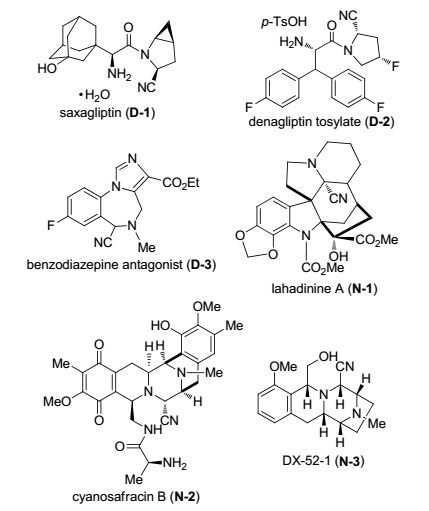
 图 1
含α-氰胺复合官能团的代表性药物和天然产物
Figure 1.
Representative drugs and natural products containing α-aminonitrile functional group
图 1
含α-氰胺复合官能团的代表性药物和天然产物
Figure 1.
Representative drugs and natural products containing α-aminonitrile functional group
引用本文:
高燕娇, 肖振华, 刘良先, 黄培强. 含未保护羟基2-吡咯烷酮衍生物的直接还原氰基化:N-甲基-2-别-Bulgecinine的立体选择性合成[J]. 有机化学,
2017, 37(5): 1189-1197.
doi:
10.6023/cjoc201703024

Citation: Gao Yanjiao, Xiao Zhenhua, Liu Liangxian, Huang Peiqiang. Direct Reductive Cyanation of A 2-Pyrrolidinone Chiral Building Block Bearing An Unprotected Hydroxyl Group: A Stereoselective Synthesis of N-Methyl-2-epi-bulgecinine[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1189-1197. doi: 10.6023/cjoc201703024
Citation: Gao Yanjiao, Xiao Zhenhua, Liu Liangxian, Huang Peiqiang. Direct Reductive Cyanation of A 2-Pyrrolidinone Chiral Building Block Bearing An Unprotected Hydroxyl Group: A Stereoselective Synthesis of N-Methyl-2-epi-bulgecinine[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1189-1197. doi: 10.6023/cjoc201703024
含未保护羟基2-吡咯烷酮衍生物的直接还原氰基化:N-甲基-2-别-Bulgecinine的立体选择性合成
摘要:
报道手性合成砌块(4S,5R)-N-苄基-4-苄氧基-5-羟甲基-2-吡咯烷酮(3a)的直接还原氰基化及产物的立体化学与转化研究.含未保护羟基的内酰胺用LiAlH4/KCN体系直接还原氰基化,生成比例为69:31的2,5-反式/顺式非对映立体异构体.与文献类似结果对比显示,氰基负离子对5-羟甲基-D-1-吡咯啉鎓中间体的加成主要受立体电子效应和C(5)位取代基(羟甲基)与进攻试剂间烯丙型1,3-位阻控制.该混合物在碱性条件下水解,生成比例为10:90的2,5-反式/2,5-顺式-bulgecinine衍生物.这一结果表明氰基水解反应伴随着在C(2)位发生了有合成价值的串联差向异构化.由此建立了立体选择性地合成2,5-顺式(-)-N-甲基-2-别-bulgecinine的简便方法.
English
Direct Reductive Cyanation of A 2-Pyrrolidinone Chiral Building Block Bearing An Unprotected Hydroxyl Group: A Stereoselective Synthesis of N-Methyl-2-epi-bulgecinine
Abstract:
The direct reductive cyanation of N-benzyl-4-benzyloxy-5-hydroxymethyl-2-pyrrolidinone (3a), a lactam bearing a free hydroxyl group, has been achieved with the LiAlH4/KCN combination. The reaction afforded 2, 5-trans-2-cyano-5-hydroxylmethyl-4-benzyloxy-pyrrolidine (5a) and its cis-diastereomer 5b in a ratio of 69:31 with a combined yield of 63%. The observed 2, 5-trans-stereoselectivity is suggested to be resulted from both stereoelectronic effect and allylic 1, 3-strain between the hydroxymethyl group at C(5) and the incoming cyanide anion on the presumed Δ-1 pyrrolinium ion intermediate. The subsequent hydrolysis of the cyano group of the diastereomeric mixture 5a/5b (trans:cis=69:31) under basic conditions afforded the corresponding 5-hydroxymethyl-4-benzyloxyproline with 2, 5-cis-diastereomer as the major diastereomer (trans:cis=10:90). This result implies that a synthetically useful epimerization at C(2) has occurred concomitantly. This unexpected result afforded a concise and highly stereoselective synthesis of 2, 5-cis-(-)-N-methyl-2-epi-bulgecinine.
-
Key words:
- amides
- / nitriles
- / iminium ion
- / stereoselective synthesis
- / epimerization
-
α-氰胺是一类多用途的复合官能团[1], 其多样的化学已在Husson著名的氰基调控构型方法学中全面展示[2].此外, α-氰胺结构单元存在于许多药物和活性天然产物及其类似物中[3](图 1), 例如, 沙格列汀(Saxagliptin, D-1)和地格列汀甲苯磺酸盐(Denagliptin tosylate, D-2)均为四型二肽酶(DPP Ⅳ)抑制剂, 前者作为高效地治疗二型糖尿病的药物于2009年获批准上市[4], 后者正在被开发为治疗二型糖尿病的药物[5].化合物D-3是一种苯并二氮杂䓬受体拮抗剂[3]. Lahadinine A (N-1)[6]和Cyanosafracin B (N-2)均为天然产物, 后者被用作合成抗癌药Ecteinascidin 743工业生产的原料[7]. DX-52-1 (N-3)表现出显著的抗肿瘤活性[1b].
图 1
含α-氰胺复合官能团的代表性药物和天然产物
Figure 1.
Representative drugs and natural products containing α-aminonitrile functional group
传统上, α-氰胺结构单元可通过Strecker反应制备, 包括氰基对亚胺或亚胺鎓的加成或胺、醛、氰化钾的三组分缩合[8].从胺出发经C—H官能化引入α-氰基的方法最近也引起了关注[9].另一对药物和天然产物合成有重要意义的方法是酰胺的还原氰基化.然而, 由于酰胺的高稳定性, 这一反应往往难以控制, 因而不具有普适性[10].随着近年对温和条件下酰胺直接转化[11]的重视, Movassaghi[12], Charette[13], Bélanger[14], 王彦广[15], 姚祝军[16], Maulide[17], Chida/Sato[18], Pace[19], Dixon[20]和本课题组[21]等研究小组报道了许多有应用价值的酰胺直接还原官能化的方法.新近, Dixon小组[3]报道了酰胺经铱催化直接还原氰基化的普适性方法.这促使我们报道本实验室在这方面取得的一些结果.
1 结果与讨论
多年前, 我们在本实验室发展的两个手性合成砌块1和2[22](图 2)的基础上, 发展了一种间接的区域和立体选择性还原羟烷基方法[23].结合我们对于生物碱合成的兴趣[24]以及Rapoport等[25]报道的4的衍生物可被用作手性合成砌块, 我们认为化合物5或许可用于(-)-bulgecinine (6)的合成.特种氨基酸(-)-bulgecinine (6)是天然抗生素糖肽bulgecins A, B, C的共同组成部分[26].因此, (-)-bulgecinine (6)及其类似物和立体异构体的全合成吸引了诸多研究组的兴趣[27, 25c].
 图 2
本文涉及的手性合成砌块与bulgecins的结构
Figure 2.
Structures of the chiral building blocks discussed in this paper and those of bulgecins
图 2
本文涉及的手性合成砌块与bulgecins的结构
Figure 2.
Structures of the chiral building blocks discussed in this paper and those of bulgecins
我们设计了三条把内酰胺3a还原羧基化合成6的路径.路线a是基于Fowler及其合作者[28]报道的内酰胺直接还原芳基化方法, 以乙烯基作为羧基的等效体.路线b是利用Dötc等[29]发展的Fischer型氨基卡宾络合物8, 经光诱导引入甲氧羰基.路线c是以比较传统但普适性并不高的酰胺直接还原氰基化方法为基础[10](Scheme 1).
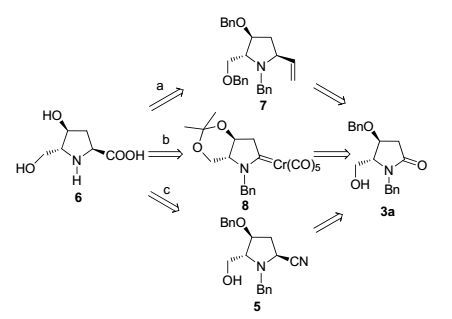 图 图式1
(-)-Bulgecinine (6)的逆合成分析
Figure 图式1.
Retrosynthetic analysis of (-)-bulgecinine (6)
图 图式1
(-)-Bulgecinine (6)的逆合成分析
Figure 图式1.
Retrosynthetic analysis of (-)-bulgecinine (6)
我们首先对本实验室先前报道的把手性合成砌块1转化为3a的四步反应[23]中产率低于90%的9的脱水反应进行优化.通过把反应物浓度从0.1 mol•L-1提高到0.2 mol•L-1, 同时把4-二甲氨基吡啶(DMAP)的用量从0.005 mol%增加到0.5 mol%, 反应时间可缩短到12 h, 而产率则从74%提高到94%.这样, 1转化为3a的总产率提高到75.6% (Scheme 2).
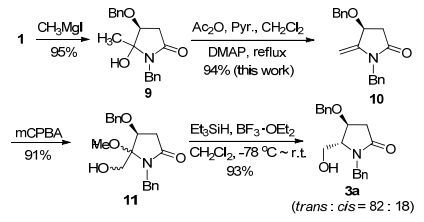 图 图式2
手性合成砌块3a的改良合成
Figure 图式2.
An improved synthesis of the chiral building block 3a
图 图式2
手性合成砌块3a的改良合成
Figure 图式2.
An improved synthesis of the chiral building block 3a
我们先探索路线a, 3a经NaH去质子化, 然后与苄溴作用, 得到全保护的吡咯烷酮12 (Scheme 3).然而, 参照Fowler等[28]的条件进行12的直接还原乙烯基化(乙烯基锂, LiAlH4还原)未能得到7.在不同的温度下进行反应时, 薄层色谱(TLC)跟踪反应均显示产物很复杂, 于是放弃这条路线.接着我们试探路线b, 遗憾的是在进行3a的O-去苄基化后, 试图用丙酮对所生成的二醇13进行保护未获成功.这一结果出乎意料, 因为Rapoport等[25]小组报道类似的吡咯烷并环体系可顺利形成双环丙酮叉缩醛.未能生成化合物14的原因可归因于化合物13中2-吡咯烷酮与吡咯烷构象柔性不同, 前者表现出一定的刚性, 不利于缩醛环系的形成.
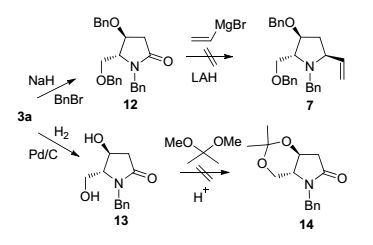 图 图式3
手性合成砌块3a的未成功转化
Figure 图式3.
Unsuccessful transformations of chiral building block 3a
图 图式3
手性合成砌块3a的未成功转化
Figure 图式3.
Unsuccessful transformations of chiral building block 3a
鉴于许多含α-氰胺复合官能团的药物和天然产物为环状结构[3](图 1), 以及5可望被转化为6, 我们转而研究3a的还原氰基化反应.常用的酰胺还原氰基化方法一般涉及多步反应, 即首先转化为酰胺的活化形式, 然后进行部分还原和氰基取代[1b, 30].有一些用LiAlH2-(OEt)2, DIBAL-H, LiAlH4还原氰基化的例子, 但却限于无官能团或仅含稳定官能团的底物, 且收率受底物的结构影响大[10].例如, Larchevêque小组[10f]成功地用DIBAL-H/KCN体系进行3-硅氧基2-吡咯烷酮的还原氰基化.遗憾的是, 当采用其反应条件对3a进行还原氰基化时, 我们只以34%的收率得到副产物吡咯衍生物15 (Eq. 1).
本实验室[10g]在2004年报道过3-羟基-2-吡咯烷酮用LiAlH4/KCN体系还原氰基化.这一方法的优点是羟基无需保护.按照当时优化的条件来进行3a的还原氰基化时, 发现虽有产物生成, 但产率极低, 只有3%, 回收得到大量原料(56%).通过调整反应温度, 延长反应时间, 加大还原剂LiAlH4用量, 最终摸索出合适的反应条件(表 1).在优化条件下, 以63%的产率得到还原氰基化产物5a和5b.其比例通过对其混合物进行HPLC分析确定为69:31[色谱条件为:色谱柱Shim-Pack VP-ODS (150 mm×4.6 mm), 洗脱剂为V(CH3CN)/V(H2O)=55/45, 流速为1.0 mL/min, λ=254 nm, t1(5a)=7.6 min, t2(5b)=8.1 min].
 表 1
3a直接还原氰基化的反应条件筛选
Table 1.
Screening of reaction conditions for the direct reductive cyanation of 3a
表 1
3a直接还原氰基化的反应条件筛选
Table 1.
Screening of reaction conditions for the direct reductive cyanation of 3a

序号 温度/℃ LiAlH4/ equiv. 反应时间 产率a/% 原料回收率a/% 1 -35 1.5 20 min 3 56 2 -15 1.5 1 h 31 6 3 -15 1.5 3 h 33 2 4 -17 1.5 3 h 38 16 5 -18 1.5 3 h 38 25 6 -19 1.5 3 h 39 37 7 -20 1.5 3 h 49 22 a分离产率. 表 1 3a直接还原氰基化的反应条件筛选
Table 1. Screening of reaction conditions for the direct reductive cyanation of 3a由表 1可知: (1) 温度太低(-35 ℃)时, 反应较慢, 若反应时间短, 则绝大部分原料剩余, 产率很低. (2) 在-15 ℃下反应较快, 但若反应时间短, 则有部分原料剩余; 延长反应时间虽可使绝大部分原料转化, 但产率依然不高. (3) 在-20℃下反应时反应速度中等, 辅以过量的氢化锂铝, 可将绝大部分原料转化为产物, 产率较好.
所得到的非对映异构体5a和5b不但对酸和碱不稳定, 而且室温下自发相互转化.其中, 次要异构体5b在柱层析分离纯化时总有杂质伴随, 无法得到纯品.主要异构体5a在冰箱中放置6 d后发生了差向异构化.
非对映立体异构体5a和5b的立体化学系通过核磁共振确定.化合物5a中氢的归属由1H-1H相关关系(图 3)确定.从1H-1H COSY谱中可以看出, H(3α) (δ 2.14) 与H2 (δ 3.84), H(4) (δ 4.09) 有较强的偶合关系(相关峰), 而H(3β)与H(2), H(4) 均没有明显的偶合(相关峰), 由此确定H(3α)与H(2), H(4) 处于顺式的位置, 即C(2) 位的氰基与C(5) 位的羟甲基为反式取向, C(2) 的构型为S. NOESY实验的结果进一步证实了这一推论(图 4): H(3α)与H(2), H(4) 间有显著的NOE相关峰, 而H(3β)与H(2), H(4) 均没有观察到明显的NOE相关.
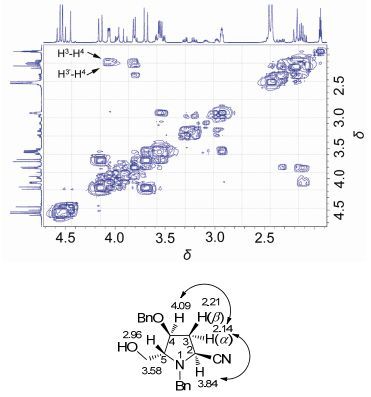 图 3
化合物5a的1H-1H相关COSY谱图(局部)
Figure 3.
1H-1H COSY spectrum (in part) of compound 5a
图 3
化合物5a的1H-1H相关COSY谱图(局部)
Figure 3.
1H-1H COSY spectrum (in part) of compound 5a
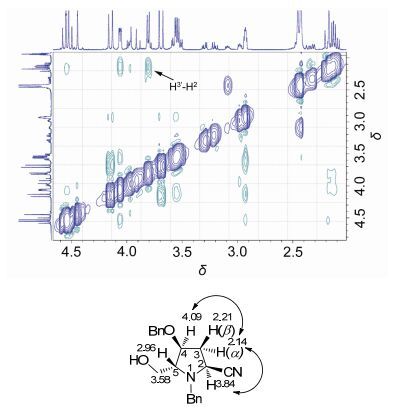 图 4
化合物5a的NOESY谱图(局部)
Figure 4.
NOESY spectrum (in part) of compound 5a
图 4
化合物5a的NOESY谱图(局部)
Figure 4.
NOESY spectrum (in part) of compound 5a
有趣的是, 内酰胺3a还原氰基化的立体化学结果(5a/5b: 2, 5-反式:顺式=69:31) 分别与Terashima[1b]和Tanaka[30]等相关反应的结果高度吻合(L-2: 2, 5-反式:顺式=69:31; L-4: 2, 5-反式:顺式=70:30, Scheme 4).三个体系吡咯烷环上的取代基各不相同(3a, L-1和L-3), 但2, 5-反式立体选择性的结果高度一致, 表明: (1) 三个反应都经历亚胺鎓中间体; (2) 反应均受立体电子控制[31]; (3) 氰基负离子对亚胺鎓离子的进攻主要受N-α-碳上而非β-碳上的取代基控制.参考Tanaka的立体化学模型[30], 我们提出3a经LiAlH4部分还原后形成的亚胺鎓中间体存在A, B两种可能的构象(Scheme 5).氰基负离子的进攻受立体电子控制, 在构象A从β-面进攻有利, 而在构象B易从α-面进行.与Tanaka不同的是, 立体选择性主要受烯丙型1, 3-位阻(allylic 1, 3-strain)[32]控制.即构象A虽然存在烯丙型1, 2-位阻(allylic 1, 2-strain)较构象B不利, 然而由于在构象B, 进攻试剂(氰基负离子)与C(5) 位羟甲基存在显著的烯丙型1, 3-位阻(allylic 1, 3-strain)[32], 因而反应性较低.净结果是氰基负离子从构象A的β-面进攻占优, 因而2, 5-反式立体异构体5a为主产物, 但2, 5-反式/2, 5-顺式立体异构体的比例只有69:31.
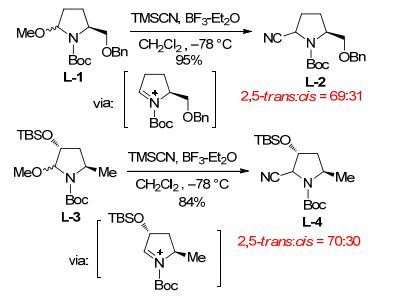 图 图式4
Terashima和Tanaka报道的经由亚胺鎓中间体的2, 5-反式-立体选择性氰基加成反应
Figure 图式4.
Terashima and Tanaka's stereoselective addition reactions of cyanide anion to iminium ion intermediates
图 图式4
Terashima和Tanaka报道的经由亚胺鎓中间体的2, 5-反式-立体选择性氰基加成反应
Figure 图式4.
Terashima and Tanaka's stereoselective addition reactions of cyanide anion to iminium ion intermediates
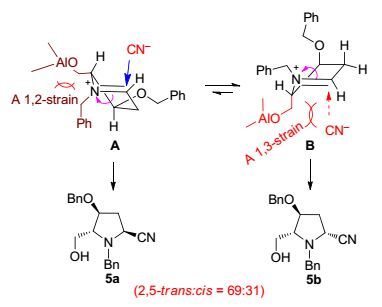 图 图式5
氰基负离子对亚胺鎓中间体加成的构象分析
Figure 图式5.
Conformation analysis of the cyanide anion addition to iminium ion intermediate
图 图式5
氰基负离子对亚胺鎓中间体加成的构象分析
Figure 图式5.
Conformation analysis of the cyanide anion addition to iminium ion intermediate
接下来, 我们研究5a/5b向(-)-bulgecinine的转化.首先是氰基的水解.由于在酸性条件下水解只得到16%的部分水解的2, 5-顺式酰胺(参见辅助材料), 我们转而尝试在碱性条件下水解.可喜的是, 把还原氰基化所得的异构体混合物(2, 5-反式:2, 5-顺式=69:31) 与NaOH/H2O2[33]在50~70 ℃下反应, 然后酸化, 以50%~60%的收率得到N, O-二苄基-2-别-bulgecinine (16b)与N, O-二苄基-bulgecinine (16a), 比例为90:10 (Scheme 6).
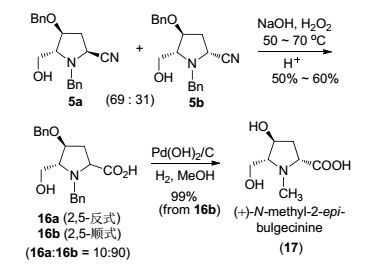 图 图式6
(+)-N-甲基-2-别-bulgecinine的立体选择性合成
Figure 图式6.
Stereoselective synthesis of (+)-N-methyl-2-epi-bulgecinine
图 图式6
(+)-N-甲基-2-别-bulgecinine的立体选择性合成
Figure 图式6.
Stereoselective synthesis of (+)-N-methyl-2-epi-bulgecinine
值得注意的是, 主要异构体的结构经单晶X射线衍射分析确证为2, 5-顺式-16b(图 5, CCDC 1537394)[34].次要异构体经二维核磁共振确定为2, 5-反式-16a.在1H-1H COSY上可以看出H(3α) (δ 2.70) 与H(2) (δ 3.98), H(4) (δ 4.26) 有较强的偶合关系(相关峰), 而H(3β)与H(2) 有较弱的偶合(相关峰), 与H(4) 没有明显的偶合(相关峰), 由此确定H(3α)与H(2), H(4) 处于顺式的位置, 即C(2) 位的羧基与C(5) 位的羟甲基处于trans相对位置, C(2) 的构型为S. NOESY实验的结果进一步证实了这一推论(图 6): H(3α)与H(2), H(4) 间有显著的NOE相关峰, 而H(3β)与H(4) 间有一弱相关峰, 与H2则没有相关.这一结果表明在反应过程中发生了串联C(2) 位差向异构化[35].得益于这一有用的差向异构化, 可从非对映立体异构体混合物5a/5b (2, 5-trans:2, 5-cis=69:31) 直接高立体选择性地得到(+)-2-别-bulgecinine衍生物16b (2, 5-trans:2, 5-cis=10:90). α-氰胺高立体选择性差向异构化的运用文献已有记载[36].
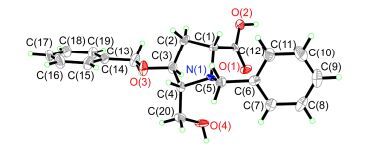 图 5
化合物16b的单晶X射线衍射图
Figure 5.
Single crystal X-ray diffraction structure of compound 16b
图 5
化合物16b的单晶X射线衍射图
Figure 5.
Single crystal X-ray diffraction structure of compound 16b
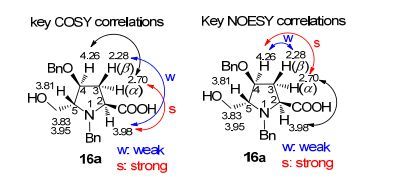 图 6
确定化合物16a C(2) 位立体化学的1H-1H COSY和NOESY谱中的关键相关关系
Figure 6.
Key correlations shown in 1H-1H COSY and NOESY spectra for the assignment of stereochemistry at C(2) of compound 16a
图 6
确定化合物16a C(2) 位立体化学的1H-1H COSY和NOESY谱中的关键相关关系
Figure 6.
Key correlations shown in 1H-1H COSY and NOESY spectra for the assignment of stereochemistry at C(2) of compound 16a
经多次重结晶得到的2, 5-顺式异构体16b在甲醇中, Pd(OH)2或Pd/C催化下氢解意外得到N, O-去苄基化-N-甲基化产物17(N-甲基-2-别-bulgecinine).关于这一反应我们在前文已报道[37].正是基于这一意外发现, 我们发展了一种高效、高化学选择性的串联N, O-去苄基化-N-烷基化方法, 其中醇为烷基化试剂[37].
2 结论
改良了手性合成砌块3a的合成, 并确定了其直接还原氰基化的条件.值得一提的是, 含未保护羟基内酰胺的直接还原氰基化文献中只有个别例子, 且不存在芳构化问题.本法能够有效避免芳构化, 而且取得与相关体系同样的1, 3-不对称诱导效果, 表明反应条件温和.结合文献结果总结出的5-取代或4, 5-二取代-2-吡咯烷酮还原氰基化的立体化学规律, 对相关体系可望有预测价值.由于α-氰胺结构单元存在于许多药物和活性天然产物中, 因此, 本法的建立对寻找新的活性分子具有意义.随后的串联氰基水解-差向异构化提供了高立体选择性地合成2, 5-顺式(+)-N-甲基-2-别-bulgecinine (17)的有效方法.化合物17所具有的2, 4-反式、2, 5-顺式立体化学特征与另一天然特种氨基酸3-羟基-5-甲基-脯氨酸一致[30].此外, 通过对16b的钯碳催化氢解去苄基化反应的研究, 发现了一个“绿色”的以醇为烷基化试剂的串联[38]N, O-去苄基化-N-烷基化反应.
3 实验部分
3.1 仪器与试剂
旋光用Perkin-Elmer 341型旋光仪测定; 核磁共振氢谱和碳谱用Bruker公司AvanceⅡ-400型核磁共振仪(400 MHz/100 MHz)测定, 化学位移以Me4Si为内标.红外谱图由Nicolet Avatar 360型FT-IR测定(KBr盐片); 质谱由Bruker公司的Dalton ESquire 3000 plus LC-MS测定; 高分辨质谱由Finnigan公司Mat-LCQ型质谱仪测定; 元素分析由Vario RL型元素分析仪测定; 熔点由Yanaco MP-500型熔点仪测定(温度未校正).柱色谱采用300~400目硅胶(烟台江友硅胶开发有限公司); 洗脱剂采用乙酸乙酯和60~90 ℃石油醚; 所用其他溶剂均用标准方法纯化后使用.
3.2 实验方法
3.2.3 N-苄基-1H-2-吡咯甲醇(15)的合成
在30 ℃下向3a (94 mg, 0.30 mmol)的四氢呋喃(0.60 mL)溶液中缓慢加入二异丁基氢化铝(DIBAL-H) (1 mol•L-1正己烷溶液, 0.60 mL, 0.60 mmol).继续反应30 min后, 升温至-15 ℃, 加入甲醇(0.30 mL), 随后在1 min之内加入KCN (78 mg, 1.2 mmol)的水溶液(0.30 mL).升至室温, 剧烈搅拌1 h后, 加入乙酸乙酯和水.水相用乙酸乙酯萃取(5 mL×3).合并有机相, 用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物通过硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:9]分离纯化得33 mg白色固体15, 产率34%. m.p. 59~61 ℃ (EtOAc/PE); 1H NMR (400 MHz, CDCl3) δ: 1.38 (s, 1H, OH, D2O exchangeable), 4.50 (s, 2H, NCH2Ph), 5.20 (s, 2H, H-6), 6.12 (dd, J=2.6, 3.5 Hz, 1H, H-4), 6.17 (dd, J=1.8, 3.5 Hz, 1H, H-3), 6.71 (dd, J=1.8, 2.6 Hz, 1H, H-5), 7.03~7.10 (m, 2H, ArH), 7.22~7.35 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3)δ 50.5, 56.8, 107.3, 109.4, 123.3, 126.5, 127.5, 128.7, 131.8, 138.3; IR (film) ν: 3369, 3088, 3059, 3029, 2919, 2849, 2370, 2345, 1599, 1495, 1453, 1357, 1300, 1072 cm-1; MS (ESI) m/z: 210 (M+Na)+; HRMS calcd for C12H13NO 187.0992, found 187.0999.
3.2.5 (2S, 4S, 5R)-N-苄基-4-苄氧基-5-羟甲基脯氨酸(16a)和(2R, 4S, 5R)-N-苄基-4-苄氧基-5-羟甲基脯氨酸(16b)的合成
向5a和5b的混合物(50 mg, 0.16 mmol, dr=61:39) 的四氢呋喃(1.0 mL)溶液中缓慢加入3 mol•L-1 NaOH (aq.)溶液(1.1 mL)和30% H2O2溶液(1.0 mL).反应混合物升温至50 ℃加热2 h, 60 ℃加热6 h, 70 ℃下加热15 h.冷却至0 ℃, 加入6 mol•L-1 HCl溶液直至pH达到2~3. CH2CH2萃取(5 mL×10).合并有机相, 用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物通过硅胶柱层析[V(乙酸乙酯):V(石油醚)=10:1]分离纯化获得3.2 mg白色固体化合物16a, 产率6%;及28.6 mg无色针状晶体16b, 产率54%.
16a: m.p. 177~179 ℃ (EtOAc/MeOH); [α]D20 -6.0 (c 0.42, MeOH); 1H NMR (400 MHz, CD3OD) δ: 2.28 (ddd, J=2.2, 4.2, 14.0 Hz, 1H, H-3), 2.69 (ddd, J=5.9, 10.1, 14.0 Hz, 1H, H-3), 3.78~3.82 (m, 1H, H-5), 3.82 (dd, J=3.6, 12.7 Hz, 1H, H-6, overlapped with H-5), 3.94 (dd, J=3.4, 12.7 Hz, 1H, H-6, overlapped with H-2), 3.98 (dd, J=4.2, 10.1 Hz, 1H, H-2), 4.25 (ddd, J=2.2, 2.3, 5.9 Hz, 1H, H-4), 4.36 (d, J=13.1 Hz, 1H, NCH2Ph), 4.47 (d, J=11.8 Hz, 1H, OCH2Ph), 4.55 (d, J=11.8 Hz, 1H, OCH2Ph), 4.56 (d, J=13.1 Hz, 1H, NCH2Ph), 7.23~7.55 (m, 10H, ArH); 13C NMR (100 MHz, CD3OD) δ: 35.5, 54.5, 59.0, 67.6, 71.0, 72.1, 80.9, 128.8, 128.9, 129.4, 130.1, 130.2, 130.9, 134.0, 139.1, 174.1; IR (film) ν: 3386, 2921, 2851, 1598, 1383, 1312, 1121, 1034 cm-1; MS (ESI) m/z: 364 (M+Na+); HRMS calcd for C20H24NO4 [M+H]+ 342.1700, found 342.1700.
16b[37]: m.p. 200~202 ℃ (EtOAc/MeOH); [α]D20 +71.5 (c 0.88, 0.1 mol•L-1 NaOH in MeOH); 1H NMR (400 MHz, DMSO-d6) δ:1.90 (ddd, J=5.0, 10.2, 12.8 Hz, 1H, H-3), 2.12 (ddd, J=1.0, 6.6, 12.8 Hz, 1H, H-3), 2.93~2.99 (m, 1H, H-5), 3.04 (dd, J=8.0, 10.7 Hz, 1H, H-6), 3.15 (dd, J=4.1, 10.7 Hz, 1H, H-6), 3.55 (dd, J=6.6, 10.2 Hz, 1H, H-2), 3.75 (d, J=13.6 Hz, 1H, NCH2Ph), 3.88~3.93 (m, 1H, H-4), 3.95 (d, J=13.6 Hz, 1H, NCH2Ph), 4.44 (t, J=11.8 Hz, 2H, OCH2Ph), 7.22~7.37 (m, 10H, ArH); 13C NMR (100 MHz, DMSO-d6)δ: 34.4, 57.7, 62.1, 64.2, 69.3, 70.8, 80.1, 126.9, 127.2, 127.3, 128.0, 128.1, 129.1, 138.2, 138.6, 174.5; IR (film) ν: 3370, 2984, 2922, 2851, 1593, 1384, 1317, 1118, 1035 cm-1; MS (ESI) m/z: 364 (M+Na)+; HRMS calcd for C20H24NO4[M+H]+ 342.1700, found 342.1700. Anal. calcd for C20H23NO4: C 70.36, H 6.79, N 4.10; found C 70.01, H 6.59, N 3.80.
3.2.6 (2R, 4S, 5R)-N-甲基-4-羟基-5-羟甲基-脯氨酸(N-甲基-2-别-bulgecinine, 17)的合成
将16b (16 mg, 0.047 mmol)的无水乙醇(5 mL)溶液和20% Pd(OH)2/C (50 mg)混合, 反应体系置换为氢气氛, 在室温下反应15 h.通过硅藻土滤除钯碳, 滤液减压浓缩得8.1 mg无色油状化合物17, 产率99%.[α]D20 +55.2 (c 0.63, MeOH); 1H NMR (400 MHz, D2O) δ: 2.40 (ddd, J=5.9, 8.3, 13.9 Hz, 1H, H-3), 2.49 (ddd, J=5.0, 8.6, 13.9 Hz, 1H, H-3), 3.13 (s, 3H, NCH3), 3.55 (app. dd, J=4.8, 9.2 Hz, 1H, H-5), 3.92 (dd, J=5.1, 12.9 Hz, 1H, H-6), 4.03 (dd, J=4.0, 12.9 Hz, 1H, H-6), 4.25 (t, J=8.5 Hz, 1H, H-2), 4.39 (app. dd, J=5.3, 10.6 Hz, 1H, H-4); 13C NMR (100 MHz, D2O)δ: 36.5, 42.4, 56.7, 69.9, 70.2, 76.3, 172.7; IR (film) ν: 3317, 1629, 1383, 1334, 1093, 1023 cm-1; MS (ESI) m/z: 198 (M+Na)+; HRMS calcd for C7H14NO4[M+H]+ 176.0917, found 176.0937.
辅助材料(Supporting Information) 化合物5a发生差向异构化的1H NMR谱图及其局部放大图; 化合物5a, 16a和18的1H-1H COSY和NOESY谱; 化合物16b的单晶X射线晶体衍射图与数据; 非对映异构体5a和5b混合物的HPLC色谱图; 5a/5b在酸性条件下水解实验步骤及所得产物2, 5-顺式-5-羟甲基脯氨酰胺(18)的鉴定数据; 新化合物5a, 12, 13, 15, 16a, 16b, 17, 18的核磁共振氢谱和碳谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
3.2.2 (4S, 5R)-N-苄基-4-羟基-5-羟甲基-2-吡咯烷酮(13)的合成
将3a (953 mg, 3.06 mmol)的95%乙醇(31 mL)溶液与10% Pd/C (974 mg)混合后, 反应体系置换为氢气氛, 室温反应24 h.通过硅藻土滤除钯碳, 滤液减压浓缩.粗产物通过硅胶柱层析[V(乙酸乙酯):V(石油醚)=15:1]分离纯化得553 mg无色油状化合物13, 产率82%.[α]D20+29.2 (c 1.01, CHCl3); 1H NMR (400 MHz, CDCl3)δ:2.29 (dd, J=2.2, 17.4 Hz, 1H, H-3), 2.77 (dd, J=6.6, 17.4 Hz, 1H, H-3), 3.30~3.40 (m, 1H, H-5), 3.51~3.62 (m, 2H, H-6), 3.76 (dd, J=5.2, 6.2 Hz, 1H, OH, D2O exchangeable), 3.95 (d, J=4.0 Hz, 1H, OH, D2O exchangeable), 4.06 (d, J=15.3 Hz, 1H, NCH2Ph), 4.28~4.34 (m, 1H, H-4), 4.85 (d, J=15.3 Hz, 1H, NCH2Ph), 7.18~7.31 (m, 5H, ArH); 13C NMR (100 MHz, CDCl3) δ: 40.7, 44.3, 59.7, 67.5, 67.9, 127.7 (3C), 128.8 (2C), 135.9, 174.5; IR (film) ν: 3373, 2926, 1665, 1451, 1357, 1296, 1253, 1167, 1079 cm-1; MS (ESI) m/z: 244 (M+Na+).
3.2.4 (2S, 4S, 5R)-2-氰基-N-苄基-4-苄氧基-5-羟甲基吡咯烷(5a)和(2R, 4S, 5R)-2-氰基-N-苄基-4-苄氧基-5-羟甲基吡咯烷(5b)的合成
在-20 ℃下向3a (47 mg, 0.15 mmol)的四氢呋喃(0.30 mL)溶液中缓慢加入氢化锂铝(LAH) (22.8 mg, 0.60 mmol)的四氢呋喃(0.8 mL)悬浊液.反应3 h后, 缓慢加入乙醇(0.20 mL).然后迅速加入KCN (39 mg, 0.60 mmol)的水溶液(0.15 mL).缓慢升至室温, 剧烈搅拌1 h.然后加入乙酸乙酯和水.水相用乙酸乙酯萃取(5 mL×3).合并有机相, 用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物通过硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:6]分离纯化得31 mg黄色油状物5a和5b (5a:5b=69:31), 产率为63%.其非对映异构体比例通过HPLC分析确定, 分析条件Shim-Pack VP-ODS柱(150 mm×4.6 mm) [V(CH3CN)/V(H2O)=55/45, 1.0 mL/min, λ=254 nm]; t1=7.6 min (69%), t2=8.1 min (31%).
5a: [α]D20-62.9 (c 1.70, CHCl3); 1H NMR (400 MHz, CD3CN) δ: 2.08~2.22 (m, 2H, H-3), 2.79 (br s, 1H, OH, D2O exchangeable), 2.91~2.96 (m, 1H, H-5), 3.51~3.62 (m, 2H, H-6), 3.70 (d, J=13.6 Hz, 1H, NCH2Ph), 3.81 (d, J=7.0 Hz, 1H, H-2), 4.05~4.09 (m, 1H, H-4), 4.15 (d, J=13.6 Hz, 1H, NCH2Ph), 4.52 (d, J=11.8 Hz, 1H, OCH2Ph), 4.57 (d, J=11.8 Hz, 1H, OCH2Ph), 7.27~7.43 (m, 10H, ArH); 13C NMR (100 MHz, CD3CN) δ: 35.9, 52.9, 55.4, 62.3, 71.4, 72.0, 81.8, 118.9, 128.47, 128.49, 128.7, 129.3, 129.5, 129.8, 139.2, 139.8; IR (film) ν: 3438, 3088, 3059, 3029, 2918, 2872, 2225, 1453, 1382, 1084, 1028 cm-1; MS (ESI) m/z: 345 (M+Na+). Anal. calcd for C12H16O5: C 74.51, H 6.88, N 8.69; found C 74.31, H 6.48, N 8.41.
3.2.1 (4S, 5R)-N-苄基-4-苄氧基-5-苄氧基甲基-2-吡咯烷酮(12)的合成
将3a (50 mg, 0.16 mmol)溶于四氢呋喃(0.60 mL)中, 然后在-20 ℃下向体系中缓慢加入NaH (7.7 mg, 分散在矿物油中含量60%, 0.19 mmol)的四氢呋喃(0.80 mL)溶液.在-20 ℃下继续搅拌30 min, 再往反应体系中加入BnBr (0.03 mL, 0.24 mmol)并在此温度下搅拌25 h, 然后向体系中加入饱和的氯化铵溶液, 并加水稀释.乙酸乙酯萃取, 合并有机相.用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物通过硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:2]分离纯化得52 mg无色油状物12, 产率81%. [α]D20+27.9 (c 1.16, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 2.50 (dd, J=1.9, 17.3 Hz, 1H, H-3), 2.80 (dd, J=6.6, 17.3 Hz, 1H, H-3), 3.38 (dd, J=4.1, 10.1 Hz, 1H, H-6), 3.42 (dd, J=3.6, 10.1 Hz, 1H, H-6, overlapped with another H-6), 3.61 (ddd, J=1.6, 3.6, 4.1 Hz, 1H, H-5), 4.05 (d, J=15.6 Hz, 1H, NCH2Ph), 4.08 (ddd, J=1.7, 1.9, 6.6 Hz, 1H, H-4), 4.33 (d, J=12.0 Hz, 1H, OCH2Ph), 4.38 (d, J=12.0 Hz, 1H, OCH2Ph), 4.40 (d, J=11.8 Hz, 1H, OCH2Ph), 4.46 (d, J=11.8 Hz, 1H, OCH2Ph), 4.92 (d, J=15.6 Hz, 1H, NCH2Ph), 7.18~7.37 (m, 15H, ArH); 13C NMR (100 MHz, CDCl3) δ: 37.7, 44.3, 63.3, 67.8, 70.5, 73.2, 74.5, 127.3, 127.49, 127.50, 127.7, 127.8, 128.3, 128.4, 128.5, 136.4, 137.43, 137.44, 173.3; IR (film) ν: 3088, 3062, 3029, 2860, 1691, 1495, 1453, 1355, 1304, 1258, 1095, 1067, 1027 cm-1; MS (ESI) m/z: 424 (M+Na)+. Anal. calcd for C26H27NO3: C 77.78, H 6.78, N 3.49; found C 77.77, H 6.30, N 3.51.
-
-
[1]
(a) Otto, N.; Opatz, T. Chem. Eur. J. 2014, 20, 13064. (b) Katoh, T.; Nagata, Y.; Kobayashi, Y.; Arai, K.; Minami, J.; Terashima, S. Tetrahedron 1994, 50, 6221.
-
[2]
Husson, H. P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383. doi: 10.1039/a900153k
-
[3]
Fuentes de Arriba, Á. L.; Lenci, E.; Sonawane, M.; Formery, O.; Dixon, D. J. Angew. Chem., Int. Ed. 2017, 56, 3655. doi: 10.1002/anie.201612367
-
[4]
Savage, S. A.; Jones, G. S.; Kolotuchin, S.; Ramrattan, S. A.; Vu, T.; Waltermire, R. E. Org. Process Res. Dev. 2009, 13, 1169. doi: 10.1021/op900226j
-
[5]
Patterson, D. E.; Powers, J. D.; LeBlanc, M.; Sharkey, T.; Boehler, E.; Irdam, E.; Osterhout, M. H. Org. Process Res. Dev. 2009, 13, 900. doi: 10.1021/op900178d
-
[6]
Fleming, F. F. Nat. Prod. Rep. 1999, 16, 597. doi: 10.1039/a804370a
-
[7]
Xu, S.-H.; Wang, G.; Zhu, J.-J.; Shen, C.; Yang, Z.-Z.; Yu, J.; Li, Z.; Lin, T.-H.; Sun, X.; Zhang, F.-L. Eur. J. Org. Chem. 2017, 975.
-
[8]
(a) Wang, J.; Liu, X.; Feng, X. Chem. Rev. 2011, 111, 6947. (b) Cai, X.-H.; Xie, B. ARKIVOC 2014, part I, 205. (c) Liu, Y.-L.; Zhou, J. Synthesis 2015, 47, 1210.
-
[9]
(a) Ping, Y.; Ding, Q.; Peng, Y. ACS Catal. 2016, 6, 5989. (b) Libendi, S. S.; Demizu, Y.; Onomura, O. Org. Biomol. Chem. 2009, 7, 351.
-
[10]
(a) Gless, R. D. ; Rapoport, H. J. Org. Chem. 1979, 44, 1324. (b) Langenskiöld, T. ; Lounasmaa, M. Heterocycles 1983, 20, 671. (c) Connolly, P. J. ; Heathcock, H. C. J. Org. Chem. 1985, 50, 4135. (d) Diez, A. ; Castells, J. ; Forns, P. ; Rubiralta, M. ; Grierson, D. S. ; Husson, H. P. ; Solans, X. ; Font-Bardia, M. Tetrahedron 1994, 50, 6585. (e) Kubo, A. ; Nakai, T. ; Koizumi, Y. ; Kitahara, Y. ; Saito, N. ; Mikami, Y. ; Yazawa, K. ; Uno, J. Heterocycles 1996, 42, 195. (f) Durand, J. O. ; Larchevêque, M. ; Petit, Y. ; Tetrahedron Lett. 1998, 39, 5743. (g) Huang, P. -Q. ; Huang, H. -Y. Synth. Commun. 2004, 34, 1377. (h) Matsubara, R. ; Kawai, N. ; Kobayashi, S. Angew. Chem. , Int. Ed. 2006, 45, 3814. (i) Xia, Q. ; Ganem, B. Org. Lett. 2001, 3, 485. (j) Xia, Q. ; Ganem, B. Tetrahedron Lett. 2002, 43, 1597. (k) Nakajima, M. ; Oda, Y. ; Wada, T. ; Minamikawa, R. ; Shirokane, K. ; Sato, T. ; Chida, N. Chem. Eur. J. 2014, 20, 17565. (l) Shirokane, K. ; Kurosaki, Y. ; Sato, T. ; Chida, N. Angew. Chem. , Int. Ed. 2010, 49, 6369. (m) Yanagita, Y. ; Nakamura, H. ; Shirokane, K. ; Kurosaki, Y. ; Sato, T. ; Chida, N. Chem. Eur. J. 2013, 19, 678. (n) Huang, P. -Q. ; Huang, Y. -H. ; Xiao, K. -J. ; Wang, Y. ; Xia, X. -E. J. Org. Chem. 2015, 80, 2861. (o) Anderson, B. K. ; Livinghouse, T. J. Org. Chem. 2015, 80, 9847.
-
[11]
(a) Li, X. -J. ; Sun Y. ; Zhang L. ; Peng, B. Chin. J. Org. Chem. 2016, 36, 2530 (in Chinese). (李晓锦, 孙艳, 张磊, 彭勃, 有机化学, 2016, 36, 2530. ) (b) Kaiser, D. ; Maulide, N. J. Org. Chem. 2016, 81, 4421. (c) Pace, V. ; Holzer, W. ; Olofsson, B. Adv. Synth. Catal. 2014, 356, 3697. (d) Sato, T. ; Chida, N. Org. Biomol. Chem. 2014, 12, 3147.
-
[12]
(a) White, K. L.; Mewald, M.; Movassaghi, M. J. Org. Chem. 2015, 80, 7403. (b) Medley, J. W.; Movassaghi, M. J. Org. Chem. 2009, 74, 1341. (c) Movassaghi, M.; Hill, M. D. J. Am. Chem. Soc. 2006, 128, 14254.
-
[13]
(a) Régnier, S.; Bechara, W. S.; Charette, A. B. J. Org. Chem. 2016, 81, 10348. (b) Cyr, P.; Régnier, S.; Bechara, W. S.; Charette, A. B. Org. Lett. 2015, 17, 3386. (c) Bechara, W. S.; Pelletier, G.; Charette, A. B. Nat. Chem. 2012, 4, 228.
-
[14]
(a) Boudreault, J.; Lévesque, F.; Bélanger, G. J. Org. Chem. 2016, 81, 9247. (b) Romanens, A.; Bélanger, G. Org. Lett. 2015, 17, 322. (c) Bélanger, G.; Gauthier, L.; Ménard, F.; Nantel, M.; Barabé, F. Org. Lett. 2005, 7, 4431.
-
[15]
(a) Wang, J.; He, Z.-X.; Chen, X.-P.; Song, W.-Z.; Lu, P.; Wang, Y.-G. Tetrahedron 2010, 66, 1208. (b) Cui, S.-L.; Wang, J.; Wang, Y.-G. J. Am. Chem. Soc. 2008, 130, 13526.
-
[16]
(a) Xu, M.; Xu, K.; Wang, S. Z.; Yao, Z. J. Tetrahedron Lett. 2013, 54, 4675. (b) Xi, J.; Dong, Q.-L.; Liu, G.-S.; Wang, S.; Chen, L.; Yao, Z.-J. Synlett 2010, 1674. (c) Dong, Q.-L.; Liu, G.-S.; Zhou, H.-B.; Chen, L.; Yao, Z.-J. Tetrahedron Lett. 2008, 49, 1636.
-
[17]
(a) Tona, V.; de la Torre, A.; Padmanaban, M.; Ruider, S.; González, L.; Maulide, N. J. Am. Chem. Soc. 2016, 138, 8348. (b) Ruider, S. A.; Maulide, N. Angew. Chem., Int. Ed. 2015, 54, 13856. (c) Peng, B.; Geerdink, D.; Farès, C.; Maulide, N. Angew. Chem., Int. Ed. 2014, 53, 5462.
-
[18]
(a) Katahara, S.; Kobayashi, S.; Fujita, K.; Matsumoto, T.; Sato, T.; Chida, N. J. Am. Chem. Soc. 2016, 138, 5246. (b) Nakajima, M.; Sato, T.; Chida, N. Org. Lett. 2015, 17, 1696. (c) Shirokane, K.; Wada, T.; Yoritate, M.; Minamikawa, R.; Takayama, N.; Sato, T.; Chida, N. Angew. Chem., Int. Ed. 2014, 53, 512.
-
[19]
(a) Pace, V.; de la Vega-Hernández, K.; Urban, E.; Langer, T. Org. Lett. 2016, 18, 2750. (b) Pace, V.; Pelosi, A.; Antermite, D.; Rosati, O.; Curini, M.; Holzer, W. Chem. Commun. 2016, 52, 2639. (c) Pace, V.; Castoldi, L.; Mamuye, A. D.; Holzer, W. Synthesis 2014, 46, 2897.
-
[20]
Gregory, A. W.; Chambers, A.; Hawkins, A.; Jakubec, P.; Dixon, D. J. Chem. Eur. J. 2015, 21, 111. doi: 10.1002/chem.201405256
-
[21]
(a) Huang, P. -Q. ; Ou, W. ; Han, F. Chem. Commun. 2016, 52, 11967. (b) Huang, P. -Q. ; Lang, Q. -W. ; Hu, X. -N. J. Org. Chem. 2016, 81, 10227. (c) Huang, P. -Q. ; Huang, Y. -H. ; Geng, H. ; Ye, J. -L. Sci. Rep. 2016, 6, 28801. (d) Lang, Q. -W. ; Hu, X. -N. ; Huang, P. -Q. Sci. China, Chem. 2016, 59, 1638. (e) Zheng, J. -F. ; Xie, Z. -Q. ; Chen, X. -J. ; Huang, P. -Q. Acta Chim. Sinica 2015, 73, 705 (in Chinese). (郑剑峰, 谢志强, 陈欣健, 黄培强, 化学学报, 2015, 73, 705. ) (f) Wang, A. -E. ; Chang, Z. ; Liu, Y. -P. ; Huang, P. -Q. Chin. Chem. Lett. 2015, 24, 1055.
-
[22]
Huang, P.-Q. Synlett 2006, 1133.
-
[23]
Zhou, X.; Zhang, P.-Y.; Ye, J.-L.; Huang, P.-Q. C. R. Chim. 2008, 11, 5. doi: 10.1016/j.crci.2007.02.018
-
[24]
(a) Guo, L. -D. ; Huang, X. -Z. ; Luo, S. -P. ; Cao, W. -S. ; Ruan, Y. -P. ; Ye, J. -L. ; Huang, P. -Q. Angew. Chem. , Int. Ed. 2016, 55, 4064. (b) Huang, P. -Q. ; Mao, Z. -Y. ; Geng, H. Chin. J. Org. Chem. 2016, 36, 315 (in Chinese). (黄培强, 茅中一, 耿辉, 有机化学, 2016, 36, 315. ) (c) Mao, Z. -Y. ; Geng, H. ; Zhang, T. -T. ; Ruan, Y. -P. ; Ye, J. -L. ; Huang, P. -Q. Org. Chem. Front. 2016, 3, 24.
-
[25]
(a) Hart, B. P.; Rapoport, H. J. Org. Chem. 1999, 64, 2050. (b) Hart, B. P.; Verma, S. K.; Rapoport, H. J. Org. Chem. 2003, 68, 187. (c) Khalaf, J. K.; Datta, A. J. Org. Chem. 2004, 69, 387.
-
[26]
Shinagawa, S.; Kashara, F.; Wada, Y.; Harada, S.; Asai, M. Tetrahedron 1984, 40, 3465. doi: 10.1016/S0040-4020(01)91497-8
-
[27]
(a) Show, K.; Upadhyay, P. K.; Kumar, P. Tetrahedron: Asymmetry 2011, 22, 1234. (b) Das, B.; Kumar, D. N. Synlett 2011, 1285. (c) Best, D.; Wang, C.; Weymouth-Wilson, A. C.; Clarkson, R. A.; Wilson, F. X.; Nash, R. J.; Miyauchi, S.; Kato, A.; Fleet, G. W. J. Tetrahedron: Asymmetry 2010, 21, 311. (d) Evano, G.; Couty, F.; Toumi, M. Tetrahedron Lett. 2008, 49, 1175. (e) Chandrasekhar, S.; Chandrasekhar, G.; Vijeender, K.; Sarma, G. D. Tetrahedron: Asymmetry 2006, 17, 2864. (f) Trost, B. M.; Horne, D. B.; Woltering, M. J. Chem. Eur. J. 2006, 12, 6607. (g) Chavan, S. P.; Praveen, C.; Sharma, P.; Kalkote, U. R. Tetrahedron Lett. 2005, 46, 439. (h) Chavan, S. P.; Praveen, C. Tetrahedron Lett. 2004, 45, 421.
-
[28]
Hwang, Y. C.; Chu, M.; Flower, F. W. J. Org. Chem. 1985, 50, 3885. doi: 10.1021/jo00220a040
-
[29]
Klumpe, M.; Dötz, K. H. Tetrahedron Lett. 1998, 39, 3683. doi: 10.1016/S0040-4039(98)00620-0
-
[30]
Tanaka, K.; Sawanishi, H. Tetrahedron 1998, 54, 10029. doi: 10.1016/S0040-4020(98)00604-8
-
[31]
Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry, Pergamon, New York, 1983.
-
[32]
Hoffmann, H. R. Chem. Rev. 1989, 89, 1841. doi: 10.1021/cr00098a009
-
[33]
Davis, F. A.; Reddy, G. V.; Chen, B. C.; Kumar, A.; Haque, M. S. J. Org. Chem. 1995, 60, 6148. doi: 10.1021/jo00124a030
-
[34]
Crystallographic data for 16b have been deposited at the Cambridge Crystallographic Data Center as supplementary publication number CCDC 1537394.
-
[35]
(a) Wang, D.; Crowe, W. E. Chin. Chem. Lett. 2015, 26, 238. (b) Ren, R.-G.; Ma, J.-Y.; Mao, Z.-Y.; Liu, Y.-W.; Wei, B.-G. Chin. Chem. Lett. 2015, 26, 1209.
-
[36]
Sakurai, R.; Suzuki, S.; Hashimoto, J.; Baba, M.; Itoh, O.; Uchida, A.; Hattori, T.; Miyano, S.; Yamaura, M. Org. Lett. 2004, 6, 2241. doi: 10.1021/ol049256q
-
[37]
Xu, C.-P.; Xiao, Z.-H.; Zhuo, B.-Q.; Wang, Y.-H.; Huang, P.-Q. Chem. Commun. 2010, 46, 7834. doi: 10.1039/c0cc01487g
-
[38]
(a) Hong, B. -C. ; Raja, A. ; Sheth, V. M. Synthesis 2015, 47, 3257. (b) Wang, F. -J. ; Lu, A. -L. ; Huang, D. -F. ; Su, Y. -P. ; Xia, X. -W. ; Xü, Y. -L. ; Wang, K. -H. ; Hu, Y. -L. Chin. J. Org. Chem. 2015, 35, 1046 (in Chinese). (王凤娇, 陆爱玲, 黄丹凤, 苏瀛鹏, 夏晓文, 徐艳丽, 王克虎, 胡雨来, 有机化学, 2015, 35, 1046. ) (c) Niu, S. -X. ; Tang, Z. -C. ; Wang, T. -L. ; You, Q. -D. ; Xiang, H. Chin. J. Org. Chem. 2015, 35, 2150 (in Chinese). (牛绍雄, 唐智超, 刘畅, 王天麟, 尤启冬, 向华, 有机化学, 2015, 35, 2150. ) (d) Li, G. -C. ; Yang, A. -M. ; Mao Z. -Y. ; Zhou, Z. ; Wei, B. -G. Chin. J. Org. Chem. 2015, 35, 2157 (in Chinese). (李国成, 杨爱梅, 毛卓亚, 周祝, 魏邦国, 有机化学, 2015, 35, 2157. ) (e) Yu, K. ; Gao, B. -L. ; Ding, H. -F. Acta Chim. Sinica 2015, 73, 410 (in Chinese). (余宽, 高北岭, 丁寒锋, 化学学报, 2015, 73, 410. ) (f) He, L. ; Gu, M. -D. ; Wang, D. -X. ; Zhao, L. ; Wang, M. -X. Acta Chim. Sinica 2015, 73, 1018 (in Chinese). (何玲, 顾梦迪, 王德先, 赵亮, 王梅祥, 化学学报, 2015, 73, 1018. )
-
[1]
-

图 1 含α-氰胺复合官能团的代表性药物和天然产物
Figure 1 Representative drugs and natural products containing α-aminonitrile functional group

图 2 本文涉及的手性合成砌块与bulgecins的结构
Figure 2 Structures of the chiral building blocks discussed in this paper and those of bulgecins

图式4 Terashima和Tanaka报道的经由亚胺鎓中间体的2, 5-反式-立体选择性氰基加成反应
Scheme 4 Terashima and Tanaka's stereoselective addition reactions of cyanide anion to iminium ion intermediates

图式5 氰基负离子对亚胺鎓中间体加成的构象分析
Scheme 5 Conformation analysis of the cyanide anion addition to iminium ion intermediate

图式6 (+)-N-甲基-2-别-bulgecinine的立体选择性合成
Scheme 6 Stereoselective synthesis of (+)-N-methyl-2-epi-bulgecinine

图 5 化合物16b的单晶X射线衍射图
Figure 5 Single crystal X-ray diffraction structure of compound 16b

图 6 确定化合物16a C(2) 位立体化学的1H-1H COSY和NOESY谱中的关键相关关系
Figure 6 Key correlations shown in 1H-1H COSY and NOESY spectra for the assignment of stereochemistry at C(2) of compound 16a
表 1 3a直接还原氰基化的反应条件筛选
Table 1. Screening of reaction conditions for the direct reductive cyanation of 3a
序号 温度/℃ LiAlH4/ equiv. 反应时间 产率a/% 原料回收率a/% 1 -35 1.5 20 min 3 56 2 -15 1.5 1 h 31 6 3 -15 1.5 3 h 33 2 4 -17 1.5 3 h 38 16 5 -18 1.5 3 h 38 25 6 -19 1.5 3 h 39 37 7 -20 1.5 3 h 49 22 a分离产率.  下载: 导出CSV
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 1356
- HTML全文浏览量: 177
 下载:
下载:
