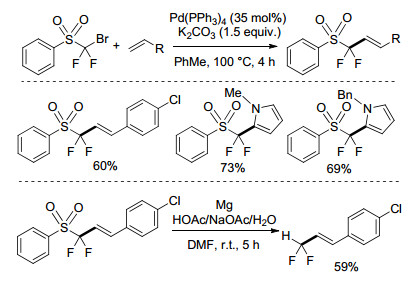
 图式 1
钯促进的(溴二氟甲基)-磺酰基苯的Heck反应
Scheme1.
Palladium-promoted Heck-type reactions of [(bromodifluoromethyl)-sulfonyl]benzene
图式 1
钯促进的(溴二氟甲基)-磺酰基苯的Heck反应
Scheme1.
Palladium-promoted Heck-type reactions of [(bromodifluoromethyl)-sulfonyl]benzene
引用本文:
周文俊, 张逸寒, 曹光梅, 刘惠东, 余达刚. 钯催化卤代烷烃参与的自由基型转化反应[J]. 有机化学,
2017, 37(6): 1322-1337.
doi:
10.6023/cjoc201702051

Citation: Zhou Wen-Jun, Zhang Yihan, Cao Guangmei, Liu Huidong, Yu Da-Gang. Palladium-Catalyzed Radical-Type Transformations of Alkyl Halides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1322-1337. doi: 10.6023/cjoc201702051
Citation: Zhou Wen-Jun, Zhang Yihan, Cao Guangmei, Liu Huidong, Yu Da-Gang. Palladium-Catalyzed Radical-Type Transformations of Alkyl Halides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1322-1337. doi: 10.6023/cjoc201702051
钯催化卤代烷烃参与的自由基型转化反应
English
Palladium-Catalyzed Radical-Type Transformations of Alkyl Halides
Abstract:
Palladium-catalyzed cross-coupling reactions have been developed for decades as useful methods in organic synthesis. Compared to aryl and alkenyl halides, alkyl halides are more challenging to be applied in cross-coupling reactions. This mainly arises from the difficulty in oxidative addition of alkyl halides to palladium catalyst, sluggish reductive elimination and competitive side reactions, such as β-H elimination and protonation, of the resulting alkylpalladium intermediates. These challenges have partly been overcome with the significant development of novel palladium catalysis involving single election transfer. A variety of cross couplings of alkyl halides have been developed. In this review the recent palladium-catalyzed radical alkylation using alkyl halides with the order of different types of coupling partners is summarized.
-
Key words:
- palladium catalysis
- / alkyl halide
- / free radical
-
过渡金属催化的反应之中, 卤代烷烃参与的偶联反应是一个具有挑战性的课题.这是因为与芳基或烯基卤代物相比, 卤代烷烃参与的偶联反应具有众多的挑战: (1) C(sp3)—X比C(sp2)—X键更富电子, 更难发生氧化加成, 特别是二级、三级烷基卤代物; (2) 发生氧化加成后生成的烷基金属物种由于缺少π电子对中心金属的稳定作用, 反应活性更高, 很容易发生质子化或β-H消除等副反应; (3) 涉及sp3碳的还原消除困难, 难以得到预期的目标产物.由于卤代烷烃是重要的有机合成中间体, 其种类繁多, 数量庞大, 结构多样, 化学家们一直致力于该领域的研究.自Tamura, Suzuki和Knochel等[1]的开创性工作以来, 越来越多的体系被成功用于卤代烷烃参与的偶联反应[2].在这些体系中, 金属钯构建的体系发挥着重要作用[3].一般认为卤代烷烃参与的偶联反应有两种启动模式: (1) 双电子氧化加成.在这种模式中, 烷基C(sp3)—X键一般只能提供能量较高的σ*轨道(活化的卤代烷烃除外)与低价钯物种作用, 能垒较高, 氧化加成活性低, 由于空间位阻原因, 二级、三级卤代烷烃更难发生氧化加成. (2) 单电子转移(SET).单电子转移过程也是卤代烷烃常见的催化循环启动模式.在此模式中, 低价钯物种首先与烷基C(sp3)—X键发生单电子转移, 形成卤素负离子的同时生成烷基自由基.由于烷基C(sp3)不直接与金属成键, 这一过程相对比较容易.近年来, 越来越多的报道证明卤代烷烃参与的化学转化经历自由基历程, 通过该过程能够高效地构建碳碳键和碳杂原子键.本文按照反应底物的种类对近年来钯催化卤代烷烃参与的自由基型化学转化进行综述, 主要包括与烯烃、炔烃、芳烃、一氧化碳(CO)、腙以及胺等的反应.
1 与烯烃的反应
1.1 分子间的Heck反应
钯催化的Heck反应能够实现广谱性的烯烃与芳基或烯基卤代物的偶联, 但将Heck反应拓展到简单烷基卤代物具有一定的挑战.早在1986年, Ishihara课题组[4]报道了钯催化的全氟碘代烷烃与碳碳不饱和键的烷基化加成反应.文中提到反应涉及自由基过程.随后, Curran课题组[5]报道了零价钯促进的不饱和α-碘代羰基化合物的环化反应, 文中提出反应经历了“原子转移(Atom Transfer, AT)”过程, 其中金属络合物起到引发自由基链式反应的作用, 而非传统金属催化剂作用.该反应中Pd(PPh3)4, (Bu3Sn)2均可以有效促进反应的进行.
1992年, 胡宏纹课题组[6]报道了钯催化苄基卤代物的烯基化反应(Eq. 1).反应以Pd(OAc)2为催化剂, 而三苯基膦(PPh3)以及联吡啶(bpy)等配体的加入会明显抑制反应.虽然该反应仅对带有吸电子基团的活化烯烃才具有较高的反应活性, 但能够将烷基氯代物成功应用到Heck反应之中, 在20世纪90年代是非常重要的突破.
Reutrakul课题组[7]于2012年报道了钯促进的(溴二氟甲基)-磺酰基苯的Heck型反应(Scheme 1).通过Pd(PPh3)4催化, (溴二氟甲基)-磺酰基苯与苯乙烯类底物或杂芳环化合物在100 ℃的甲苯溶剂中进行偶联, 高效地实现α-烯基和杂芳环取代的苯磺酰二氟甲基化, 所得到的含PhSO2CF2结构的产物也可转化为具有生物活性的二氟甲基取代的烯烃.
图式 1
钯促进的(溴二氟甲基)-磺酰基苯的Heck反应
Scheme1.
Palladium-promoted Heck-type reactions of [(bromodifluoromethyl)-sulfonyl]benzene
2014年, Zhou课题组[8]以Pd(0)(dppf)为催化剂, Cy2NMe为碱, 在110 ℃的三氟甲苯中实现了非活化烷基卤代物与简单烯烃的分子间Heck反应(Scheme 2).经过研究, 作者发现添加LiI可提高溴代物的转换产率.对于大多数的一级烷基卤代物, Pd(dba)2搭配dppf能有效抑制烷基卤代物消除等副反应的发生[9].烷基氯代物(RCl)也能很好地参与反应, 作者推断是RCl与LiI原位生成的RI为活性的偶联试剂.该反应具有较好的底物兼容性, 容易衍生化, 而且能实现克级反应.机理研究中发现当疑似中间体C加到CyI与苯乙烯的Heck反应中, C被完全消耗, 但是只得到41%的消除产物(Scheme 2).主要的副反应为苄基自由基的自偶联.因此, C干涉了CyI参与的Heck反应, 使其产率从>70%减少到13% (Scheme 2), 由此作者排除了原子转移自由基加成形成苄基碘代物的可能(Scheme 2), 并提出了反应可能的机理: (dppf)Pd(0) 与烷基卤代物通过单电子转移历程形成的烷基自由基, 进而与苯乙烯发生自由基加成, 形成的苄基自由基物种和Pd(Ⅰ)重组形成烷基钯物种(dppf)XPd(Ⅱ), 再经β-H消除得到产物(Scheme 2, path Ⅰ).
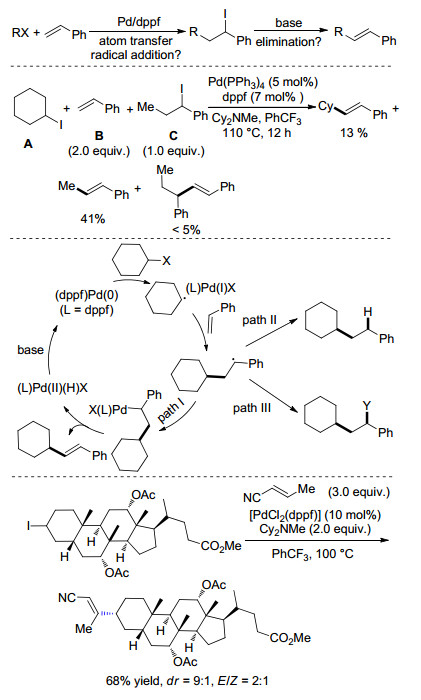 图式 2
钯催化非活化烷基碘代物的Heck-型偶联反应
Scheme2.
Palladium-catalyzed Heck-type cross-couplings of unactivated alkyl iodides
图式 2
钯催化非活化烷基碘代物的Heck-型偶联反应
Scheme2.
Palladium-catalyzed Heck-type cross-couplings of unactivated alkyl iodides
通过反应条件控制, 由烷基自由基与烯烃加成形成的自由基物种还可以实现别的转化, 如还原(Scheme 2, path Ⅱ), 或者可以被别的基团捕获生成相应的产物(Scheme 2, path Ⅲ), 各种反应途径为实现该类反应的多样性提供了可能.
几乎同时, Alexanian课题组[10]报道了类似的工作.该反应以PdCl2(dppf)为催化剂, 以K3PO4或Cy2NMe为碱, 在三氟甲苯中100 ℃的反应条件下, 不同电性的烯烃都可以参与反应, 作者还将该反应应用到了天然产物修饰上, 高立体选择性地得到相应的产物(Scheme 2).
1.2 分子内的Heck反应
一般而言, 分子内反应比分子间反应速度要快得多, 同时由于分子内反应在合成环状化合物方面的优势而得到化学家们的青睐.通过烷基卤代物参与的分子内Heck反应也能实现环状化合物的合成.
Alexanian课题组[11]于2011年报道了钯催化的烷基碘代物参与的自由基型5-exo环化Heck反应(Scheme 3).反应以Pd(PPh3)4为催化剂, 1, 2, 2, 6, 6-五甲基哌啶(PMP)为碱, 在110 ℃, 1.01 MPa CO氛围下进行.该反应底物适用性较广, 以中等到较高产率得到Heck类型的五元环产物.作者发现, 对于一级烷烃碘代物若将反应中的CO氛围换成Ar氛围, 会有副产物产生.对于二级碘代物, CO的影响则不显著.尽管CO的作用还不是很清楚, 但作者推断在高压的CO中生成的Pd(PPh3)x(CO)y物种是促进金属-自由基过程的活性物种[12].
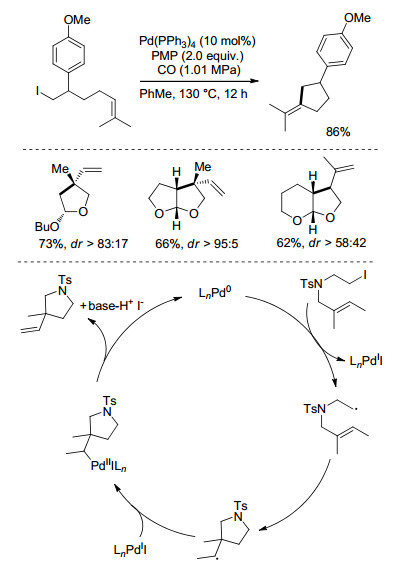 图式 3
钯催化的分子内环化反应
Scheme3.
Palladium-catalyzed 5-exo cyclization
图式 3
钯催化的分子内环化反应
Scheme3.
Palladium-catalyzed 5-exo cyclization
2016年, 刘会和刘青课题组[13]通过巧妙的底物设计, 利用芳基的位阻效应, 成功实现了第一例钯催化非活化的烷基卤代物选择性的6-endo烷基化Heck反应(Scheme 4).该反应以PdCl2作催化剂, Cy2NMe作碱, 于110 ℃的甲苯溶液中高选择性地得到了5-芳基-1, 2, 3, 6-四氢吡啶类衍生物.反应底物适用范围广, 芳环上带有给电子基或吸电子基官能团的底物都有着较高的收率.
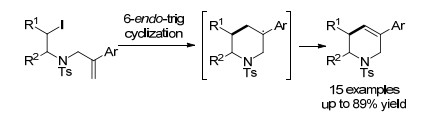 图式 4
钯催化的6-endo烷基化反应
Scheme4.
Palladium-catalyzed 6-endo cyclization
图式 4
钯催化的6-endo烷基化反应
Scheme4.
Palladium-catalyzed 6-endo cyclization
1.3 还原Heck反应
2016年初, Ryu课题组[14]报道了在钯-光催化体系下的活化烯烃的加氢烷基化的Giese型反应(Scheme 6).该反应在氙灯保护箱中, 以PdCl2作催化剂, Et3N作碱, Hantzsch酯为氢源, 在80 ℃苯中反应16 h即可实现烯烃的加氢烷基化.该方法无需使用有毒的氢化锡, 在高效反应的同时实现反应的绿色化.另外, 该反应实现了C(sp3)—I与C(sp2)—X (X=Br or I)之间的化学选择性, 为一锅法实现对两类C—X键的分别官能团化提供了有效途径.作者提出了反应的可能机理:首先在钯-光作用下发生SET过程产生烷基自由基, 进而与烯烃加成另一个烷基自由基, 由于体系中没有强碱的存在, 该自由基不经β-H消除, 而是从Hantzsch酯的获得一个氢原子得到还原的目标产物(Scheme 5).
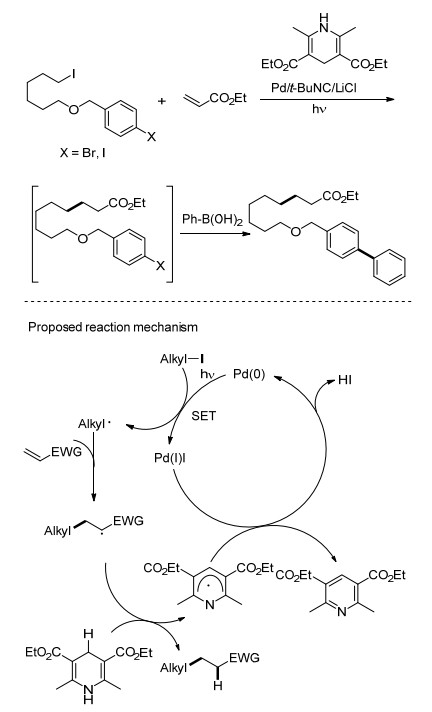 图式 5
钯/光体系下的烯烃加氢烷基化反应
Scheme5.
Hydroalkylation of alkenes under Pd/light system
图式 5
钯/光体系下的烯烃加氢烷基化反应
Scheme5.
Hydroalkylation of alkenes under Pd/light system
1.4 烯烃双官能团化反应
除了以上转化之外, 烷基卤代物与烯烃加成形成的烷基自由基中间体还可以参与其他化学键构建(如碳卤键和碳碳键), 实现烯烃的双官能团化反应. 2012年, 姜雪峰课题组[15]发展了Pd/dppf催化非活化烷基碘代物参与的原子转移自由基环化反应(atom transfer radical cyclization, ATRC) (Scheme 6).该反应在130 ℃下反应24 h, 得到了一系列五元环化产物.作者通过机理研究证明该过程涉及烷基自由基过程, 并认为在该无碱体系中, 烷基自由基和双键反应新形成的碳自由基很有可能直接与Pd(Ⅰ)I发生原子转移实现碳碘键的构建.
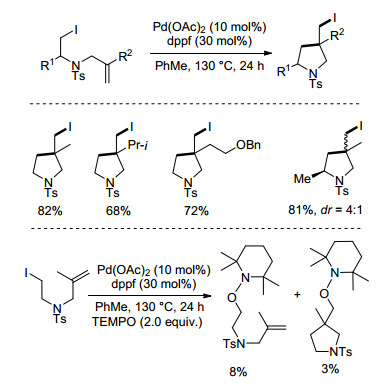 图式 6
钯催化烷基碘代物的原子转移自由基环化反应
Scheme6.
Palladium-catalyzed atom transfer radical cyclization of alkyl iodide
图式 6
钯催化烷基碘代物的原子转移自由基环化反应
Scheme6.
Palladium-catalyzed atom transfer radical cyclization of alkyl iodide
2014年, 李金恒课题组[16]报道了一种新型钯催化的烯烃双官能团化反应(Scheme 7).该反应以PdCl2(MeCN)2作催化剂, dppe作配体, Ag2CO3作氧化剂, 于100 ℃甲苯中反应12 h得到了系列二氢吲哚-2-酮类化合物.此方法可应用于不同类型的N-芳基烯烃以及一级、二级、三级、α-羰基烷基溴代物.作者提出了可能的反应路径:中间体A加成到烯烃碳碳双键上, 产生自由基B, 接着B环化形成自由基中间体C, C再被Pd(Ⅱ)Ln物种氧化得到阳离子中间体D, 最后经脱质子化形成产物.此外不能排除Pd(Ⅱ)/Pd(0) 的催化历程.
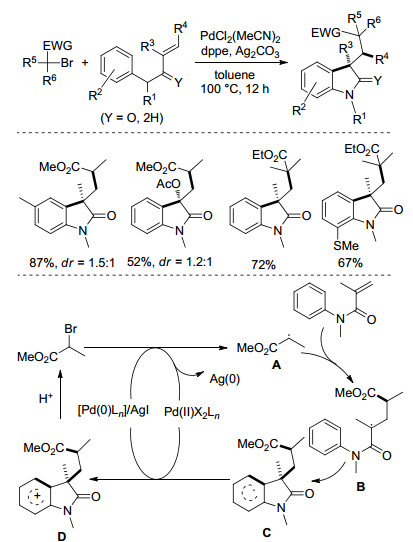 图式 7
自由基型烯烃双官能团化反应
Scheme7.
Radical alkene difunctionalizations
图式 7
自由基型烯烃双官能团化反应
Scheme7.
Radical alkene difunctionalizations
2015年, 童晓峰课题组[17]报道了钯催化N-烯丙基-α-氯代物的原子转移自由基环化反应(Eq. 2), 高选择性地合成了一系列γ-内酰胺衍生物.该反应利用Pd(cod)-Cl2作为催化剂前体, IMes为配体, NaI为碘源, 在135 ℃的邻二甲苯中下高效地实现了N-烯丙基-α-氯代物的环化反应.
2016年初, 段新华课题组[18]实现了钯催化非活化烷基卤代物与丙烯酰胺与的串联反应(Eq. 3).反应以PdCl2作催化剂, dppf为配体, K3PO4·3H2O为碱, 在100 ℃的二乙二醇二甲醚中, 芳基丙烯酰胺能顺利地与烷基卤化物发生反应, 高效地实现二氢吲哚-2-酮类化合物的合成.该反应无需加入氧化剂, 底物适应性广, 官能团兼容性好.值得注意的是, 当使用二级卤代烷烃进行环化反应时, 必须在反应中添加NaI才能得到相应产物.作者推断, 较为活泼的二级溴代烷烃在反应中可能涉及了一个溴碘交换的过程. 2, 2, 6, 6-四甲基哌啶氧化物(TEMPO)和2, 6-二叔丁基-4-甲基苯酚(BHT)抑制实验表明该反应可能涉及自由基过程.此外, 动力学同位素效应(KIE)实验结果表明碳氢键的活化不是反应的决速步, 由此作者认为反应经历了一个自由基过程.
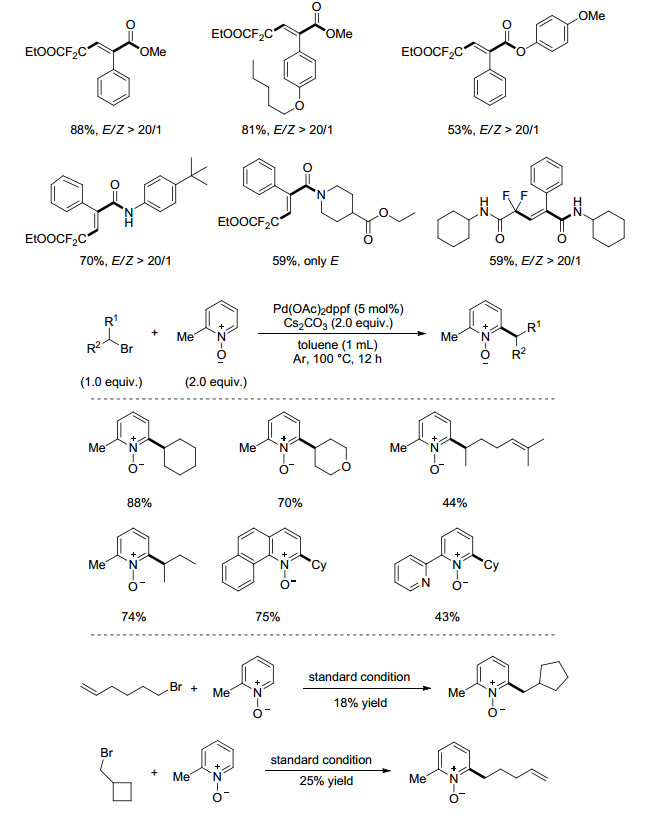
作为药物分子中常见的结构, 含氟官能团的引入方法一向引人注目.二氟烷基溴代物作为一种常见的二氟甲基化试剂, 被广泛应用于自由基化学中[16]. 2016年, 夏晓峰课题组[19]利用钯催化丙烯酰胺与二氟溴代乙酸乙酯的自由基型二氟烷基化/环化反应(Eq. 4), 合成了一类含氟的异喹啉二酮骨架分子.全氟代烷基碘化合物也能在该体系下进行反应.
2 与炔烃的反应
相对于烯烃而言, 由于炔烃中sp碳原子的电负性比烯烃sp2碳原子的电负性强, 使得π电子云不易极化, 因此自由基对炔烃亲电加成条件较为苛刻. 1986年, Ishihara课题组[4]曾报道, 在Pd(PPh3)4为催化剂, 正己烷为溶剂, 炔可以与全氟烷基碘代物在60~67 ℃反应生成烯基碘代物.通过1, 4-二硝基苯作为自由基抑制剂的实验证明该反应可能涉及自由基机理.
2013年, Cook课题组[20]报道钯催化下炔与非活化的烷基碘代物的插入、还原反应(Scheme 8), 以此来合成一系列三取代的烯烃.该反应具有反应条件温和、官能团容忍性好等特点.初步的机理探究过程中, 作者发现底物为2-碘代乙基二苯乙炔, 其产物是以E/Z比为1:3的异构体存在, 作者推测该反应可能经历烯基钯异构化过程, 但膦配体的位阻和电性对该反应E/Z比值没有明显影响.随后作者推测该反应可能经历了烯基自由基异构化过程, 自由基控制实验中, TEMPO抑制剂对反应有一定的抑制作用, BHT对产率没有影响等实验结果.综上, 作者提出该反应可能经历一个杂化的有机钯物种和烯基自由基过程.
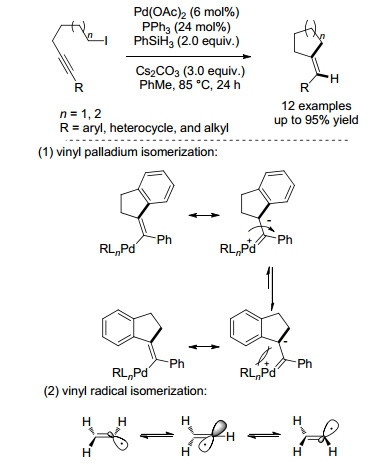 图式 8
钯催化下炔的插入/还原反应和可能的机理
Scheme8.
Palladium-catalyzed alkyne insertion/reduction reactions and proposed mechanism
图式 8
钯催化下炔的插入/还原反应和可能的机理
Scheme8.
Palladium-catalyzed alkyne insertion/reduction reactions and proposed mechanism
随后, 该课题组[21]报道了β-H存在下钯催化分子内碘原子迁移反应(Scheme 9).该反应以Pd(PPh3)4为催化剂, Cs2CO3为碱, 甲苯为溶剂, 50 ℃条件下, 一系列非环状的二级烷基碘代物能以较高的收率转变为含碘甲基的二奎烷类化合物.反应具有原子经济性高、官能团兼容性好、产物易于衍生化等特点.作者提出的可能反应机理为:碘代物在零价钯作用下生成烷基自由基, 同时产生的Pd(Ⅰ)与端烯发生配位作用, 随后与叁键发生分子内自由基加成反应得到烯基自由基, 再与Pd(Ⅰ)-烯烃络合物反应得到目标产物.
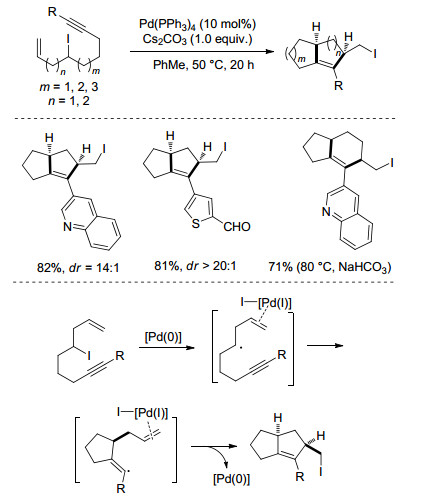 图式 9
β-H存在下钯催化分子内碘原子迁移反应
Scheme9.
Palladium-catalyzed intramolecular iodine-transfer reactions in the presence of β-hydrogen atoms
图式 9
β-H存在下钯催化分子内碘原子迁移反应
Scheme9.
Palladium-catalyzed intramolecular iodine-transfer reactions in the presence of β-hydrogen atoms
2015年, Nevado课题组[22]报道了钯催化下端炔双官能团化反应(Scheme 10).在PdCl2(PPh3)2催化下, K2CO3为碱, 二氯甲烷和水作为溶剂, 芳基或烯基硼酸和全氟碘代物同时与端炔进行反应, 以较高区域选择性及立体选择性合成系列三取代的烯烃化合物.作者通过控制实验提出可能的反应机理:二价钯在硼酸的作用下原位生成零价钯, 进而与全氟碘代烷发生单电子转移形成具有亲电性的全氟烷基自由基, 该自由基与炔烃反应形成烯基自由基, 再经过单电子转移(path Ⅰ)或与硼酸的转金属化(path Ⅱ)形成烯基钯物种, 最后经过还原消除得到目标产物.
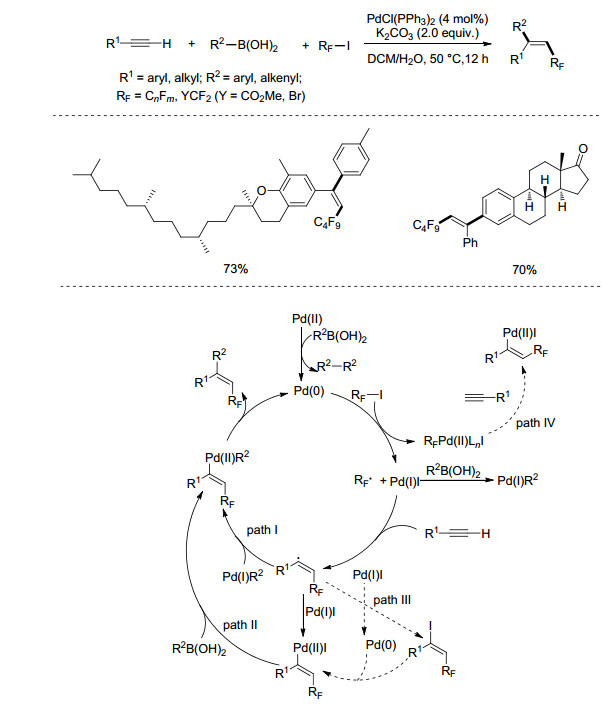 图式 10
钯催化下端炔、全氟碘代烷烃和硼酸三组分反应
Scheme10.
Palladium-catalyzed three-component reactions of terminal alkynes with iodoperfluoroalkanes and boronic acids
图式 10
钯催化下端炔、全氟碘代烷烃和硼酸三组分反应
Scheme10.
Palladium-catalyzed three-component reactions of terminal alkynes with iodoperfluoroalkanes and boronic acids
与此同时, 梁永民课题组[23]报道了类似的三组分反应(Eq. 5).在氩气氛围中, 80 ℃条件下, 以Pd(PPh3)4为催化剂, 通过一锅法高效地实现烯基双官能团化产物的合成.在该体系中烷基炔也能够参与反应, 具有较广的底物范围.基于控制实验及已有的文献报道, 作者认为该反应经历二氟烷基自由基加成机理, 与上述报道path Ⅱ相同.
2016年, Chaładaj课题组[24]报道了一种钯络合物催化下炔烃的烷基化反应(Scheme 11).一系列炔烃(端位和非端位)、芳基硼酸、全氟碘代物通过一锅法反应, 以较高的收率、高区域选择性、高立体选择性得到三取代或四取代的烯烃.作者推测可能的反应机理包含两个独立的催化循环, 分别是自由基型Pd(0)/Pd(Ⅰ)和典型的Suzuki偶联机理Pd(0)/Pd(Ⅱ), 其间以零价钯和烯基碘代物作为桥梁.
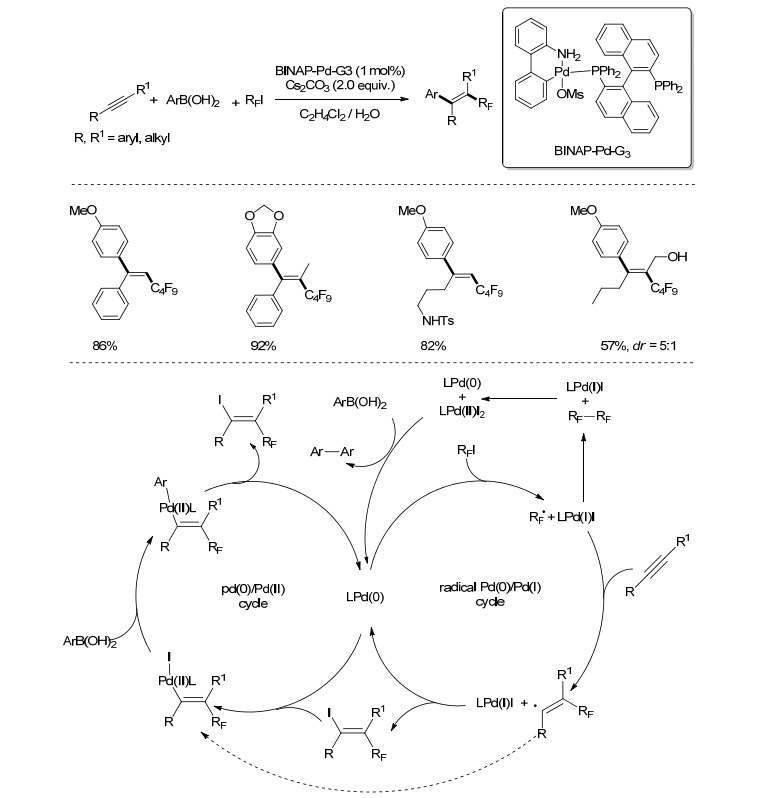 图式 11
钯催化下炔的全氟烷基化
Scheme11.
Palladium-catalyzed carboperfluoroalkylation of alkynes
图式 11
钯催化下炔的全氟烷基化
Scheme11.
Palladium-catalyzed carboperfluoroalkylation of alkynes
多组分反应是绿色化学的重点之一, 因为其较高的原子经济性和步骤经济性而备受关注. 2016年, 梁永民课题组[25]基于之前钯催化下端炔的芳基化和多氟烷基化的研究, 发展了钯催化下端炔的二氟烷基化和羰基化反应(四组分反应)(Eq. 6).反应以PdCl2(PPh3)2为催化剂, DPEphos为配体, 碳酸钾为碱, 一系列端炔、二氟碘代乙酸乙酯, 醇/胺与一氧化碳(101 kPa)在一锅法条件下, 高区域选择性地合成了β-二氟烷基不饱和酯/酰胺.通过系列控制实验, 作者认为二氟烷基自由基的形成对该反应至关重要.
3 与芳烃的反应
相对于烯烃和炔烃, 芳烃的自由基取代或者加成反应由于可能涉及到去芳构化/芳构化的过程而显得极具挑战性. 2013年, 傅尧课题组[26]实现了一系列非活化二级溴代烷烃参与钯催化的吡啶氮氧化物邻位烷基化反应(Scheme 12).在最优条件下, 几乎所有底物均有较好的收率, 并且可兼容多种官能团.吡啶氮氧化衍生物表现出很好的区域选择性, 只在邻位发生烷基化. 6-溴-1-己烯和溴甲基环丁烷的自由基验证实验说明反应可能经历了自由基历程.尽管作者没有提出明确的反应机理, 但是在芳烃直接烷基化领域向前迈出了一大步.
 图式 12
钯催化的吡啶N-氧化物与二级烷基溴的偶
Scheme12.
Palladium-catalyzed C—H activation/cross-coupling of pyridine N-oxides with secondary alkyl bromides
图式 12
钯催化的吡啶N-氧化物与二级烷基溴的偶
Scheme12.
Palladium-catalyzed C—H activation/cross-coupling of pyridine N-oxides with secondary alkyl bromides
Zhou课题组[27]发展了一种钯催化的杂芳烃与二、三级卤代烃的区域选择性烷基化方法(Scheme 13).在Pd(PPh3)4为催化剂、dppp为配体、Cs2CO3为碱的条件下, 1, 3-苯并噁唑、咪唑、吡咯、呋喃、噻吩等杂环化合物可以实现与二级、三级碘代烷烃的烷基化反应.添加NaI后, 溴代烷烃在上述条件下也可以很好的反应, 同时在体系中检测到了少量的碘代烷烃, 说明参与反应的可能是碘代烷烃.值得一提的是, 该体系不仅适用于一级、二级烷基卤代物, 就连位阻更大的三级烷基卤代物也能参与转化.综合理论计算与实验结果, 零价钯首先与烷基卤代烃发生SET过程, 生成Pd(Ⅰ)和烷基自由基, 然后自由基与杂芳环发生自由基加成, 随后芳基自由基物种与[(dppp)Pd(Ⅰ)X]发生单电子转移, 再经去质子化生成最终产物, 零价钯参与下一次的催化循环.此外, 张扬会课题组[28]在2015年也报道了类似的钯催化富电子杂芳烃的二氟甲基化反应.
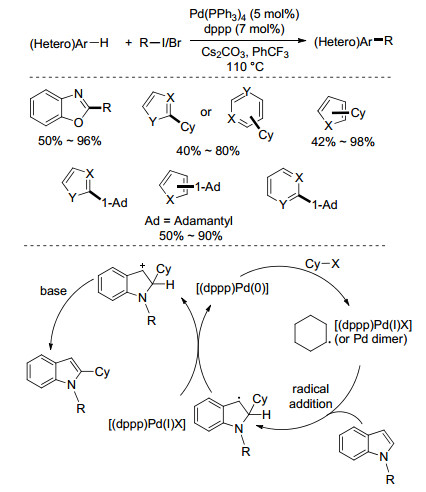 图式 13
杂芳烃与烷基卤代物的偶联
Scheme13.
Couplings of heteroarene and alkyl halides
图式 13
杂芳烃与烷基卤代物的偶联
Scheme13.
Couplings of heteroarene and alkyl halides
Alexanian课题组[29]在2015年发展了一种钯催化非活化烷基卤代烃与芳烃的分子内环化反应(Scheme 14).以Pd(PPh3)4为催化剂, PMP为碱, 苯基叔丁烷为溶剂, 130 ℃反应48 h(以碘代烷烃为底物, 反应条件更为温和), 高产率地得到一系列四氢萘、二氢茚、氮取代的吲哚啉、喹啉以及异喹啉等环化产物.作者给出了可能的反应机理, 零价钯与卤代烷烃先通过SET过程生成Pd(Ⅰ)物种和碳自由基中间体, 进而通过自由基加成得到环己二烯自由基中间体, 再通过单电子氧化和去质子化得最终产物.邻位取代的芳基底物在生成产物后, 可能会发生1, 2-烷基迁移生成环己二烯阳离子, 再芳构化形成重排产物.
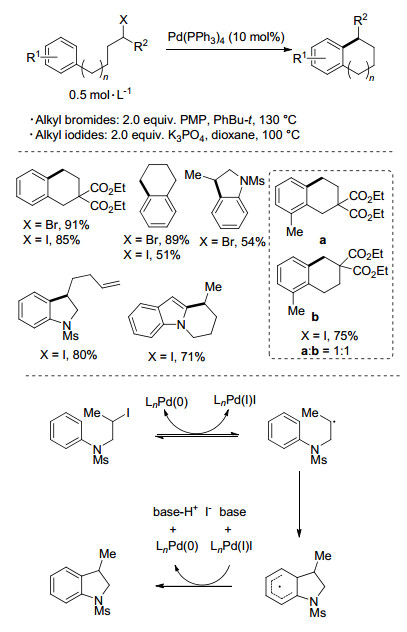 图式 14
非活化烷基卤代烃与芳烃的环化反应
Scheme14.
Synthesis of diverse cyclic heterocycles using the catalytic C—H alkylati
图式 14
非活化烷基卤代烃与芳烃的环化反应
Scheme14.
Synthesis of diverse cyclic heterocycles using the catalytic C—H alkylati
2016年, 王官武课题组[30]通过钌钯共催化和邻位金属化的策略, 实现了芳基间位的二氟甲基化(Eq. 7).研究发现, 以[RuCl2(p-cymene)]2和Pd(PPh3)4作共催化剂, Na2CO3作碱, Ba(OAc)2作添加剂, 以中高产率得到一系列二氟甲基化、单氟甲基化或者非氟代甲基化产物.本篇工作拓宽了近年来氟甲基化和钌催化的间位C—H键活化的范围.通过导向基团与钌催化剂形成环金属物种, 从而增强了芳环间位的电子云密度, 进而与亲电性的二氟甲基自由基反应(由二氟甲基化试剂与钯催化剂通过单电子转移生成), 再经单电子氧化和去质子化等步骤得到目标产物(Scheme 15).
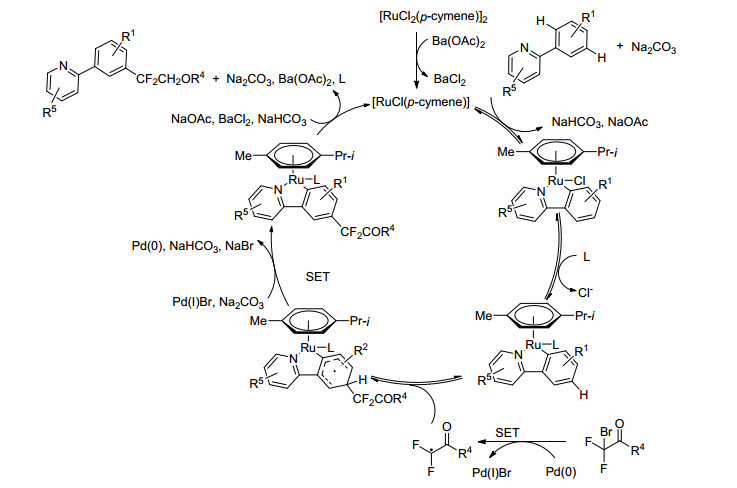 图式 15
钌钯共催化芳基二氟甲基化及其机理
Scheme15.
Pd and Ru co-catalyzed meta-difluoromethylation of arenes and plausible mechanism
图式 15
钌钯共催化芳基二氟甲基化及其机理
Scheme15.
Pd and Ru co-catalyzed meta-difluoromethylation of arenes and plausible mechanism
4 与CO的反应
近年来出现了许多CO参与的自由基型反应, 同烷基自由基与不饱和烃的反应类似, 烷基自由基同样来源于烷基卤代物与0价钯的原子转移反应, 同时产生Pd(Ⅰ)物种, 启动自由基反应[31].不同之处在于, 生成的烷基自由基在CO的存在下优先与之加成生成较稳定的酰基自由基, 酰基自由基物种随后被Pd(Ⅰ)捕获发生后续反应.这种类型的反应被称为原子转移羰基化(ATC)反应[2d].活性自由基物种产生后, 其往往会与体系中的其他活性物质发生复杂而精美的连锁反应, 因此这种类型的反应又被称为多米诺反应[2e].
早在1985年, Tsuji课题组[32]就发展了一种钯催化CO参与的自由基型反应(Eq. 8).反应在4.04 MPa CO压力下, 使用EtOH为溶剂和反应物, 以Pd(OAc)2为催化剂, PPh3为配体, K2CO3为碱.在这一反应中, CCl4或CCl3Br作为卤代烷烃, 与0价钯发生单电子转移反应, 生成烷基自由基, 随后与端位烯烃、CO反应, 再经醇解生成相应的目标产物.尽管烷基自由基与烯烃加成后直接猝灭的产物与羰基化产物在体系中共存, 且反应时间长, 底物范围窄, 但这一反应开创了原子转移羰基化反应的先河.
随后, Fuchikami课题组[33]发展了全氟代烷基碘代物的羰基化、双羰化反应(Eq. 9).反应用胺类作为亲核试剂, 使用5.05 MPa的CO, 在PdCl2(PPh3)2的催化下实现.该反应的产物可方便地转化为α-氨基酸.然而, 反应的选择性差依然是其一大缺陷.
1991年, Suzuki及其合作者[34]首次将钯催化原子转移羰基化反应与Suzuki反应结合, 发展了一种新型的酮类化合物合成方法(Eq. 10).反应以Pd(PPh3)4作为催化剂, K3PO4为碱, 在101 kPa CO以及光照下便可在室温实现烷基碘代物、CO和9-烷基-9-BBN的温和偶联, 具有较好的官能团容忍性, 生物活性分子也可以得到兼容.
在此之后, Miyaura等[35]进一步发展了Suzuki的酮类合成策略, 使用碘代烯烃与CO及9-烷基-9-BBN反应, 历经环化与羰基化过程生成酮类偶联产物(Eq. 11).这种温和的反应条件同样造就了其良好的底物容忍性, 各类底物反应顺利, 产物收率较高.
2002年, 原子转移羰基化反应这一概念被Ryu与Komatsu等[36]所提出, 同时, 一种钯催化, 光促进的连锁自由基多羰基化反应模式得到发展(Eq. 12).反应使用Pd(PPh3)4作为催化剂, Et3N作为碱, 4-二甲氨基吡啶(DMAP)作为添加剂, 在500 W氙灯照射, 4.04 MPa CO压力和8~40 equiv.醇类底物的存在下实现反应.这一钯-光催化体系在15个底物中实现了高达83%的产率.
在这一反应中, 卤代物首先与Pd(0) 发生原子转移, 生成烷基自由基和Pd(Ⅰ).烷基自由基随后与CO反应并发生5-exo分子内环化, 再一次捕获CO, 生成酰基自由基.这一酰基自由基与先前产生的Pd(Ⅰ)物种结合, 生成酰基Pd(Ⅱ)物种, 在R1OH的存在下, 还原消除生成最终产物(Scheme 16).
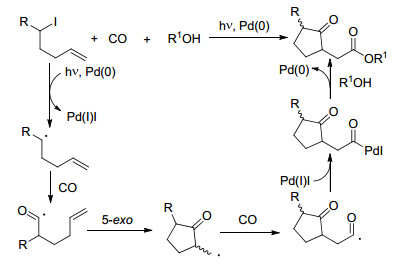 图式 16
钯-光催化羰基化机理
Scheme16.
Mechanism of Pd/photo-catalyzed carbonylation reactions
图式 16
钯-光催化羰基化机理
Scheme16.
Mechanism of Pd/photo-catalyzed carbonylation reactions
在此之后, Ryu课题组[37]对光-钯催化体系的潜在应用价值进行了深入挖掘. 2006年, 不含烯基的烷基碘代物的原子转移羰基化反应得以发展(Scheme 17).在这一反应中, 醇和胺都能以较好的产率生成最终产物.
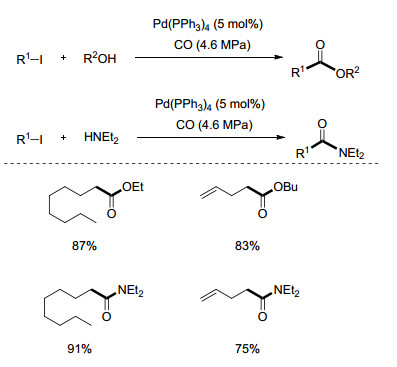 图式 17
碘代烷烃的钯-光催化羰基化反应
Scheme17.
Pd/light catalyzed carbonylation reactions of iodoalkanes
图式 17
碘代烷烃的钯-光催化羰基化反应
Scheme17.
Pd/light catalyzed carbonylation reactions of iodoalkanes
2010年, Alexanian课题组[38]发展了一种Heck反应类型的钯催化羰基化反应(Eq. 13).反应运用Pd(PPh3)4作为催化剂, 2 equiv. i-Pr2NEt作为碱, 在5.07 MPa CO压力、130 ℃实现.在这一方法中, 一级和二级卤代物都能顺利发生反应, 并生成应用广泛的烯酮产物.通过TEMPO实验, 涉及自由基的反应历程得到确认.
与此同时, Ryu课题组[39]将其钯-光催化体系进一步发展(Eq. 14), 使之能与端炔反应生成生物活性物质中间体烷基炔基酮类化合物.一级、二级和三级碘代烷烃都能顺利完成反应.
2011年和2012年, Ryu及其合作者[40]实现并完善了钯-光催化的四组分原子转移羰基化反应(Eq. 15), 通过烷基碘代物、CO、烯烃和醇的共同反应生成酯.当烯基醇类物质作为反应物时, 反应则为三组分反应, 生成内酯产物(a).反应选择性好, 满足多组分体系反应高效性的基本要求.此外, 除了常用的(PPh3)2PdCl2催化剂以外, 二聚体[Pd2(MeCN)6](PF6)2在多组分反应中展现了优异的催化能力[40b].
随后, 钯-光催化体系的应用被发挥到了极致[41].在2012年到2015年中, Ryu课题组先后实现了多种钯-光催化在体系下的偶联反应. 2012年, 碘代乙酸酯与CO和胺类化合物反应首先得到实现(Eq. 16)[41a].随后, 芳基硼酸的反应得到兼容, 反应生成烷基芳基酮产物(Eq. 17)[41b]. 2015年, Ryu课题组再次证明其催化体系同样适用于Heck类型的反应(Eq. 18)[41c]以及Suzuki类型的反应(Eq. 19)[41d].
最近, 张新刚课题组[42]发展了一种钯催化的二氟甲基羰基化方法(Eq. 20).反应使用1:2的芳基硼酸和BrCF2CO2R或BrCF2CO2Et, 在PdCl2(PPh3)2, Xantphos, Cu(hfac)2共同催化下, 在二氧六环中反应24 h实现.此方法将传统钯催化烷基卤代物的羰基化反应与新型二氟甲基试剂结合起来, 大大丰富了CO参与的羰基化反应.反应条件温和, 仅使用101 kPa CO进行反应.
同年, Alexanian课题组[43]实现了非活化的二级烷基卤代物低压下的氧烷基羰基化反应(Eq. 21).非活化的二级烷基溴代物在强供电卡宾配位的钯的催化下实现羰基化, 并被作为混合溶剂的醇捕获生成酯类产物.反应有较好的产率和容忍性, 这一温和的转化方式同样兼容生物活性物质.
5 其他反应
5.1 胺类C—H键烷基化反应
2016年, Hartwig课题组[44]报道了利用三烷基膦Cy2(t-Bu)P(叔丁基二环己基膦)的位阻效应, 实现钯催化的非活化二级或三级卤代烃与二苯甲酮亚胺的偶联反应(Eq. 22).该反应以(Cy2t-BuP)2PdHBr为催化剂, Cs2CO3为碱, 叔戊醇为溶剂, 80 ℃反应24 h, 高产率地得到一系列偶联的产物.实验结果说明, 反应可能含有烷基自由基中间体.作者认为, 溴代烷烃先与Pd(0) 反应生成烷基自由基和Pd(Ⅰ)物种, 然后可能经历三种不同的途径(Scheme 19).其一, Pd(Ⅰ)物种与亚胺进行配体交换生成亚胺钯络合物, 紧接着亚胺钯络合物再与烷基自由基形成C—N键, 同时生成零价钯参与下一次的催化循环(path Ⅰ).其二, 烷基自由基和Pd(Ⅰ)物种进行自由基加成, 生成的Pd(Ⅱ)络合物在碱的作用下与亚胺发生配体交换, 最后通过还原消除构建C—N键(path Ⅱ).其三, 通过烷基自由基与亚胺进行自由基加成形成C—N键, 同时形成的碳自由基与Pd(Ⅰ)物种加成生成Pd(Ⅱ)配合物, 进而通过还原消除得到最终产物, 同时产生的Pd(Ⅱ)物种与碱作用生成零价钯参与下次催化循环(path Ⅲ).
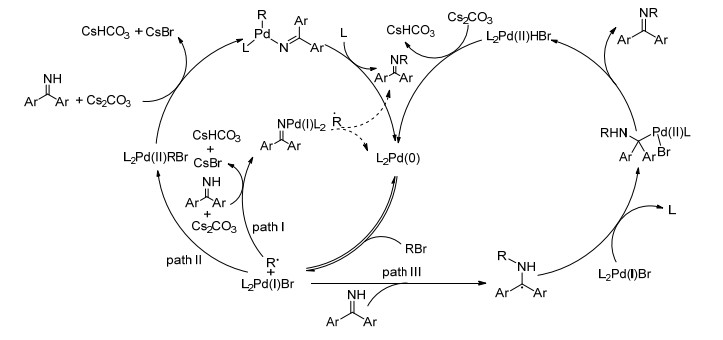 图式 19
亚胺N-烷基化机理
Scheme19.
Plausible mechanisms for palladium-catalyzed N-alkylation of imines
图式 19
亚胺N-烷基化机理
Scheme19.
Plausible mechanisms for palladium-catalyzed N-alkylation of imines
5.2 腙C—H键烷基化反应
考虑含氟分子的独特性质, Bouyssi和Monteiro课题组[45]在2015年报道了醛腙与溴代二氟乙酸乙酯的偶联反应(Eq. 23).该反应用KOAc作碱, [Pd2(dba)3]作催化剂, t-BuXantphos作配体, 得到了一系列C—C偶联产物.该反应底物适应性广泛, 各类芳香醛腙、吡啶醛腙、喹啉醛腙、吡唑醛腙等均能很好的参与到反应之中.硝基、氰基、酯基、醛基和卤素等官能团也可以兼容.自由基验证实验表明, 该反应经历了自由基历程.作者提出的反应历程与上述Hartwig课题组二级或三级卤代烃与二芳基甲酮亚胺的偶联反应机理类似(Scheme 19).
6 总结与展望
综上所述, 金属钯构建的催化体系在卤代烷烃参与的自由基型化学转化之中起到了举足轻重的作用.随着反应体系的不断完善, 越来越多的卤代烷烃被成功用于该类转化之中, 也为该类反应的发展提供了许多新的机遇, 但是从目前来看, 该类反应目前依然存在一些问题: (1) 成键类型较集中, 目前较多的工作集中在C(sp3)—C(sp2), C(sp3)—C(sp)键的构建, 有关C(sp3)—C(sp3)键构建的报道较少; (2) 反应类型较少, 仅局限于与烯烃、炔烃、一氧化碳等活性较高的物种反应, 反应类型有待完善.烷基C—H键作为最广泛存在的亲核试剂, 然而截至目前, 并没有出现该领域的成功报道; (3) 大多数反应需要在大位阻富电子膦配体、氮杂卡宾配体的辅助下才能完成, 这些配体大都价格昂贵, 合成路线繁琐.由此可见, 在发展传统催化体系实现卤代烷烃自由基转化的同时, 探寻更加简单、高效、高选择性的催化体系, 如通过引入光、电等其他形式能量, 以实现反应类型更加丰富、产品结构更加多样性的化学转化仍然是一个具有挑战性和发展潜力的方向.
-
-
[1]
(a) Kochi, J. K.; Tamura, M. J. Am. Chem. Soc. 1971, 93, 1483.
(b) Tamura, M.; Kochi, J. K. J. Organomet. Chem. 1972, 42, 205.
(c) Ishiyama, T.; Abe, S.; Miyaura, N.; Suzuki, A. Chem. Lett. 1992, 21, 691.
(d) Devasagayaraj, A.; Stüdemann, T.; Knochel, P. Angew. Chem., Int. Ed. 1996, 34, 2723. -
[2]
(a) Ryu, I. Chem. Soc. Rev. 2001, 30, 16.
(b) Frisch, A. C. ; Beller, M. Angew. Chem. , Int. Ed. 2005, 44, 674.
(c) Ackermann, L. Chem. Commun. 2010, 46, 4866.
(d) Sumino, S. ; Fusano, A. ; Fukuyama, T. ; Ryu, I. Acc. Chem. Res. 2014, 47, 1563.
(e) Sebren, L. J. ; Devery, J. J. ; Stephenson, C. R. J. ACS Catal. 2014, 4, 703.
(f) Zhang, W. ; Dai, J. ; Xu, H. Chin. J. Org. Chem. 2015, 35, 1820 (in Chinese).
(张文曼, 戴建军, 许华建, 有机化学, 2015, 35, 1820. ) -
[3]
Kambe, N.; Iwasaki, T.; Terao, J. Chem. Soc. Rev. 2011, 40, 4937. doi: 10.1039/c1cs15129k
-
[4]
Ishihara, T.; Kuroboshi, M.; Okada, Y. Chem. Lett. 1986, 15, 1895. doi: 10.1246/cl.1986.1895
-
[5]
Curran, D. P.; Chang, C.-T. Tetrahedron Lett. 1990, 31, 933. doi: 10.1016/S0040-4039(00)94396-X
-
[6]
Yi, P.; Zhang, Z.; Hu, H. Synth. Commun. 1992, 22, 2019. doi: 10.1080/00397919208021336
-
[7]
Surapanich, N.; Kuhakarn, C.; Pohmakotr, M.; Reutrakul, V. Eur. J. Org. Chem. 2012, 2012, 5943. doi: 10.1002/ejoc.v2012.30
-
[8]
Zou, Y.; Zhou, J. Chem. Commun. 2014, 50, 3725. doi: 10.1039/C4CC00297K
-
[9]
Bissember, A. C.; Levina, A.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 14232. doi: 10.1021/ja306323x
-
[10]
McMahon, C. M.; Alexanian, E. J. Angew. Chem., Int. Ed. 2014, 53, 5974. doi: 10.1002/anie.201311323
-
[11]
Bloome, K. S.; McMahen, R. L.; Alexanian, E. J. J. Am. Chem. Soc. 2011, 133, 20146. doi: 10.1021/ja2091883
-
[12]
(a) Hidai, M.; Kokura, M.; Uchida, Y. J. Organomet. Chem. 1973, 52, 431.
(b) Ozawa, F.; Sugimoto, T.; Yuasa, Y.; Santra, M.; Yamamoto, T.; Yamamoto, A. Organometallics 1984, 3, 683.
(c) Cavinato, G.; Toniolo, L.; Vavasori, A. J. Mol. Catal. A: Chem. 2004, 219, 233. -
[13]
Dong, X.; Han, Y.; Yan, F.; Liu, Q.; Wang, P.; Chen, K.; Li, Y.; Zhao, Z.; Dong, Y.; Liu, H. Org. Lett. 2016, 18, 3774. doi: 10.1021/acs.orglett.6b01787
-
[14]
Sumino, S.; Ryu, I. Org. Lett. 2016, 18, 52. doi: 10.1021/acs.orglett.5b03238
-
[15]
Liu, H.; Qiao, Z.; Jiang, X. Org. Biomol. Chem. 2012, 10, 7274. doi: 10.1039/c2ob25990g
-
[16]
Fan, J.-H.; Wei, W.-T.; Zhou, M.-B.; Song, R.-J.; Li, J.-H. Angew. Chem., Int. Ed. 2014, 53, 6650. doi: 10.1002/anie.201402893
-
[17]
Liu, Q.; Chen, C.; Tong, X. Tetrahedron Lett. 2015, 56, 4483. doi: 10.1016/j.tetlet.2015.05.094
-
[18]
Wang, H.; Guo, L.-N.; Duan, X.-H. J. Org. Chem. 2016, 81, 860. doi: 10.1021/acs.joc.5b02433
-
[19]
Xia, X.-F.; Zhu, S.-L.; Li, Y.; Wang, H. RSC Adv. 2016, 6, 51703. doi: 10.1039/C6RA05744F
-
[20]
Fruchey, E. R.; Monks, B. M.; Patterson, A. M.; Cook, S. P. Org. Lett. 2013, 15, 4362. doi: 10.1021/ol4018694
-
[21]
Monks, B. M.; Cook, S. P. Angew. Chem., Int. Ed. 2013, 52, 14214. doi: 10.1002/anie.201308534
-
[22]
Li, Z.; García-Domínguez, A.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 11610. doi: 10.1021/jacs.5b07432
-
[23]
He, Y.-T.; Wang, Q.; Li, L.-H.; Liu, X.-Y.; Xu, P.-F.; Liang, Y.-M. Org. Lett. 2015, 17, 5188. doi: 10.1021/acs.orglett.5b02512
-
[24]
Domański, S.; Chaładaj, W. ACS Catal. 2016, 6, 3452. doi: 10.1021/acscatal.6b00777
-
[25]
Wang, Q.; He, Y.-T.; Zhao, J.-H.; Qiu, Y.-F.; Zheng, L.; Hu, J.-Y.; Yang, Y.-C.; Liu, X.-Y.; Liang, Y.-M. Org. Lett. 2016, 18, 2664. doi: 10.1021/acs.orglett.6b01038
-
[26]
Xiao, B.; Liu, Z.-J.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 616. doi: 10.1021/ja3113752
-
[27]
Wu, X.; See, J. W. T.; Xu, K.; Hirao, H.; Roger, J.; Hierso, J.-C.; Zhou, J. Angew. Chem., Int. Ed. 2014, 53, 13573. doi: 10.1002/anie.201408355
-
[28]
Shao, C.; Shi, G.; Zhang, Y.; Pan, S.; Guan, X. Org. Lett. 2015, 17, 2652. doi: 10.1021/acs.orglett.5b01024
-
[29]
Venning, A. R. O.; Bohan, P. T.; Alexanian, E. J. J. Am. Chem. Soc. 2015, 137, 3731. doi: 10.1021/jacs.5b01365
-
[30]
Li, Z.-Y.; Li, L.; Li, Q.-L.; Jing, K.; Xu, H.; Wang, G.-W. Chem. Eur. J. 2017, 23, 3285. doi: 10.1002/chem.201700354
-
[31]
Liu, Q.; Dong, X.; Li, J.; Xiao, J.; Dong, Y.; Liu, H. ACS Catal. 2015, 5, 6111. doi: 10.1021/acscatal.5b01469
-
[32]
Tsuji, J.; Sato, K.; Nagashima, H. Tetrahedron 1985, 41, 5003. doi: 10.1016/S0040-4020(01)96745-6
-
[33]
Urata, H.; Ishii, Y.; Fuchikami, T. Tetrahedron Lett. 1989, 30, 4407. doi: 10.1016/S0040-4039(00)99373-0
-
[34]
Ishiyama, T.; Miyaura, N.; Suzuki, A. Tetrahedron Lett. 1991, 32, 6923. doi: 10.1016/0040-4039(91)80445-C
-
[35]
Ishiyama, T.; Murata, M.; Suzuki, A.; Miyaura, N. J. Chem. Soc., Chem. Commun. 1995, 295.
-
[36]
Ryu, I.; Kreimerman, S.; Araki, F.; Nishitani, S.; Oderaotoshi, Y.; Minakata, S.; Komatsu, M. J. Am. Chem. Soc. 2002, 124, 3812. doi: 10.1021/ja017315e
-
[37]
Fukuyama, T.; Nishitani, S.; Inouye, T.; Morimoto, K.; Ryu, I. Org. Lett. 2006, 8, 1383. doi: 10.1021/ol060123+
-
[38]
Bloome, K. S.; Alexanian, E. J. J. Am. Chem. Soc. 2010, 132, 12823. doi: 10.1021/ja1053913
-
[39]
Fusano, A.; Fukuyama, T.; Nishitani, S.; Inouye, T.; Ryu, I. Org. Lett. 2010, 12, 2410. doi: 10.1021/ol1007668
-
[40]
(a) Fusano, A.; Sumino, S.; Fukuyama, T.; Ryu, I. Org. Lett. 2011, 13, 2114.
(b) Fusano, A.; Sumino, S.; Nishitani, S.; Inouye, T.; Morimoto, K.; Fukuyama, T.; Ryu, I. Chem. Eur. J. 2012, 18, 9415. -
[41]
(a) Sumino, S.; Fusano, A.; Fukuyama, T.; Ryu, I. Synlett 2012, 23, 1331.
(b) Sumino, S.; Ui, T.; Ryu, I. Org. Lett. 2013, 15, 3142.
(c) Sumino, S.; Ui, T.; Hamada, Y.; Fukuyama, T.; Ryu, I. Org. Lett. 2015, 17, 4952.
(d) Sumino, S.; Ui, T.; Ryu, I. Org. Chem. Front. 2015, 2, 1085. -
[42]
Zhao, H.-Y.; Feng, Z.; Luo, Z.; Zhang, X. Angew. Chem., Int. Ed. 2016, 55, 10401. doi: 10.1002/anie.201605380
-
[43]
Sargent, B. T.; Alexanian, E. J. J. Am. Chem. Soc. 2016, 138, 7520. doi: 10.1021/jacs.6b04610
-
[44]
Peacock, D. M.; Roos, C. B.; Hartwig, J. F. ACS Cent. Sci. 2016, 2, 647. doi: 10.1021/acscentsci.6b00187
-
[45]
Prieto, A.; Melot, R.; Bouyssi, D.; Monteiro, N. Angew. Chem., Int. Ed. 2016, 55, 1885. doi: 10.1002/anie.201510334
-
[1]
-

图式 1 钯促进的(溴二氟甲基)-磺酰基苯的Heck反应
Scheme 1 Palladium-promoted Heck-type reactions of [(bromodifluoromethyl)-sulfonyl]benzene

图式 2 钯催化非活化烷基碘代物的Heck-型偶联反应
Scheme 2 Palladium-catalyzed Heck-type cross-couplings of unactivated alkyl iodides

图式 6 钯催化烷基碘代物的原子转移自由基环化反应
Scheme 6 Palladium-catalyzed atom transfer radical cyclization of alkyl iodide

图式 8 钯催化下炔的插入/还原反应和可能的机理
Scheme 8 Palladium-catalyzed alkyne insertion/reduction reactions and proposed mechanism

图式 9 β-H存在下钯催化分子内碘原子迁移反应
Scheme 9 Palladium-catalyzed intramolecular iodine-transfer reactions in the presence of β-hydrogen atoms

图式 10 钯催化下端炔、全氟碘代烷烃和硼酸三组分反应
Scheme 10 Palladium-catalyzed three-component reactions of terminal alkynes with iodoperfluoroalkanes and boronic acids

图式 12 钯催化的吡啶N-氧化物与二级烷基溴的偶
Scheme 12 Palladium-catalyzed C—H activation/cross-coupling of pyridine N-oxides with secondary alkyl bromides

图式 14 非活化烷基卤代烃与芳烃的环化反应
Scheme 14 Synthesis of diverse cyclic heterocycles using the catalytic C—H alkylati

图式 15 钌钯共催化芳基二氟甲基化及其机理
Scheme 15 Pd and Ru co-catalyzed meta-difluoromethylation of arenes and plausible mechanism

图式 17 碘代烷烃的钯-光催化羰基化反应
Scheme 17 Pd/light catalyzed carbonylation reactions of iodoalkanes
-


 下载:
下载:

















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 122
- 文章访问数: 8779
- HTML全文浏览量: 2679
 下载:
下载:
