 图 图式1
磷配体对于醛-炔-胺三组分反应的影响
Figure 图式1.
Effects of phosphorus ligands on the three component reaction of aldehyde-alkyne-amine
图 图式1
磷配体对于醛-炔-胺三组分反应的影响
Figure 图式1.
Effects of phosphorus ligands on the three component reaction of aldehyde-alkyne-amine
引用本文:
杨军, 付婷, 龙洋, 周向葛. 水相催化碳氢活化反应[J]. 有机化学,
2017, 37(5): 1111-1116.
doi:
10.6023/cjoc201702045

Citation: Yang Jun, Fu Ting, Long Yang, Zhou Xiangge. Progress in Catalytic C-H Activation Reactions in Water[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1111-1116. doi: 10.6023/cjoc201702045
Citation: Yang Jun, Fu Ting, Long Yang, Zhou Xiangge. Progress in Catalytic C-H Activation Reactions in Water[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1111-1116. doi: 10.6023/cjoc201702045
水相催化碳氢活化反应
English
Progress in Catalytic C-H Activation Reactions in Water
Abstract:
C-H bond functionalization is one of the hot spots in the research field of organic chemistry, and selective C-H activation is a challenging project. Among these reactions, organic solvent is normally used as reaction media. Using cheap, environmentally friendly water as reaction solvent would be in line with the requirements of "green chemistry" and low-carbon sustainable development. This paper reviews the recent progress of aqueous catalyzed C-H functionalization reactions, including hybridized sp-, sp2-, and sp3-C-H bonds.
-
Key words:
- catalysis
- / C-H activation
- / water
- / green chemistry
-
偶联反应是有机合成中形成C—C键的重要方法之一, 人们熟知的Stille、Suzuki-Miyaura和Negishi等偶联反应具有良好的底物兼容性, 以及高效、条件温和等优点, 使它们具有很大的应用价值.但这类反应也存在一些不足.比如, 步骤繁琐, 需要对反应物的C—H键进行预先活化, 以此来降低反应的难度.而预先活化就对底物有一定的要求, 这就极大地限制了反应底物的拓展.同时, 繁琐的步骤还导致了反应的原子经济性不好.反之, 如果省略预先活化的过程, 直接活化C—H键进行偶联反应, 就可以减少实验步骤, 提高反应的原子经济性.近年来, 金属催化的直接C—H键的功能化反应成为了有机化学的研究热点之一.诸如钯[1~9]、铑[10~16]、钌[17~26]等过渡金属已成功应用于多种反应.
另一方面, 随着社会的发展, 绿色环保和可持续发展的理念逐渐得到人们的认可, 发展对环境友好的“绿色”化学是化学研究的一个重要方向.目前, 在大多数的碳氢活化反应中, 通常使用的是有机溶剂, 而水作为一种廉价、无毒无害、蕴藏丰富的反应介质来代替毒性大、易挥发的有机溶剂, 进行C—H键功能化反应是符合当前所倡导的“绿色”化学和低碳可持续发展的理念.由于反应底物和催化剂的水溶性, 对水的敏感性, 以及产物的分离纯化等问题, 水相中进行C—H键的功能化仍然是一个挑战.本文对近年来的水相中的C—H活化(包括底物配位导向和非导向的sp-, sp2-, sp3-杂化的C— H键的活化)进展进行介绍.
1 sp杂化的碳氢键活化
针对水相中的sp杂化的碳氢键活化的研究, 也就是对端炔的C—H键功能化, 主要是Li课题组进行的.
2002年, 该课题组[27]使用苯乙炔和亚胺作为底物, 三氟甲磺酸铜作为催化剂, 手性二苯基噁唑啉吡啶为配体, 水作为溶剂, 进行了C—H/C—H的功能化反应, 形成新的C(sp2)—C(sp)键.该反应采用了手性氮配体.将端炔加成到亚胺, 取得了较高的对映选择性, ee值可达96% (Eq. 1).
2003年, 他们[28]在前期工作基础上, 使用苯乙炔、醛和二级胺作为底物, 三组分“一锅法”合成了相应的产物.该反应使用金盐作为催化剂, 水作为溶剂, 不需要配体的辅助, 以接近百分之百的转化率, 得到外消旋产物(Eq. 2).
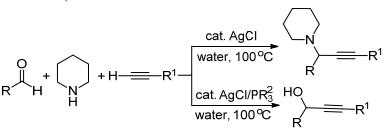
2005年, 该课题组[29]继续以哌啶代替二级胺, 和苯甲醛以及苯乙炔进行水相中的银催化三组分反应, 在没有膦配体的情况下, 仍然可以得到醛-炔-胺偶联的产物(Scheme 1).
图 图式1
磷配体对于醛-炔-胺三组分反应的影响
Figure 图式1.
Effects of phosphorus ligands on the three component reaction of aldehyde-alkyne-amine
同时, 他们还进行了苯甲醛和苯乙炔的偶联反应, 使用银作为催化剂, 有机膦为配体, 水作为溶剂, 得到了相应的醇炔产物.这个反应进一步扩展了端炔的偶联反应.
在反应机理研究中, 他们还使用苯乙炔银和醛在水相中进行反应, 得到相应产物, 证实了反应的机理是银先活化端炔的C—H键(Scheme 2).
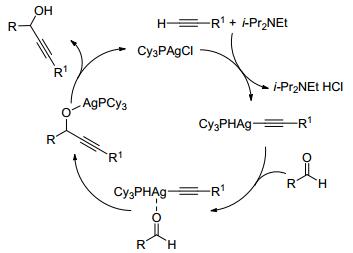 图 图式2
银催化三组分反应的机理.
Figure 图式2.
Mechanism of Ag-catalyzed three component reaction
图 图式2
银催化三组分反应的机理.
Figure 图式2.
Mechanism of Ag-catalyzed three component reaction
2006年该课题组[30]使用邻炔基苯甲醛和苯乙炔成功进行了成环反应, 该反应使用了带有膦配体的金配合物作为催化剂, 水作为溶剂.根据反应机理, 金先和苯乙炔配位, 然后进攻醛基的碳原子.同时, 醛基的氧原子和邻位的炔基关环, 形成新的C—O键, 完成反应(Eq. 3).
2 sp2杂化的碳氢键活化
2.1 配位导向的sp2杂化的碳氢键活化
2010年, Dixneuf等[31]使用钌配合物为催化剂, 吡啶作为配位导向基, 氯苯作为芳基来源, 碳酸钾作为碱, 行邻位C—H键活化芳基化反应.反应在水相中取得了很好的效果, 对于单、双的取代选择性也比在有机溶剂中的效果好.同时, 对于该反应对于其它多种含氮导向基团也有良好的兼容性(Eq. 4).
2012年, Ackermann等[32]使用含膦配体的钌催化剂在水相中催化嘧啶苯胺类化合物和二苯乙炔进行的炔基化关环反应, 成功合成了吲哚衍生物.嘧啶基团的引入实现了导向作用, 提高反应的选择性.反应中, 导向基先和钌形成六元环, 随后炔基插入, 以及催化剂的脱除.作者成功地将炔基化和成环反应以及水相结合在一起(Eq. 5).
同年, 该课题组[33]还使用吡唑作为导向基, 苯酚作为芳基来源, 钌配合物作为催化剂, 碳酸钾为碱, 在水相中实现邻位C—H功能化芳基化.这个反应在钌催化剂中引入羧酸配体, 有助于增加其水溶性.此外, 吡唑和噁唑等导向基以及芳基来源苯酚也具有一定的水溶性, 促进了该反应在水相中的进行(Eq. 6).
2013年Zhou课题组[34]使用邻碘苯甲酸作为底物, 苯酚作为芳基来源, 在水相中反应得到苯甲酸的邻位芳基化产物.该工作以碘代苯甲酸的羧基作为导向基, 醋酸钯作为催化剂, 三氟乙酸银在反应中同时作为氧化剂和碘的淬灭剂.反应条件温和, 25℃就可以得到良好的收率.当采用对甲基苯酚时, 可以得到关环的内酯产物(Scheme 3).
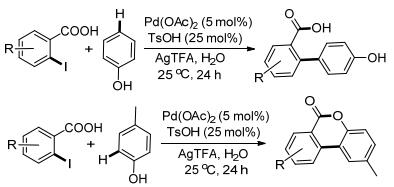 图 图式3
钯催化邻碘苯甲酸与苯酚反应.
Figure 图式3.
Pd-catalyzed the reaction between 2-iodobenzoic acid and phenol
图 图式3
钯催化邻碘苯甲酸与苯酚反应.
Figure 图式3.
Pd-catalyzed the reaction between 2-iodobenzoic acid and phenol
另外, 当直接使用邻甲基苯甲酸和苯酚反应时, 可以直接对C—H/C—H键进行功能化, 得到两种产物, 一种是邻位芳基化的产物, 另一种是关环的产物(Eq. 7).
同年, Zhou等[35]还使用苯甲酸为底物, 具有水溶性的二芳基碘盐作为芳基来源, 醋酸钯作为催化剂, 在水相中进行邻位芳基化反应.该反应的特点是使用了具有水溶性的芳基来源, 对不同阴离子的高价碘盐均具有良好的效果.反应不需要额外使用氧化剂, 也避免了昂贵的银盐的使用(Eq. 8).
2014年, Loh等[36]使用吲哚嘧啶作为底物, 带有环戊二烯基配体的铑作为催化剂, 三甲氧基苯基硅作为芳基来源, 选择性实现吲哚2位的芳基化.嘧啶导向基的加入提高了反应的选择性.氟化银在反应中起到了活化C—Si键的作用.当使用纯水作为溶剂时, 可以得到65%的产率, 当使用四氢呋喃和水的体积比为1:1的混合溶剂, 可以得到92%的产率(Eq. 9).
2015年, Zhou课题组[37]使用具有一定水溶性的乙酰苯胺作为底物, 苯甲醇作为酰基来源, TBHP作为氧化剂, 醋酸钯作为催化剂, 在水相中进行邻位酰基化反应.三氟乙酸的加入可能有利于钯中间体的稳定, 促进反应的进行(Eq. 10).
2015年, Yu课题组[38]使用噁唑啉导向基进行邻位羟基化反应, 碳酸钠作为碱, 乙酸铜作为催化剂, 在DMSO和水的混合溶剂中进行.氧气作为氧化剂, 促进铜完成催化循环(Eq. 11).
同年, Li课题组[39]在水相中进行了苯甲酸的自偶联反应.反应中使用了降冰片烯铑配合物催化剂.二氧化锰氧化剂的加入, 使铑完成一价到三价的催化循环.该方法为抗癌药物鞣花酸的合成提供了一种新的思路(Eq. 12).
2015年, Deng课题组[40]使用酮肟等含氮基团底物, 苯甲醛作为酰基来源, 醋酸钯作为催化剂, 过氧化叔丁醇(TBHP)作为氧化剂, 在水相中进行邻位的酰基化反应.相转移试剂十二烷基硫酸钠(SDS)的加入促进了反应在水相中的进行.该反应兼容性较好, 采用酮肟、偶氮苯、2-苯基吡啶等多种含氮导向基均取得了良好的催化效果, 反应条件也相对温和.为二芳基酮化合物的合成提供了一种有效方法(Eq. 13).
2.2 非导向的sp2杂化的碳氢键活化
2008年, Greaney等[41]使用一定水溶性的2-苯基噁唑作为底物, 碘苯作为芳基来源, 钯作为催化剂, 碳酸银作为氧化剂, 对噁唑环上的C—H键进行芳基化反应, 合成2, 5-二取代的噁唑.该反应中的底物、催化剂、氧化剂等均具有一定的水溶性, 该反应可以在水相中进行.银离子同时作为氧化剂和碘离子淬灭剂, 促进反应进行(Eq. 14).
2010年, 该课题组[42]使用苯基-2H-吲唑作为底物, 碘苯作为芳基来源, 钯作为催化剂, 银盐作为氧化剂, 在水相中实现杂环的芳基化.银离子同样促进反应进行.苯基的作用在于保护N—H键, 抑制副反应的发生(Eq. 15).
2015年, Gonzalez-Gomez等[43]使用2-苯基苯甲酸进行光催化脱氢偶联关环形成内酯.反应使用了乙腈与水混合溶剂, 过硫酸铵作为氧化剂.该工作具有非金属催化及反应条件温和等优点(Eq. 16).
2016年, Pan课题组[44]使用喹啉氮氧化合物和亚磺酸钠作为底物, 溴化铜作为催化剂, 在过硫酸钾作为氧化剂的条件下, 得到喹啉邻位磺酰化的产物.反应最终选用的溶剂是硝基甲烷与二氯乙烷的混合溶剂, 当加入0.1 mL水时可以提高产率.作者认为水的加入有助于提高催化剂和过硫酸钾的溶解度.作者提出的机理是, 亚磺酸钠在铜和过硫酸钾的作用下产生磺酰基自由基, 和喹啉氮氧化物结合, 最后脱去与氮相连的氧, 得到产物(Eq. 17).
同年, Deng等[45]使用2-萘酚和苯亚磺酸钠作为底物, 通过反应条件尤其是酸碱性的控制, 分别生成硫醚和磺酰化两种不同的产物.在作者提出的机理中, 酸性条件下, 苯亚磺酸钠会脱去氧, 最后得到硫醚化产物; 在碱性条件下, 则会得到苯磺酰化产物.碘的作用是和硫结合, 形成硫碘键, 促进反应的进行(Scheme 4).该方法的特点是无金属催化, 而且使用了环境友好的水作为溶剂.
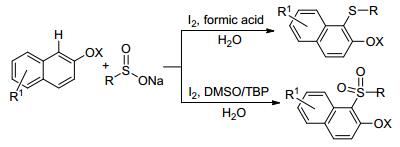 图 图式4
无金属催化2-萘酚与苯亚磺酸钠的反应.
Figure 图式4.
Reaction between 2-naphthol and sodium benzenesulfinate without metal catalyst
图 图式4
无金属催化2-萘酚与苯亚磺酸钠的反应.
Figure 图式4.
Reaction between 2-naphthol and sodium benzenesulfinate without metal catalyst
2016年, Nallasamy等[46]采用钯催化剂, 将乙酰苯胺的邻位芳基化反应和2-苯基苯胺的关环形成咔唑的反应结合在一起.这个反应只需0.01%催化剂用量, 碳酸钾作为碱.多种溶剂经过对比, 发现水相的反应效果最好, 收率为93%.反应条件也很温和, 只需要60 ℃.而且后处理十分简单, 只需萃取和洗涤, 即可得到纯净固体产物(Eq. 8).
3 sp3杂化的碳氢键活化
3.1 配位导向的sp3杂化的碳氢活化
关于水相sp3的碳氢功能化例子较少.在2015年, Rao等[47]报道了首例水作为羟基来源, 钯催化的C(sp3)—H键羟基化的反应.该反应使用高价碘盐作为氧化剂, 对8-氨基喹啉酰胺化衍生物的β位C—H键进行羟基化反应.根据同位素实验显示, 产物中的氧原子来自于溶剂水.机理研究表明, 钯催化剂首先和双齿导向基配位, 随后经历高价钯过渡态, 最后, 通过还原消去, 羟基转移至β位(Eq. 19).
2016年, Zhou课题组[48]使用8-氨基喹啉的丁酰化衍生物和碘苯在水相中进行羰基β位的sp3-C—H活化芳基化反应.发现通过筛选不同银盐可以提高产率, 研究表明脂肪族银盐中特戊酸银效果较好, 可以得到93%的收率.特戊酸银起到了碘离子淬灭剂以及可能的相转移试剂的功能(Eq. 20).
3.2 非导向的sp3杂化的碳氢活化
目前, 水相中非导向的sp3-C—H功能化反应主要集中于苄基的氧化反应.
2008年, Kita等[49]对乙苯的苄基位进行氧化反应, 使乙基苯转化为苯乙酮.反应所使用的氧化剂为高价碘盐, 水作为溶剂.该反应具有良好的兼容性, 对含杂原子的化合物亦有良好的反应效果.该反应可能的机理是, 高价碘盐作为自由基引发剂, 引发苄基生成自由基, 和同时产生的溴的自由基结合, 随后在水的作用下, 进行水解反应, 得到相应的酮类产物(Eq. 21).
2009年, Lee课题组[50]使用钌的配合物作为催化剂, TBHP为氧化剂, 在水相中进行苄基位的氧化反应, 将乙基苯转化为苯乙酮.和之前的催化反应相比较, 反应条件更为温和(Eq. 21).
2014年, Hilinski等[51]使用三氟甲基苯甲酮作为催化剂对叔碳进行氧化反应.该反应使用16 equiv.过氧化氢作为氧化剂.如果使用水和有机溶剂的混合溶剂进行反应, 可以得到30%的收率(Eq. 23).
4 结论与展望
综上所述, 水相中的碳氢功能化反应在近年来取得了一些成果, 这些反应大多采用具有一定水溶性的底物或者催化剂, 或者在反应体系中加入相转移试剂.但是距离在工业中有实际应用价值的绿色的水相催化体系仍然还有相当的距离, 也有许多的问题需要解决.比如, 酰胺导向基团在水中的水解, 反应的后处理经常还需要大量的有机溶剂等问题.此外, 一些底物以及金属催化剂对水的敏感性, 造成许多在有机相能正常进行的反应不能拓展至水相中.因此能在水相中进行的反应类型较少, 能进行催化剂的回收和循环利用的体系也很少.因此, 发展更加高效、可循环利用的、后处理简单的催化体系仍然是目前水相催化的重要任务.
-
-
[1]
廖港, 史炳锋, 化学学报, 2015, 73, 1283.Liao, G.; Shi, B. Acta Chim. Sinica 2015, 73, 1283 (in Chinese).
-
[2]
张博, 管晗曦, 刘斌, 史炳锋, 有机化学, 2014, 34, 1487.Zhang, B.; Guan, H.; Liu, B.; Shi, B. Chin. J. Org. Chem. 2014, 34, 1487 (in Chinese).
-
[3]
Hull, K.; Sanford, M. J. Am. Chem. Soc. 2007, 129, 11904.
-
[4]
Desai, L.; Stowers, K.; Sanford, M. J. Am. Chem. Soc. 2008, 130, 13285.
-
[5]
Joo, J.; Guo, P.; Sames, D. J. Org. Chem. 2013, 78, 738.
-
[6]
Tan, J.; Chen, Y.; Li, H.; Yasuda, N. J. Org. Chem. 2014, 79, 8871.
-
[7]
Batchu, H.; Bhattacharyya, S.; Kant, R.; Batra, S. J. Org. Chem. 2015, 80, 7360.
-
[8]
Brahim, M.; Smari, I.; Ammar, H.; Hassine, B.; Soulé, J.; Doucet, H. Org. Chem. Front. 2015, 2, 917.
-
[9]
Testa, C.; Roger, J.; Scheib, S.; Fleurat-Lessard, P.; Hierso, J. Adv. Synth. Catal. 2015, 357, 2913.
-
[10]
Umeda, N.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2009, 74, 7094.
-
[11]
Kim, M.; Kwak, J.; Chang, S. Angew. Chem., Int. Ed. 2009, 48, 8935.
-
[12]
Gong, T.; Xiao, B.; Cheng, W.; Su, W.; Xu, J.; Liu, Z.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 10630.
-
[13]
Reddy, V.; Qiu, R.; Iwasaki, T.; Kambe, N. Org. Lett. 2013, 15, 1290.
-
[14]
Wang, H.; Schrçder, N.; Glorius, F. Angew. Chem., Int. Ed. 2013, 52, 5386.
-
[15]
Xie, F.; Qi, Z.; Yu, S.; Li, X. J. Am. Chem. Soc. 2014, 136, 4780.
-
[16]
Zhang, P.; Hong, L.; Li, G.; Wang, R. Adv. Synth. Catal. 2015, 357, 345.
-
[17]
Asaumi, T.; Matsuo, T.; Fukuyama, T.; Ie, Y.; Kakiuchi, F.; Chatani, N. J. Org. Chem. 2004, 69, 4433.
-
[18]
Ackermann, L.; Althammer, A.; Born, R. Angew. Chem., Int. Ed. 2006, 45, 2619.
-
[19]
Oi, S.; Sasamoto, H.; Funayama, R.; Inoue, Y. Chem. Lett. 2008, 37, 994.
-
[20]
Kumar, P.; Jeyachandran, R.; Ackermann, L. J. Org. Chem. 2013, 78, 4145.
-
[21]
Ackermann, L.; Vicente, R.; Potukuchi, H.; Pirovano, V. Org. Lett. 2010, 12, 5032.
-
[22]
Arockiam, P.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. Green Chem. 2011, 13, 3075.
-
[23]
Muralirajan, K.; Parthasarathy, K.; Cheng, C. Org. Lett. 2012, 14, 4262.
-
[24]
Hashimoto, Y.; Hirano, K.; Satoh, T.; Kakiuchi, F.; Miura, M. J. Org. Chem. 2013, 78, 638.
-
[25]
Schinkel, M.; Marek, I.; Ackermann, L. Angew. Chem., Int. Ed. 2013, 52, 3977.
-
[26]
Gonell, S.; Peris, E. ACS Catal. 2014, 4, 2811.
-
[27]
Wei, C.; Li, C. J. Am. Chem. Soc. 2002, 124, 5638.
-
[28]
Wei, C.; Li, C. J. Am. Chem. Soc. 2003, 125, 9584.
-
[29]
Yao, X.; Li, C. Org. Lett. 2005, 7, 4395.
-
[30]
Yao, X.; Li, C. Org. Lett. 2006, 8, 1953.
-
[31]
Arockiam, P.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. Angew. Chem., Int. Ed. 2010, 49, 6629.
-
[32]
Ackermann, L.; Lygin, A. Org. Lett. 2012, 14, 764.
-
[33]
Ackermann, L.; Pospech, J.; Potukuchi, H. Org. Lett. 2012, 14, 2146.
-
[34]
Wu, Z. Luo, F.; Chen, S.; Li, Z.; Xiang, H.; Zhou, X. Chem. Commun. 2013, 49, 7653.
-
[35]
Wu, Z.; Chen, S.; Hu, C.; Li, Z.; Xiang, H.; Zhou, X. ChemCatChem 2013, 5, 2839.
-
[36]
Lu, M.; Lu, P.; Xu, Y.; Loh, T. Org. Lett. 2014, 16, 2614.
-
[37]
Luo, F.; Yang, J.; Li, Z.; Xiang, H.; Zhou, X. Eur. J. Org. Chem. 2015, 2463.
-
[38]
Sun, S.; Shang, M.; Wang, H.; Lin, H.; Dai, H.; Yu, J. J. Org. Chem. 2015, 80, 8843.
-
[39]
Gong, H.; Zeng, H.; Zhou, F.; Li, C. Angew. Chem., Int. Ed. 2015, 54, 5718.
-
[40]
Xiao, F.; Chen, S.; Huang, H. Deng, G. Eur. J. Org. Chem. 2015, 7919.
-
[41]
Ohnmacht, S.; Mamone, P.; Culshaw, A. Greaney, M. Chem. Commun. 2008, 1241.
-
[42]
Ohnmacht, S.; Culshaw, A.; Greaney, M. Org. Lett. 2010, 12, 224.
-
[43]
Ramirez, N.; Bosque, I.; Gonzalez-Gomez, J. Org. Lett. 2015, 17, 4550.
-
[44]
Du, B.; Qian, P.; Wang, Y.; Mei, H.; Han, J.; Pan, Y. Org. Lett. 2016, 18, 4144.
-
[45]
Xiao, F.; Chen, S.; Tian, J.; Huang, H.; Liu, Y.; Deng, G. Green Chem. 2016, 18, 1538.
-
[46]
Arumugam, V.; Kaminsky, W.; Nallasamy, D. Green Chem. 2016, 18, 3295.
-
[47]
Hu, J.; Lan, T.; Sun, Y.; Chen, H.; Yao, J.; Rao, Y. Chem. Commun. 2015, 51, 14929.
-
[48]
罗飞华, 龙洋, 李正凯, 周向葛, 化学学报, 2016, 74, 805.Luo, F.; Long, Y.; Li, Z.; Zhou, X. Acta Chim. Sinica 2016, 74, 805 (in Chinese).
-
[49]
Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365.
-
[50]
Yi, C.; Kwon, K.; Lee, D. Org. Lett. 2009, 11, 1567.
-
[51]
Pierce, C.; Hilinski, M. Org. Lett. 2014, 16, 6504.
-
[1]
-

图式1 磷配体对于醛-炔-胺三组分反应的影响
Scheme 1 Effects of phosphorus ligands on the three component reaction of aldehyde-alkyne-amine

图式3 钯催化邻碘苯甲酸与苯酚反应.
Scheme 3 Pd-catalyzed the reaction between 2-iodobenzoic acid and phenol
-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 27
- 文章访问数: 2882
- HTML全文浏览量: 427
 下载:
下载:
