 图 1
Nocathiacin Ⅰ尾巴部分的化学半合成修饰
Figure 1.
Semisynthetic modifications of nocathiacin Ⅰ tails
图 1
Nocathiacin Ⅰ尾巴部分的化学半合成修饰
Figure 1.
Semisynthetic modifications of nocathiacin Ⅰ tails
引用本文:
王守锋, 郑庆飞, 段盼盼, 刘文. 硫肽类抗生素类似物合成进展[J]. 有机化学,
2017, 37(7): 1653-1666.
doi:
10.6023/cjoc201702017

Citation: Wang Shoufeng, Zheng Qingfei, Duan Panpan, Liu wen. Progress in Synthesis of Thiopeptide Antibiotics Analogues[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1653-1666. doi: 10.6023/cjoc201702017
Citation: Wang Shoufeng, Zheng Qingfei, Duan Panpan, Liu wen. Progress in Synthesis of Thiopeptide Antibiotics Analogues[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1653-1666. doi: 10.6023/cjoc201702017
硫肽类抗生素类似物合成进展
摘要:
硫肽类抗生素是一类富含硫元素且被高度修饰的聚噻(噁)唑多肽类天然产物,该家族化合物以其复杂的分子结构、良好的生物活性以及新颖的抗菌作用模式而成为研究热点.近年来,对于硫肽类抗生素类似物的合成研究发展迅速.综述了通过化学半合成、组合生物合成以及前体导向突变生物合成方法获得的硫肽类抗生素类似物的研究进展.
-
关键词:
- 硫肽类抗生素类似物
- / 化学半合成
- / 组合生物合成
- / 前体导向突变生物合成
English
Progress in Synthesis of Thiopeptide Antibiotics Analogues
Abstract:
Thiopeptide antibiotics, which are a growing class of sulfur-rich and highly modified polyazolyl peptide natural products, have been appreciated because of their complex structures, potent biological activities and unusual modes of action. Recently, a great deal of effort has been devoted to the development of various approaches for the efficient synthesis of thiopeptide antibiotics analogues. This review summarizes synthetic approaches towards thiopeptide antibiotics analogues via semisynthesis, combinatorial biosynthesis and precursor-directed mutasynthesis.
-
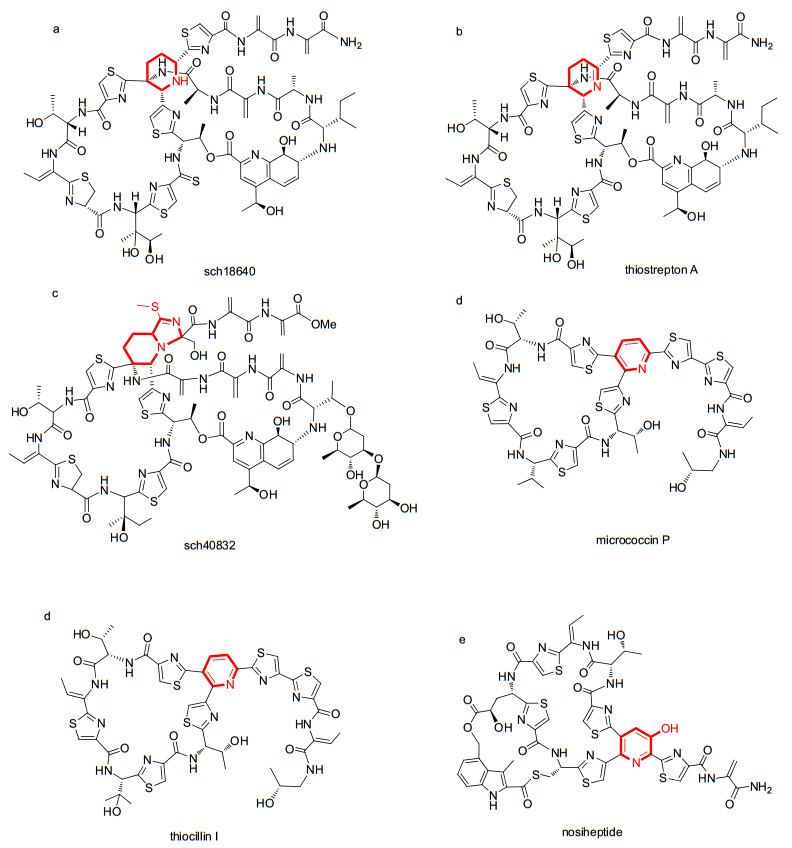
硫肽类抗生素是一类由微生物次级代谢产生的环肽类天然产物[1].自1948年第一例硫肽类抗生素micrococcin P被发现以来, 越来越多的硫肽类抗生素成员被报道, 到目前为止该家族已包含超过一百个成员[2].该家族化合物结构复杂, 富含多个噻唑(噁唑)或噻唑啉(噁唑啉)、多个脱水氨基酸及该家族化合物特有的氮六元杂环.根据其核心氮杂六元环取代方式及氧化程度的差异, Hensens等[3]首次将硫肽类抗生素分为a, b, c, d, e五个亚家族. a:哌啶类, 如sch18640; b:脱氢哌啶类, 如thiostreptons; c:二氢咪唑哌啶类, 如sch40832; d:三取代吡啶类, 如micrococcins和thiocillins; e:羟化吡啶类, 如nosiheptide(图 1).
图 1
Nocathiacin Ⅰ尾巴部分的化学半合成修饰
Figure 1.
Semisynthetic modifications of nocathiacin Ⅰ tails
硫肽类抗生素大多具有良好的抗革兰氏阳性菌生理活性, 尤其是对多种耐药致病菌如耐新青霉素金黄色葡萄球菌(MRSA), 耐青霉素肺炎链球菌(PRSP)和多耐药性粪肠球菌(MREF)等具有很好的抑制作用.研究发现, 硫肽类抗生素是以核糖体为靶点抑制蛋白质的合成从而达到抑菌的效果[4~6], 但其作用模式与目前临床上应用的抗生素均不同, 因此具有很大的成药潜力.尽管硫肽类抗生素具有良好的抗菌活性, 但是其水溶性差、生物利用度低, 从而限制了它们在临床上的应用.为了克服硫肽类抗生素成药性方面的不足, 人们尝试用各种化学合成和生物合成方法制备硫肽类抗生素类似物, 以便从中筛选活性好、水溶性好的潜在成药分子.
1 化学半合成
通过对硫肽类抗生素分子的局部基团进行化学修饰获得其类似物是硫肽类抗生素类似物化学半合成中普遍使用的方法.硫肽类抗生物分子结构中存在的活泼反应位点和官能团可以作为发展其类似物的切入点.化学半合成方法在nocathiacin Ⅰ, GE2270A及thiostrepton A类似物的合成中取得了丰硕的成果.
1.1 Nocathiacin Ⅰ类似物的化学半合成
Nocathiacin Ⅰ是一个高度后修饰的硫肽类抗生素, 其可发生化学修饰的位点如图 2.
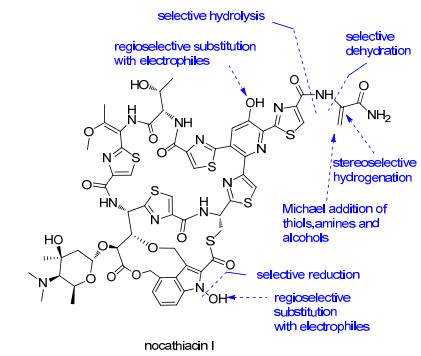 图 2
Nocathiacin Ⅰ的化学半合成修饰
Figure 2.
Semisynthetic modifications of nocathiacin Ⅰ
图 2
Nocathiacin Ⅰ的化学半合成修饰
Figure 2.
Semisynthetic modifications of nocathiacin Ⅰ
目前, 对该分子的化学修饰主要集中在空间位阻小的分子末端.最常用的修饰是亲核试剂对末端脱水丙氨酸(dehydroalanine, Dha)的Michael加成[7]. 2005年, Naidu小组[8]通过这种方法得到了胺基修饰化合物6和含硫醚化合物7, 化合物6可以进一步被修饰得到含尿素基团的化合物8, 而化合物7可以进一步转化为含砜基的化合物9. 2006年, Naidu小组[9]将末端双键立体专一性地还原获得化合物10, 化合物10可以进一步脱氨得末端羧酸化合物11, 在化合物11的基础上又可以引入水溶性基团得到化合物12 (Scheme 1).化合物8和9的抗菌活性相比于nocathiacin Ⅰ都有所降低, 降低为nocathiacin Ⅰ的十分之一.化合物12在抗菌活性保持的情况下水溶性提高了近10倍(表 1).
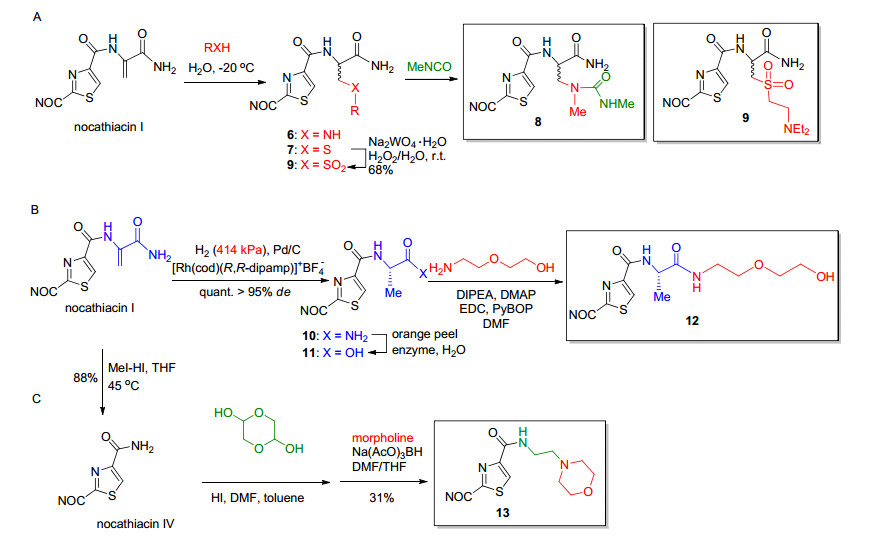 图式1
Nocathiacin Ⅰ尾巴部分的化学半合成修饰
图式1.
Semisynthetic modifications of nocathiacin Ⅰ tails
图式1
Nocathiacin Ⅰ尾巴部分的化学半合成修饰
图式1.
Semisynthetic modifications of nocathiacin Ⅰ tails
 表 1
Nocathiacin Ⅰ类似物的抑菌活性a
Table 1.
Inhibitory activity of nocathiacin Ⅰ analogues
表 1
Nocathiacin Ⅰ类似物的抑菌活性a
Table 1.
Inhibitory activity of nocathiacin Ⅰ analogues
化合物 Nocathiacin Ⅰ Nocathiacin Ⅳ 8 9 13 12 14 15 MICa/(μg•mL-1) 0.007 0.03 0.06 0.06 0.015 ND ND ND PD50b/(mg•kg-1) 0.8 NDb ND ND ND 1 3 0.15 aPD50 : dose required to cure 50% of the animals infected; MIC: minimum inhibitory concentration; bND: not test. 此外, nocathiacin Ⅰ的末端还可以选择性地降解获得两种类型的产物:一种是诺卡噻唑菌酸(nocathiacin acid), 另一种是相对应的末端酰胺化的诺卡噻唑菌素(nocathiacin Ⅳ)[10, 11]. 2007年, Liu小组[12, 13]报道了诺卡噻唑菌酸可以通过与含有胺基基团的化合物缩合获得进一步修饰的类似物. 2002年, Ueda小组[14]报道了诺卡噻唑菌素Ⅳ可以通过一锅法N-烷基化/还原胺化引入长的烷基链, 如化合物13 (Scheme 1).这些末端修饰获得了一些水溶性改善且具有良好生物活性的类似物, 有些对万古霉素抗性的致病菌有较好的活性(表 1)[15].
在对其环骨架修饰方面, Naidu等[16, 17]报道了两例大环上2-羟基吡啶和侧环上N-羟基吲哚的衍生, 其中有膦酰氧基甲基修饰的化合物14和15 (Eq. 1) 在提高了水溶性的同时都保持了良好的体内体外活性[16].由于膦酰氧基甲基醚能够被磷酸酶催化水解并释放nocathiacin Ⅰ, 因此这两种化合物具有成为nocathiacin Ⅰ前药的潜力[18].
1.2 GE2270A类似物的化学半合成
GE2270A(图 3)的结构中不包含有脱水氨基酸残基, 因此没有Michael加成的受体, 尽管如此, 该分子却是硫肽类抗生素成员中化学半合成研究最为详尽的硫肽分子之一[19], 这主要归结于该分子结构具有特殊的末端结构.其另外一个反应位点是大环骨架上的苯丝氨酸侧链[20, 21], 该结构单元可以被修饰甚至被转变成甘氨酸残基.由于对大环的修饰不能获得令人感兴趣的分子, 因此大部分对GE2270A的化学半合成工作集中在末端尾巴的修饰.
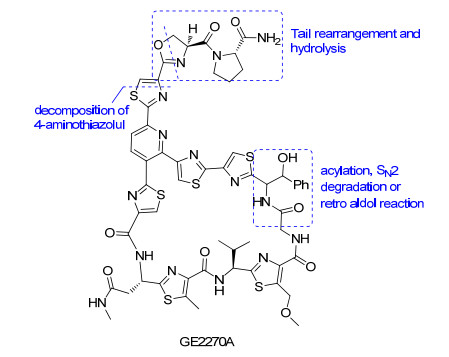 图 3
GE2270A的化学半合成修饰
Figure 3.
Semisynthetic modifications of GE2270A
图 3
GE2270A的化学半合成修饰
Figure 3.
Semisynthetic modifications of GE2270A
合成化学家对GE2270A末端的修饰主要是降解, 降解后的产物可以进一步发生官能团修饰(Scheme 2).对GE2270A的酸处理可以获得末端重排化合物17, 化合物17在碱性条件下可以水解形成化合物18或者被NaBH4还原为化合物19.化合物18可以与胺或者肽发生缩合, 化合物19也可与羧酸发生缩合反应.通过这种方法人们获得了大量的类似物, 但是仅有少数保留了活性并具有好的水溶性.
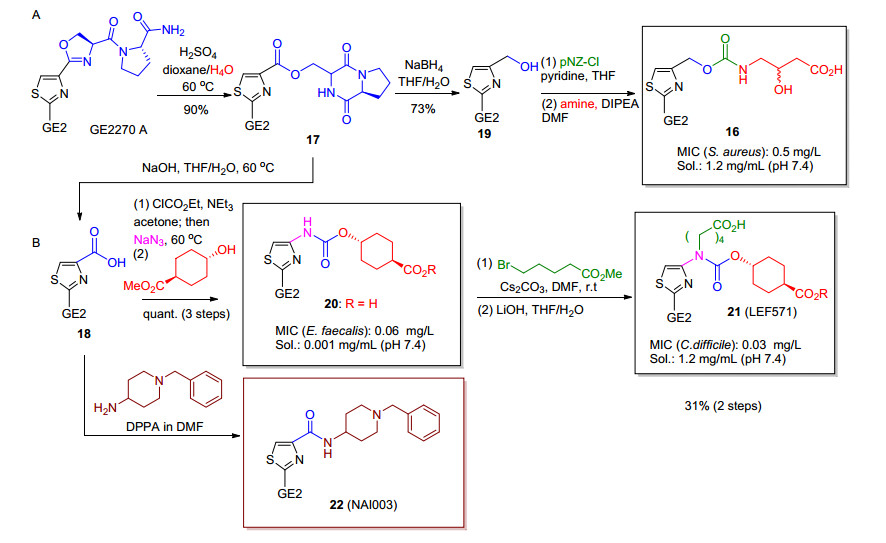 图式2
GE2270A尾巴部分的化学半合成修饰
图式2.
Semisynthetic modifications of GE2270A tails
图式2
GE2270A尾巴部分的化学半合成修饰
图式2.
Semisynthetic modifications of GE2270A tails
2011年以来, LaMarche等[22~24]对GE2270A末端的结构改造工作, 取得了显著的成果.通过Curtius重排, 他们将降解掉末端尾巴的18转化为4-胺基噻唑类似物.他们对4-胺基噻唑类似物的胺基进一步修饰, 得到的吡啶酰胺及咪唑类似物活性都不理想[25], 而从酰胺或胺基甲酸酯修饰的4-胺基噻唑类似物中筛选出的环己醇酸系列类似物, 如化合物20, 则表现出很好的活性[22]. 2012年, 该小组在化合物20的基础上做了进一步修饰, 在胺基甲酸酯的胺基上引入末端含有羧基的烷基链以进一步提高水溶性.在此基础上获得的GE2270A类似物LEF571(化合物21)是第一例用于临床试验的硫肽类化合物, 它能有效地抑制由艰难梭状芽(Clostridium difficile)引起的肠道感染[24].由于其渗透膜能力差, 因此口服LEF571主要是通过粪便排出, 血液中浓度很低.正是由于这一特性, 使得LEF571成为治疗由Clostridium difficile引起的肠道感染的潜力药[26, 27], 目前LEF571已经完成了临床Ⅱ期试验.
2015年, Fabbretti小组[28]对GE2270A的末端修饰获得NAI003 (22), 该化合物的修饰没有引入距噻唑至少五个原子的羧酸模体.根据先前其他小组对化学半合成的GE2270A类似物的SAR研究, NAI003的抗菌活性相比于其母体GE2270A不会有大的提高.生物活性测试发现其抗菌谱相比于其母体化合物明显缩小, 仅对几个革兰氏阳性菌有活性且抗菌活性不如其母体GE2270A.对NAI003敏感的细菌包括痤疮丙酸杆菌(Propioni-bacterium acnes, P. acnes)家族中的21种测试菌.虽然NAI003抗痤疮丙酸杆菌的活性不如GE2270A, 但其窄的抗菌谱使得它在对痤疮丙酸杆菌发挥作用的同时不致杀死皮肤表面有利的共生菌.这一优点使NAI003具有成为临床上治疗痤疮新药物的潜力, 目前NAI003已经完成了临床Ⅰ期试验.
1.3 Thiostrepton A类似物的化学半合成
Thiostrepton A的分子结构更为复杂, 其结构中可修饰的位点(尤其是大环和侧环结构中)比nocathiacin Ⅰ和GE2270A更多.另外, thiostrepton A除了具有前两种化合物具有的抗菌活性外, 还具有抗肿瘤和抗疟活性, 因此也被一些合成化学家选作母体化合物进行化学修饰, 其可化学修饰的位点如图 4.这些获得的thiostrepton A类似物主要用于研究其抗菌的构效关系(SAR)及其作用机制、抗肿瘤的SAR及其作用机制.
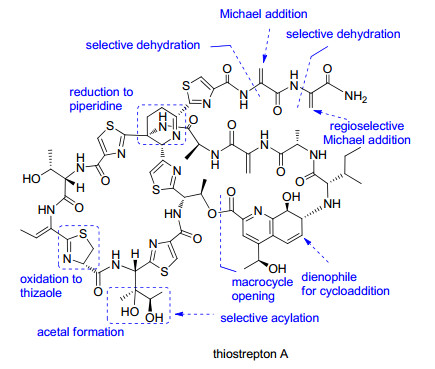 图 4
Thiostrepton A的化学半合成修饰
Figure 4.
Semisynthetic modifications of thiostrepton A
图 4
Thiostrepton A的化学半合成修饰
Figure 4.
Semisynthetic modifications of thiostrepton A
在对thiostrepton A的抗菌的SAR和作用机制研究方面, 人们主要集中对其末端尾巴的修饰(双键的Michael加成或末端尾巴的降解). 2009年, Arndt小组[29]通过Michael加成, 在截短的化合物25的末端双键上引入荧光基团获得化合物28, 荧光标签的引入使得他们发现双环类硫肽thiostrepton A在与23S rRNA/L11蛋白相互作用的过程中, 23S rRNA上A1067和A1095对形成复合体起着至关重要的作用[30]. 2010年, Honek小组[31]通过Michael加成到thiostrepton A的末端尾巴引入硫酸基团获得化合物30, 化合物30在体外能很好地抑制革兰氏阴性菌蛋白合成, 但是却对革兰氏阴性菌无抗菌活性.这一结果证实硫肽分子对抗革兰氏阴性菌无体内抗菌活性是由于其无法穿透革兰氏阴性菌的细胞壁.在对大环骨架修饰方面, Jonker等[32]选择性地将硫链丝菌素(TSR)核心脱氢哌啶还原可以获得化合物29, 模拟计算发现化合物29与23r RNA/L11蛋白的结合能力比thiostrepton A更强, 这预示着天然thiostrepton A可能并不是其靶点的最适底物(Scheme 3).
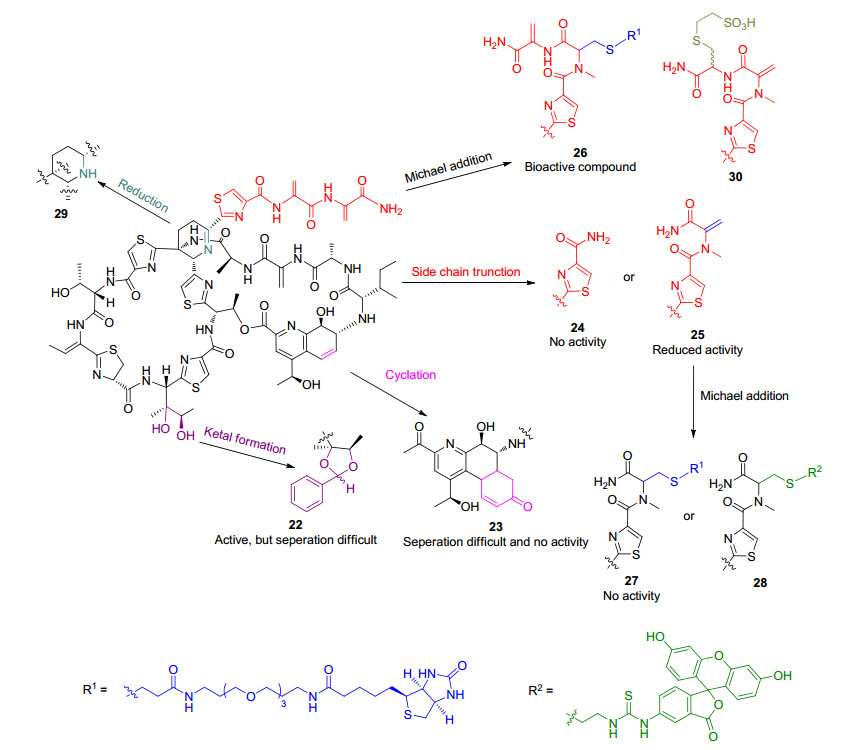 图式3
Thiostrepton尾巴和环的半合成修饰
图式3.
Semisynthetic modifications of thiostrepton A tails and macrocycles
图式3
Thiostrepton尾巴和环的半合成修饰
图式3.
Semisynthetic modifications of thiostrepton A tails and macrocycles
在对thiostrepton A抗肿瘤的SAR和机制研究方面, 人们对thiostrepton A结构上的多个可修饰位点进行了尝试(Scheme 3).在大环骨架修饰方面: 2011年, Balasubramanian小组[33]利用异亮氨酸残基上的双羟基合成了具有缩醛结构的化合物22, 但是该结构不稳定, 无法分离.在侧环修饰方面:对喹萘啶酸(quinaldic acid, QA)上的双键环加成获得化合物23, 但化合物23的抗肿瘤机制活性完全消失.在末端尾巴修饰方面:末端降解掉两个脱氢丙氨酸的化合物24, 降解掉一个脱氢丙氨酸的化合物25及在其末端双键引入生物素获得的化合物27, 不再具有抗肿瘤活性或抗肿瘤活性明显降低.在末端倒数第二个脱氢丙氨酸双键上引入生物素获得的化合物26保持了抗肿瘤活性.以带有生物素探针的化合物26对其抗肿瘤机制研究发现: thiostrepton A是通过直接与FOXM1结合, 从而抑制FOXM1与基因组作用位点结合, 进而降低FOXM1下游基因的表达.
2 组合生物合成
近年来, 对硫肽类抗生素的生物合成研究为获取硫肽类似物提供了新的思路.自2009年硫肽生物合成基因簇被报道以来, 硫肽类抗生素的生物合成途径是核糖体的来源已经被多个研究组阐明.核糖体来源途径意味着在硫肽抗生素的生物合成中, 前体肽的合成遵循中心法则, 即分子结构中氨基酸序列是由基因编码的.这意味着可以通过引入非天然氨基酸或者前体肽氨基酸的定点突变来获取硫肽类抗生物的类似物.另外, 许多硫肽类抗生素成员的前体肽基因和其后修饰基因位于同一个生物合成基因簇中, 这意味着可以通过基因敲除的手段获得其类似物.
2.1 非天然氨基酸的引入获取类似物
2016年, Young小组[34]报道了在thiocillin体系中引入非天然氨基酸的工作(图 5).他们把正交琥珀抑制基因(pyrrolysyl-tRNA合成酶/tRNApylCUA)引入到thiocillin产生菌B. cereusz中, 并分别将3位、4位、6位、8位和13位氨基酸对应的密码子突变成为终止密码子TAG, 在发酵体系中加入BocK, AlocK或ProcK, 最终获得45种引入非天然氨基酸的thiocillin类似物.引入的非天然氨基酸ProcK的末端连有炔基, 因此可以通过Click反应引入其他基团. Click反应特异性地发生在炔基上, 因此不会与结构中其他官能团发生副反应, 这一点优于化学半合成中通过Michael加成(因为硫肽结构中一般包含有多个双键)引入其他基团.他们通过Click反应引入荧光基团获得了化合物31.更有意义的是, 带有生物素的活性探针32为证明thiocillin和L11蛋白之间的直接相互作用提供了有力的证据.因此, 硫肽中引入非天然氨基酸不仅能够获得多种硫肽类似物, 探针的引入还能为研究人类微生物群落中微生物-微生物间以及微生物-宿主间相互作用、未知的抗肿瘤靶行点提供可行的工具.
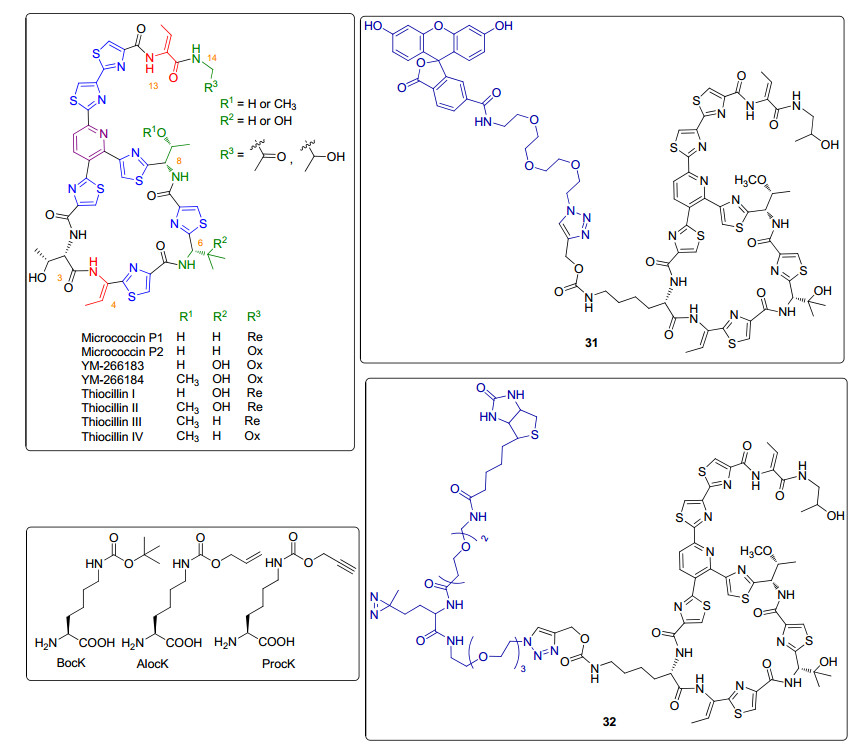 图 5
非天然氨基酸引入thiocillin中获得其类似物
Figure 5.
Production of thiocillin analogues by incorporation of noncanonical amino acids
图 5
非天然氨基酸引入thiocillin中获得其类似物
Figure 5.
Production of thiocillin analogues by incorporation of noncanonical amino acids
2.2 基因缺失获取类似物
由于生物合成途径的连续性可能会导致被敲除基因之后的后修饰酶因为底物不识别而不能得到预期产物, 因此, 只能敲除分子成熟过程中较靠后的后修饰基因才有可能获得成熟的硫肽类似物. 2011年, 刘文小组[35]在thiostrepton A体系中敲除TsrB(酯水解酶)后获得了末端甲酯化thiostrepton A类似物, 敲除TsrC(末端酰胺化酶)后获得末端为羧酸的thiostrepton A类似物(图 6).抗菌活性及水溶性测试表明:甲酯化类似物活性提高了8倍, 但溶解度降低为thiostrepton A的1/16;羧酸类似物活性降低为thiostrepton A的1/8, 但溶解度提高了1.5倍.
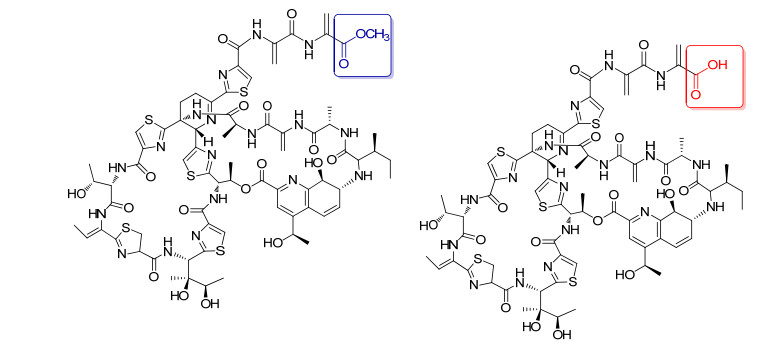 图 6
基因缺失获得thiostrepton A类似物
Figure 6.
Production of thiostrepton A analogues by gene deletion
图 6
基因缺失获得thiostrepton A类似物
Figure 6.
Production of thiostrepton A analogues by gene deletion
2.3 前体肽的定点突变获取类似物
在thiocillin体系中, 2010年Walsh小组[36]将其大环骨架上所有氨基酸进行了突变(图 7A). Thiocillin结构中14个氨基酸分别突变为丙氨酸(Ala)的结果表明:除了Cys11及与核心氮杂六元环形成相关的Ser1和Ser10的突变没能获得成熟thiocillin类似物外, 其他氨基酸的突变都能获得成熟的thiocillin类似物.获得的这些thiocillin类似物中除了Val6Ala, Thr8Ala, Thr13Ala及Thr14Ala保留了thiocillin的生物活性外, 其余位点突变获得的成熟化合物均不再具有生物活性. Thiocillin骨架上Cys分别突变为丝氨酸(Ser)的结果表明: Cys11的突变没能获得thiocillin类似物, Cys5的突变获得了丝氨酸没有发生修饰的thiocillin类似物, Cys2, Cys7, Cys9及Cys12的突变获得了丝氨酸没有发生修饰及修饰为噁唑(噁唑啉)共存的thiocillin类似物.获得的这些thiocillin类似物都不再具有抗菌活性. Thr3, Thr4, Val6, Thr8位点突变成丙氨酸(Ala)外的其他氨基酸结果表明:虽然这些位点的突变能够获得thiocillin类似物, 但它们均不再具有抗菌活性. 2012年, Bowers等[37]还采取了两种不同的策略来获得不同环大小的thiocillin类似物:一种是通过删除Thr3 (3位苏氨酸)或在Thr3和Thr4之间引入1个到3个额外的甘氨酸获得了环大小为23, 29, 32和35个原子的thiocillin类似物.另外一种策略是在体内对六元环形成的区域通过置换或者插入的方式引入丝氨酸以获得不同环大小的类似物.生物活性测试表明, 通过这两种策略获得的thiocillin类似物都不再具有抗菌活性.根据thiocillin中氨基酸突变的结果可以得出其构效关系的规律: Cys11及与核心氮杂六元环形成相关的Ser1和Ser10的突变不能获得成熟的化合物; 大环结构上Cys2, Thr3, Thr4, Cys5, Cys7及Cys9的突变虽然能获得成熟的化合物, 但均不再具有抗菌活性; 大环结构上Val6, Thr8及末端尾巴Cys12, Thr13Ala, Thr14Ala的突变能获得抗菌活性保持的thiocillin类似物.
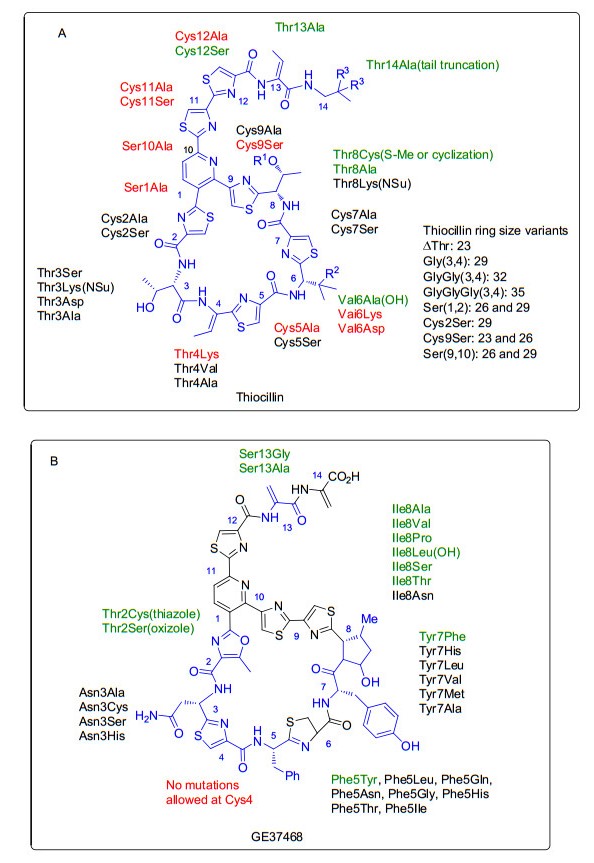 图 7
前体肽突变获得的thiocillin和GE37468的类似物
Figure 7.
Production of thiocillin and GE37468 analogues by pre-peptide mutagenesis
图 7
前体肽突变获得的thiocillin和GE37468的类似物
Figure 7.
Production of thiocillin and GE37468 analogues by pre-peptide mutagenesis
在GE37468体系(图 7B)中, 2012年Walsh小组[38]通过密码子随机突变的方法对GE37468中7个位点(Thr2, Asn3, Cys4, Phe5, Tyr7, Ile8, Ser13) 进行点突变共获得了133种突变株, 并利用高通量MALDI-TOF检测手段对133种突变株的发酵产物进行了检测.结果表明:将Thr2分别突变成半胱氨酸(Cys)或丝氨酸(Ser)后得到的产物获得了2位形成噻唑或噁唑的GE37468类似物, 并且抗菌活性得到了保持. Phe5只有突变成与其性质相近的酪氨酸(Tyr)后才能获得抗菌活性保持的产物, 突变成其他氨基酸获得的是抗菌活性丧失的产物.类似地, Tyr7只有突变成与其性质相近的Phe才能获得活性保持的产物. Ile8的突变获得的成熟分子中除Ile8Asn不再具有抗菌活性外, 其余6种成熟分子的抗菌活性都得到了保持. Cys4的突变未能获得成熟的分子. Asn3的突变获得的GE37468类似物均不再具有抗菌活性, 预示着3位天门冬氨酸是GE37468发挥抗菌活性的关键位点.在29环硫肽类抗生素GE2270A, GE37468, baringolin及thiomuracin的3位也都是保守的天门冬氨酸, 这表明3位天门冬氨酸在29环硫肽类抗生素发挥抗菌活性上起着重要的作用.总之, 它们从133种突变株中共检测到29种成熟的GE37468类似物, 其中12种类似物的抗菌活性得到保持, 1种类似物(Thr2 Cys(thiozole))的抗菌活性有所提高.
在thiostrepton A体系中, 2014年刘文小组[39]通过对1位氨基酸的突变及2位氨基酸的突变获得了抗菌活性提高的Ile1Val和抗菌活性略有降低的Ala2Dha.进一步, 他们对thiostrepton A结构肽1位的异亮氨酸进行了饱和点突变.点突变结果表明: 1位异亮氨酸可以突变为非极性氨基酸(Phe, Leu, Met, Ala, Trp, Pro)并产生相应的thiostrepton A类似物.相反, 当1位异亮氨酸突变成极性氨基酸中的中性氨基酸(Ser, Thr, Gly, Tyr, Cys, Gln, Asn)、酸性氨基酸(Asp, Glu)以及碱性氨基酸(Lys, Arg, His)后, 突变株不再产生硫肽分子.在此基础上, 他们通过双突变(结构肽1位异亮氨酸突变成缬氨酸, 结构肽2位丙氨酸突变成丝氨酸)以期实现盐屋霉素(siomycin)在thiostrepton A宿主菌中的产生. Siomycin作为thiostrepton A的天然类似物比thiostrepton A具有更好的药用生物活性.有意思的是, 双突变突变株SL-Ile1Ala-Val2Ser不仅产生了预期的siomycin, 还产生了2位丝氨酸未发生脱水的新型硫肽类抗生素OH-siomycin (OH-SIO)(图 8).水溶性和抗菌活性测试表明: TSR-Ile1Met, TSR-Ile1Ala和OH-SIO的水溶性有了很大的提高, 其中OH-SIO的水溶性相比于thiostrepton A提高了近一倍.除siomycin外, 其余5种thiostrepton A类似物对测试的耐药菌的抗菌活性都有了明显降低, 这表明thiostrepton A结构肽1位氨基酸残基在其抗菌活性中扮演未知的重要角色[40].
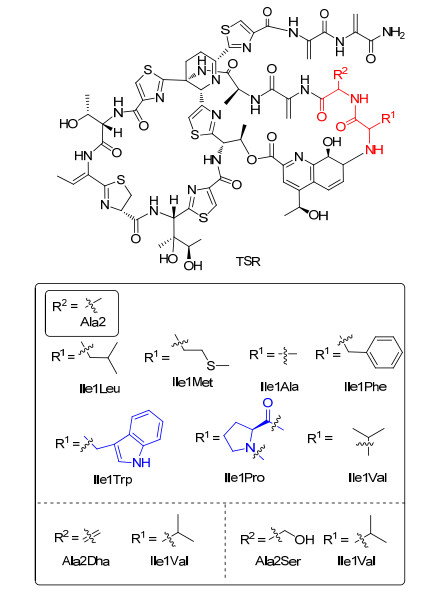 图 8
前体肽突变获得thiostrepton A的类似物(刘文小组)
Figure 8.
Production of thiostrepton A analogues by pre-peptide mutagenesis (Liu et al.)
图 8
前体肽突变获得thiostrepton A的类似物(刘文小组)
Figure 8.
Production of thiostrepton A analogues by pre-peptide mutagenesis (Liu et al.)
2015年, Zhang小组[41]在Ala4的饱和点突变实验中发现, 除Ala4突变为Arg, Asp, Glu, Lys和Pro没有得到成熟的化合物外, 突变成其余14种天然氨基酸共获得了16种成熟化合物(图 9).其中Ala4突变为半胱氨酸(Cys)后, 获得了4位半胱氨酸上巯基与17位脱氢丙氨酸加成的产物Ala4Cys F1和Ala4Cys F2. Ala4突变为丝氨酸(Ser)后获得了丝氨酸没有发生修饰及发生脱水修饰共存的化合物, 而Ala4突变为苏氨酸(Thr)后仅获得苏氨酸发生脱水修饰的化合物.生物活性测试发现, 16种化合物除Ala4Cys F1和Ala4Cys F2的抗菌活性丧失外, 其余14种化合物的抗菌活性都得到了一定程度上的保持; 16种化合物的体外抑制蛋白翻译活性却相当. 2016年, Wendy小组在对Ala2的饱和点突变中发现[42]:将Ala2突变成Gly, Met, Phe, Tyr, Ser, Thr, Ile和Val能获得8种成熟的化合物.其中Ala2突变成丝氨酸(Ser)和苏氨酸(Thr)后都是获得发生脱水修饰的化合物.将Ala2突变成异亮氨酸(Ile)或缬氨酸(Val)后, 得到的是1位异亮氨酸缺失的缩环产物Ala2Ile-ΔIle1和Ala2Val-ΔIle1.生物活性测试发现: 8种化合物除Ala2Ile-ΔIle1和Ala2Val-ΔIle1的抗菌活性丧失外, 其余6种化合物的抗菌活性都得到了一定程度上的保持; 体外抑制蛋白翻译活性Ala2Ile-ΔIle1和Ala2Val-ΔIle1明显降低而其余6种化合物得到保持.
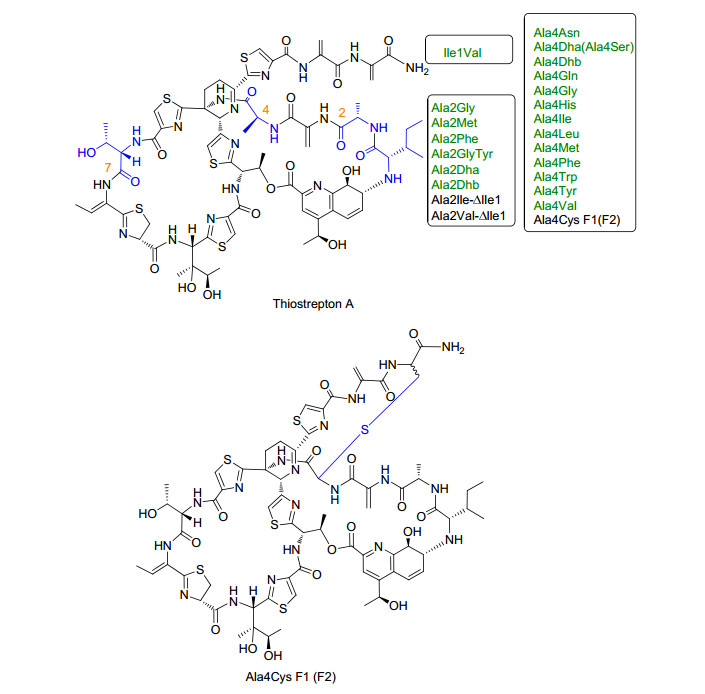 图 9
前体肽突变获得thiostrepton A的类似物(Wendy小组)
Figure 9.
Production of thiostrepton A analogues by pre-peptide mutagenesis (Wendy et al.)
图 9
前体肽突变获得thiostrepton A的类似物(Wendy小组)
Figure 9.
Production of thiostrepton A analogues by pre-peptide mutagenesis (Wendy et al.)
3 前体导向的生物合成和突变生物合成
传统的前体导向生物合成指的是在培养基中加入前体小分子并利用野生菌株来产生次级代谢产物, 该方法得到的大多是次级代谢产物及其结构类似物的多组分混合物.前体导向的突变生物合成首先构建前体生物合成途径被阻断的突变菌株, 然后在培养基中加入前体小分子或其类似物, 利用突变菌株来产生次级代谢产物, 该方法得到的是次级代谢产物或其结构类似物的单一组分.
很多双环硫肽的侧环中包含有吲哚酸(MIA)或者喹萘啶酸(QA), 生物合成研究表明这两个模块是由色氨酸经修饰后引入的, 他们独立于前体肽氨基酸序列之外, 因此通过喂养色氨酸类似物来获取硫肽类似物是可行的[43, 44].在nosiheptide的类似物合成中, 2011年, 刘文小组[45]将5-F-色氨酸喂养到nosiheptide产生菌S. actuosus中, 利用前体导向的生物合成方法获得了氟代的nosiheptide(图 10, A).在thiostrepton A的类似物合成中, 2015年, 刘文小组[46]通过化学合成了5'-F-QA, 6'-F-QA及12'-Me-QA, 并将这三种QA类似物喂养到QA生物合成阻断的tsrT突变株中, 利用前体导向的突变生物合成方法获得了5'-F-TSR, 6'-F-TSR和12'-Me-TSR(图 10, B).对这三个thiostrepton A类似物抗菌活性测试发现, 三者对多种革兰氏阳性菌的抗菌活性都优于thiostrepton A.这表明侧环上QA结构的微调(取代基的引入)不会影响其整体抗菌活性并且还有可能获得抗菌活性提高的thiostrepton A类似物.他们在研究这三种thiostrepton A类似物对胞内菌M. marinum的抑制机制时发现:它们能够分别对M. marinum及其宿主发挥作用而表现为双功能作用模式(MOA).即它们除了能够通过直接与M. marinum的50S核糖体亚基结合(即TSR的抗菌机制)抑制M. marinum外, 还能够诱导M. marinum的宿主细胞发生由内质网应(ER-stress)介导的自噬, 从而达到抑菌效果[47].至此, thiostrepton A是目前发现的唯一一例主动对胞内菌及其宿主细胞同时发挥作用以达到抗菌效果的抗生素.这一发现为发展抑制胞内菌感染的抗生素提供了一种新思路.进一步, 2016年, 刘文小组[48]通过将5'-F-QA, 6'-F-QA及12'-Me-QA喂养到tsrB突变株中, 获得了5'-F-cmTSR, 6'-F-cmTSR和12'-Me-cmTSR, 这些类似物的抗菌活性较thiostrepton A提高10倍以上, 而且对于艰难梭状芽(Clostridium difficile)的抗菌效果较万古霉素提高50倍以上.在随后的研究过程中, 他们基于同样的策略使用不同的合成前体对相同的突变菌株进行化学喂养, 却发现了与预期产物不一致的硫肽类似物.在使用前体7-F-QA喂养突变株ΔtsrT时, 它们发现了一个侧环结构不完整的氟代thiostrepton A类似物(图 10, C), 其中喹萘啶酸(QA)结构单元没有发生相应的氧化后修饰.以此种具备二酮结构特征的TSR分流产物(shunt product)作为预期, 他们研究了TSR生物合成途径与分子骨架氧化后修饰相关的基因功能, 通过基因敲除/回补、化学喂养、体外酶学测活等手段, 最终确定P450蛋白TsrP负责了thiostrepton A侧环形成过程中QA结构单元的环氧化反应[49].在最近的研究中, 他们继续沿用前体导向突变生物合成的策略, 通过优化6'-F-QA作为前体喂养突变株ΔtsrT后的发酵时间, 成功分离、鉴定了一个侧环尚未关闭但QA基团已经发生环氧化的关键氟代中间体(图 10, D).正是由于氟原子的引入, 降低了催化侧环形成的关键蛋白的酶活性, 从而导致了这样一个中间体的短暂积累; 而继续延长发酵时间, 该中间体会被转化为最终成熟的6'-F-TSR分子.该中间体的分离和鉴定为thiostrepton A侧环生物合成途径中的酶学机制研究奠定了重要基础.通过生物信息学分析, 他们锁定了thiostrepton A生物合成基因簇中一个功能尚未得到归属的基因tsrI, 其编码的蛋白TsrI隶属于α/β水解酶超家族.在此基础上综合使用基因敲除/回补、化学半合成模拟底物制备、体外酶学测活、化学喂养等手段, 证实了TsrI是thiostrepton A侧环生物合成过程中的关键蛋白, 该蛋白同时负责了先导肽的切除和TSR侧环关闭过程的大环化反应.关于TsrI功能的阐明, 不但解决了长期以来困扰人们的双环硫肽抗生素侧环如何关闭的问题, 更拓展了人们对于α/β水解酶这一超家族蛋白生化功能的进一步认识和理解, 同时为基于合成生物学策略改造双大环硫肽类抗生素分子结构的研究奠定了基础[50].
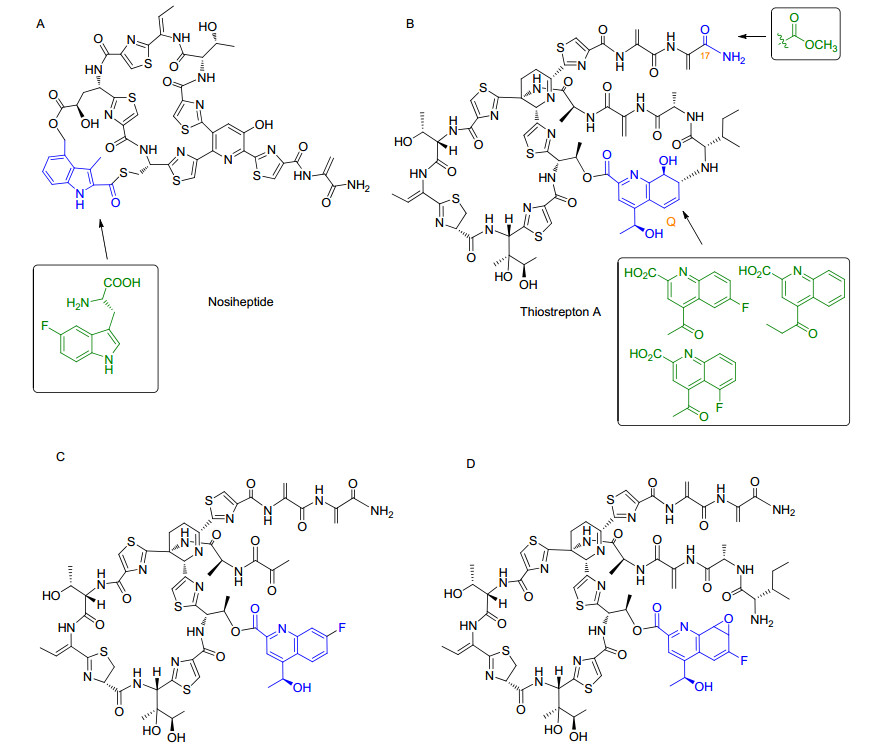 图 10
前体导向的生物合成和突变生物合成获得的硫肽类似物
Figure 10.
Production of analogues by precursor-directed biosynthesis and mutational biosynthesis
图 10
前体导向的生物合成和突变生物合成获得的硫肽类似物
Figure 10.
Production of analogues by precursor-directed biosynthesis and mutational biosynthesis
4 总结与展望
随着致病菌临床耐药性的日益严峻, 硫肽类抗生素优良的生物活性以及独特的构效关系和作用机制重新引起了化学家和生物学家的研究兴趣, 但是硫肽类分子本身并不适于直接作为药物或者化学探针.近年来化学家们通过化学半合成等手段获得了一些生物活性或水溶性有所提高的硫肽衍生物(如LEF571和NAI003), 但是由于受反应条件及可修饰位点的限制, 目前人们对硫肽类抗生素的化学半合成仍然只是集中在其C端尾部.与其他常见的临床抗生素相比, 通过硫肽类抗生素“模板化”的生物合成途径对其进行分子改造显得尤为简洁, 即通过对于其前体肽序列的定点突变即可获得一系列的结构衍生物.因此, 在了解了硫肽抗生素生物合成途径中的化学逻辑和酶学机制后, 有助于人们通过基于合成生物学的策略进一步拓展硫肽分子结构和功能的多样性.
本文对于目前常用的制备硫肽类抗生素衍生物的方法进行了归纳和总结, 从化学和生物学两方面进行了比较.我们认为, 在未来的研究中, 通过“化学辅助的合成生物学”手段能够进一步构建成员更多的以硫肽为代表的复杂天然产物文库, 即通过合成生物学构筑具有某种特定生物活性的复杂分子骨架, 通过化学半合成或使用合成的前体喂养相应突变株, 从而进行骨架侧链的修饰以及生物活性的“微调”.我们相信, 在不久的将来基于硫肽类抗生素分子骨架的新型药物和化学探针将会在临床治疗和基础研究中大展神威.
-
-
[1]
Bagley, M. C.; Dale, J. W.; Merritt, E. A.; Xiong, X. Chem. Rev. 2005, 105, 685. doi: 10.1021/cr0300441
-
[2]
Li, C.; Kelly, W. L. Nat. Prod. Rep. 2010, 27, 153. doi: 10.1039/B922434C
-
[3]
Hensens, O. D.; Albers-Schönberg, G. Tetrahedron Lett. 1978, 19, 3649.
-
[4]
Naidu, B. N.; Sorenson, M. E.; Zhang, Y.; Kim, O. K.; Matiskella, J. D.; Wichtowski, J. A.; Connolly, T. P.; Li, W.; Lam, K. S.; Bronson, J. J.; Pucci, M. J.; Clark, J. M.; Ueda, Y. Bioorg. Med. Chem. Lett. 2004, 14, 5573. doi: 10.1016/j.bmcl.2004.08.058
-
[5]
Zhang, C.; Herath, K.; Jayasuriya, H.; Ondeyka, J. G.; Zink, D. L.; Occi, J.; Birdsall, G.; Venugopal, J.; Ushio, M.; Burgess, B.; Masurekar, P.; Barrett, J. F.; Singh, S. B. J. Nat. Prod. 2009, 72, 841. doi: 10.1021/np800783b
-
[6]
Zhang, C.; Occi, J.; Masurekar, P.; Barrett, J. F.; Zink, D. L.; Smith, S.; Onishi, R.; Ha, S.; Salazar, O.; Genilloud, O.; Basilio, A.; Vicente, F.; Gill, C.; Hickey, E. J.; Dorso, K.; Motyl. M.; Singh, S. B. J. Am. Chem. Soc. 2008, 130, 12102. doi: 10.1021/ja803183u
-
[7]
Bower, J.; Drysdale, M.; Hebdon, R.; Jordan, A.; Lentzen, G.; Matassova, N.; Murchie, A.; Powles, J.; Roughley, S. Bioorg. Med. Chem. Lett. 2003, 13, 2455. doi: 10.1016/S0960-894X(03)00495-5
-
[8]
Naidu, B. N.; Sorenson, M. E.; Bronson, J. J.; Pucci, M. J.; Clark, J. M.; Ueda, Y. Bioorg. Med. Chem. Lett. 2005, 15, 2069. doi: 10.1016/j.bmcl.2005.02.046
-
[9]
Naidu, B. N.; Sorenson, M. E.; Matiskella, J. D.; Li, W.; Sausker, J. B.; Zhang, Y.; Connolly, T. P.; Lam, K. S.; Bronson, J. J.; Pucci, M. J.; Yang, H.; Ueda, Y. Bioorg. Med. Chem. Lett. 2006, 16, 3545. doi: 10.1016/j.bmcl.2006.03.079
-
[10]
Regueiro-Ren, A.; Ueda, Y. J. Org. Chem. 2002, 67, 8699. doi: 10.1021/jo0261698
-
[11]
Connolly, T. P.; Regueiro-Ren, A.; Leet, J. E.; Springer, D. M.; Goodrich, J.; Huang, X.; Pucci, M. J.; Clark, J. M.; Bronson, J. J.; Ueda, Y. J. Nat. Prod. 2005, 68, 550. doi: 10.1021/np040225d
-
[12]
Xu, L.; Farthing, A. K.; Shi, Y.; Meinke, P. T.; Liu, K. J. Org. Chem. 2007, 72, 7447. doi: 10.1021/jo071115p
-
[13]
Xu, L.; Farthing, A. K.; Dropinski, J. F.; Meinke, P. T.; McCallum, C.; Leavitt, P. S.; Hickey, E. J.; Colwell, L.; Barrett, J.; Liu, K. Bioorg. Med. Chem. Lett. 2009, 19, 3531. doi: 10.1016/j.bmcl.2009.04.144
-
[14]
Hrnciar, P.; Ueda, Y.; Huang, S.; Leet, J. E.; Bronson, J. J. J. Org. Chem. 2002, 67, 8789. doi: 10.1021/jo020385z
-
[15]
Pucci, M. J.; Bronson, J. J.; Barrett, J. F.; DenBleyker, K. L.; Discotto, L. F.; Fung-Tomc, J. C.; Ueda, Y. Antimicrob. Agents Chemother. 2004, 48, 3697. doi: 10.1128/AAC.48.10.3697-3701.2004
-
[16]
Regueiro-Ren, A.; Naidu, B. N.; Zheng, X.; Hudyma, T. W.; Connolly, T. P.; Matiskella, J. D.; Zhang, Y.; Kim, O. K.; Sorenson, M. E.; Pucci, M. J.; Clark, J.; Bronson, J. J.; Ueda, Y. Bioorg. Med. Chem. Lett. 2004, 14, 171. doi: 10.1016/j.bmcl.2003.09.061
-
[17]
Naidu, B. N.; Sorenson, M. E.; Hudyma, T.; Zheng, X.; Zhang, Y.; Bronson, J. J.; Pucci, M. J.; Clark, J. M.; Ueda, Y. Bioorg. Med. Chem. Lett. 2004, 14, 3743. doi: 10.1016/j.bmcl.2004.04.102
-
[18]
Golik, J.; Wong, H. S. L.; Chen, S. H.; Doyle, T. W.; Wright, J. J. K.; Knipe, J.; Rose, W. C.; Casazzam, A. M.; Vyas, D. M. Bioorg. Med. Chem. Lett. 1996, 6, 1837. doi: 10.1016/0960-894X(96)00321-6
-
[19]
Tavecchia, P.; Gentili, P.; Kurz, M.; Sottani, C.; Bonfichi, R.; Selva, E.; Lociuro, S.; Restelli, E.; Ciabatti, R. Tetrahedron 1995, 51, 4867. doi: 10.1016/0040-4020(95)00171-4
-
[20]
Tavecchia, P.; Kruz, M.; Colombo, L.; Bonfichi, R.; Selva, E.; Lociuro, S.; Marzorati, E.; Ciabatti, R. Tetrahedron 1996, 52, 8763. doi: 10.1016/0040-4020(96)00417-6
-
[21]
Lociuro, S.; Tavecchia, P.; Marzorati, E.; Goldstein, B. P.; Denaro, M.; Ciabatti, R. J. Antibiot. 1997, 50, 344.. doi: 10.7164/antibiotics.50.344
-
[22]
LaMarche, M. J.; Leeds, J. A.; Amaral, K.; Brewer, J. T.; Bushell, S. M.; Dewhurst, J. M.; Dzink-Fox, J.; Gangl, E.; Goldovitz, J.; Jain, A.; Mullin, S.; Neckermann, G.; Osborne, C.; Palestrant, D.; Patane, M. A.; Rann, E. M.; Sachdeva, M.; Shao, J.; Tiamfook, S.; Whitehead, L.; Yu, D. J. Med. Chem. 2011, 54, 8099. doi: 10.1021/jm200938f
-
[23]
LaMarche, M. J.; Leeds, J. A.; Dzink-Fox, J.; Mullin, S.; Patane, M. A.; Rann, E. M.; Tiamfook, S. Bioorg. Med. Chem. Lett. 2011, 21, 3210. doi: 10.1016/j.bmcl.2011.04.048
-
[24]
LaMarche, M. J.; Leeds, J. A.; Amaral, A.; Brewer, J. T.; Bushell, S. M.; Deng, G.; Dewhurst, J. M.; Ding, J.; Dzink-Fox, J.; Gamber, G.; Jain, A.; Lee, K.; Lee, L.; Lister, T.; McKenney, D.; Mullin, S.; Osborne, C.; Palestrant, D.; Patane, M. A.; Rann, E. M.; Sachdeva, M.; Shao, J.; Tiamfook, S.; Trzasko, A.; Whitehead, L.; Yifru, A.; Yu, D.; Yan, W.; Zhu, Q. J. Med. Chem. 2012, 55, 2376. doi: 10.1021/jm201685h
-
[25]
LaMarche, M. J.; Leeds, J. A.; Dzink-Fox, J.; Gunderson, K.; Krastel, P.; Memmert, K.; Patane, M. A.; Rann, E. M.; Schmitt, E.; Tiamfook, S.; Wang, B. J. Med. Chem. 2011, 54, 2517. doi: 10.1021/jm101602q
-
[26]
Trzasko, A.; Leeds, J. A.; Praestgaard, J.; Lamarche, M. J.; McKenney, D. Antimicrob. Agents Chemother. 2012, 56, 4459. doi: 10.1128/AAC.06355-11
-
[27]
Ting, L. S.; Praestgaard, J.; Grunenberg, N.; Yang, J. C.; Leeds, J. A.; Pertel, P. Antimicrob. Agents Chemother. 2012, 56, 5946. doi: 10.1128/AAC.00867-12
-
[28]
Fabbretti, A.; He, C. G.; Gaspari, E.; Maffioli, S.; Brandi, L.; Spurio, R.; Sosio, M.; Jabes, D.; Donadio, S. Antimicrob. Agents Chemother. 2015, 59, 4560. doi: 10.1128/AAC.05155-14
-
[29]
Schoof, S.; Baumann, S.; Ellinger, B.; Arndt, H. D. ChemBioChem 2009, 10, 242. doi: 10.1002/cbic.v10:2
-
[30]
Baumann, S.; Schoof, S.; Bolten, M.; Haering, C.; Takagi, M.; Shin-ya, K.; Arndt, H. D. J. Am. Chem. Soc. 2010, 132, 6973. doi: 10.1021/ja909317n
-
[31]
Myers, C. L.; Hang, P. C.; Ng, G.; Yuen, J.; Honek, J. F. Bioorg. Med. Chem. 2010, 18, 4231. doi: 10.1016/j.bmc.2010.04.098
-
[32]
Jonker, H. R.; Baumann, S.; Wolf, A.; Schoof, S.; Hiller, F.; Schulte, K. W.; Kirschner, K. N.; Schwalbe, H.; Arndt, H. D. Angew. Chem., Int. Ed. Engl. 2011, 50, 3308. doi: 10.1002/anie.201003582
-
[33]
Hegde, N. S.; Sanders, D. A.; Rodriguez, R.; Balasubramanian, S. Nat. Chem. 2011, 3, 725. doi: 10.1038/nchem.1114
-
[34]
Luo, X.; Zambaldo, C.; Liu, T.; Zhang, Y.; Xuan, W.; Wang, C.; Reed, S. A.; Yang, P. Y.; Wang, R. E.; Javahishvili, T.; Schultz, P. G.; Young, T. S. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3615. doi: 10.1073/pnas.1602733113
-
[35]
Liao, R.; Liu, W. J. Am. Chem. Soc. 2011, 133, 2852. doi: 10.1021/ja1111173
-
[36]
Bowers, A. A.; Acker, M. G.; Koglin, A.; Walsh, C. T. J. Am. Chem. Soc. 2010, 132, 7519. doi: 10.1021/ja102339q
-
[37]
Bowers, A. A.; Acker, M. G.; Young, T. S.; Walsh, C. T. J. Am. Chem. Soc. 2012, 134, 10313. doi: 10.1021/ja302820x
-
[38]
Young, T. S.; Dorrestein, P. C.; Walsh, C. T. Chem. Biol. 2012, 19, 1600. doi: 10.1016/j.chembiol.2012.10.013
-
[39]
Guo, H.; Wang, J.; Li, Y.; Yu, Y.; Zheng, Q.; Wu, J.; Liu, W. Chem. Sci. 2014, 5, 240. doi: 10.1039/C3SC52015C
-
[40]
Duan, P.; Zheng, Q.; Lin, Z.; Wang, S.; Chen, D.; Liu, W. Org. Chem. Front. 2016, 3, 1254. doi: 10.1039/C6QO00320F
-
[41]
Zhang, F.; Kelly, W. L. ACS Chem. Biol. 2015, 10, 998. doi: 10.1021/cb5007745
-
[42]
Zhang, F.; Li, C.; Kelly, W. L. ACS Chem. Biol. 2016, 11, 415. doi: 10.1021/acschembio.5b00731
-
[43]
Zhang, Q.; Chen, D.; Lin, J.; Liao, R.; Tong, W.; Xu, Z.; Liu, W. J. Biol. Chem. 2011, 286, 21287. doi: 10.1074/jbc.M111.224832
-
[44]
Duan, L.; Wang, S.; Liao, R.; Liu, W. Chem. Biol. 2012, 19, 443. doi: 10.1016/j.chembiol.2012.02.008
-
[45]
Zhang, Q.; Li, Y.; Chen, D.; Yu, Y.; Duan, L.; Shen, B.; Liu, W. Nat. Chem. Biol. 2011, 7, 154. doi: 10.1038/nchembio.512
-
[46]
Wang, S.; Zheng, Q.; Wang, J.; Zhao, Z.; Li, Q.; Yu, Y.; Wang, R.; Liu, W. Org. Chem. Front. 2015, 2, 106. doi: 10.1039/C4QO00288A
-
[47]
Zheng, Q.; Wang, Q.; Wang, S.; Wu, J.; Gao, Q.; Liu, W. Chem. Biol. 2015, 22, 1002. doi: 10.1016/j.chembiol.2015.06.019
-
[48]
Wang, S.; Zheng, Q.; Wang, J.; Chen, D.; Yu, Y.; Liu, W. Org. Chem. Front. 2016, 3, 496. doi: 10.1039/C5QO00433K
-
[49]
Zheng, Q.; Wang, S.; Liao, R.; Liu, W. ACS Chem. Biol. 2016, 11, 2673. doi: 10.1021/acschembio.6b00419
-
[50]
Zheng, Q.; Wang, S.; Duan, P.; Liao, R.; Chen, D.; Liu, W. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14318. doi: 10.1073/pnas.1612607113
-
[1]
-

图 1 Nocathiacin Ⅰ尾巴部分的化学半合成修饰
Figure 1 Semisynthetic modifications of nocathiacin Ⅰ tails

图式1 Nocathiacin Ⅰ尾巴部分的化学半合成修饰
Scheme 1 Semisynthetic modifications of nocathiacin Ⅰ tails

图式3 Thiostrepton尾巴和环的半合成修饰
Scheme 3 Semisynthetic modifications of thiostrepton A tails and macrocycles

图 5 非天然氨基酸引入thiocillin中获得其类似物
Figure 5 Production of thiocillin analogues by incorporation of noncanonical amino acids

图 6 基因缺失获得thiostrepton A类似物
Figure 6 Production of thiostrepton A analogues by gene deletion

图 7 前体肽突变获得的thiocillin和GE37468的类似物
Figure 7 Production of thiocillin and GE37468 analogues by pre-peptide mutagenesis

图 8 前体肽突变获得thiostrepton A的类似物(刘文小组)
Figure 8 Production of thiostrepton A analogues by pre-peptide mutagenesis (Liu et al.)

图 9 前体肽突变获得thiostrepton A的类似物(Wendy小组)
Figure 9 Production of thiostrepton A analogues by pre-peptide mutagenesis (Wendy et al.)

图 10 前体导向的生物合成和突变生物合成获得的硫肽类似物
Figure 10 Production of analogues by precursor-directed biosynthesis and mutational biosynthesis
表 1 Nocathiacin Ⅰ类似物的抑菌活性a
Table 1. Inhibitory activity of nocathiacin Ⅰ analogues
化合物 Nocathiacin Ⅰ Nocathiacin Ⅳ 8 9 13 12 14 15 MICa/(μg•mL-1) 0.007 0.03 0.06 0.06 0.015 ND ND ND PD50b/(mg•kg-1) 0.8 NDb ND ND ND 1 3 0.15 aPD50 : dose required to cure 50% of the animals infected; MIC: minimum inhibitory concentration; bND: not test.  下载: 导出CSV
下载: 导出CSV
-














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 21
- 文章访问数: 3987
- HTML全文浏览量: 654
 下载:
下载:
