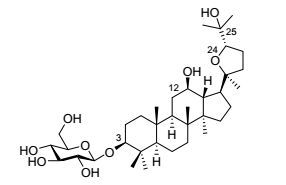
 图 1
拟人参皂苷HQ (PHQ, 1)结构式
Figure Figure1.
Chemical structure of pseudoginsenoside HQ (PHQ, 1)
图 1
拟人参皂苷HQ (PHQ, 1)结构式
Figure Figure1.
Chemical structure of pseudoginsenoside HQ (PHQ, 1)
引用本文:
杨刚强, 李阳, 杨青, 岳馨, 姚雷, 姜永涛. 拟人参皂苷HQ的简明高效合成[J]. 有机化学,
2017, 37(6): 1530-1536.
doi:
10.6023/cjoc201703006

Citation: Yang Gangqiang, Li Yang, Yang Qing, Yue Xin, Yao Lei, Jiang Yongtao. Simple and Efficient Synthesis of Pseudoginsenoside HQ[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1530-1536. doi: 10.6023/cjoc201703006
Citation: Yang Gangqiang, Li Yang, Yang Qing, Yue Xin, Yao Lei, Jiang Yongtao. Simple and Efficient Synthesis of Pseudoginsenoside HQ[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1530-1536. doi: 10.6023/cjoc201703006
拟人参皂苷HQ的简明高效合成
摘要:
拟人参皂苷HQ(PHQ),化学名称3β-O-β-D-吡喃葡萄糖基-(20S,24S)-环氧达玛-12β,25-二醇,是一种生物活性较高的稀有人参皂苷Rh2在体内的主要代谢产物,具有潜在的药用价值.目前报道的合成线路复杂且总收率较低,是因为关键的苷元C-3位糖苷化需要合理的保护策略才能实现.通过奥克梯隆型皂苷元C-3位的糖苷化条件探索,首次发现以Ag2CO3为促进剂,免保护策略,即可实现苷元C-3位选择性糖苷化制备PHQ.从商品20(S)-原人参二醇出发,经氧化环化、选择性糖苷化和对糖基脱苯甲酰基保护三步完成PHQ的合成.本方法为PHQ及其衍生物的制备提供了一条简明高效途径.
English
Simple and Efficient Synthesis of Pseudoginsenoside HQ
Abstract:
Pseudoginsenoside HQ[PHQ, 3β-O-β-D-glucopyranosyl-(20S, 24S)-epoxydammarane-12β, 25-diol] is the main in vivo metabolite of 20(S)-ginsenoside Rh2 which has high biological activities, with potential medicinal value. The reported chemical synthesis of PHQ suffered complex synthetic route and low overall yield because the rational protecting group strategies were required for the key glycosylation on C-3 position of sapogenins. In this paper, the glycosylation conditions on C-3 position of ocotillol type sapogenins were investigated, and it was found that it could realize the selective glycosylation on C-3-position of sapogenins to prepare PHQ under the Ag2CO3 promoter without the protecting group strategy. A new synthesis of PHQ was achieved in three steps via epoxidation, selective glycosylation and debenzoylation reaction of glycon using commercially available 20(S)-protopanoxadiol as the starting material. This method provides a simple and efficient way for the preparation of PHQ and its derivatives.
-
Key words:
- pseudoginsenoside HQ
- / glycosylation
- / ocotillol type ginsenoside
- / chemical synthesis
-
20(S)-人参皂苷Rh2属于红参中的极微量组分, 为原人参二醇组皂苷.药理学研究证实Rh2具有良好的肿瘤细胞生长抑制作用[1], 已进入临床研究阶段.其主要作用机制是调节机体免疫功能[1a], 诱导肿瘤细胞凋亡[1c], 对化学抗癌药物具有增效减毒作用[2].此外, 在抗过敏、抗炎、耐缺氧等方面均有作用.近年来, 体内、外的代谢研究表明20, 24-环氧的奥克梯隆型代谢产物可能是C-20位不含糖基的人参皂苷(如人参皂苷Rg3、Rh2、Rg2和Rh1) 及其苷元(如原人参二醇(PPD)和原人参三醇(PPT))在体内发挥作用的真正有效成分[3].
拟人参皂苷HQ (PHQ, 1)是Rh2在体内的主要代谢产物和潜在的真正有效成分[4], 化学名称为3β-O-β-D-吡喃葡萄糖基-(20S, 24S)-环氧达玛-12β, 25-二醇, 属于四环三萜类奥克梯隆(Ocotillol)型人参皂苷(图 1).活性研究表明, PHQ具有较好的肿瘤细胞生长抑制作用[5].目前, PHQ主要通过天然来源极其稀少的Rh2氧化环化制备获得[4~6], 来源问题严重制约了与PHQ相关的活性和机制等方面的深入研究.因此, 有课题组研究了PHQ的合成方法, 主要包括: (1) 俞飙课题组[7]报道了以20(S)-PPD为原料, 先差异化保护C-12位和C-3位的羟基, 再氧化环化, 并通过C-3位羟基脱保护分离出C-24位差向异构体, 然后再依次C-3位和C-25位羟基差异化保护后, C-3位羟基选择性脱保护, 用Ph3PAuNTf2促进剂与邻炔基苯甲酸酯型糖基供体糖苷化, 最后脱保护所有羟基合成PHQ的线路, 总收率仅有28.2%, 且促进剂Ph3PAuNTf2的成本较高, 以及邻炔基苯甲酸酯型糖基供体的制备较为复杂(Scheme 1, 线路A); (2) 李平亚课题组[6]和Yang等[5]分别报道了以原人参二醇组皂苷和Rh2为原料, 经氧化环化合成PHQ的线路, 由Rh2合成PHQ的已知最高产率为54.6%[4], 但Rh2本身也是难得的天然产物资源, 其化学合成从原料20(S)-PPD开始, 需要C-12位羟基先保护再糖苷化, 然后脱保护制得[8], 产率约在47.4%[9], 因此本线路的总产率也仅有25.9% (Scheme 1, 线路B).
图 1
拟人参皂苷HQ (PHQ, 1)结构式
Figure Figure1.
Chemical structure of pseudoginsenoside HQ (PHQ, 1)
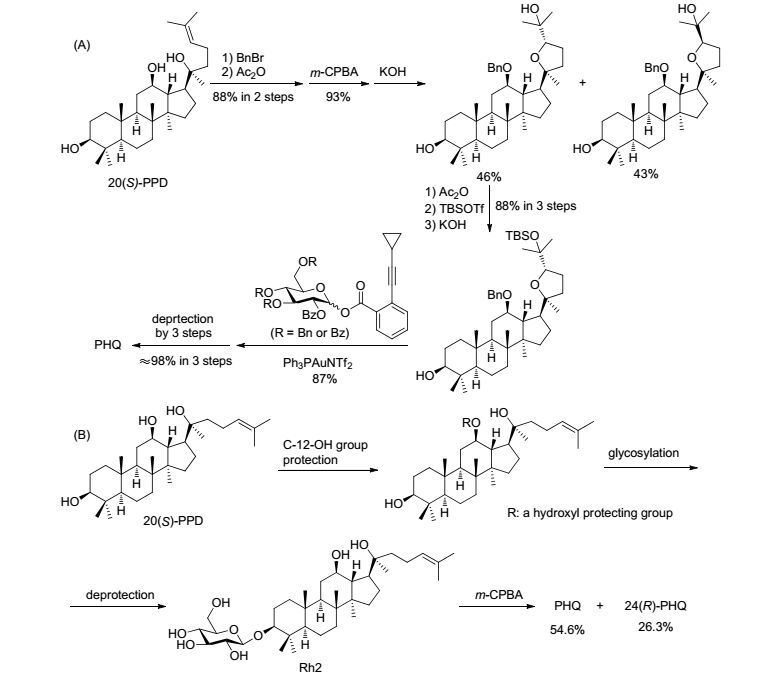 图式1
已报道的拟人参皂苷HQ合成方法
Scheme1.
Reported approaches for the synthesis of pseudoginsenoside HQ
图式1
已报道的拟人参皂苷HQ合成方法
Scheme1.
Reported approaches for the synthesis of pseudoginsenoside HQ
综合分析发现, 皂苷元的C-3位糖苷化是化学合成PHQ的关键步骤.糖苷化可以在初始的原人参二醇阶段或者在形成奥克梯隆型皂苷元后发生.然而, 目前的糖苷化条件中, 均需要选择性保护C-12位或者C-12位和C-25位的羟基后才能有效地C-3位糖苷化[7, 8], 增加了反应步骤, 合成效率也因此低下.且在奥克梯隆型皂苷元上直接糖苷化探索研究中, 目前的糖苷化条件对C-3位糖苷化无选择性[7, 10].
本文通过多种糖苷化条件的探索, 发现了以碳酸银为促进剂, 其他羟基免保护, 即可实现在奥克梯隆型皂苷元C-3位羟基选择性糖苷化, 从而实现简明高效地合成PHQ.
1 结果与讨论
本文以商品易得的20(S)-PPD为起始原料, 经氧化环化得到奥克梯隆型皂苷元(20S, 24S)-PDQ (2)和(20S, 24R)-PDQ (3) (Scheme 2), 他们的绝对构型通过图谱数据与已知文献对比确认[11].此反应条件在工业化放大生产中, (20S, 24S)构型产物2可达78.3%[12].
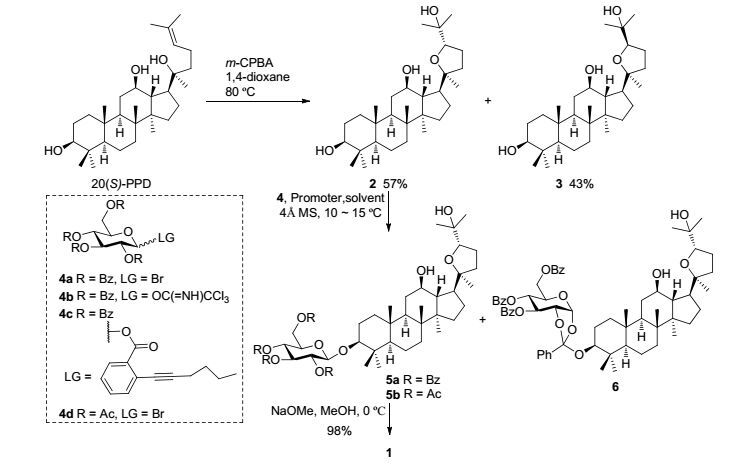 图式2
本文的拟人参皂苷HQ合成方法
Scheme2.
Our synthesis of Pseudoginsenoside HQ
图式2
本文的拟人参皂苷HQ合成方法
Scheme2.
Our synthesis of Pseudoginsenoside HQ
以化合物2为糖苷化受体(acceptor), 进行C-3位选择性糖苷化反应是本研究的关键步骤.我们认为:虽然C-12位仲羟基与呋喃环上的氧原子存在分子内氢键而增强了C-12位氧的亲核能力[13], 但C-12位仲羟基和C-25位叔羟基存在不利于糖苷化反应的较大空间位阻, 因此, 在合适的条件下, C-3位仲羟基可以有更好的反应选择性.首先, 我们探讨了经典的Koenigs-Knorr法(表 1, Entries 1~7), 以碳酸银为促进剂, 在10~15 ℃的反应温度下, 反应缓慢进行, 糖苷化选择性地发生在C-3位仲羟基上, 生成目标糖苷化产物5a (33%)和原酸酯型的糖苷化产物6 (43%) (Entry 1).提高反应温度降低了目标产物5a的产率, 同时促进了其他糖苷化产物的产生(Entry 2).溶剂极性增加使糖苷化反应无法进行(Entries 3~4), 利用混合促进剂(碳酸银和三氟甲基磺酸银)[14], 通过TLC跟踪反应发现, 反应在1 h左右结束, 但糖苷化C-3位选择性显著降低(Entry 5).增加碳酸银的用量至10~15 equiv., 糖苷化C-3位选择性不受影响, 而原酸酯型糖苷化产物6显著降低, 以正常的糖苷化产物5a生成为主(Entries 6~7).其次, 我们探讨了常用的三氯乙酰亚胺酯法(表 1, Entries 8~12), 其中, 以TMSOTf为促进剂的反应有更好的C-3位糖苷化选择性和更高的转化率(Entries 8~9).同样, 在以TMSOTf为促进剂的反应中, 反应温度的提高和溶剂极性的增加, 均使反应副产物增多(Entries 10~11).再次, 我们探讨了Au(Ⅰ)催化的糖基邻炔基苯甲酸酯型给体法[15], 以Ph3PAuNTf2为催化剂, 转化率达到了92%, 然而几乎无目标产物5a获得(Entry 13), 说明本反应条件对C-3位糖苷化没有选择性, 文献亦有类似报道[7].最后, 我们探讨了全乙酰基保护的糖基供体(donor)对C-3位糖苷化选择性反应的影响, 对比全苯甲酰基保护糖基供体的糖苷化反应(Entry 6), C-3位糖苷化产物5b的产率明显降低(Entry 14), 文献亦有类似无选择性的报道[10e].
 表 1
奥克梯隆型皂苷元2的C-3位糖苷化条件探索a
Table 1.
Glycosylation at C-3 of ocotillol type sapogenins 2
表 1
奥克梯隆型皂苷元2的C-3位糖苷化条件探索a
Table 1.
Glycosylation at C-3 of ocotillol type sapogenins 2

以上结果显示:在上述条件中, 以苯甲酰基保护糖基供体, 10 equiv.碳酸银为促进剂, 二氯甲烷为溶剂, 在10~15 ℃反应的条件最佳, 本反应可获得最好的C-3位糖苷化选择性和产率.
根据实验反应的现象和结果, 在表一中所述最佳条件对C-3位糖苷化选择性的可能机理进行阐述(Scheme 3).在非极性溶剂中, 碳酸银作用下, 糖基供体形成氧碳鎓正离子中间体7, 邻位效应形成相对稳定的氧碳鎓正离子中间体8, 糖基供体上苯甲酰基的位阻效应与C-12位仲羟基和C-25位叔羟基的位阻效应, 选择性地使位阻相对较小的C-3位仲羟基反式进攻中间体8形成目标β糖苷产物5a.
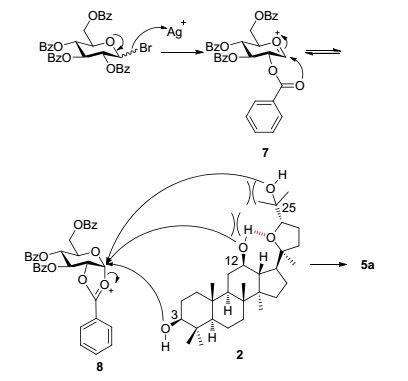 图式3
奥克梯隆型皂苷元2的C-3位选择性糖苷化机理
Scheme3.
Plausible mechanism for selective glycosylation at C-3 of ocotillol type sapogenins 2
图式3
奥克梯隆型皂苷元2的C-3位选择性糖苷化机理
Scheme3.
Plausible mechanism for selective glycosylation at C-3 of ocotillol type sapogenins 2
最后, 糖苷化产物5a经脱苯甲酰基保护高收率地转化为PHQ (1), 所得产物经核磁共振氢谱、碳谱和高分辨质谱确证.其中, 糖端基质子的耦合常数J1', 2'为7.8 Hz可以判断所合成化合物1的糖苷键为β构型; 糖苷化前后C-3位的化学位移向低场移动, 由δC 78.8移至δC 90.6, C-12位与C-25位的化学位移无变化, 可以判断所合成化合物1的糖苷键连接在C-3位上.且化合物1与文献报道PHQ在氘代吡啶中的图谱数据基本一致[4, 5, 7].
2 结论
本文以商品易得的20(S)-PPD为原料, 经3步反应简明、高效地合成具有高活性的稀有人参皂苷Rh2在体内的主要代谢产物PHQ, 总收率32%(回收产率45%).该合成方法较之已报道合成PHQ的方法, 具有步骤少, 糖苷化免保护策略等优点, 可为深入研究其药理活性及结构改造奠定物质基础.
3 实验部分
3.1 仪器与试剂
NMR用Bruker VANCE-400核磁共振仪测定, 以TMS作为内标; 高分辨质谱(HRMS)在Thermo Scientific Q Exactive质谱仪上测定; 旋光采用SGW-3自动旋光仪测定.薄层色谱(TLC)采用HSGF254(烟台化学工业研究所), 柱层析使用硅胶为200~300目(烟台化学工业研究所).所用试剂和溶剂为市售化学纯或分析纯; 要求无水的溶剂均用分子筛干燥处理.
3.2 实验方法
3.2.2 3β-O-(2', 3', 4', 6'-四-O-苯甲酰基-β-D-吡喃葡萄糖基)-(20S, 24S)-环氧达玛-12β, 25-二醇(5a)的合成
化合物2 (20 mg, 0.042 mmol)、4a (56 mg, 0.085 mmol)和4Ả分子筛(200 mg)悬浮于无水DCM (1 mL)中, 氩气保护下搅拌0.5 h后, 加入碳酸银(116 mg, 0.42 mmol)于10~15 ℃反应2 d.加TMSOTf (2 μL, 0.01 mmol)后结束反应, 用硅藻土层过滤后浓缩, 经柱层析得到白色固体5a (25 mg, 0.024 mmol, 57%, 回收产率81%), 回收原料2 (6 mg, 0.0013 mmol, 30%), m.p. 110~125 ℃; [α]D17+18.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.02 (dd, J=8.4, 1.2 Hz, 2H), 7.95 (dd, J=8.4, 1.2 Hz, 2H), 7.92 (dd, J=8.4, 1.2 Hz, 2H), 7.83 (dd, J=8.4, 1.2 Hz, 2H), 7.58 (tt, J=7.4, 1.2 Hz, 1H), 7.52~7.48 (m, 2H), 7.44~7.34 (m, 7H), 7.28 (t, J=7.9 Hz, 2H), 5.91 (t, J=9.7 Hz, 1H), 5.82 (s, 1H), 5.59 (dd, J=9.7, 4.5 Hz, 1H), 5.57 (dd, J=9.7, 2.8 Hz, 2H), 4.85 (d, J=7.9 Hz, 1H), 4.60 (dd, J=11.9, 3.4 Hz, 1H), 4.54 (dd, J=12.0, 6.7 Hz, 1H), 4.16 (ddd, J=10.0, 6.7, 3.4 Hz, 1H), 3.88 (dd, J=11.4, 4.9 Hz, 1H), 3.51 (dt, J=10.6, 5.0 Hz, 1H), 3.07 (dd, J=11.7, 4.5 Hz, 1H), 2.25 (td, J=10.2, 9.6, 3.9 Hz, 1H), 2.07~0.60 (m, 21H), 1.27 (s, 3H), 1.25 (s, 3H), 1.11 (s, 3H), 0.96 (s, 3H), 0.87 (s, 3H), 0.81 (s, 3H), 0.67 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.0, 165.8, 165.3, 165.0, 133.4, 133.3, 133.2, 133.0, 129.8 (2C), 129.7 (2C), 129.7 (2C), 129.6 (2C), 129.5, 129.3, 128.8, 128.7, 128.4 (4C), 128.2 (4C), 103.2, 90.6, 87.4, 87.1, 72.9, 72.1, 71.9, 70.5, 70.2, 70.1, 63.4, 56.1, 52.1, 50.1, 48.8, 48.7, 39.6, 38.9, 38.7, 36.7, 34.6, 32.1, 31.6, 31.6, 28.8, 28.5, 28.0, 27.5, 25.9, 25.0, 24.1, 18.0, 17.7, 16.1, 15.9, 15.4. HRMS (ESI, positive) calcd for C64H78-O13Na [M+Na]+ 1077.5335, found 1077.5368.
3.2.1 (20S, 24S)-达玛-20, 24-环氧-3β, 12β, 25-三醇(2)和(20S, 24R)-达玛-20, 24-环氧-3β, 12β, 25-三醇(3)的合成
20(S)-PPD (41 mg, 0.089 mmol)溶于1, 4-二氧六环(1 mL)中, 滴加溶于1, 4-二氧六环(0.5 mL)的间氯过氧苯甲酸(m-CPBA, 28 mg, 0.12 mmol), 在80 ℃下反应4 h, 用饱和Na2SO3猝灭反应, 加饱和NaHCO3溶液, 用乙酸乙酯萃取, 合并的有机相经饱和食盐水洗涤后, 无水硫酸钠干燥, 浓缩.经柱层析得到白色固体2 (24 mg, 0.050 mmol, 57%)和3 (18 mg, 0.038 mmol, 43%).
化合物2[11c, 11d]: m.p. 190~193 ℃; [α]D20+4.3 (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.78 (s, 1H), 3.88 (dd, J=10.7, 5.3 Hz, 1H), 3.52 (td, J=10.3, 4.7 Hz, 1H), 3.20 (dd, J=11.3, 4.8 Hz, 1H), 2.25 (td, J=10.5, 4.3 Hz, 1H), 2.10~1.46 (m, 16H), 1.31~1.10 (m, 4H), 1.27 (s, 3H), 1.23 (s, 3H), 1.11 (s, 3H), 1.01 (s, 3H), 0.97 (s, 3H), 0.91 (s, 3H), 0.88 (s, 3H), 0.78 (s, 3H), 0.76~0.73 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 87.3, 87.1, 78.8, 70.5, 70.0, 55.9, 52.1, 50.2, 48.8, 48.7, 39.7, 38.9, 38.8, 37.1, 34.7, 32.2, 31.6, 31.6, 28.8, 28.5, 28.0, 28.0, 27.4, 25.0, 24.1, 18.3, 17.7, 16.3, 15.4, 15.3. HRMS (ESI, positive) calcd for C30H52O4Na [M+Na]+ 499.3758, found 499.3745.
化合物3[11c, 11d]: m.p. 223~226 ℃; [α]D20+16.1 (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 3.84 (dd, J=8.8, 6.8 Hz, 1H), 3.51 (td, J=10.5, 4.6 Hz, 1H), 3.18 (dt, J=9.9, 4.5 Hz, 1H), 2.19 (td, J=10.9, 3.6 Hz, 1H), 2.08~1.82 (m, 4H), 1.72~1.39 (m, 10H), 1.34~1.26 (m, 3H), 1.28 (s, 3H), 1.27 (s, 3H), 1.14~0.96 (m, 3H), 1.09 (s, 3H), 0.98 (s, 3H), 0.97 (s, 3H), 0.90 (s, 3H), 0.85 (s, 3H), 0.77 (s, 3H), 0.73~0.71 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 86.5, 85.4, 78.8, 71.0, 70.1, 55.9, 52.0, 50.5, 49.3, 47.9, 39.7, 38.9, 38.9, 37.1, 34.8, 32.6, 31.3, 31.2, 28.6, 28.0, 27.9, 27.6, 27.4, 26.1, 25.0, 18.2, 18.1, 16.3, 15.3, 15.3. HRMS (ESI, positive) calcd for C30H52O4Na [M+Na]+ 499.3758, found 499.3748.
3.2.4 3', 4', 6'-三-O-苯甲酰基-α-D-吡喃葡萄糖-1', 2'-[(20S, 24S)-环氧-12β, 25-二羟基-达玛烷-3β-氧基原苯甲酸酯] (6)的合成
化合物2 (26 mg, 0.055 mmol)、4a (73 mg, 0.110 mmol)和4 Ả分子筛(260 mg)悬浮于无水CH2Cl2 (1 mL)中, 氩气保护下搅拌0.5 h后, 加入碳酸银(31 mg, 0.11 mmol)于10~15 ℃反应2 d.用硅藻土层过滤后浓缩, 经柱层析得到白色固体5a (19 mg, 0.018 mmol, 33%)和6 (25 mg, 0.024 mmol, 43%), 回收原料2 (2 mg, 0.0042 mmol, 8%).
化合物6: m.p. 111~123 ℃; [α]D17+0.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.09 (dd, J=8.4, 1.2 Hz, 2H), 7.94 (dd, J=8.4, 1.2 Hz, 2H), 7.88 (dd, J=8.4, 1.2 Hz, 2H), 7.77 (dd, J=7.6, 1.8 Hz, 2H), 7.61 (tt, J=7.4, 1.2 Hz, 1H), 7.56 (tt, J=7.4, 1.2 Hz, 1H), 7.50 (tt, J=7.4, 1.2 Hz, 1H), 7.45 (t, J=7.8 Hz, 2H), 7.42~7.37 (m, 5H), 7.29 (t, J=7.8 Hz, 2H), 6.02 (d, J=5.2 Hz, 1H), 5.71 (dd, J=3.0, 1.4 Hz, 1H), 5.68 (s, 1H), 5.40 (d, J=8.9 Hz, 1H), 4.78~4.75 (m, 1H), 4.45 (dd, J=12.1, 2.7 Hz, 1H), 4.30 (dd, J=12.1, 5.3 Hz, 1H), 3.97 (ddd, J=8.3, 5.2, 2.8 Hz, 1H), 3.86 (dd, J=10.8, 5.3 Hz, 1H), 3.47 (td, J=10.0, 4.7 Hz, 1H), 3.10 (dd, J=11.2, 4.4 Hz, 1H), 2.22 (td, J=10.6, 4.3 Hz, 1H), 2.09~1.26 (m, 17H), 1.26 (s, 3H), 1.22 (s, 3H), 1.15~1.09 (m, 1H), 1.09 (s, 3H), 1.06~0.98 (m, 1H), 0.97 (s, 3H), 0.94~0.88 (m, 1H), 0.86 (s, 3H), 0.84 (s, 3H), 0.79 (s, 3H), 0.75 (s, 3H), 0.67~0.62 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 165.9, 165.1, 164.5, 137.4, 133.6, 133.4, 133.0, 130.0 (2C), 129.9 (2C), 129.7 (2C), 129.6, 129.1, 129.0, 128.5 (2C), 128.4, 128.3 (2C), 128.2 (2C), 127.9 (2C), 126.2 (2C), 122.2, 97.4, 87.3, 87.1, 81.3, 72.6, 70.5, 70.0, 69.3, 68.3, 67.6, 64.0, 56.3, 52.1, 50.1, 48.8, 48.7, 39.6, 38.9, 38.7, 36.8, 34.7, 32.1, 31.6, 31.6, 28.8, 28.5, 28.1, 28.0, 25.0, 25.0, 24.2, 18.3, 17.7, 16.3, 16.2, 15.4. HRMS (ESI, positive) calcd for C64H79O13 [M+H]+1055.5515, found 1055.5525.
3.2.3 3β-O-(2', 3', 4', 6'-四-O-乙酰基-β-D-吡喃葡萄糖基)-(20S, 24S)-环氧达玛-12β, 25-二醇(5b)的合成
化合物2 (49 mg, 0.103 mmol)、4d (85 mg, 0.207 mmol)和4Ả分子筛(161 mg)悬浮于无水CH2Cl2 (1 mL)中, 氩气保护下搅拌0.5 h后, 加入碳酸银(283 mg, 1.03 mmol)于10~15 ℃反应2 d.加TMSOTf (3.7 μL, 0.0204 mmol)后结束反应, 用硅藻土层过滤后浓缩, 经柱层析得到白色固体5b (33 mg, 0.041 mmol, 40%, 回收产率57%), 回收原料2 (15 mg, 0.031 mmol, 30%), m.p. 76~92 ℃; [α]D19+6.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.21 (t, J=9.5 Hz, 1H), 5.04 (t, J=9.2 Hz, 2H), 4.53 (d, J=7.9 Hz, 1H), 4.27 (dd, J=12.1, 5.7 Hz, 1H), 4.09 (dd, J=12.1, 2.3 Hz, 1H), 3.88 (dd, J=10.8, 5.2 Hz, 1H), 3.69 (ddd, J=9.8, 5.6, 2.3 Hz, 1H), 3.52 (td, J=10.2, 4.5 Hz, 1H), 3.07 (dd, J=11.5, 4.6 Hz, 1H), 2.24 (ddd, J=13.0, 8.8, 3.6 Hz, 1H), 2.07 (s, 3H), 2.03 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H), 1.28 (s, 3H), 1.23 (s, 3H), 1.10 (s, 3H), 1.00 (s, 3H), 0.90 (s, 6H), 0.88 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3)δ: 170.6, 170.4, 169.4, 169.1, 102.9, 90.7, 87.3, 87.1, 72.8, 71.5, 71.5, 70.5, 70.0, 68.7, 62.2, 56.2, 52.1, 50.2, 48.8, 48.7, 39.7, 38.9, 38.8, 36.8, 34.7, 32.2, 31.6, 31.5, 28.8, 28.5, 28.0, 27.6, 25.8, 25.0, 24.2, 20.7, 20.7, 20.6, 20.6, 18.1, 17.7, 16.2, 16.0, 15.4. HRMS (ESI, positive) calcd for C44H71O13[M+H]+ 807.4889, found 807.4899.
3.2.5 3β-O-β-D-吡喃葡萄糖基-(20S, 24S)-环氧达玛-12β, 25-二醇(1)的合成
化合物5a (22 mg, 0.021 mmol)溶于甲醇(1 mL), 在冰水浴上加入甲醇钠(7 mg, 0.13 mmol), 反应2 h, 用阳离子交换树脂中和反应液、过滤、浓缩.经柱层析得到白色固体1 (13 mg, 0.020 mmol, 98%). m.p. 185~190 ℃; [α]D22+9.5 (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD)δ: 4.31 (d, J=7.8 Hz, 1H), 3.83 (dd, J=12.2, 2.0 Hz, 1H), 3.80 (dd, J=10.1, 4.6 Hz, 1H), 3.65 (dd, J=11.8, 5.1 Hz, 1H), 3.49 (td, J=10.4, 4.6 Hz, 1H), 3.32 (t, J=8.0 Hz, 1H), 3.27 (t, J=8.0 Hz, 1H), 3.25~3.21 (m, 1H), 3.20~3.15 (m, 2H), 2.22 (td, J=10.7, 3.9 Hz, 1H), 2.04~1.00 (m, 20H), 1.26 (s, 3H), 1.17 (s, 3H), 1.10 (s, 3H), 1.04 (s, 6H), 0.93 (s, 3H), 0.92 (s, 3H), 0.85 (s, 3H), 0.88~0.80 (m, 1H); 13C NMR (100 MHz, CD3OD) δ: 106.7, 90.6, 88.9, 88.5, 78.3, 77.7, 75.7, 72.1, 71.7, 71.3, 62.8, 57.6, 53.3, 51.6, 50.2, 49.8, 41.0, 40.4, 40.3, 38.0, 35.9, 33.4, 32.8, 32.7, 29.4, 29.1, 28.4, 27.2, 26.5, 26.2, 26.1, 19.3, 18.3, 17.0, 16.8, 15.9. HRMS (ESI, positive) calcd for C36H63O9 [M+H]+639.4467, found 639.4477.
辅助材料(Supporting Information)中间体及最终产物的氢谱和碳谱图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) Si, J. G. ; Tian, C. E. Lishizhen Med. Mater. Med. Res. 2016, 27, 1190 (in Chinese).
(司建功, 田长恩, 时珍国医国药, 2016, 27, 1190. )
(b) Yang, Z. Q. ; Zhao, T. T. ; Liu, H. L. ; Zhang L. D. Sci. Rep. 2016, 6, 19383.
(c) Li, B. H. ; Zhao, J. O. ; Wang, C. Z. ; Searle, J. ; He, T. C. ; Yuan, C. S. ; Du, W. Cancer Lett. 2011, 301, 185. -
[2]
Zhang, J. W.; Zhou, F.; Wu, X. L.; Zhang, X. X.; Chen, Y. C.; Zha, B. S.; Niu, F.; Lu, M.; Hao, G.; Sun, Y.; Sun, J. G.; Peng, Y.; Wang, G. J. Br. J. Pharmacol. 2012, 165, 120. doi: 10.1111/j.1476-5381.2011.01505.x
-
[3]
杨洁, 博士论文, 吉林大学, 长春, 2016.Yang, J. Ph.D. Dissertation, Jilin University, Changchun, 2016 (in Chinese)
-
[4]
Li, L.; Chen, X. Y.; Zhou, J. L.; Zhong, D. F. Drug Metab. Dispos. 2012, 40, 2041. doi: 10.1124/dmd.112.046995
-
[5]
Yang, J.; Li, X. W.; Sun, T.; Gao, Y.; Chen, Y. X.; Jin, Y. R.; Li, Y. Steroids 2016, 106, 26. doi: 10.1016/j.steroids.2015.12.005
-
[6]
刘金平, 博士论文, 沈阳药科大学, 沈阳, 2005.Liu, J. Ph.D. Dissertation, Shenyang Pharmaceutical University, Shenyang, 2016 (in Chinese).
-
[7]
Shen, R. Z.; Xin, C.; Laval, S.; Sun, J. S.; Yu, B. J. Org. Chem. 2016, 81, 10279. doi: 10.1021/acs.joc.6b01265
-
[8]
杨宁, 杨世林, 赵余庆, 现代药物与临床, 2014, 29, 574.Yang, N.; Yang, S. L.; Zhao, Y. Q. Drugs Clinic 2014, 29, 574 (in Chinese)
-
[9]
Liao, J. X.; Sun, J. S.; Niu, Y. M.; Yu, B. Tetrahedron Lett. 2011, 52, 3075. doi: 10.1016/j.tetlet.2011.04.003
-
[10]
(a) Atopkina, L. N. Uvarova, N. I. Chem. Nat. Compd. 1981, 17, 254.
(b) Samoshina, N. F.; Novikov, V. L.; Denisenko, V. A.; Uvarova, N. I. Chem. Nat. Compd. 1983, 19, 299.
(c) Atopkina, L. N.; Novikov, V. L.; Denisenko, V. A.; Uvarova, N. I. Chem. Nat. Compd. 1985, 21, 674.
(d) Atopkina, L. N.; Uvarova, N. I. Chem. Nat. Compd. 1986, 22, 421.
(e) Atopkina, L. N.; Samoshina, N. F.; Denisenko, V. A.; Pokhilo, N. D.; Uvarova, N. I. Chem. Nat. Compd. 1986, 22, 415. -
[11]
(a) Yosioka, I.; Yamauchi, H.; Kitagawa, I. Chem. Pharm. Bull. 1972, 20, 502.
(b) Appendino, G.; Gariboldi, P.; Wollenweber, E. Sironi, A.; Molinari, H. Phytochemistry 1992, 31, 923.
(c) Bi, Y.; Tian, J. W.; Wang, L.; Zhao, F. L.; Zhang, J. F.; Wang, N.; Sun, H. J.; Meng, Q. G. J. Med. Plant. Res. 2011, 5, 2424.
(d) Li, L.; Chen, X. Y.; Li, D.; Zhong, D. F. Drug Metab. Dispos. 2011, 39, 472. -
[12]
任媛媛, 硕士论文, 吉林大学, 长春, 2012.Ren Y. Y. M.S. Thesis, Jilin University, Changchun, 2012 (in Chinese).
-
[13]
Meng, Q. G.; Tan, W. J.; Hou, G. G.; Zhang, X. Y.; Hu, X. Y.; Yang, F.; Bai, G. J.; Zhu, W. W.; Cai, Y.; Bi, Y. J. Mol. Struct. 2013, 1054–1055, 1.
-
[14]
Ueda, M.; Yang, G. Q.; Ishimaru, Y.; Itabashi, T.; Tamura, S.; Kiyota, H.; Kuwahara, S.; Inomata, S.; Shoji, M.; Sugai, T. Bioorg. Med. Chem. 2012, 20, 5832. doi: 10.1016/j.bmc.2012.08.003
-
[15]
Li, Y.; Yang, X. Y.; Liu, Y. P.; Zhun, C. S.; Yang, Y.; Yu, B. Chem.-Eur. J. 2010, 16, 1871. doi: 10.1002/chem.v16:6
-
[1]
-

图式1 已报道的拟人参皂苷HQ合成方法
Scheme 1 Reported approaches for the synthesis of pseudoginsenoside HQ

图式3 奥克梯隆型皂苷元2的C-3位选择性糖苷化机理
Scheme 3 Plausible mechanism for selective glycosylation at C-3 of ocotillol type sapogenins 2
-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 1685
- HTML全文浏览量: 248
 下载:
下载:
