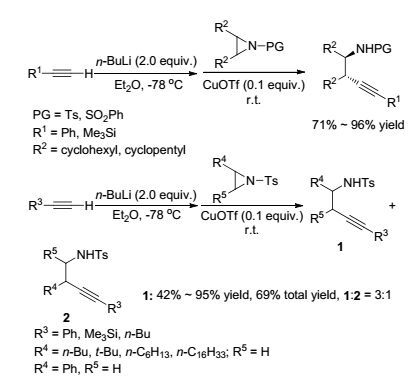
 图式 1
Cu(OTf)2促进的炔对氮杂环丙烷的开环反应
Scheme1.
Cu(OTf)2-promoted ring-opening reactions of aziridines by alkynes
图式 1
Cu(OTf)2促进的炔对氮杂环丙烷的开环反应
Scheme1.
Cu(OTf)2-promoted ring-opening reactions of aziridines by alkynes
引用本文:
褚旭, 常宏宏, 高文超, 魏文珑, 李兴. 碳亲核试剂对氮杂环丙烷的开环反应研究进展[J]. 有机化学,
2017, 37(10): 2569-2589.
doi:
10.6023/cjoc201702033

Citation: Chu Xu, Chang Honghong, Gao Wenchao, Wei Wenlong, Li Xing. Research Progress in the Ring-Opening Reactions of Aziridines by Carbon Nucleophiles[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2569-2589. doi: 10.6023/cjoc201702033
Citation: Chu Xu, Chang Honghong, Gao Wenchao, Wei Wenlong, Li Xing. Research Progress in the Ring-Opening Reactions of Aziridines by Carbon Nucleophiles[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2569-2589. doi: 10.6023/cjoc201702033
碳亲核试剂对氮杂环丙烷的开环反应研究进展
English
Research Progress in the Ring-Opening Reactions of Aziridines by Carbon Nucleophiles
Abstract:
The recent progress in ring-opening reactions of aziridines by various carbon nucleophiles, such as alkynes, nitriles, arenes, heteroarenes, active methylene compounds, organometallic reagents and so on, is reviewed. Moreover, the prospects of future development are also discussed.
-
Key words:
- aziridines
- / ring-opening reactions
- / carbon nucleophiles
-
氮杂环丙烷是有机合成中一类非常重要的合成砌块和中间体, 它被广泛地应用于有机合成, 尤其是含氮天然产物的合成中.由于氮杂环丙烷具有较高的环张力, 使得其反应活性较高, 在一定的条件下可与各种各样偶极试剂发生环加成反应来制备一些新的含氮杂环化合物[1~4].此外, 氮杂环丙烷还可与各种各样的亲核试剂, 比如含氮、氧、硫、卤素和碳等原子的亲核试剂[5~13], 发生杂原子参与的高立体选择性和高区域选择性的亲核开环反应, 生成含有不同官能团的邻氨基类化合物如二胺、氨基醇、氨基硫醇、卤代胺、氨基酸以及其它具有生物活性的化合物.其中, 各种杂原子对其进行的开环反应已有许多报道[14~17], 且对其进行的相关研究也日趋成熟.而碳亲核试剂对其进行的开环反应近几年才得到了一定的发展, 也出现了一些相关报道, 常用的亲核试剂如氰化物、有机金属试剂、活性亚甲基化合物和不饱和碳原子等.本文将对近十来年碳亲核试剂对氮杂环丙烷进行的亲核开环反应研究进行综述.
1 sp杂化的碳原子对氮杂环丙烷的开环反应
1.1 炔对氮杂环丙烷的开环反应
一直以来炔与氮杂环丙烷发生的环加成反应受到人们的重视, 发展地较为成熟[1].炔烃也是很好的亲核试剂, 还可对氮杂环丙烷发生碳开环反应.
2004年, Hou课题组[18]报道了在溶剂乙醚中于-78 ℃条件下, CuOTf催化炔对氮杂环丙烷的开环反应.该反应需要先加入2 equiv.的n-BuLi, 于-78 ℃条件下对炔进行活化, 再加入氮杂环丙烷并与之反应.广普性考察发现, 对于多种环烯衍生的氮杂环丙烷反应均能获得71%~96%的收率, 得到反式立体异构体.对于多种链烯衍生的氮杂环丙烷, 反应所用时间更短, 反应仅得到于取代较少碳上发生开环反应的产物1, 均能获得42%~95%的收率, 苯乙烯氮杂环丙烷则得到总收率为69%的两种异构体(Scheme 1).
图式 1
Cu(OTf)2促进的炔对氮杂环丙烷的开环反应
Scheme1.
Cu(OTf)2-promoted ring-opening reactions of aziridines by alkynes
2005年, 该课题组[19]报道了在1.5 equiv. t-BuOK作用下, 端基炔对氮杂环丙烷的开环反应(Scheme 2).在最优反应条件下, 对氮杂环丙烷氮上保护基以及氮杂环丙烷和炔上多种取代基进行了考察, 除苯甲酰基作保护基外(29%), 其它保护基以及各种官能团取代的氮杂环丙烷和炔烃均能得到中等到优秀的收率(55%~98%).反应表现出专一的区域选择性, 均于取代较少的碳上发生开环反应, 得到单一的异构体产物.所得到的开环产物可进一步在I2、K2CO3和AgOAc的作用下用于合成吡咯衍生物, 为吡咯合成提供了一种简单有效的方法.相对于该课题组[18]2004年发表的CuOTf催化炔对氮杂环丙烷的开环反应, 该反应条件更温和、操作更简单, 具有更高的实用价值.
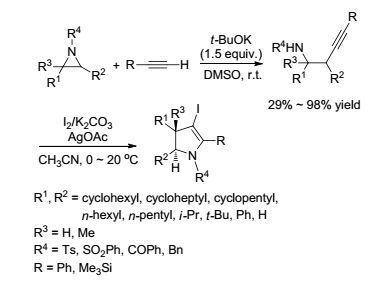 图式 2
炔对氮杂环丙烷的开环反应
Scheme2.
Ring-opening reactions of aziridines by alkynes
图式 2
炔对氮杂环丙烷的开环反应
Scheme2.
Ring-opening reactions of aziridines by alkynes
2009年, Pineschi课题组[20]报道了有机铝试剂对氮杂环丙烷的开环反应(Eq. 1).研究发现溶剂种类对原料转化率起决定性作用:当溶剂为四氢呋喃或乙醚时, 转化率较低; 而在二氯甲烷或甲苯中, 原料则能完全转化, 反应最终在二氯甲烷中进行可取得最好的效果.广普性考察发现对于环烯和链烯衍生的氮杂环丙烷分别与烷基、烯基以及炔基衍生的有机铝试剂反应均能得到45%~92%的收率和88/12~>95/<5的区域选择性, 当环己二烯衍生的氮杂环丙烷与苯乙炔铝反应时, 得到7/93的区域选择性.
1.2 氰基对氮杂环丙烷的开环反应
氰基不仅能够与偶极化合物发生环加成反应, 而且也能与亲电试剂发生偶联反应生成各种腈类化合物; 腈类化合物在官能团转化中具有重要的意义, 其中, 氰基与氮杂环丙烷经开环制得的β-氨基腈可进一步转化为一系列的药物中间体, 因此受到人们的重视.然而, 此类反应所用氰基试剂主要为TMSCN和某些无机盐, 氰基来源有一定的局限性.
2000年, Hou课题组[21]报道了四丁基氟化铵(TBAF)在溶剂四氢呋喃(THF)中于40 ℃条件下催化TMSCN对氮杂环丙烷的开环反应(Eq. 2).该反应具有高的立体选择性, 均得到反式结构的异构体产物; 此外该反应也表现出高的区域选择性, 均于氮杂环丙烷取代较少的碳上发生开环反应.广普性考察发现对于多种官能团取代的氮杂环丙烷反应均能获得79%~>99%的收率, 对氮杂环丙烷氮上保护基考察发现仅当氮上连有吸电子保护基时, 才能得到相应的开环产物.
2005年, Shibasaki课题组[22]报道了手性镉配合物催化剂催化TMSCN对内消旋氮杂环丙烷开环得到立体构型为(2S, 3S)开环产物的反应(Eq. 3).镉配合物催化剂Cat. 1是由三异丙醇镉与相应配体L1 [1, 5-脱水-2, 6-双脱氧-4-邻-(4, 5-二氟-2-羟基甲基-)-6-(二苯基膦酰基)-D-阿糖基己糖醇]按2:3配合而成, 反应需添加2, 6-二甲基苯酚和三氟乙酸作添加剂来提高产物的对映选择性.广普性考察表明, 该催化体系对环烷烃、杂环烷烃和脂肪链烃取代的氮杂环丙烷均表现出广泛的适用性, 都能得到81%~94%的收率和82%~99%的对映选择性.通过ESI-MS对催化剂研究发现, 加入三氟乙酸后, 配合物与三氟乙酸结合产生新的催化活性物质3, 三氟乙酸可能起到桥联两个镉中心的作用, 增加了催化活性物质立体结构的稳定性.此外, 三氟乙酸也起到了增强镉的路易斯酸酸性和调整两个镉原子间相对位置的作用, 这样就能增强催化活性物质的立体选择性.
2005年, Komatsu课题组[23]报道了路易斯碱N, N, N′, N′-四甲基乙二胺(TMEDA)催化TMSCN对氮杂环丙烷的开环反应.广普性考察发现, 多种单取代的氮杂环丙烷与TMSCN反应均能获得47%~93%的收率, 且均于取代较少的碳上发生开环(Eq. 4).多种2, 3-二取代氮杂环丙烷与TMSCN发生反应时, 需要加入1 equiv.的KCN以加速反应的完成.当采用环戊基和环己基取代的氮杂环丙烷作底物时, 反应分别获得57%和97%的收率.对于考察的两个不对称二取代氮杂环丙烷底物, 反应主要于空间位阻较小的碳上发生开环, 分别得到67:33和80:20的区域选择性(Eq. 5).截至2005年, 这是首篇采用路易斯碱催化三甲基硅基试剂对氮杂环丙烷的开环反应, 在此之前所报道的该反应中, 多数需使用过渡金属作催化剂, 且大部分面临着区域选择性较低这一难题.
2006年, 该课题组[24]进一步开发了另一新型手性镉配合物催化剂来催化TMSCN对氮杂环丙烷的开环反应(Eq. 6).催化剂采用的还是一类由葡萄糖衍生的手性配体L2(2-[3-(二苯基膦酰基)-2-羟基环己基-氧]-4, 5-二氟苯酚)与三异丙醇镉按1.5:1配合形成的具有多个金属催化中心的镉配合物.广普性考察发现无论是环烷烃还是脂肪链烃取代的氮杂环丙烷, 均有较好的适用性, 均可得到良好到优秀的收率(83%~99%)和对映选择性(87%~99%).
2006年, Komatsu课题组[25]报道了在水中于80 ℃条件下, 硅胶促进的KCN对氮杂环丙烷的开环反应(Eq. 7).反应具有高的区域选择性, 均于取代较少的碳上发生开环.广普性考察发现, 对于多种官能团取代的氮杂环丙烷, 反应均能获得24%~88%的收率.硅胶经分离后重复使用2次, 催化效果几乎没有改变.
2009年, Matsukawa课题组[26]报道了于50 ℃下在DMF溶剂中, 采用三(2, 4, 6-三甲氧基苯基)膦(TTMPP)作催化剂催化TMSCN对氮杂环丙烷的开环反应(Eq. 8).三(2, 4, 6-三甲氧基苯基)膦可通过与亲核试剂TMSCN中的硅形成O—Si和C—Si键来活化TMSCN.对于多数单取代和环上两个碳均被脂肪族链烃取代的氮杂环丙烷, 反应均获得了中等到优秀的收率(68%~99%).尤其对单官能团(包括苯基)取代的氮杂环丙烷, 反应表现出专一的区域选择性, 均于取代较少的碳上发生开环反应.相对于2005年Komatsu开发的方法, 该反应采用了具有更强路易斯碱性的有机膦(TTMPP)作催化剂, 无需加入KCN作为促进剂同时也降低有机碱的用量, 反应体系更为简单.
2009[27]和2014[28]年, RajanBabu课题组报道了钇盐与联萘胺和邻羟基苯甲醛衍生制得的Salen配体形成的催化剂Cat. 2催化TMSCN在二氯乙烷溶剂中于室温条件下对2, 3-二取代氮杂环丙烷的开环反应(Eq. 9).广普性考察发现对于三种环烯衍生的氮杂环丙烷反应均能获得85%~87%的收率和92%~99%的对映选择性; 而2, 3-二正丙基取代的氮杂环丙烷反应仅获得47%的收率和82%的对映选择性.
2012年, Matsukawa课题组[29]报道了聚苯乙烯负载有机碱1, 5, 7-三氮杂二环[4, 4, 0]5-十烯(TBD)作催化剂(PS-TBD)催化TMSCN对氮杂环丙烷的开环反应(Eq. 10).该反应用5 mol%的PS-TBD在N, N-二甲基甲酰胺(DMF)溶剂中于80 ℃下进行, 对于单官能团取代的氮杂环丙烷反应表现出优秀的区域选择性(>98:2), 开环发生在位阻较小的碳上(包括苯基), 且得到良好到优秀的收率(85%~95%).对于2位和3位二取代的氮杂环丙烷, 除2, 3-二苯基取代的氮杂环丙烷获得12%的收率外, 其他都能得到89%~95%的收率.实验表明这种聚合物负载的催化剂经由简单的分离纯化而重复使用4次, 催化活性没有显著下降.
2 sp2杂化的碳原子对氮杂环丙烷的开环反应
2.1 苯及其衍生物对氮杂环丙烷的开环反应
苯环及其衍生物也可以对氮杂环丙烷进行开环, 自1996年Mann课题组[30]之后, 许多课题组就此类化合物对氮杂环丙烷的开环反应进行了研究, 其中导向碳氢活化开环反应尤其受到人们的关注.
2001年, Yadav课题组[31]报道了首例芳环碳对2-芳基氮杂环丙烷的开环反应(Eq. 11).该反应采用In(OTf)3作催化剂, 室温下即能快速得到芳环碳对氮杂环丙烷的开环产物.亲核试剂芳环上取代基位阻较小时, 则主要于氮杂环丙烷苄基位开环, 产物的选择性为93:7~100:0;而当亲核试剂芳环上取代基位阻较大时, 在生成苄基位开环产物同时有大量的亚甲基位碳开环产物生成, 区域选择性有所下降, 得到中等到良好的选择性(70:30~90:10).该反应对于芳环上带有吸电子取代基的氮杂环丙烷, 虽然需要更长的反应时间; 而对于未活化的芳环, 尽管需要增加催化剂用量, 但对于所考察的各种取代基均具有良好的适用性均能得到中等到优秀的收率(68%~92%).
2004年, Bergmeier课题组[32]报道了BF3·Et2O作促进剂促进的分子内含π键官能团的亲核试剂在二氯甲烷溶剂中于0 ℃下对氮杂环丙烷的开环反应.当亲核试剂为苯基衍生物时, 开环发生在氮杂环丙烷取代基较多的碳上; 当亲核试剂为烯基双键时, 则开环发生在取代基较少的碳上.普适性考察表明反应对含π键官能团亲核试剂上多种取代基均具有较好的适用性, 芳环上碳作为亲核碳原子时能得到一般到优秀的收率(45%~98%) (Eq. 12), 而烯基碳作为亲核碳原子时, 收率会有所下降得到9%~75%的收率(Eq. 13).
2007年, Roy课题组[33]报道了AgPF6催化芳环碳对2-芳基取代氮杂环丙烷的开环反应, 反应仅发生芳环或芳杂环的2位碳对氮杂环丙烷的苄基位开环, 且反应具有良好的收率(75%~85%) (Eq. 14).研究发现可能由于水或醇等含氧类溶剂中氧的配位作用, 当反应体系中有水或醇存在时导致反应不能发生.广普性考察发现, 对于苯环上用对甲基或对氯官能团取代的氮杂环丙烷作底物, 当采用给电子取代基活化的芳环或芳杂环作亲核试剂时, 反应均表现出良好的兼容性, 取得良好的反应收率, 得到的结果都比采用2-苯基氮杂环丙烷作底物或用未活化的芳环或芳杂环作亲核试剂时的好.
2007年, Wu课题组[34]报道了使用AuCl3和AgOTf作共催化剂在溶剂硝基甲烷中于室温条件下, 芳环碳对氮杂环丙烷的开环反应(Eq. 15).广普性考察发现脂肪族取代基取代的氮杂环丙烷在该条件下不能得到相应的产物, 仅芳香族取代基取代的氮杂环丙烷能够获得64%~95%的收率且仅得到于苄基位开环的产物.对亲核试剂芳烃的考察发现富电子芳烃均能得到64%~95%的收率, 而带有吸电子官能团的芳烃无法得到相应的产物.杂环芳烃呋喃得到呋喃环2位参与反应的产物, 收率为64%.相比于之前报道的芳烃对氮杂环丙烷的开环反应, 该方法操作简单, 所需催化剂量少但催化活性高, 反应可在2 min内完成.
2008年, Wu课题组[35]又报道了采用FeCl3作催化剂在硝基甲烷溶剂中于室温条件下催化芳环碳对芳基取代氮杂环丙烷高选择性于苄基位的开环反应(Eq. 16).研究发现当使用的催化剂量较小时, 由于产物本身的碱性, 延缓了反应的顺利进行.广普性研究表明对于缺电子芳烃和某些杂环芳烃, 相关反应不能得到相应的产物或仅得到中等收率的产物(56%~77%), 而富电子取代基取代的芳环则能得到较好的收率(82%~90%).对氮杂环丙烷上取代基的考察表明; 2位苯基上的取代基对反应结果没有显著影响, 对氮上保护基的考察发现当氮上保护基换成硝基苯磺酰基时, 也能得到相应的产物, 收率为78%.当保护基为苄基时, 则不能得到相应的产物.相比于该课题组2007年报道的方法, 该报道所用催化剂更为廉价环保且能够获得相似的高催化活性.
2009年, Dineen课题组[36]报道了采用BF3·Et2O作促进剂于-78 ℃在甲苯溶剂中促进2, 6-二取代碘苯与正丁基锂试剂原位生成的锂亲核试剂对单取代氮杂环丙烷发生的碳开环反应(Scheme 3).对氮杂环丙烷取代基的研究发现, 反应对于苯基以及其他空间位阻不同的取代基均能得到良好的收率(79%~85%).此外, 对于不同的氮杂环丙烷保护基以及碘苯衍生物上的取代基进行了考察, 反应也能获得一般到良好的收率(30%~89%).反应生成的邻溴苯乙胺在Pd(OAc)2的催化作用下, 可进一步关环生成吲哚啉, 具有一定的合成应用价值.
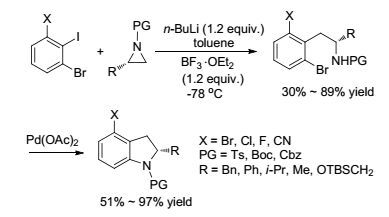 图式 3
碘苯对氮杂环丙烷的开环反应
Scheme3.
Ring-opening reactions of aziridines by iodobenzene
图式 3
碘苯对氮杂环丙烷的开环反应
Scheme3.
Ring-opening reactions of aziridines by iodobenzene
2009年, Zhu课题组[37]报道了使用溴化铜作催化剂, 在氯化锂与氢化二异丁基铝(DIBAL)的共同作用下, 利用芳基溴试剂于四氢呋喃中原位生成的有机镁试剂对2-酯基取代氮杂环丙烷的开环反应(Eq. 17).开环发生在取代较少的碳上, 反应具有高度的立体选择性并可取得80%的收率.
2010年, Roy课题组[38]报道了Ag[(COD)2]PF6催化芳香族化合物芳环碳对2-芳基取代氮杂环丙烷的开环反应(Scheme 4).在温和的条件下, 一系列取代芳基与氮杂环丙烷反应, 均得到80%左右的收率, 活化的芳香族化合物参与反应时, 反应时间短, 收率也高(80%~88%).反应也表现出专一的区域选择性, 氮杂环丙烷开环位置集中在苄基位上.此外, 该反应条件也适用于呋喃和噻吩等芳香杂环类化合物, 反应取得75%~85%的收率, 脂肪取代基取代的氮杂环丙烷却得不到相应产物.作者又利用NMR (H, P, Ag)和ESI-MS对催化剂对底物活化的本质和断键成键过程进行了研究, 提出反应关键步骤在于催化剂经过解离得到Ag(COD)2+, 并进一步得到活性中间体Ag(COD)+, 它去螯合氮杂环丙烷和芳环从而来稳固反应过渡态.
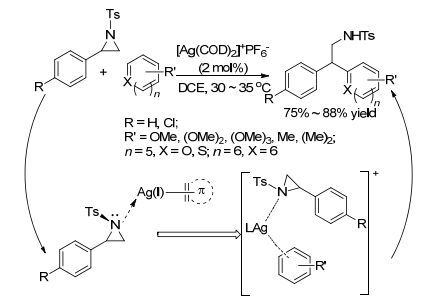 图式 4
Ag[(COD)2]PF6催化芳香族化合物对氮杂环丙烷的开环反应
Scheme4.
Ag[(COD)2]PF6-catalyzed ring-opening reactions of aziridines by aromatic compounds
图式 4
Ag[(COD)2]PF6催化芳香族化合物对氮杂环丙烷的开环反应
Scheme4.
Ag[(COD)2]PF6-catalyzed ring-opening reactions of aziridines by aromatic compounds
2012年, Ha课题组[39]报道了CuI作促进剂参与的格氏试剂对2-取代氮杂环丙烷的开环反应(Scheme 5).由于氮杂环丙烷氮上的保护基为供电子取代基, 所以需要经过三氟甲磺酸甲酯的活化, 才能得到氮原子上四取代季铵盐型氮杂环丙烷8, 该中间体再与格氏试剂发生开环反应.反应表现出专一的区域和对映选择性, 当氮杂环丙烷被烷基取代时, 开环反应发生在取代较少的碳原子上.当取代基是烷氧酰基或乙烯基时, 开环反应发生在取代较多的碳原子上.反应广普性考察表明烷基、烷氧酰基或烯基取代的氮杂环丙烷与各种格氏试剂反应均能得到中等到优秀的收率(43%~90%).该反应虽然采用了活性稍逊于吸电子官能团的烷基保护基保护的氮杂环丙烷, 然而通过三氟甲磺酸酯的活化作用, 也能得到相应的开环产物, 在一定程度上丰富了氮杂环丙烷的种类.
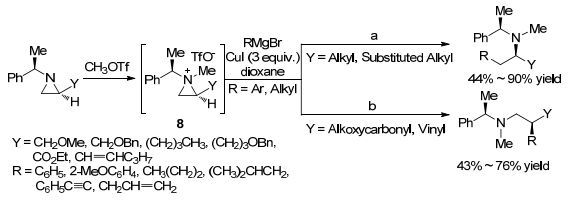 图式 5
CuI促进的格氏试剂对氮杂环丙烷的开环反应
Scheme5.
CuI-promoted ring-opening reactions of aziridines by Grignard reagents
图式 5
CuI促进的格氏试剂对氮杂环丙烷的开环反应
Scheme5.
CuI-promoted ring-opening reactions of aziridines by Grignard reagents
2013年, Doyle课题组[40]报道了镍催化剂催化的有机锌金属试剂对N-2-(反-2-(甲氧羰基)乙烯基)苯磺酰基保护的2位官能团取代的氮杂环丙烷的开环反应(Eq. 18).该研究发现添加大量的LiCl能够显著提高开环产物的收率, 这可能是由于LiCl能与氮杂环丙烷反应生成β-氯胺中间体, 而该中间体更容易与亲核试剂偶联生成相应产物.底物扩展实验表明, 除2-甲基喹诺啉有机锌试剂得到26%的收率外, 其他有机锌试剂均能得到中等到良好的收率(50%~80%).对于多种官能团取代的氮杂环丙烷研究发现环己基和苯基取代的氮杂环丙烷仅得到39%和43%的收率, 而链状烷烃取代基取代的氮杂环丙烷得到中等的收率(53%~69%).除苯基取代的氮杂环丙烷主要得到支链产物(1:>20) 外, 其他氮杂环丙烷都主要生成直链产物并具有一般到良好的选择性(1.5:1~3.6:1).
2013年, Jain课题组[41]报道了富电子芳环及芳香杂环在Zn(OTf)2和Sc(OTf)3的联合催化作用下对氮杂环丙烷的开环反应, 芳环主要对苄基位的碳发生亲核进攻(Eq. 19).对氮杂环丙烷氮上保护基和碳上各种取代基的考察发现, 保护基和取代基的种类对反应结果几乎没有影响, 反应均可得到65%~97%的收率.对亲核试剂的考察发现, 除芳香杂环取得的收率较低(65%~75%)外, 芳环亲核试剂均能得到良好到优秀的收率(85%~97%).作者还考察了使用单一异构体的手性氮杂环丙烷作底物时反应的情况, 反应仅取得43%~83%的对映选择性.进一步研究发现, 原料和产物在反应过程中均会发生消旋化, 因此导致产物选择性下降.当使用反式C-2, C-3二取代氮杂环丙烷的单一异构体作底物时, 原料不会发生消旋化, 反应能得到相应单一异构体的产物.本文是截止2013年报道的首例芳香碳亲核试剂对氮杂环丙烷发生的立体选择性开环反应, 反应具有中等到良好的对映选择性.
2013年, Michael课题组[42]报道了双(二亚苄基丙酮)钯[Pd(dba)2]与配体三(1-萘基)膦配合形成的催化剂催化2-烷基取代N-对硝基苯磺酰基氮杂环丙烷与芳基硼酸的开环反应(Eq. 20).反应表现出优秀的区域选择性:开环主要发生在取代较少的碳原子上.研究发现, 当反应中加入三(1-萘基)膦(PNp3)作配体时, 其空间位阻能够抑制活性中间态发生β-氢消除使反应向生成开环产物的方向进行.该反应不同于需要加入当量级碱的传统偶联反应, 由于离去基团磺酰胺的碱性, 此反应仅需加入催化量的Cs2CO3即可.值得一提的是, 间氯苯酚对于提高反应收率是不可或缺的.广普性研究发现富电子基取代的苯硼酸比缺电子基取代的苯硼酸能取得更高的收率, 且反应对苯硼酸上许多敏感取代基如羰基、酰胺、酯和酚羟基等均表现出良好的兼容性, 得到43%~87%的收率.对于未取代和单取代的氮杂环丙烷, 反应得到63%~94%的收率和高的区域选择性(>20:1).相比于同年Doyle课题组[40]报道的有机锌试剂对氮杂环丙烷的开环反应, 该条件下反应能够获得更高的区域选择性.
2013年, Li课题组[43]报道了(CpRhCl2)2/AgSbF6催化剂体系催化的利用吡啶及其衍生物作为导向官能团构建的2-芳基吡啶化合物芳环碳对2-芳基取代氮杂环丙烷的开环反应(Eq. 21).广普性实验考察发现, 反应对于芳基吡啶类底物吡啶环上的吸电子或供电子取代基均具有较好的兼容性, 得到中等到优秀的收率(60%~90%).对于芳基吡啶类底物苯环上间位、对位的吸电子或给电子取代基也具有较好的兼容性, 得到中等到良好的收率(61%~84%).对于间位上的取代基, 除F外, 其它均于空间位阻较小的邻位发生反应, 而当F在间位时则由于其具有较强的配位作用, 主要于位阻较大的邻位发生反应.当苯环上邻位有取代基时, 由于位阻较大几乎得不到相应产物.当苯环换成五元芳杂环时也能得到61%和74%收率的相应开环产物.此外, 导向基团并不局限于吡啶, 其他氮杂芳基导向基也能同样活化邻位C—H键, 并取得58%~63%的收率.对氮杂环丙烷氮上保护基和2位芳基考察表明反应条件对烷基磺酰基和芳基磺酰基保护的氮杂环丙烷均适用, 反应均发生在苄基位碳原子上, 得到48%~84%的收率.尽管在此之前已有多篇关于芳环与氮杂环丙烷的开环反应, 但仅限于富电子芳烃, 本文报道了缺电子芳烃在此类反应中的应用, 一定程度上丰富了此类反应的应用前景.
2014年, Yoshikai课题组[44]报道了利用CoCl2、IPr·HCl和t-BuCH2MgBr原位生成的催化剂催化2-芳基吡啶的芳环邻位碳对2-芳基氮杂环丙烷的开环反应, 反应主要发生在氮杂环丙烷苄基位碳上(Eq. 22).反应中2-芳基吡啶与氮杂环丙烷的比例为1:2, CoCl2、IPr·HCl与t-BuCH2MgBr的比例为1:1:36.实验发现如果降低氮杂环丙烷或t-BuCH2MgBr的量都会导致收率的明显下降.对2-芳基吡啶考察发现, 苯环对位给电子官能团取代的底物仅能得到较差到一般的收率(21%~54%).位阻效应对反应有较大的影响, 苯环上C-3位甲基取代的底物主要发生苯环位阻较小邻位碳参与的反应, 苯环邻位取代的底物和吡啶C-3位取代的底物均只得到痕量产物, 而吡啶环C-4位取代基对反应效果没有影响.电子效应的考察表明吸电子取代基取代的芳环底物得不到相应产物.对氮杂环丙烷考察发现, 对于带有供电子或中性取代基取代的芳基反应能得到63%~67%的收率, 而吸电子取代基取代的芳基底物则需要较长的反应时间, 收率最高才能达到46%.对氮杂环丙烷氮上保护基的考察表明, 除了烷基和磺酰基保护的氮杂环丙烷得不到相应产物外, 其它各类芳基保护基对反应结果都没有显著影响.竞争实验研究发现, 氮杂环丙烷芳环取代基上供电子取代基取代的底物活性优于吸电子取代基取代的底物, 同样值得注意的是钴倾向于活化酸性强的C—H键.
2014年, Minakata课题组[45]报道了利用氮杂环卡宾(NHC)作配体与氯化钯盐形成的催化剂催化芳基硼酸对2-芳基氮杂环丙烷的开环反应, 生成手性2-二芳基乙基胺衍生物(Eq. 23).反应中所使用的配体对反应结果有显著影响:使用三烷基膦作配体时主要生成了β-H消除产物, 使用NHC与钯盐形成的催化剂则能够抑制β-H的消除反应只得到目标的开环产物.反应广普性考察表明, 在使用Cat. 3的最优条件下, 对位带有供电子或中性取代基的苯硼酸能够得到手性转变的产物且具有高的对映选择性(99%), 收率为74%~98%.该条件也适用于某些间位取代的苯硼酸以及稠环与芳香杂环的硼酸试剂, 收率为91%~97%, 选择性均为99%.对于2位取代和2, 3位二取代的氮杂环丙烷, 均表现出高的区域选择性, 得到收率为62%~91%和对映选择性均为99%的开环产物.然而, 缺电子的苯硼酸反应慢收率低, 对反应条件再优化发现钯络合的配体对反应有显著影响, 且当采用氮杂环卡宾与预先耦合了烯丙基配体的钯配合物新配合形成的Cat. 4时有最好的效果可得46%~84%的收率, 98%~99%的对映选择性.
2015年, Bari课题组[46]报道了在路易斯酸Cu(O-Tf)2、InCl3和FeCl3分别催化作用下, 芳香族化合物对氮杂环丙烷的开环反应制得各种丙氨酸酯衍生物(Eq. 24).反应广普性考察表明, 三种路易斯酸催化剂均能催化该反应.对于多种芳香杂环和各种取代基取代的芳环均表现出一定的适用性. InCl3催化该反应能得到中等到良好的收率(60%~88%), 而Cu(OTf)2和FeCl3催化该反应则得到稍微低的反应收率.实验中所使用的氮杂环丙烷由丝氨酸制备, 经一定的反应过程实现了由丝氨酸合成丙氨酸酯.
2.2 吲哚对氮杂环丙烷的开环反应
吲哚对氮杂环丙烷发生开环反应得到的色胺类化合物是一类重要的合成中间体, 被广泛地用于药物和天然产物的合成中.近年来, 人们越来越重视吲哚环上特定位点对氮杂环丙烷的开环反应研究, 同时也构建了多种新方法, 这些方法可用于对药物中间体的修饰.
2003年, Farr课题组[47]报道了在1.1 equiv. BF3·Et2O的促进作用下, 2-芳基吲哚3位碳对2-官能团取代氮杂环丙烷的开环反应, 反应表现出优秀的区域选择性和对映选择性, 得到单一异构体的β-胺类化合物(Eq. 25).广普性考察表明, 苯基取代的氮杂环丙烷反应时间短, 可得到66%的收率和92%的对映选择性, 脂肪官能团取代的氮杂环丙烷反应时间较长, 其中取代基α位带有支链时反应时间尤其长且收率较差(<48%), 然而可取得优秀的对映选择性(>99%).
2008年, Tse课题组[48]报道了利用活化硅胶作催化剂催化吲哚衍生物对由吲哚衍生的氮杂环丙烷选择性苄基位开环生成反式不对称双吲哚化合物的反应(Eq. 26).该反应不需要溶剂, 使用活化的硅胶作非均相催化剂就能催化反应, 且在开环的同时氮杂环丙烷上两个氮的保护基叔丁氧羰基可以被脱去.广普性考察表明吲哚环上被多种官能团如卤素、甲基、烷氧基或酯基等取代基取代时, 均能得到中等到优秀的收率(57%~99%).
2013年, Hutton课题组[49]报道了在1 equiv.路易斯酸Sc(OTf)3作用下吲哚对N-酯基保护的2-酯基取代氮杂环丙烷开环生成色氨酸衍生物的反应(Eq. 27).对一系列路易斯酸的筛选研究表明, 亲氧路易斯酸促进剂有利于开环反应, 亲氮路易斯酸促进剂则有利于扩环反应, 且Sc(OTf)3最利于开环反应, 然而会生成两种开环产物.
2014年, Wang课题组[50]报道了利用二丁基镁与手性喹啉(L3)作配体形成的催化剂在对二甲苯溶剂中于30 ℃下催化吲哚3位碳对N-(2-吡啶羰基)保护的氮杂环丙烷发生的不对称开环反应(Scheme 6).底物扩展表明, 当吲哚4, 5, 6位被取代时, 无论取代基为吸电子基还是供电子基, 反应均能得到74%~94%的收率和75%~94%的对映选择性, 而7位上被甲基取代时仅能获得36%的收率, 但仍可得到80%的对映选择性.五元和六元环并氮杂环丙烷分别能得到74%与80%的收率和91%与95%的对映选择性, 而七元环并氮杂环丙烷由于容易发生扩环反应, 收率仅为31%, 对映选择性为85%; 2, 3-二取代的非环氮杂环丙烷均能得到良好的收率(71%~83%)和良好到优秀的对映选择性(81%~93%).对氮上保护基中吡啶环上官能团考察表明除5位溴取代时会导致对映选择性显著下降(70%)外, 不同电子性质和取代位置的官能团均能得到一般到良好的收率(63%~88%)和良好到优秀的对映选择性(80%~94%).作者采用合适的手性配体完成了至2014年首例吲哚对内消旋氮杂环丙烷的不对称开环反应, 所得产物在一定条件下可进一步发生环加成反应得到吡咯吲哚啉.
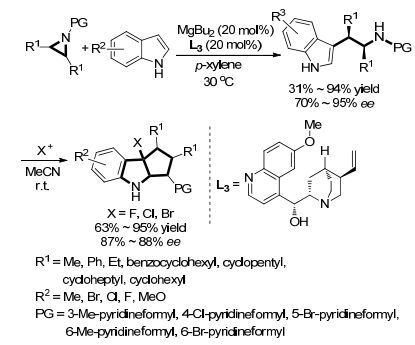 图式 6
吲哚对氮杂环丙烷的开环反应
Scheme6.
Ring-opening reactions of aziridines by indoles
图式 6
吲哚对氮杂环丙烷的开环反应
Scheme6.
Ring-opening reactions of aziridines by indoles
2014年, Hanamoto课题组[51]报道了在1.3 equiv. Et2Zn的作用下于甲苯溶剂中100 ℃条件下, 吲哚3位碳对2-三氟甲基氮杂环丙烷选择性于取代较少碳上发生的开环反应(Eq. 28).广普性考察发现对于多种官能团取代的吲哚, 除羟基取代的底物不能得到相应产物外, 其它均能得到94%~99%的收率.对吲哚氮上保护基的考察发现, 当氮上是甲基取代时不能得到相应产物, 因此氮上无保护基对反应的顺利进行至关重要.该反应操作简单、收率高、对多种官能团均有一定的兼容性, 且合成的三氟甲基取代的色胺衍生物在药物合成上有一定的应用价值.
2014年, You课题组[52]报道了使用Sc(OTf)3作催化剂在甲苯溶剂中于室温条件下, 2, 3-二取代吲哚对氮杂环丙烷的开环反应(Eq. 29).主要得到吲哚6位碳对氮杂环丙烷的开环产物9, 此外还有吲哚5位碳开环的产物10, 控制实验发现所得到的开环产物均是由环加成产物转变而来.广普性考察表明对于多种芳基取代的氮杂环丙烷和多种官能团取代的吲哚, 反应均能得到62%~93%的收率和2.5:1~>20:1的区域选择性, 而烷基取代的氮杂环丙烷则不能发生反应.
2015年, Tokuyama课题组[53]报道了使用MgBr2作催化剂, 格氏试剂MeMgBr作添加剂在乙醚溶剂中于室温条件下, 各种官能团取代的吲哚3位碳对氮杂环丙烷的开环反应, 生成色氨醇衍生物(Scheme 7).对反应广普性研究表明, 吲哚上官能团的电子性质和取代位置对反应收率的影响较小, 所考察的底物均能得到50%~73%的收率, 且该反应条件还适用于某些较为敏感的官能团, 如氯、溴和碘等, 也都能取得中等的收率(59%~70%).
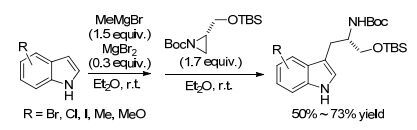 图式 7
吲哚对氮杂环丙烷的开环反应
Scheme7.
Ring-opening reactions of aziridines by indoles
图式 7
吲哚对氮杂环丙烷的开环反应
Scheme7.
Ring-opening reactions of aziridines by indoles
2016年, Majee课题组[54]报道了利用咪唑形成的两性季铵盐4-(3-甲基咪唑)丁烷磺酸盐(MBS)作催化剂催化吲哚及其衍生物3位碳对芳香氮杂环丙烷发生选择性的苄基位开环反应(Eq. 30).广普性考察发现氮上甲基保护的吲哚能得到81%的收率, 对吲哚苯环上多种官能团, 反应均能获得80%~84%的收率.对于2-位取代的氮杂环丙烷反应也均能得到79%~84%的收率, 而且对于3-羟甲基与3-苯甲酰基取代的氮杂环丙烷也可以得到良好的收率(76%~82%).该反应不需要使用溶剂, 且反应条件适用于多种亲核试剂对氮杂环丙烷的开环反应, 具有一定的实用性.
2016年, Morgan课题组[55]报道了使用1 equiv. BF3·Et2O作促进剂在二氯甲烷溶剂中于-78 ℃条件下, 吲哚3位碳对氮杂环丙烷的开环反应(Eq. 31).反应采用了外消旋的N-对硝基苄氧酰基-2-甲基氮杂环丙烷和N-对硝基苄氧酰基环己烯氮杂环丙烷两种底物, 均能与多种官能团取代的吲哚发生反应得到48%~90%的收率, 且对于同一吲哚, N-对硝基苄氧酰基-2-甲基氮杂环丙烷能够得到更高的收率.所得产物可在温和条件下脱除氮上保护基得到β-取代的色胺衍生物.作者引入对硝基苄氧酰基作为氮杂环丙烷氮上的保护基, 不仅能活化氮杂环丙烷、所得产物也不易发生重排且能在温和条件下脱去, 具有较高的合成应用价值.
2016年, Jia课题组[56]报道了以Cu(CH3CN)4BF4作路易斯酸, L4作配体形成的催化剂在溶剂甲苯中于室温条件下, 吲哚3位碳对2-芳基取代氮杂环丙烷的开环反应(Eq. 32).广普性考察发现对于多种官能团取代的氮杂环丙烷, 反应均能获得65%~88%的收率和80%~95%的对映选择性, 除4-乙酰氧基苯基、2-甲基苯基和2-萘基取代的氮杂环丙烷主要得到苄基位开环产物且区域选择性为5:1~13:1外, 其余官能团取代的氮杂环丙烷仅得到苄基位开环的产物.考察还发现吸电子官能团取代的氮杂环丙烷能够获得更高的对映选择性, 然而取代基的空间位阻越大反应的对映选择性越低.氮杂环丙烷氮上保护基对对映选择性也有显著影响, 当保护基为对甲苯磺酰基时, 反应能获得最高的对映选择性.对吲哚的考察发现多种官能团取代的吲哚反应均能获得75%~89%的收率、86%~>99%的对映选择性和专一地于苄基位开环的产物.
2016年, Rossi课题组[57]报道了Au(JohnPhos)NTf2作催化剂在二氯甲烷溶剂中于80 ℃条件下, 吲哚对氮杂环丙烷的开环反应.广普性考察发现对于多种官能团取代的吲哚和所考察的氮杂环丙烷反应能够获得30%~98%的收率, 其中未取代的氮杂环丙烷, 反应能够获得62%~71%的收率(Eq. 33).当使用手性氮杂环丙烷作为底物时, 反应能够得到手性反转的相应产物(Eq. 34).
2016年, Hyland课题组[58]报道了Pd(CH3CN)Cl2作路易斯酸, 1, 4-苯醌作配体催化吲哚3位碳对氮杂环丙烷的开环反应, 反应发生在环的苄基位上.首先, 作者考察了2-苯基氮杂环丙烷与吲哚的开环反应, 优化条件时发现, 加入30 mol%的1, 4-苯醌能够显著提高反应收率.广普性考察发现对于多种芳基官能团取代的氮杂环丙烷和吲哚, 反应均能获得50%~86%的收率(Eq. 35), 吡咯与2-苯基氮杂环丙烷的开环反应仅获得15%的收率.对2, 2-二取代氮杂环丙烷与吲哚的开环反应, 广普性考察发现多种官能团取代的吲哚反应均能获得44%~93%的收率(Eq. 36).
2.3 萘对氮杂环丙烷的开环反应
2015年, Wang课题组[59]报道了利用由邻羟基苯乙酸衍生制得的2-(4, 5-二氢-4-(1-甲基乙基)-2-噁唑基)苯酚(Box-OH)作配体与MgBu2以1:1比例原位生成的镁催化剂, 催化β-萘酚羟基邻位碳对N-吡啶酰基保护的2, 3-二取代氮杂环丙烷的开环反应(Eq. 37).对氮杂环丙烷的扩展发现, 除七元环并氮杂环丙烷由于环张力导致收率较低(48%), 其他氮杂环丙烷均能得到优秀的收率(93%~99%)、对映选择性(>99%)和非对映选择性(>20:1).对于β-萘酚考察发现, 其1位上取代基的空间位阻仅对非对映选择性有一定的影响, 非对映选择性降至11:1~18:1, 但仍能得到97%~99%的收率和97%~>99%的对映选择性.当3位官能团为氢和氯时, 相应产物的收率(59%~74%)低于其他官能团取代时的收率(80%~99%), 且当官能团为氢和苯乙炔基时, 相应产物的非对映选择性(4.6:1~14:1) 低于其他官能团取代时的非对映选择性(18:1~>20:1), 但所有情况下均能获得91%~>99%的对映选择性.通过用1H NMR对反应过程进行研究发现, 反应中配体的酚羟基和β-萘酚都会与镁发生配位, 镁配合物则作为路易斯酸在配位后进一步活化氮杂环丙烷.通过对手性配体和产物ee值的对比, 得到它们存在正非线性关系, 它揭示了去芳基化过程是在单一异构体的镁催化剂二聚合物作用下发生的, 而非单一异构体的镁复合物由于键合太紧而致催化活性过低以至于不能催化反应.
2.4 杂芳香碳对氮杂环丙烷的开环反应
2002年, Yadav课题组[60]报道了温和条件下, InCl3分别催化吲哚、吡咯、呋喃和噻吩等芳香杂环化合物对氮杂环丙烷的开环反应(Eq. 38).对催化剂的筛选发现, 三氟甲磺酸盐作催化剂会导致吡咯和呋喃等的分解, 金属卤化物中, InCl3有更好的催化活性.反应广普性考察表明对于脂肪取代基取代的氮杂环丙烷, 开环反应发生在亚甲基位碳上收率为70%~85%, 当环烯衍生制得的氮杂环丙烷作底物时, 仅得到芳香杂环化合物3位碳参与的开环反应产物, 而由链烯制备的氮杂环丙烷作底物时, 则得到芳香杂环化合物2位与3位碳上同时反应的产物, 且以2位碳上反应产物为主, 区域选择性为60:40~90:10.芳香取代基取代的氮杂环丙烷主要得到氮杂环丙烷苄基位碳与芳香杂环化合物2位碳参与反应的产物, 收率为80%~90%.该报道是作者在2001年报道的基础上, 进一步将铟盐催化剂的应用范围扩展到多种芳香杂环对氮杂环丙烷的开环反应中.
2.5 双键对氮杂环丙烷的开环反应
2010年, Yadav课题组[61]报道了在四氢呋喃和叔丁醇组成的混合溶剂中于室温条件下由Cat. 5和1, 8-二氮杂桥[5.4.0]十二-7-烯(DBU)组成的催化剂共同催化α, β-不饱和烯基醛对2-芳基取代氮杂环丙烷的开环反应, 仅能得到于氮杂环丙烷取代较少碳上的开环产物(Eq. 39).广普性考察发现对于多种官能团取代的氮杂环丙烷和α, β-不饱和烯基醛反应均能获得76%~94%的收率.
2016年, Yang课题组[62]报道了以Bu2Mg和L5作催化剂在溶剂甲苯中于30 ℃条件下γ-丁内酰胺对氮杂环丙烷的开环反应(Eq. 40).广普性考察发现, 反应使用R型或S型配体均能得到相应的产物, 且能获得相似的收率, 然而产物的非对映选择性相反.对于多种氮杂环丙烷, 当使用S型配体L5时, 反应均能获得30%~83%的收率和90:10~98.5:1.5的非对映选择性; 当使用R型配体L5时, 反应均能获得30%~89%的收率和11.5:88.5~3:97的非对映选择性.该反应是第一例羰基α位sp2杂化碳原子对氮杂环丙烷的开环反应, 扩大了碳亲核试剂的种类.
3 sp3杂化的碳原子对氮杂环丙烷的开环反应
3.1 甲基亲核试剂对氮杂环丙烷的开环
2011年, Hou课题组[63]报道了在n-BuLi和六甲基膦酰胺(HMPA)作用下在溶剂甲基叔丁基醚(MTBE)中于-78 ℃条件下酮肟醚α-碳对氮杂环丙烷的开环反应.优化条件中发现单独使用n-BuLi时反应不发生, 而加入碱性较弱的HMPA后能够得到相应的产物, 可能原因是具有更强螯合能力的HMPA能够破坏过于稳定的中间体结构从而提高中间体的亲核性.广普性考察发现, 对于多种官能团取代的氮杂环丙烷和苯乙酮肟醚, 反应均能获得41%~78%的收率.除苯乙烯氮杂环丙烷主要得到于取代较少碳上的开环产物且有2.1:1的区域选择性外, 其余氮杂环丙烷均于取代较少碳上发生开环反应(Eq. 41).此外对于四氢萘酮肟醚分别与1-己烯和1-辛烯衍生氮杂环丙烷的反应也能获得45%和50%的收率(Eq. 42).
3.2 活泼亚甲基亲核试剂对氮杂环丙烷的开环
活泼亚甲基作为一种重要的潜手性中心砌块, 是一类具有较强亲核性的反应试剂, 可与多种亲电试剂发生反应制备手性化合物.其对氮杂环丙烷的开环反应产物中所具有的C—C—C—N结构是许多药物和天然化合物的重要结构组成部分.
2004年, Kimpe课题组[64]报道了有机铜锂试剂在醚类溶剂中对N-芳磺酰基保护的2-溴甲基氮杂环丙烷的开环反应(Scheme 8).该氮杂环丙烷有三个亲电中心, 所得开环产物受有机铜锂试剂用量的控制:当铜锂试剂用量为1 equiv.时, 仅得到氮杂环丙烷取代亚甲基上的溴被取代的产物, 而当其用量大于2 equiv.时, 则在发生对溴的取代反应后, 进一步得到烷基铜锂试剂对取代后氮杂环丙烷的开环产物收率为68%~79%.
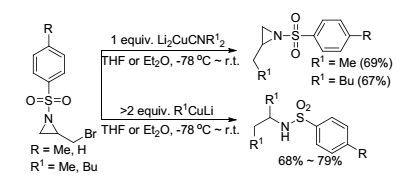 图式 8
有机铜锂试剂对氮杂环丙烷的开环反应
Scheme8.
Ring-opening reactions of aziridines by lithium organocuprate reagents
图式 8
有机铜锂试剂对氮杂环丙烷的开环反应
Scheme8.
Ring-opening reactions of aziridines by lithium organocuprate reagents
2007年, Yudin课题组[65]报道了用潜手性亲核试剂芳香杂环取代的活泼亚甲基化合物对环烷烃取代的氮杂环丙烷在强碱和相转移催化剂作用下的碳开环反应(Eq. 43).前期实验中, 作者发现通过碱和温度的调节, 反应都会得到多种开环产物, 目标产物的收率仅为17%, 而当在反应体系中加入相转移催化剂时能显著把目标产物的收率提高到94%, 在该条件下, 能成功避免目标开环反应过程中的副反应.广普性考察发现对于多种芳香杂环取代的碳亲核试剂, 反应能得到收率为30%~96%反式结构的产物, 并且具有良好到优秀的非对映选择性80:20~>99:1.对碳上无取代基的氮杂环丙烷, 反应也可得到61%的收率.此外, 该反应产物的立体选择性受动力学控制, 且在反应条件下, 产物不会发生差向异构化.
2011年, Yadav课题组[66]报道了于四氢呋喃溶剂中在1.1 equiv. t-BuOK的作用下, 芳基乙腈的亚甲基碳对氮杂环丙烷的开环反应.底物扩展表明, 对于2-取代和2, 3-二取代的氮杂环丙烷反应均能得到95%~>99%的收率(Eq. 44), 当使用单一异构体的氮杂环丙烷作底物时, 能得到相应的单一异构体开环产物且选择性为98%~>99% (Eq. 45).
2011年, Shibasaki课题组[67]报道了利用La(O-i-Pr)3、Yb(OTf)3与席夫碱以1:1:1的比例形成的双中心催化剂催化氮杂环丙烷与丙二酸酯的不对称开环反应(Eq. 46).研究发现两种稀土金属对该双核催化剂的活性不可或缺, 且表现为它们的路易斯酸性和阴离子的Brønsted碱性越强, 收率越高.对于环烷烃、脂肪链烃和杂环烷烃取代的氮杂环丙烷与多种丙二酸酯, 反应均能得到63%~99%的收率和97%~99.5%的立体选择性.
2012年, 我们课题组[68]报道了KOH在丙酮溶剂中于35 ℃下催化马来酸酯的羰基α-碳对氮杂环丙烷的开环反应(Eq. 47).广普性考察表明, 脂肪取代基取代的氮杂环丙烷只在亚甲基碳上发生开环, 反应收率为82%~99%.对芳香取代基苯环上多种官能团的考察表明官能团的电子效应和空间位阻对收率有显著影响, 含有供电子官能团的芳基氮杂环丙烷能得到65%~84%的收率, 而含有吸电子官能团的芳基取代氮杂环丙烷能获得73%~96%的收率, 苯环对位官能团取代的氮杂环丙烷(89%~96%)比苯环邻位和间位含有官能团的氮杂环丙烷也可获得更高的收率(65%~84%).这些反应都可取得良好到优秀的区域选择性(19:81~7:93), 反应优先于苄基位进行.实验结果证明该反应条件也适用于芳香杂环取代的底物和乙酰乙酸乙酯等其他的亚甲基化合物, 并取得较高的收率(83%~85%).
2014年, Matsunaga课题组[69]报道了利用路易斯酸Y(OTf)3、La(O-i-Pr)3与席夫碱以1:1:1的比例形成的双核催化剂催化丙二酸酯中的α-碳对外消旋氮杂环丙烷的开环反应(Scheme 9).反应中需要加入4-二甲胺基吡啶(DMAP)来抑制路易斯酸进一步催化开环产物重排产生噁唑啉以此提高开环产物收率.在不同的反应条件下, 氮杂环丙烷C-2、C-3位的开环反应都有可能发生, 分别生成相应的开环产物.研究发现结构为(2R, 3S)的氮杂环丙烷参与反应时, 选择性生成产物16, 而立体结构为(2S, 3R)的氮杂环丙烷参与反应时, 则专一性地生成产物17.广普性研究发现, 该反应对于丙二酸酯的空间位阻较为敏感, 随着位阻的增大, 反应时间显著, 增加收率也有所下降, 然而仍能获得优秀的对映选择性(99%~>99.5%).环烷烃、2-烷基和2, 3-二烷基取代的氮杂环丙烷都只分别得到较差到一般的收率(36%~49%), 但都能获得优秀的对映选择性(96%~>99.5%).该反应是在2011年[63]的基础上, 将该催化体系应用到丙二酸酯对外消旋氮杂环丙烷的开环反应中.
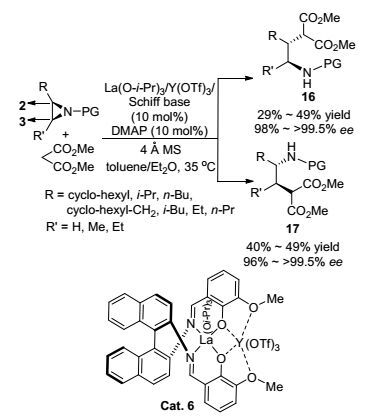 图式 9
腈的α-碳对氮杂环丙烷的开环反应
Scheme9.
Ring-opening reactions of aziridines by α-carbon of nitriles
图式 9
腈的α-碳对氮杂环丙烷的开环反应
Scheme9.
Ring-opening reactions of aziridines by α-carbon of nitriles
2015年, Krische课题组[70]报道了由[Ir(cod)Cl]2、乙酸烷基酯、4-氰基-3-硝基苯甲酸在四氢呋喃溶剂中于60 ℃下与(R)-甲氧基-2, 2′-二(二苯基膦基)-1, 1′-二苯基腈(BIPHEP)制备的手性铱催化剂来催化N-对硝基苯磺酰基保护的乙烯基氮杂环丙烷, 分别与一级醇和醛发生的碳开环反应(Eq. 48).研究发现该催化剂能有效避免醇羟基的烷基化, 且在该体系中, 不添加K3PO4会导致收率显著下降.广普性研究表明, 对多种官能团取代的伯醇反应都得到中等到优秀的收率(64%~96%), 优秀的非对映选择性(4:1~9:1) 和对映选择性(94%~99%); 对多种官能团取代的醛, 反应也得到中等到优秀的收率(63%~96%)和高的非对映选择性(4:1~9:1) 和对映选择性(91%~99%) (Eq. 49).作者还进一步对二元醇进行了探讨, 发现需要延长反应时间, 在某些情况下还需要调整反应条件, 反应才能得到中等的收率(60%~68%)和较好的非对映选择性(4:1~7:1) (Eq. 50).
3.3 有机金属试剂对氮杂环丙烷的开环
许多科研工作者很早就对有机金属试剂对氮杂环丙烷的开环反应进行过一些探究, 如1995年Ibuka课题组[71]、1996年Fan课题组[72]、2001年Simpson课题组[73]等都对此类反应做出了一定的贡献, 开发出了铜、镁、锂、锌等多种金属有机试剂, 然而反应普遍在低温下才能进行.近年来, 常温下有机金属试剂参与的反应受到人们的关注, 其中有机锌试剂尤为重要.
2012年, Doyle课题组[74]报道了用NiCl2·glyme作路易斯酸, 反丁烯二酸二甲酯作配体, 有机锌试剂对2-芳基取代氮杂环丙烷的开环反应(Eq. 51).前期实验发现缺电子烯烃能加速催化剂与氮杂环丙烷络合物的还原消除反应, 从而促进有机锌试剂对氮杂环丙烷的亲核进攻生成目标产物.底物扩展表明:该法不适用于脂肪基取代的氮杂环丙烷, 然而对于芳环取代基, 当苯环间位和对位上有吸电子或供电子官能团时, 反应取得了55%~84%的收率, 即使是某些较为敏感的官能团如氨基、醛基和酮等, 也都能获得58%~80%的收率.当采用2, 3-二取代的氮杂环丙烷作底物时, 增加催化剂量后, 反应也能获得52%的收率, 对映选择性为99%的顺式主产物(Eq. 52).当使用苯基溴化锌作试剂与苯乙烯衍生的氮杂环丙烷反应仅能取得48%的收率.
2014年, Jamison课题组[75]报道了利用NiCl2与配体(四甲基邻菲罗啉)形成的催化剂催化烷基溴化锌在对2-烷基取代氮杂环丙烷发生的碳开环反应(Eq. 53).该反应于26 ℃下在二氯乙烷与四氢呋喃的混合溶剂中进行.作者选用NiCl2作路易斯酸, 四甲基邻菲罗啉作配体, 反应能得到令人满意的效果.实验发现NiCl2和配体按照不同的比例加入在不同的温度下络合能够得到不同的催化剂, 且对反应有显著影响.加入过量的配体得到的催化剂有利于实验结果, 而加入过量的金属盐得到的催化剂则不利于实验结果.广普性考察表明, 对于各种脂肪基甚至取代基上含有某些敏感官能团(如氯、酰胺和酯等)取代的氮杂环丙烷, 都表现出良好的适用性, 均能得到中等到优秀(66%~97%)的收率和优秀的区域选择性(>20:1), 各种有机溴化锌的反应也能得到70%~95%的收率.
2015年, Doyle课题组[76]报道了利用Ni(acac)2与反丁烯二酸酯(Fro-DO)配合形成的催化剂催化有机锌试剂对2, 2-二取代氮杂环丙烷区域选择性地于取代较多的苄基位碳上发生开环反应(Eq. 54).该反应于23 ℃下在DMA中进行, 广普性研究发现反应条件对于氮杂环丙烷C-2上取代芳基芳环的间位和对位官能团均有良好的普适性, 可取得57%~74%的收率, 且对于某些含有敏感官能团如氯或酰胺等的底物也能给出47%~74%的收率, 4-三氟甲基吡啶取代的底物也能得到71%的收率, 然而官能团在取代基邻位时由于位阻效应则结果令人失望(<5%~25%); 烷基溴化锌均能得到52%~86%的收率, 值得注意的是, 对某些有合成价值官能团如乙缩醛基和氯等均有较好的兼容性(52%~86%), 邻甲苯基溴化锌作底物时收率为31%, 而当采用三氟甲基苯基和间二甲基苯基溴化锌作底物时, 收率分别为51%和56%.该课题组已经报道了多篇镍盐与含有双键的配体形成的催化剂催化有机锌试剂对多种单取代、2, 3-二取代和2, 2-二取代氮杂环丙烷的开环反应, 此催化体系对多种氮杂环丙烷具有较为广泛地应用性.
3.4 活泼次甲基亲核试剂对氮杂环丙烷的开环
活泼次甲基在一定条件下易脱除氢离子再对氮杂环丙烷进行开环就可引入手性中心, 可得到含有多个手性中心的具有C—C—C—N结构的化合物, 在天然产物和药物的不对称合成中有独特的应用价值.
2008年, Dixon课题组[77]报道了α-茚酮甲酸叔丁酯在相转移催化剂Cat. 7作用下, 于K2HPO4溶液中对碳上没有取代基的氮杂环丙烷发生的开环反应(Eq. 55).前期实验发现反应转化率受氮杂环丙烷氮上保护基的影响较大, 将氮杂环丙烷氮上保护基由4-吗啉乙基磺酰基换成邻三氟苯基磺酰基后, 能够显著提高原料转化率.反应广普性考察表明, 多种官能团取代的α-茚酮甲酸酯均能得到中等到良好的收率(78%~92%)和良好到优秀的对映选择性(82%~>95%).对于2, 2-二脂肪基取代的氮杂环丙烷开环选择性发生在取代较少的碳上, 且能得到72%~95%的收率和9:2~30:1的非对映选择性(Eq. 56).本文是首例手性相转移催化剂催化活泼次甲基对氮杂环丙烷发生的开环反应, 反应条件温和操作简单具有一定的应用前景.
同年, 该课题组[78]还报道了利用碱2-叔丁亚胺基-2-二乙胺基-1, 3-二甲基全氢-1, 3, 2-二氮杂膦(BEMP)在四氢呋喃溶剂中催化含活泼次甲基的亲核试剂对氮杂环丙烷的开环反应(Eq. 57).对碱的研究发现得益于BEMP较强的碱性和弱的亲核性, 它可作为有效的催化剂来利用.对氮杂环丙烷氮上保护基的考察表明, 对甲苯磺酰基和均三甲苯磺酰基作保护基时, 开环反应能顺利地发生, 且无氮杂环丙烷聚合反应的发生.该反应条件对于含有酯基、内酰胺基和氰基等官能团的活泼次甲基化合物均表现出一定的兼容性, 可得到44%~99%的收率.
2013年, Ooi课题组[79]报道了利用1, 2, 3-三氮唑溴化铵盐作催化剂催化3-取代-2-羰基吲哚的3位碳对2, 2-二取代氮杂环丙烷的开环反应来制得多手性中心的开环产物(Eq. 58), 开环反应选择性地发生在氮杂环丙烷苄基位碳上.对催化剂的考察表明:三氮唑环C-4位上相连苯环的对位和三氮唑环N-3位上苄基苯环带有强吸电子官能团如三氟甲基时, 能促进反应的发生并提高反应的非对映选择性和对映选择性.广普性考察发现, 对于2位芳基苯环上多种官能团取代的氮杂环丙烷和多种官能团取代的2-羰基吲哚, 反应均得到71%~97%的收率, 11:1~>20:1的非对映选择性以及99%的对映选择性.尽管随着吲哚衍生物取代基空间位阻的不断增大, 收率会下降, 然而仍能保持高的对映选择性.动力学研究表明, 该反应对催化剂表现出伪一级反应, 对氮杂环丙烷和吲哚表现出零级反应, 增加K2CO3的量, 反应表现为一级反应, 而不加催化剂时则不发生反应, 意味着控速步骤不是形成碳碳键而是形成烯醇钾盐这一步.
2013年, Dixon课题组[80]报道了于25 ℃下在四氢呋喃溶剂中使用BEMP催化活泼次甲基对氮杂环丙烷开环生成连于季碳的乙撑胺产物(Eq. 59).通过对氮杂环丙烷保护基的筛选发现反应采用均三甲苯磺酰基作保护基时能得到最好的收率(98%).广普性实验表明, 反应条件对酯、内酰胺和酰胺等许多敏感基团取代的亲核试剂均有良好的适用性, 取代基的电子效应对反应也没有显著影响, 均能得到73%~99%的收率.基于以上结果, 该研究又使用手性氯代季铵盐Cat. 7作催化剂催化氮杂环丙烷的不对称开环反应, 所考察的反应都得到中等到良好的收率(78%~88%)和良好到优秀的对映选择性(82%~97%) (Eq. 60).对于2, 2-二取代的氮杂环丙烷开环选择性发生在取代较少的碳上且能得到72%~95%的收率和9:2~30:1的非对映选择性(Eq. 61).
2015年, Wang课题组[81]还报道了利用Bu2Mg和(R)-联萘酚(L7)原位形成的镁催化剂来催化3-芳基-2-羰基苯并呋喃对2-吡啶酰基保护氮杂环丙烷的开环反应(Eq. 62).实验发现该反应不同于多数其他反应中联萘酚(BINOL)的规律即位阻越大, 选择性越高, 当该反应中使用3, 3′-二取代的BINOL时会明显降低反应的选择性.另外, 当反应中加入痕量的水时, 收率和对映选择性都会发生显著的降低.此外, 基于对比N-苯甲酰基保护氮杂环丙烷的实验, 发现2-吡啶酰基的二齿配位能力在反应中起到重要作用.对一系列环烷烃或非环烷烃取代的氮杂环丙烷考察表明, 反应均得到中等到良好的收率(44%~81%), 良好到优秀的非对映选择性(9:1~>20:1) 和对映选择性(87:13~99.5:0.5).对苯并呋喃酮扩展发现C-3上芳基取代的底物会导致非对映选择性的显著下降(5:1), 而C-5和C-7上的取代基则具有较好的兼容性可取得优秀的非对映选择性(12:1~>20:1), C-3, C-5, C-7位取代的苯并呋喃酮也能得到50%~80%的收率和78:22~96:4的对映选择性.
同年, 该课题组[82]采用Bu2Mg和手性3, 3′-二氟-(R)-联萘酚(L8)原位生成的Mg催化剂催化3-异硫氰酸基氧化吲哚3位碳对氮杂环丙烷的开环反应(Scheme 10).对配体的考察发现BINOL的C-3和C-3′位上取代基位阻小比较好, 当取代基为F时, 标准反应能得到96%的收率, >20:1的非对映选择性和97%的对映选择性, 作者认为这是由于F取代的BINOL配体具有更强的路易斯酸性所致.对底物的扩展实验表明, 除底物为五元环并氮杂环丙烷收率较差(63%)外, 其它反应均能得到90%~96%的收率, 但所有底物均能获得>20:1的非对映选择性和88%~98%的对映选择性.所得到的开环产物, 由于含有异硫氰酸酯, 可用于合成肽衍生物, 也可用于合成脲类衍生物.
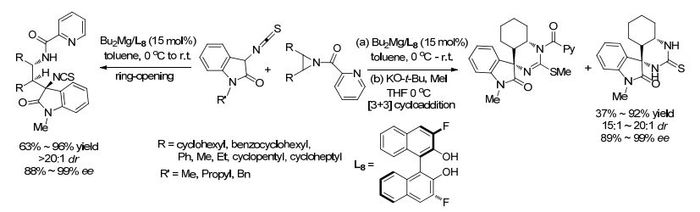 图式 10
氧化吲哚对氮杂环丙烷的开环反应
Scheme10.
Ring-opening reactions of aziridines by oxindoles
图式 10
氧化吲哚对氮杂环丙烷的开环反应
Scheme10.
Ring-opening reactions of aziridines by oxindoles
2016年, 该课题组[83]利用Bu2Mg与3, 3′-二氟-(R)-联萘酚(L8)原位生成的镁催化剂催化3-芳基氧化吲哚对氮杂环丙烷的开环反应, 生成具有多手性中心的3-烷基-3-芳基氧化吲哚衍生物(Eq. 63).对多种芳酰基保护的氮杂环丙烷的考察发现, 仅当芳基为吡啶时, 由于吡啶能够促进底物与催化剂镁中心的螯合从而促进不对称开环来得到相应的产物.广普性考察发现五元、七元和苯并六元氮杂环丙烷参与反应时需要升高温度至60 ℃, 其余取代的氮杂环丙烷室温下即能反应, 均能得到50%~86%的收率, >20:1的非对映选择性和96~>99%的对映选择性; 氧化吲哚C-3位上多种官能团如烯丙基、苄基和苯基等对反应的对映选择性没有影响, C-5, C-6, C-7位上官能团取代的底物也均能得到44%~94%的收率, >20:1的非对映选择性和72%~>99%的对映选择性.
4 结论与展望
氮杂环丙烷的碳亲核开环反应在碳碳键偶联和合成含有氮原子的化合物方面有其独特的价值, 近年来越来越受到人们的重视, 所用碳亲核试剂也向着带有多样化官能团的方向发展, 用于构建具有更广泛应用价值的功能分子.整体而言, 芳烃以及杂环芳烃类化合物对氮杂环丙烷的开环反应得到了更为广泛的关注, 反应向着导向催化、碳氢活化以及合成带有手性中心化合物的方向发展.饱和碳对氮杂环丙烷的开环反应更多地集中于使用有机金属试剂, 氰基对氮杂环丙烷的开环反应也得到了发展, 主要用于构建多种光学活性的腈类化合物.但是, 非金属试剂的饱和烷烃、烯烃类、炔烃类试剂则由于诸多因素的限制, 发展相对比较缓慢, 而且用它们来构建手性化合物的应用就更少.如今, 开发各种各样亲核试剂对氮杂环丙烷的碳开环反应是构建许多功能性有机分子的必要补充, 尤其是在手性催化剂作用下, 构建不对称碳开环反应更是发展的重点和难点.随着有机化学的不断发展, 会有许多亲核试剂被开发出来用于对氮杂环丙烷的开环反应中来制取各种多官能团的化合物.
-
-
[1]
Feng, J. J.; Zhang, J. L. ACS catal. 2016, 6, 6651. doi: 10.1021/acscatal.6b02072
-
[2]
王清宇, 常宏宏, 魏文龙, 刘强, 高文超, 李彦威, 李兴, 有机化学, 2016, 36, 939. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345388.shtmlWang, Q. Y.; Chang, H. H.; Wei, W. L.; Liu, Q.; Gao, W. C.; Li, Y. W.; Li, X. Chin. J. Org. Chem. 2016, 36, 939(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345388.shtml
-
[3]
Cardoso, L. A.; Pinhoe Melo, M. V. D. T. Eur. J. Org. Chem. 2012, 33, 6479.
-
[4]
Subrahmanyam, G.; Adhya, A. J. Sci. Ind. Res. Indian 1974, 33, 308.
-
[5]
Ha, H. J.; Jung, J. H.; Lee, W. K. Asian J. Org. Chem. 2014, 3, 1020. doi: 10.1002/ajoc.v3.10
-
[6]
Luginina, J.; Turks, M. Chem. Heterocycl. Compd. 2016, 10, 773.
-
[7]
Pineschi, M. Synlett 2014, 1817.
-
[8]
Turks, M.; Posevins, D. Eur. J. Org. Chem. 2016, 2016, 1760. doi: 10.1002/ejoc.v2016.9
-
[9]
Benjamin, L.; Richard, G.; Markus, L.; Riccardo, M. M. Angew. Chem., Int. Ed. 2014, 53, 7063. doi: 10.1002/anie.201400169
-
[10]
Hammond, B. G.; Xu, B.; Mashuta, S. M.; Okoromoba, E. O. Chem. Commun. 2016, 52, 13353. doi: 10.1039/C6CC07855A
-
[11]
Wang, R.; Jiang, X. X.; Liu, X.; Li, D. Chem.-Eur. J. 2016, 22, 17141. doi: 10.1002/chem.201603898
-
[12]
Murty, M. S. R.; Yadav, J. S. Tetrahedron Lett. 2005, 46, 6385. doi: 10.1016/j.tetlet.2005.06.165
-
[13]
Feng, X. M.; Lin, L. L.; Liu, X. H.; Li, J. Chem. Commun. 2014, 50, 6672. doi: 10.1039/c4cc02206h
-
[14]
李彦威, 苏继娟, 常宏宏, 李兴, 魏文龙, 广州化工, 2011, 39, 12. doi: 10.3969/j.issn.1001-9677.2011.05.005Li, Y. W.; Su, J. J.; Chang, H. H.; Li, X.; Wei, W. L. Chem. Ind. 2011, 39, 12(in Chinese). doi: 10.3969/j.issn.1001-9677.2011.05.005
-
[15]
Schneider, C. Angew. Chem., Int. Ed. 2009, 48, 2082. doi: 10.1002/anie.v48:12
-
[16]
Hu, X. E. Tetrahedron 2004, 60, 2701. doi: 10.1016/j.tet.2004.01.042
-
[17]
McCoull, W.; Davis, F. A. Synthesis 2000, 1347.
-
[18]
Ding, C. H.; Dai, L. X.; Hou, X. L. Synlett 2004, 1691.
-
[19]
Ding, C. H.; Dai, L. X.; Hou, X. L. Tetrahedron 2005, 61, 9586. doi: 10.1016/j.tet.2005.07.076
-
[20]
Bertolini, F.; Woodward, S.; Crotti, S.; Pineschi, M. Tetrahedron Lett. 2009, 50, 4515. doi: 10.1016/j.tetlet.2009.05.081
-
[21]
Wu, J.; Hou, X. L.; Dai, L. X. J. Org. Chem. 2000, 65, 1344. doi: 10.1021/jo9913816
-
[22]
Mita, T.; Fujimori, I.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 11252. doi: 10.1021/ja053486y
-
[23]
Minakata, S.; Okada, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2005, 7, 3509. doi: 10.1021/ol051186f
-
[24]
Fujimori, I.; Mita, T.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 16438. doi: 10.1021/ja067003h
-
[25]
Minakata, S.; Hotta, T.; Oderaotoshi, Y.; Komatsu, M. J. Org. Chem. 2006, 71, 7471. doi: 10.1021/jo061239m
-
[26]
Matsukawa, S.; Tsukamoto, K. Org. Biomol. Chem. 2009, 7, 3792. doi: 10.1039/b908322g
-
[27]
Wu, B.; Gallucci, C. J.; Parquette, R. J.; RajanBabu, V. T. Angew. Chem., Int. Ed. 2009, 48, 1126. doi: 10.1002/anie.v48:6
-
[28]
Wu, B.; Gallucci, C. J.; Parquette, R. J.; RajanBabu, V. T. Chem. Sci. 2014, 5, 1102. doi: 10.1039/C3SC52929K
-
[29]
Matsukawa, S.; Harada, T.; Yasuda, S. Org. Biomol. Chem. 2012, 10, 4886. doi: 10.1039/c2ob25435b
-
[30]
Schneider, M. R.; Mann, A.; Taddei, M. Tetrahedron Lett. 1996, 37, 8493. doi: 10.1016/0040-4039(96)01975-2
-
[31]
Yadav, J. S.; Reddy, S. V. B.; Nagaiah, K. Tetrahedron Lett. 2001, 42, 8067. doi: 10.1016/S0040-4039(01)01699-9
-
[32]
Bergmeier, C. S.; Katz, J. S.; Donoghue, J. P.; Reed, D. D. Tetrahedron Lett. 2004, 45, 5011. doi: 10.1016/j.tetlet.2004.05.009
-
[33]
Bera, M.; Roy, S. Tetrahedron Lett. 2007, 48, 7144. doi: 10.1016/j.tetlet.2007.07.200
-
[34]
Sun, X. Y.; Sun, W.; Fan, R. H.; Wu, J. Adv. Synth. Catal. 2007, 349, 2151. doi: 10.1002/(ISSN)1615-4169
-
[35]
Wang, Z. Y.; Sun, X. Y.; Wu, J. Tetrahedron 2008, 64, 5013. doi: 10.1016/j.tet.2008.03.081
-
[36]
Michaelis, J. D.; Dineen, A. T. Tetrahedron Lett. 2009, 50, 1920. doi: 10.1016/j.tetlet.2009.02.047
-
[37]
Wu, Y. C.; Zhu, J. P. Org. Lett. 2009, 11, 5558. doi: 10.1021/ol9024919
-
[38]
Bera, M.; Roy, S. J. Org. Chem. 2010, 75, 4402. doi: 10.1021/jo100260k
-
[39]
Yoon, D. H.; Lee, K. W.; Kim, Y.; Ha, H. J. Org. Lett. 2012, 14, 429. doi: 10.1021/ol202683k
-
[40]
Nielsen, K. D.; Huang, C. Y.; Doyle, G. A. J. Am. Chem. Soc. 2013, 135, 13605. doi: 10.1021/ja4076716
-
[41]
Ghorai, K. M.; Tiwari, P. D.; Jain, N. J. Org. Chem. 2013, 78, 7121. doi: 10.1021/jo401028j
-
[42]
Duda, L. M.; Michael, E. F. J. Am. Chem. Soc. 2013, 135, 18347. doi: 10.1021/ja410686v
-
[43]
Li, X. W.; Yu, S. J.; Wan, B. S.; Yu, X. Z. Angew. Chem., Int. Ed. 2013, 52, 2577. doi: 10.1002/anie.v52.9
-
[44]
Gao, K.; Paira, R.; Yoshikai, N. Adv. Synth. Catal. 2014, 356, 1486. doi: 10.1002/adsc.v356.7
-
[45]
Takeda, Y.; Ikeda, Y.; Kuroda, A.; Minakata, S. J. Am. Chem. Soc. 2014, 136, 8544. doi: 10.1021/ja5039616
-
[46]
Chaudhari, P.; Bari, S. Synth. Commun. 2015, 45, 391. doi: 10.1080/00397911.2014.965328
-
[47]
Farr, N. R.; Alabaster, J. R.; Johnson, A. S.; Grabowski, J. J. E. Tetrahedron:Asymmetry 2003, 14, 3503. doi: 10.1016/j.tetasy.2003.08.038
-
[48]
Kaiser, M. H.; Lo, F. W.; Beller, M. Tse, K. M. Org. Lett. 2006, 8, 5761. doi: 10.1021/ol062338p
-
[49]
Tirotta, I.; Fifer, L. N.; Hutton, A. C. Tetrahedron Lett. 2013, 54, 618. doi: 10.1016/j.tetlet.2012.11.139
-
[50]
Yang, D. X.; Wang, L. Q.; Han, F. X.; Wang, R. Chem.-Eur. J. 2014, 20, 16478. doi: 10.1002/chem.201404354
-
[51]
Hirotaki, K.; Yamada, Y.; Hanamoto, T. Asian J. Org. Chem. 2014, 3, 285. doi: 10.1002/ajoc.v3.3
-
[52]
Liu, H.; Zheng, C.; You, S. L. J. Org. Chem. 2014, 79, 1047. doi: 10.1021/jo402511b
-
[53]
Noji, T.; Okano, K.; Tokuyama, H. Tetrahedron 2015, 71, 3833. doi: 10.1016/j.tet.2015.04.015
-
[54]
Ghosal, C. N.; Santra, S.; Das, S.; Majee, A. Green Chem. 2016, 18, 565. doi: 10.1039/C5GC01323B
-
[55]
Kidd, J.; Maiden, K.; Morgan, B. J. Tetrahedron 2016, 72, 3802. doi: 10.1016/j.tet.2016.03.031
-
[56]
Ge, C.; Liu, R. R.; Gao, J. R.; Jia, Y. X. Org. Lett. 2016, 18, 3122. doi: 10.1021/acs.orglett.6b01317
-
[57]
Rossi, E.; Abbiati, G.; Dell'Acqua, M.; Negrato, M.; Paganoni, A.; Pirovano, V. Org. Biomol. Chem. 2016, 14, 6095. doi: 10.1039/C6OB00672H
-
[58]
Yin, J. X.; Hyland, J. T. C. Asian J. Org. Chem. 2016, 5, 1368. doi: 10.1002/ajoc.v5.11
-
[59]
Yang, D. X.; Wang, L. Q.; Han, F. X.; Wang, R. Angew. Chem., Int. Ed. 2015, 54, 2185. doi: 10.1002/anie.201410257
-
[60]
Yadav, J. S.; Reddy, S. V. B.; Nagaiah, K. Tetrahedron Lett. 2002, 43, 1565. doi: 10.1016/S0040-4039(02)00015-1
-
[61]
Yadav, D. S. L.; Rai, K. V.; Singh, S.; Singh, P. Tetrahedron Lett. 2010, 51, 1657. doi: 10.1016/j.tetlet.2010.01.058
-
[62]
Li, D.; Wang, Y. J.; Wang, L. Q.; Wang, J.; Wang, P. X.; Wang, K. Z.; Lin, L.; Liu, D. S.; Jiang, X. X.; Yang, D. X. Chem. Commun. 2016, 52, 9640. doi: 10.1039/C6CC02877B
-
[63]
陈冬冬, 丁昌华, 侯雪龙, 戴立信, 高等学校化学学报, 2011, 32, 694.Chen, D. D.; Ding, C. H.; Hou, X. L.; Dai, L. X. Chem. J. Chin. Univ. 2011, 32, 694(in Chinese).
-
[64]
D'hooghe, M.; Kerkaert, I.; Rottiers, M.; Kimpe, D. N. Tetrahedron 2004, 60, 3637. doi: 10.1016/j.tet.2004.02.059
-
[65]
Blyumin, V. E.; Gallon, J. H.; Yudin, K. A. Org. Lett. 2007, 9, 4677. doi: 10.1021/ol7015302
-
[66]
Ghorai, K. M.; Nanaji, Y.; Yadav, K. A. Org. Lett. 2011, 13, 4256. doi: 10.1021/ol2016077
-
[67]
Xu, Y. J.; Lin, L. Q.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2011, 133, 5791. doi: 10.1021/ja201492x
-
[68]
Li, X.; Su, J. J.; Chang, H. H. Wei, W. L. J. Mol. Catal. A:Chem. 2012, 363~364, 446.
-
[69]
Xu, Y. J.; Shibasaki, M.; Matsunaga, S. J. Am. Chem. Soc. 2014, 136, 9190. doi: 10.1021/ja5039165
-
[70]
Wang, G.; Franke, J.; Krische, J. M. J. Am. Chem. Soc. 2015, 137, 7915. doi: 10.1021/jacs.5b04404
-
[71]
Fujii, N.; Nakai, K.; Tamamura, H.; Otaka, A.; Mimura, N.; Yamamoto, Y.; Ibuka, T. J. Chem. Soc., Pekin Trans. 1 1995, 1359.
-
[72]
Hudlicky, T.; Tian, X. R.; Konisberger, K.; Maurya, R. Fan, B. J. Am. Chem. Soc. 1996, 118, 10752. doi: 10.1021/ja960596j
-
[73]
Penkett, C. S.; Simpson, I. D. Tetrahedron Lett. 2001, 42, 1179. doi: 10.1016/S0040-4039(00)02202-4
-
[74]
Huang, C. Y.; Doyle, G. A. J. Am. Chem. Soc. 2012, 134, 9541. doi: 10.1021/ja3013825
-
[75]
Jensen, L. K.; Standley, A. E.; Jamison, F. T. J. Am. Chem. Soc. 2014, 136, 11145. doi: 10.1021/ja505823s
-
[76]
Huang, C. Y.; Doyle, G. A. J. Am. Chem. Soc. 2015, 137, 5638. doi: 10.1021/jacs.5b02503
-
[77]
Moss, A. T.; Fenwick, R. D.; Dixon, J. D. J. Am. Chem. Soc. 2008, 130, 10076. doi: 10.1021/ja8036965
-
[78]
Moss, A. T.; Alba, A.; Dixon, J. D. Chem. Commun. 2008, 21, 2474.
-
[79]
Ohmatsu, K.; Ando, Y.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 18706. doi: 10.1021/ja411647x
-
[80]
Moss, A. T.; Barber, M. D.; Dixon, J. D. Chem.-Eur. J. 2013, 19, 3071. doi: 10.1002/chem.201203825
-
[81]
Li, D.; Wang, L. Q.; Yang, D. X.; Zhang, B. Z.; Wang, R. ACS Catal. 2015, 5, 7432. doi: 10.1021/acscatal.5b02177
-
[82]
Wang, L. Q.; Yang, D. X.; Li, D.; Wang, R. Org. Lett. 2015, 17, 3004. doi: 10.1021/acs.orglett.5b01291
-
[83]
Wang, L. Q.; Li, D.; Su, W.; Wang, R. Chem. Asian J. 2016, 11, 691. doi: 10.1002/asia.201501369
-
[1]
-

图式 1 Cu(OTf)2促进的炔对氮杂环丙烷的开环反应
Scheme 1 Cu(OTf)2-promoted ring-opening reactions of aziridines by alkynes

图式 4 Ag[(COD)2]PF6催化芳香族化合物对氮杂环丙烷的开环反应
Scheme 4 Ag[(COD)2]PF6-catalyzed ring-opening reactions of aziridines by aromatic compounds

图式 5 CuI促进的格氏试剂对氮杂环丙烷的开环反应
Scheme 5 CuI-promoted ring-opening reactions of aziridines by Grignard reagents

图式 8 有机铜锂试剂对氮杂环丙烷的开环反应
Scheme 8 Ring-opening reactions of aziridines by lithium organocuprate reagents

图式 9 腈的α-碳对氮杂环丙烷的开环反应
Scheme 9 Ring-opening reactions of aziridines by α-carbon of nitriles
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 82
- 文章访问数: 5182
- HTML全文浏览量: 823
 下载:
下载:
