. 2014, 12, 1044. doi: 10.1039/C3OB42318B
-
[77]
Wang, R.; An, C. H.; Li, Y.; Zhao, Y.; Wang, T.; Li, A. Tetrahedron Lett. 2015, 56, 2077. doi: 10.1016/j.tetlet.2015.03.004
-
[78]
Arora, A.; Weaver, J. D. Org. Lett. 2016, 18, 3996. doi: 10.1021/acs.orglett.6b01718
-
[79]
Hili, R.; Yudin, A. K. Nat. Chem. Biol. 2006, 2, 284. doi: 10.1038/nchembio0606-284
-
[80]
Hu, E.; Kunz, R. K.; Chen, N.; Rumfelt, S. N.; Siegmund, A.; Andrews, K.; Chmait, S.; Zhao, S. R.; Davis, C.; Chen, H.; Zeiner, D. L.; Ma, J.; Biorn, C.; Shi, J. X.; Porter, A.; Treanor, J.; Allen, J. R.; J. Med. Chem. 2013, 56, 8781. doi: 10.1021/jm401234w
-
[81]
Reddy, K. H. V.; Anil, Kumar. B. S. P.; Reddy, V. P.; Kumar, R. U.; Nageswar, Y. V. D. RSC Adv. 2014, 4, 45579. doi: 10.1039/C4RA05447D
-
[82]
Satish, G.; Reddy, K. H. V.; Anil, B. S. P.; Shankar, J.; Kumar, R. U.; Nageswar, Y. V. D. Tetrahedron Lett. 2014, 55, 5533. doi: 10.1016/j.tetlet.2014.07.100
-
[83]
Kumar, R. U.; Reddy, K. H. V.; Anil-Kumar, B. S. P.; Satish, G.; Reddy, V. P.; Nageswar, Y. V. D. Tetrahedron Lett. 2016, 57, 637. doi: 10.1016/j.tetlet.2015.12.084
-
[84]
Demmer, C. S.; Krogsgaard-Larsen, N.; Bunch, L. Chem. Rev. 2011, 111, 7981. doi: 10.1021/cr2002646
-
[85]
Zhang, H.; Hu, R. B.; Zhang, X. Y.; Li, S. X.; Yang, S. D. Chem. Commun. 2014, 50, 4686. doi: 10.1039/C4CC01238K
-
[86]
Hou, C. D.; Ren, Y. L.; Lang, R.; Hu, X. X.; Xia, C. G.; Li, F. W. Chem. Commun. 2012, 48, 5181. doi: 10.1039/c2cc30429e
-
[87]
Zhang, H. Y.; Sun, M.; Ma, Y. N.; Tian, Q. P.; Yang, S. D. Org. Biomol. Chem. 2012, 10, 9627. doi: 10.1039/c2ob26874d
-
[88]
Golla, M.; Syed, R.; Katla, V. R.; Devineni, S. R.; Kondapalli, N.; Chamarthi, N. R. J. Chem. Sci. 2014, 126, 117. doi: 10.1007/s12039-013-0550-3
-
[89]
Chen, X. L.; Li, X.; Qu, L. B.; Tang, Y. C.; Mai, W. P.; Wei, D. H.; Bi, W. Z.; Duan, L. K.; Sun, K.; Chen, J. Y.; Ke, D. D.; Zhao, Y. F. J. Org. Chem. 2014, 79, 8407. doi: 10.1021/jo501791n
-
[90]
Zhang, H. J.; Lin, W. D.; Wu, Z. J.; Ruan, W. Q.; Wen, T. B. Chem. Commun. 2015, 51, 3450. doi: 10.1039/C4CC10017D
-
[91]
Feng, C. G.; Ye, M. C.; Xiao, K. J.; Li, S. H.; Yu, J. Q. J. Am. Chem. Soc. 2013, 135, 9322. doi: 10.1021/ja404526x
-
[92]
Luo, K.; Chen, Y. Z.; Chen, L. X.; Wu, L. J. Org. Chem. 2016, 81, 4682. doi: 10.1021/acs.joc.6b00592
-
[93]
Luo, K.; Chen, Y. Z.; Yang, W. C.; Zhu, J.; Wu, L. Org. Lett. 2016, 18, 452. doi: 10.1021/acs.orglett.5b03497
-
[94]
Li, L.; Wang, J. J.; Wang, G. W. J. Org. Chem. 2016, 81, 5433. doi: 10.1021/acs.joc.6b00786
-
[1]
-

图式1 NaHMDS催化苯并噻唑2-位烷基化
Scheme 1 NaHMDS catalyzed 2-position alkylation of benzothiazole

图式3 Pd催化2-卤代苯并噻唑和腙的烷基化反应
Scheme 3 Palladium catalyzed alkylation reactions of 2-halo-benzothiazole with hydrazones

图式4 Pd催化芳杂环的亲核加成
Scheme 4 Palladium catalyzed nucleophilic addition of aromatic heterocycles

图式6 可见光诱导的苯并噻唑和醚的反应
Scheme 6 Visible light-induced reactions of benzothiazole with ethers
-


 下载:
下载:






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 78
- 文章访问数: 4144
- HTML全文浏览量: 978
 下载:
下载:
