 图 图式1
Chan-Evans-Lam 反应可能的机理
Figure 图式1.
Proposed mechanism for Chan-Evans-Lam reaction
图 图式1
Chan-Evans-Lam 反应可能的机理
Figure 图式1.
Proposed mechanism for Chan-Evans-Lam reaction
引用本文:
马小盼, 刘凤萍, 莫冬亮. Chan-Evans-Lam偶联反应研究进展[J]. 有机化学,
2017, 37(5): 1069-1087.
doi:
10.6023/cjoc201702001

Citation: Ma Xiaopan, Liu Fengping, Mo Dongliang. Recent Advances in Chan-Evans-Lam Coupling Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1069-1087. doi: 10.6023/cjoc201702001
Citation: Ma Xiaopan, Liu Fengping, Mo Dongliang. Recent Advances in Chan-Evans-Lam Coupling Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1069-1087. doi: 10.6023/cjoc201702001
Chan-Evans-Lam偶联反应研究进展
English
Recent Advances in Chan-Evans-Lam Coupling Reaction
Abstract:
Chan-Evans-Lam reaction has been served as a powerful cross-coupling reaction in organic synthesis since its discovery in the 1990s owing to the feature of mild reaction conditions, functional group tolerance and open-flask chemistry. Up to now, it has been not only extensively utilized in carbon-heteroatom or carbon-carbon bonds formation, but also successfully applied to synthesize natural products and alkaloids. The new strategies of Chan-Evans-Lam reaction in organic synthesis as well as applications in total synthesis in recent five years are discussesed.
-
近年来, 过渡金属催化的氧化交叉偶联反应已经被证明是构建碳-碳键、碳-杂键最有效和最直接的策略之一, 在医药、农药、染料及日用化工品等领域中都起到了非常重要的作用[1, 2]. 1998年, Chan、Evans和Lam三个课题组[3]分别独立地报道了铜盐促进下不同杂原子作为亲核试剂与芳基硼酸进行偶联反应构建碳-杂键的方法.与其它过渡金属(如钯、镍等)参与的偶联反应相比, 此类反应具有铜盐价格便宜(铜金属约0.01美元/克, 而钯金属约350美元/克)、反应条件温和、不需要复杂配体、操作简便、在空气氛围中搅拌就能顺利进行等特点[4].因此, 铜盐促进不同原子类型的亲核试剂与有机硼试剂进行的偶联反应被称之为Chan-Evans-Lam偶联反应[5], 此反应由于在空气中就能进行, 又被称为open-flask Chemistry (Eq. 1).
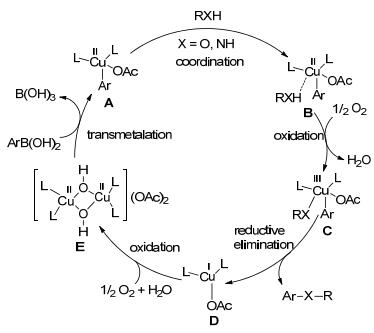
普遍认为Chan-Evans-Lam反应的机理是(Scheme 1):芳基硼酸首先与二价铜络合物E 发生转金属形成中间体A 和硼酸.中间体A 与亲核试剂(RXH)配位得到络合物B.在氧气存在下, 络合物B 被氧化形成三价铜中间体C.中间体C 发生还原消除生成偶联产物(Ar—X—R)和一价铜络合物D.络合物D 在氧气和水的作用下转化成二价铜络合物E, 从而完成催化循环.另外一种可能的反应机理则认为:亲核试剂(RXH)进攻有可能发生在二价铜与芳基硼酸转金属之前.尽管很多有机化学家如Stahl[6], Norrby[7], Das[8]等通过密度泛函理论(DFT)对反应机理做了深入的化学计算研究, 但目前Chan-Evans-Lam反应的机理仍无明确定论.
图 图式1
Chan-Evans-Lam 反应可能的机理
Figure 图式1.
Proposed mechanism for Chan-Evans-Lam reaction
自从反应被发现以来, 引起了很多有机化学家及生物化学家的广泛关注, 在有机合成中不仅被广泛地应用于构建C—N, C—O, C—S, C—C键, 也成功地应用于复杂天然产物的全合成中, 特别是在药物分子的结构修饰中占有重要的地位. Lam小组[9]在2011年和Wu小组[10]在2012年分别对Chan-Evans-Lam偶联反应的最新研究进展进行了综述, 而之后这类偶联反应的研究并无相关综述的报道.但近些年铜参与的Chan-Evans-Lam偶联反应在碳-杂键交叉偶联化学的应用中又取得了很多新的进展.因此, 本文针对2012年之后通过Chan-Evans-Lam反应构建C—N, C—O, C—S, C—C键的最新进展进行了综述, 主要涉及不同原子(氮、氧、硫和碳原子)的亲核试剂与有机硼试剂进行偶联反应构建C—N, C—O, C—S和C—C键的新策略, 以及Chan-Evans-Lam反应在复杂天然产物和生物碱全合成中的一些应用.
1 C—N键形成的偶联反应
众所周知, 含氮化合物是目前化合物种类中最庞大的一类衍生物.胺、酰胺、亚胺、烯胺以及氮杂环化合物是组成生命体的基本元素, 因此C—N键的构建一直是有机合成中一个非常重要的研究课题.近些年, Chan-Evans-Lam反应在C—N键构建领域发展迅猛, 在很多研究领域都有广泛的应用.
1.1 芳基硼试剂参与的C—N键偶联反应
1.2 烯基硼酸参与的C—N键偶联反应
1.3 烷基硼试剂参与的C—N键偶联反应
烷基硼酸稳定性较芳基硼酸和烯基硼酸都差, 同时与催化剂发生转金属后的物种也相对不稳定, 在一定程度上限制了烷基硼酸进行的Chan-Evans-Lam反应. 2013年, Watson小组[41]首次报道了酰胺与烷基硼酸的交叉偶联反应(Eq. 28).反应中, 以CuBr为催化剂, NaOSiMe3为碱, 叔丁基过氧醚(DTBP)为氧化剂, 不同酰胺底物的N-烷基化产率可以达到优秀.反应中并没有观察到双烷基化产物, 同时底物中可以兼容多种官能团如羟基、胺基等.值得一提的是, 反应可以实现克级量(67d), 产率仍然可以达到82%.
同年, Monnier和Taillefer小组[42]也发展了链状酰胺直接环丙烷化反应(Eq. 29).反应以1.0 equiv.的醋酸铜为催化剂, 吡啶作碱, 环丙基硼酸酯为偶联试剂, 在空气中反应就能发生.不同酰胺包括链状和环状都能取得中等到优秀的收率, 从而高效地在化合物中引入环丙基, 这是在药物分子修饰引入环丙基的有效策略.
1.1.2 氮杂环参与的C—N键偶联反应
2-取代咪唑啉衍生物不仅是很多生物活性分子的重要骨架, 也被认为是用于调节咪唑啉受体药理学活性的药效团[23].同时, N-芳基-2-取代咪唑啉也是一种酶抑制剂[24], 因此这类化合物的合成是非常重要而有意义. 2014年, Krasavin小组[25]通过Chan-Evans-Lam偶联反应以良好的收率合成了不同的N-芳基-2-取代咪唑啉化合物28.该反应条件温和, 在空气氛围中就能进行.反应底物适用范围较广, 但反应中醋酸铜需要加入1.5 equiv. (Eq. 10).
2014年, Onaka和Maegawa小组[26]报道了使用[Cu(OH)(TMEDA)]2Cl2为催化剂, 在二氯甲烷中, 以氧气为氧化剂首次高区域选择性实现了5-烷基四氮唑化合物29 的N-芳基化.反应中N(1) 并没有发生芳基化, 而是选择性地在N(2) 位进行芳基化.不同底物都能以良好的产率得到N-芳基四氮唑化合物30 (Eq. 11).
3-甲基水杨酸铜(Ⅰ) (CuMeSal)是市售的一种铜催化剂, 价格便宜. 2015年, Boykin小组[27]将这种催化剂用到芳香杂环化合物31与芳基硼酸的偶联反应中.反应以甲醇为溶剂, 碳酸钾或碳酸铯为碱, 空气为氧化剂, 可以以中等到良好的产率得到芳基化产物32.但对于邻位取代的芳基硼酸, 偶联产物如32b的产率比较低(Eq. 12).
2016年, Kumar小组[28]发展了一种在空气条件下亚氨基化合物33 与芳基硼酸的C—N键偶联反应.该反应在铜的催化作用下, 乙醇作为溶剂, 在室温下不需要配体和碱, 反应就能进行.亚氨基是一种比较敏感的官能团, 而在这种反应条件下N-芳基化产物可以取得较好的收率(Eq. 13).同年, Pale小组[29]也发展了一种无配体无碱参与的Chan-Evans-Lam偶联反应(Eq. 14).该反应的条件是以铜-沸石(Cu-USY)为催化剂, 在甲醇中回流, 不同的咪唑杂环化合物35 与芳基硼酸进行偶联反应, 以优秀产率得到N-芳基化的产物36.
2-喹啉酮由于分子内存在异构化平衡, 分子中的N和O原子都可以作为亲核试剂参与反应. Das小组[8]通过进一步研究, 发现以CuOTf为催化剂, 1, 10-菲咯啉(1, 10-Phen)为配体, 不需要加入碱, 在室温下就可以实现2-喹啉酮37 与芳基硼酸选择性地N-芳基化, 反应中并没有发现O-芳基化产物(Eq. 15).通过DFT化学计算研究机理表明, 无论是动力学还是热力学, 形成N-芳基化中间体以及N-芳基化产物的能量更低, 更有利于发生N-芳基化反应.
3-氨基苯并吲唑或2-氨基苯并咪唑底物中含两个不同亲核位点的氮原子, 都可以发生N-芳基化. Das课题组[30]利用Chan-Evans-Lam反应的策略, 通过铜催化3-氨基苯并吲唑或2-氨基苯并咪唑与芳基硼酸的连续N-芳基化得到二芳基取代唑类杂环化合物(Scheme 7).该课题组在不加入配体和碱的温和条件下, 实现了3-氨基苯并吲唑或2-氨基苯并咪唑化合物选择性地进行单芳基反应.通过加入无机碱CsOPiv还可以对单芳基化产物40 进行进一步N-芳基化.该方法的发现拓展了选择性的Chan-Evans-Lam反应.
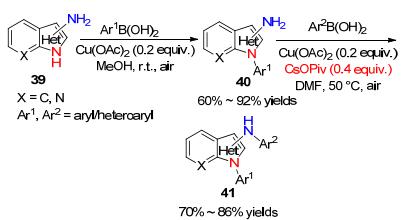 图 图式7
唑类杂环化合物选择性 N-芳基化反应
Figure 图式7.
Selective N-arylation of azole compounds
图 图式7
唑类杂环化合物选择性 N-芳基化反应
Figure 图式7.
Selective N-arylation of azole compounds
2015年, Prestat小组[31]首次利用CuI与CuII辅助的Ullmann和Chan-Evans-Lam串联反应实现了一锅法3-氨基吡唑(42)的双芳基化(Eq. 16).这种新的串联方法在小分子药物的发现中是一个重要的策略.该反应首先用碘化亚铜作催化剂, 碳酸铯作碱, (NMP)作溶剂, 3-氨基吡唑与芳基碘化物在120 ℃下反应24 h后, 直接加入芳基硼酸、四氟硼酸银、醋酸、3 分子筛, 并在80 ℃下反应23 h, 就可以直接生成二芳基化产物.对于芳基碘化合物与芳基硼酸, 芳基上是给电子和拉电子取代基时, 都可以以良好收率得到目标产物43.但以间硝基碘苯和4-甲基苯硼酸作为反应试剂时, 双芳基化产物43h 的产率只有14%.
为了实现2-氨基苯并咪唑化合物选择性芳基化. 2016年, Das课题组[32]用醋酸铜作催化剂, 2, 2-联吡啶为配体, 碳酸铯为碱, 在60 ℃条件下实现了2-氨基苯并咪唑化合物44 在氨基上进行的选择性N-芳基化, 反应中并没有观察到苯并咪唑环中氮原子进行芳基化的产物(Eq. 17). 2, 2-联吡啶对氮芳基化的选择性有非常重要的影响, 如果不加入2, 2-联吡啶配体, 反应会选择性地在苯并咪唑中的氮原子上进行N-芳基化(Eq. 18).
在此基础上, Das课题组[33]通过不同金属催化剂调控2-氨基苯并咪唑化合物的选择性N-芳基化(Scheme 8).研究表明:当使用Cu2O为催化剂, 芳基碘化物为偶联试剂时, 得到的产物46 是2-氨基苯并咪唑的1位氮原子进行芳基化, 而使用Ni(OAc)2为催化剂, 芳基硼酸为偶联试剂时, 得到的产物45 是2-氨基苯并咪唑的2位氨基芳基化.
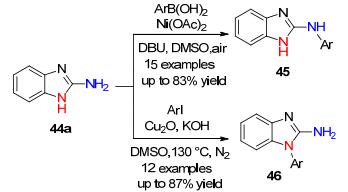 图 图式8
2-氨基苯并咪唑的选择性 N-芳基化反应
Figure 图式8.
Selective N-arylation of 2-aminobenzimidazole compounds
图 图式8
2-氨基苯并咪唑的选择性 N-芳基化反应
Figure 图式8.
Selective N-arylation of 2-aminobenzimidazole compounds
2014年, Arvidsson小组在醋酸铜为催化剂, 三乙胺作碱, 乙腈作溶剂的条件下, 实现了氮杂环取代亚氨基磺酰胺47 与芳基硼酸的N-芳基化.在亚氨基磺酰胺的反应物中, 不论是未保护的氮原子还是保护的氮原子, 都可以实现氮的选择性芳基化, 兼容不同的杂环骨架, 产物的产率从良好到优秀(Eq. 19).
1.1.1 胺类化合物参与的C—N键偶联反应
2013年, Messaoudi小组[11]报道了以醋酸铜作为催化剂, 吡啶作为碱, 空气作为氧化剂, 葡萄糖胺化合物作为反应底物, 在室温下与芳基硼酸进行偶联反应.首次实现了葡萄糖胺C(sp2)—N的构建, 反应底物的官能团兼容性较好, 以中等到良好的产率得到β-葡萄糖胺类产物3 (Eq. 2).
近年来, 无溶剂的化学合成方法也受到了化学工作者的广泛关注. 2014年, Su小组[12]发展了一种无溶剂参与的, 芳香胺4 或烷基胺5 与芳基硼酸的N-芳基化偶联反应.该反应在研磨条件下可以快速地得到N-芳基化产物, 水合醋酸铜比醋酸铜更加经济廉价, 无溶剂的反应可以减少溶剂对反应带来的负影响.在这种温和条件下, 反应可以以良好的收率得到N-芳基化产物(Scheme 2).
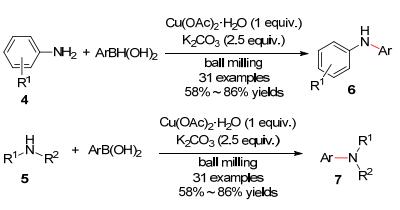 图 图式2
研磨条件下胺的 N-芳基化反应
Figure 图式2.
N-Arylation of amines under ball milling conditions
图 图式2
研磨条件下胺的 N-芳基化反应
Figure 图式2.
N-Arylation of amines under ball milling conditions
同年, Bora小组[13]利用Cu(Ⅱ)-salen配合物在室温条件下实现了苯胺衍生物4 或咪唑类化合物8 的N-芳基化(Scheme 3).该反应使用的溶剂都是低毒的.苯胺类化合物的N-芳基化以水作溶剂, 咪唑类化合物的N-芳基化是以异丙醇作溶剂, 反应底物适用性广.该方法中Cu(Ⅱ)-salen络合物易制备, 起始原料都已商业化, 为该方法的进一步应用提供良好的前景.
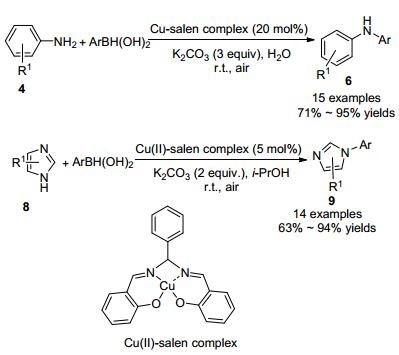 图 图式3
Cu-Salen 络合物催化的 C—N 偶联反应
Figure 图式3.
Cu-Salen complex-catalyzed C—N coupling reaction
图 图式3
Cu-Salen 络合物催化的 C—N 偶联反应
Figure 图式3.
Cu-Salen complex-catalyzed C—N coupling reaction
Das课题组[14]利用Chan-Evans-Lam反应与分子内Pd催化C—H键活化交叉偶联反应的串联策略, 实现了咔唑类衍生物12 的合成(Scheme 4).反应首先是3-碘-4-胺基苯甲酸甲酯(10)与芳基硼酸在醋酸铜的催化下进行N-芳基化得到的芳基化产物11, 化合物11 在醋酸钯催化剂下, 发生分子内的交叉偶联反应形成新的C—C键, 关环后得到咔唑化合物12.
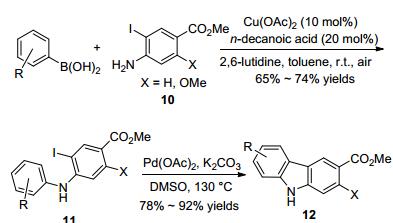 图 图式4
Chan-Evans-Lam 反应合成咔唑化合物
Figure 图式4.
Synthesis of carbazoles via the Chan-Evans-Lam
reaction
图 图式4
Chan-Evans-Lam 反应合成咔唑化合物
Figure 图式4.
Synthesis of carbazoles via the Chan-Evans-Lam
reaction
可见光作为取之不尽的清洁能源, 在有机合成化学中具有非常大的潜力.近年来, 光化学在偶联反应中的应用已经引起了许多化学工作者的广泛关注. 2015年, Kobayashi课题组[15]发展了一种可见光催化的苯胺衍生物与芳基硼酸的偶联反应(Eq. 3).反应中以醋酸铜为催化剂, 豆蔻酸为添加剂, 2, 6-二甲基吡啶为碱, 在空气中, 使用蓝色LED灯为光源, 给电子和拉电子取代基的芳基硼酸可以以中等到优秀的产率得到胺化产物.
作者对偶联反应提出了可能机理(Scheme 5):二价铜物种首先与胺底物进行配体交换、再与芳基硼酸发生转金属形成胺基铜物种F.在光和氧气作用下光敏剂15 变成高氧化态Ir物种16 和超氧, 物种F 在高价态Ir络合物作用下通过单电子氧化形成三价铜物种G, 三价铜物种G 发生还原消除反应形成N-芳基化产物14 和再生催化剂Cu(Ⅰ)物种, 完成催化循环.
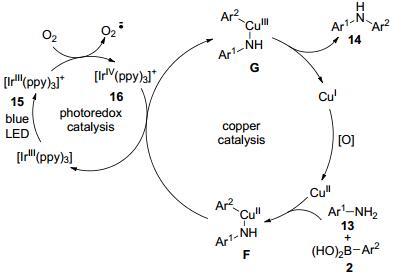 图 图式5
光介导的铜催化芳胺与芳基硼酸偶联反应机理
Figure 图式5.
Proposed mechanism for Visible-Light-Mediated copper-catalyzed coupling of arylamines and aryl boronic acids
图 图式5
光介导的铜催化芳胺与芳基硼酸偶联反应机理
Figure 图式5.
Proposed mechanism for Visible-Light-Mediated copper-catalyzed coupling of arylamines and aryl boronic acids
2015年, Das小组[16]进一步将Ullmann与Chan-Evans-Lam串联反应的策略简化, 通过铜催化分子间/分子内的两次C—N键的形成构建稠环苯并咪唑杂环化合物18 (Eq. 4).该反应产物的形成首先是经过分子间的Chan-Evans-Lam偶联反应, 再经过分子内的Ullmann反应而形成的.反应不需要加入配体就能进行, 而且稠环苯并咪唑的产率可以取得中等到优秀.这一策略为稠环苯并咪唑类杂环化合物的合成提供了简洁高效的方法.
同年, Wu课题组[17]发展了铜盐催化2-氨基吡啶(19)和芳基硼酸的偶联反应, 以良好的产率高效地合成了N-芳基吡啶-2-胺衍生物20.芳基硼酸适用于含敏感官能团如溴, 碘及酯基等(Eq. 5).与此同时, Molander小组[18]也同样利用铜催化Chan-Evans-Lam偶联反应的方法合成了N-芳基化2-氨基噻吩-3-甲酸甲酯化合物22 (Eq. 6).
2016年, Baxendale课题组[19]实现了使用氧气作为铜催化剂再生的氧化剂进行各种胺和芳基硼酸的Chan-Evans-Lam偶联反应.使用氧气作为氧化剂比使用其它氧化剂如TEMPO和过氧化苯甲酸叔丁酯具有更好的原子经济性和可操作性.反应适用于不同的芳胺、脂肪胺、杂芳香胺和氮杂环化合物等(Eq. 7).
为了进一步拓展胺的底物范围及反应的高效性, Phukan小组[20]在2016年合成了一种新颖的四方锥铜络合物[Cu(DMAP)4I]I, 该配合物的制备方法简单, 只需要CuI和DMAP在DMSO溶剂里搅拌.此反应以四方锥铜配合物为催化剂, 在甲醇中室温条件下进行偶联反应, 反应底物范围非常广泛, 芳基硼酸不但可以与伯氨, 仲氨反应, 也可以与酰胺、叠氮化物、硫醇反应, 取得中等以上的收率, 比如:当反应底物为氨时, 不论是芳基硼酸、杂环芳基硼酸还是烷基硼酸, 产率都可以达到67%以上.当使用不同的芳基硼酸与酰胺、叠氮化物反应时, 也可以得到60%以上的收率, 这表明该铜络合物具有非常高的催化活性, 为催化剂的进一步设计和开发提供了基础(Eq. 8).
2017年, Das小组[21]通过Chan-Evans-Lam反应对氨基苯酚选择性N-芳基化进行了研究(Scheme 6).研究表明:当间氨基苯酚为底物时, 以醋酸铜为催化剂, 醋酸银为添加剂, 在甲醇中室温下可以选择性地在氨基上发生偶联反应得到产物25.而当对氨基苯酚为底物时, 则以Cu(OAc)2/Cs2CO3为体系, 苯甲酸为添加剂, 在1, 4-二氧六环中90 ℃下进行氨基偶联反应得到26.
 图 图式6
氨基苯酚的选择性 N-芳基化反应
Figure 图式6.
Selective N-arylation of aminophenols
图 图式6
氨基苯酚的选择性 N-芳基化反应
Figure 图式6.
Selective N-arylation of aminophenols
Chan-Evans-Lam偶联反应中的硼试剂通常都是芳基硼酸, 而芳基硼酸频哪醇酯(BPin)由于其反应活性较芳基硼酸低, 在Chan-Evans-Lam反应中研究得比较少, 其产率通常较低, 特别是与芳香胺的反应中. 2016年, Watson小组[22]在醋酸铜作催化剂, 乙腈和乙醇为混合溶剂下发展了芳基硼酸频哪醇酯和芳香胺, 烷基胺的C—N键偶联反应, 胺化产物的产率可以达到优秀.值得注意的是, 当芳基硼酸频哪醇酯与烷基胺进行偶联反应时, 则需用乙腈作溶剂.该反应拓展了Chan-Evans-Lam偶联反应中硼试剂的范围(Eq. 9).
1.1.4 肟参与的C—N键偶联反应
2013年, Anderson小组[37]发现α, β-不饱和酮肟与芳基硼酸在醋酸酮的作用下选择性地发生N-芳基化, 得到α, β-不饱和N-芳基硝酮54 (Eq. 22).反应条件温和, α, β-不饱和酮肟与芳基硼酸底物的适用范围都较广, 芳基上是给电子与拉电子基团时, 都能以中等的产率得到硝酮.但是当芳基硼酸为邻甲基或邻溴苯硼酸时, 产物硝酮的产率比较低.结果表明芳基硼酸取代基的位阻效应对偶联反应的影响比较大.大部分官能团包括卤素、双键、硝基、酯基等都能取得较好的产率.这一策略为α, β-不饱和N-芳基硝酮的合成提供了简便的方法.
作者对进一步α, β-不饱和N-芳基硝酮的反应性质做了研究.发现在铜催化下, α, β-不饱和N-芳基硝酮可以发生氧转移反应得到环氧亚胺化合物(Eq. 23).反应中只需要催化量的CuCl和1, 10-菲咯啉(1, 10-Phen)为配体, 在乙腈溶剂中以优秀的产率得到目标产物55.环氧亚胺化合物55 中亚胺的E/Z 比例约为2:1.
1.1.3 磺酰叠氮参与的C—N键偶联反应
芳基磺酰胺类化合物因具有抗癌、抗菌等生物活性, 在药物化学中被广泛研究.因此, 芳基磺酰胺类化合物的合成尤为重要.在以往的合成中, 因磺酰胺的亲核性低, 反应都需要很高的温度或很长的时间. 2014年, Kim课题组[35]发展了一种温和、高效、快速地合成N-芳基磺酰胺类化合物的方法, 该反应不需要碱和配体, 只需在氯化亚铜作催化剂, 甲醇为溶剂, 在室温下, 就能高效实现磺酰叠氮化合物与芳基硼酸的偶联反应, 进而合成一系列的芳基磺酰胺类化合物(Eq. 20).
同时, 该小组对磺酰叠氮与芳基硼酸的偶联反应提出了可能的机理(Scheme 9):首先氯化亚铜H 被氧化成二价铜物种I.磺酰叠氮与二价铜I 配位脱掉氮气形成络合物J, 中间体J 进一步与芳基硼酸发生转金属得到中间体K, 在氧气条件下, 中间体K 被氧化成三价铜物种L, 然后还原消除形成产物50 并再生一价铜物种H, 从而实现催化循环.
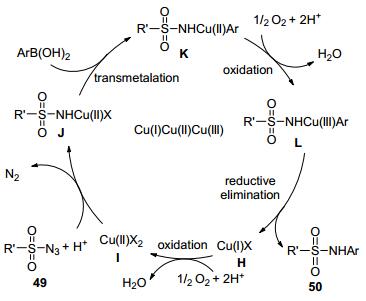 图 图式9
磺酰叠氮与芳基硼酸的偶联反应机理
Figure 图式9.
Proposed mechanism for coupling of sulfonyl azides and aryl boronic acids
图 图式9
磺酰叠氮与芳基硼酸的偶联反应机理
Figure 图式9.
Proposed mechanism for coupling of sulfonyl azides and aryl boronic acids
2016年Cai课题组[36]发展了一种环境友好的脯胺酸络合的铜络合物(MCM-41-L-Proline-CuCl)为固载催化剂合成N-芳基磺酰胺的方法, 以甲醇作溶剂在室温条件下反应就可以进行, 此反应中没有用到任何的碱或其它添加物.反应为非均相反应, 产物易于分离和纯化, 所用的催化剂廉价易得, 对环境友好.这种新的方法适用于多种芳基硼酸与磺酰基叠氮化物的偶联反应, 以优秀的产率得到N-芳基磺酰胺化合物(Eq. 21).这为Chan-Evans-Lam反应在绿色化学领域开辟新的思路.
1.2.1 肟参与的C—N键偶联反应
除了芳基硼酸参与Chan-Evans-Lam反应, 烯基硼酸由于其稳定性较差, 研究得相对较少.但烯基硼酸参与Chan-Evans-Lam反应后, 在产物中引入的双键使其具有丰富的化学转化, 因此, 烯基硼酸参与Ch an -Evans-Lam反应构建C—N键, 近年来也引起了广泛关注. 2012年, Anderson小组[38]报道了9-茐基酮肟(56)与烯基硼酸57 在化学计量的醋酸铜为催化剂, 10 equiv.的吡啶为碱下, 直接发生N-烯基化反应, 以良好的产率一步合成N-烯基硝酮.烯基硼酸的范围较广, 可以是链状单取代烯基硼酸、Z-双取代烯基硼酸以及环状烯基硼酸, 环状烯基硼酸可以兼容五、六、七元环.目前, 这一策略是合成N-烯基硝酮最有效和最直接的方法, 也为N-烯基硝酮的进一步发展开辟新的空间(Eq. 24).
在合成N-烯基硝酮后, 作者对烯基硝酮58 的热稳定性进行了研究, 发现在140 ℃下N-烯基硝酮可以发生分子内的重排形成螺茐基异噁唑啉衍生物59 (Eq.25).链状双取代的硝酮生成产物的产率比环状硝酮生成产物的产率较高, 时间也相对较短(59a, 59b vs 59c, 59d, 59e).而这一方法并不适用于单取代的烯基硝酮(59f).
为了实现铜催化的N-烯基化反应, Anderson小组[39]通过添加双烯化合物作为配体, 铜的用量可以降低到10% (Eq. 26).反应中只要加入10%的醋酸酮、1.2 equiv.的环辛二烯(COD), 以吡啶作碱, α, β-不饱和酮肟60 与烯基硼酸61 反应可以高效地得到α, β-不饱和N-烯基硝酮62.烯基硼酸可以是单取代、Z-双取代链状硼酸, 或者是不同环大小的环状烯基硼酸, 都可以以中等到良好的收率得到N-烯基硝酮.
1.2.2 酰胺参与的C—N键偶联反应
2016年, Ball小组[40]发展了以组氨酸(histidine)为导向的N-烯基化反应(Eq. 27).反应中, 以缓冲液为溶剂, 醋酸铜为催化剂, 于室温在咪唑基的导向下反应, 选择性地在酰胺的氮原子上引入烯基, N-烯基化产物的产率高达99%.这一策略为选择性偶联反应, 特别是反应位点比较多的复杂底物提供了新的思路.
2 C—O键形成的偶联反应
2.1 芳基硼试剂参与C—O键偶联反应
2.2 烯基硼试剂参与的C—O键偶联反应
2.1.1 肟参与C—O键偶联反应
O-芳基肟醚是天然产物或药物分子的重要骨架, 由于N—O键化合物可以通过N—O断裂而具有丰富的化学转化, 因此, 这类化合物的合成引起有机化学家的广泛关注[43]. C—O键的交叉偶联反应是实现这一目标最有效方法之一. 2012年, Bora小组[44]发展了一种合成O-芳基肟醚的方法(Eq. 30).该反应以醋酸铜为催化剂, 碳酸铯为碱, 在室温条件下就能发生, 偶联产物产率达到82%.当芳基硼酸为邻甲基苯硼酸时, 只能得到18%的O-芳基肟醚产物; 而当硼酸为2-噻吩硼酸时, 只能得到痕量的产物.
2014年Mulla小组[45]合成了铜盐负载催化剂(Eq. 31).底物中可兼容不同的官能团, 如醛基、硝基、酯基等.这一反应的特点是, 产物易于分离和纯化, 催化剂(CuFAP)可以回收至少6次, 且回收以后的铜催化剂催化的活性没有降低, 反应的产率仍然高达90%.
2.1.2 醇、酚参与C—O键偶联反应
2013年, Das课题组[46]分别用羟基香豆素、(羟基亚氨基)乙基香豆素与芳基硼酸进行C—O偶联反应, 得到了O-芳基化香豆素衍生物, 不同位置上取代的羟基香豆素发生O-芳基化反应的产率较好.这种铜催化的羟基香豆素作为亲核体与芳基硼酸的偶联反应是修饰香豆素的有效策略(Eqs. 32~35).
2014年, Církva小组[47]用铜催化氯代苯酚83 与氯代苯基硼酸偶联的方法合成多氯二苯醚化合物, 该反应在0 ℃下以醋酸铜为催化剂, 三乙胺为碱, 加入4 分子筛的条件下进行.当对位、间位取代芳基硼酸为试剂时, 反应产率较好; 而当邻位取代的芳基硼酸为试剂时, 偶联反应的产物产率较低(Eq. 36).
氟原子具有很强的电负性, 半径小, C—F键极化率低等性质, 有机氟化合物在有机化学中扮演着重要的角色, 可以改变化合物的性质. 2015年, Wu课题组[48]发展了一种有效合成三氟乙基芳基醚的方法.该反应以三氟乙醇和芳基硼酸或杂芳基硼酸为原料, 在一水合醋酸铜作催化剂, 4-二甲氨基吡啶(DMAP)为碱, 在40 ℃条件下进行, 三氟乙醇既作亲核试剂又作溶剂, 芳基硼酸底物范围广, 可以取得中等到良好的产率(Eq. 37).
2014年, Molander小组[49]首次报道了氨基保护的丝氨酸和苏氨酸衍生物的β-氧芳基化反应.该反应条件温和, 与各种保护基(Boc, Cbz, Tr, Fmoc)兼容.反应的硼试剂底物可以是芳基三氟硼酸钾和芳基硼酸(Eq. 38).
2016年, Clark课题组[50]在铜催化条件下实现了邻位苄胺硼酸酯和苯酚的醚化反应.这种二芳醚是具有生物活性的骨架.在反应条件下, 通过对不同取代的邻位苄胺硼酸酯和苯酚的底物拓展, 可以得到中等到良好的收率.通过对苯酚与苯胺的竞争实验显示, 在反应条件下苯酚相对于苯胺, 更具有高的选择性, 更容易发生O-芳基化反应(Eq. 39).
2.2.1 羧基参与的C—O键偶联反应
2013年, Batey小组[51]发展了一种铜催化以烯基三氟硼酸盐和羧酸或羧酸盐进行Chan-Evans-Lam反应合成烯醇酯.该反应的条件温和, 以溴化铜为催化剂, N, N-二甲基胺吡啶(DMAP)为配体, 4 分子筛为添加剂, 氧气为氧化剂, 可以以中等到优秀的产物得到烯醇酯94.反应具有一定的立体选择性, E-烯基硼酸得到E-烯醇醚, Z-烯基硼酸得到Z-烯醇醚.当反应底物为4-氯苯甲酸, 2-萘甲酸, 2-呋喃甲酸, 3-甲基丁酸和(E)-三氟己烯基硼酸钾反应时, 产率较低(Eq. 40).
2.2.2 羟胺、肟参与的C—O键偶联反应
2012年, Anderson小组[52]发展了铜介导N-羟基邻苯二甲酰亚胺的O-烯基化反应(Eq. 41).在1.0 equiv.的醋酸铜作用下, 以吡啶为碱, 在氯代溶剂中空气下进行反应.不同类型的烯基硼酸包括单取代、Z-二取代链状或环状烯基硼酸都可以发生, 产率可以达到中等到优秀.当三取代烯基硼酸置于该反应条件时, 反应并不能发生.值得一提的是, 反应也可以在20%催化量的醋酸铜下进行, 但是产率相比于1.0 equiv.的醋酸铜略低.
进一步研究O-烯基化产物96 的反应性质时, 作者发现产物96 在甲苯中加热时可以发生3, 3-重排反应, 通过水解或用苯甲酰氯保护就能得到α-羟基酮或苯甲酰基保护的α-羟基酮(Eq. 42).可以看出, 对于环状底物, 当环已基的4位与3位上有取代基时, 主要得到顺式重排产物(97j~97l, 97n).而环已基的2位有甲基时, 主要得到反式重排产物(97m).实验表明3, 3-重排的非对映选择性受到六元环上取代基的影响较明显.这一策略为α-羟基酮类化合物提供一条简易的合成方法.
2014年, Anderson小组[53]进一步将这一策略应用到N-芳基苯甲酰基羟肟酸的O-烯基化反应中(Eq. 43).作者发现不能得到偶联反应的产物, 而是直接得到3, 3-重排反应的产物99.反应表明, 当使用五水硫酸铜为铜盐时, 只要体系中加入锌粉, 五水硫酸铜的量可以降低到20%;而如果是Cu Br时, 铜盐的量需要加到1. 0 equiv., 反应才能取得较好的产率. Anderson小组发展的Chan-Lam偶联反应与3, 3-重排串联反应策略为邻取代芳香胺类化合物的合成提供了很好的方法.
肟化合物与芳基硼酸的偶联反应构建O-芳基化已经有很多报道, 但对于肟的O-烯基化反应研究则报道得相对较少. Anderson小组[54]在2013年就实现了肟的O-烯基化反应(Eq. 44).发现二苯甲酮肟与烯基硼酸在2-噻酚甲酸铜(CuTC)为催化剂, DABCO为碱, AgClO4为添加剂, 在空气中可以直接发生O-烯基化.烯基硼酸上可以兼容不同的官能团, 如硅基、卤素、腈基、酯基等, 但是烯基硼酸只适用于单取代链状烯基硼酸, 双取代的烯基硼酸则不发生反应.
作者进一步研究发现, O-烯基化产物101 在加热条件下可以发生1, 3-重排生成α-胺基醛化合物102 (Scheme 10).由于α-胺基醛化合物稳定性差, 进一步经过Horner-Wadsworth-Emmons烯化反应以优秀的产率将醛转化成更稳定的烯丙基胺类化合物103.作者通过一锅法策略, 方便地合成了γ-亚胺-α, β-不饱和酯化合物.
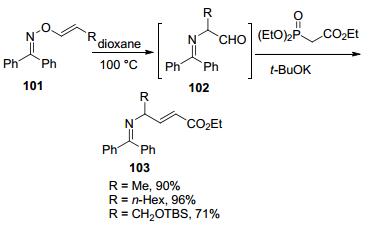 图 图式10
O-烯基化产物的1,3-重排反应
Figure 图式10.
1,3-Rearrangement of O-vinylation products
图 图式10
O-烯基化产物的1,3-重排反应
Figure 图式10.
1,3-Rearrangement of O-vinylation products
3 C—S键形成的偶联反应
3.1 硫酚参与的C—S键偶联反应
芳基硫化物在制药工业和材料科学方面有广泛的应用, 同时也是有机合成中重要的中间体, 但是C—S键的形成并没有受到化学工作者很大的关注, 因为在C—S键形成的过程中, 硫物种的存在可能使催化剂快速不可逆地失活. 2012年, Feng小组[55]报道了在室温条件下芳基硫醇与芳基硼酸的偶联反应, 通过S-芳基化高效地构建C—S键.反应中只需5 mol%的铜催化剂, 1, 10-菲咯啉(1, 10-Phen)为配体, 并且以环境友好的乙醇作溶剂.反应底物范围较广, S-芳基化的产率从中等到优秀(Eq. 45).
2016年, Zhao课题组[56]合成了一种铜络合物负载催化剂(MCM-41-1, 10-Phen-CuSO4), 催化高效绿色的非均相反应合成不对称二芳基硫化物.该反应在乙醇中以硫醇和芳基硼酸为底物, n-Bu4NOH作碱, 在室温条件下进行S-芳基化反应.这种新的非均相铜催化剂可以通过简单的方法从商品化和廉价的原料制备, 并循环使用至少8次而没有损失催化活性.使用可回收的铜催化剂是对传统C—S偶联反应的强有力补充(Eq. 46).
3.2 二硫键化合物参与的C—S键偶联反应
2015年, Jiang课题组[57]发现二氧化碳可以促进氧化交叉偶联反应构建C—S键(Eq. 47).作者以硫代芳基磺酸钠为底物, Cu(OTf)2为催化剂, 1, 10-菲咯啉(1, 10-Phen)为配体, 碳酸银为添加剂, 过氧叔丁醚(DTBP)为氧化剂, 在二氧化碳的氛围中, 以良好的收率得到二芳基硫醚化合物109.反应底物范围广, 产率中等到优秀.进一步通过热重分析实验证实, 二氧化碳的存在可以抑制二硫化物的形成, 并对偶联反应起到加速作用.
在此基础上, Jiang课题组[58]通过Chan-Evans-Lam偶联反应, 发展了一种易制备的二硫化试剂110.在温和的条件下二硫化试剂110 和芳基硼酸反应形成新的C —S键(Eq. 48).作者发现反应体系的pH值大小是选择性地断裂C—S键而非S—S键的关键.这一方法将为在药物及天然产物的后期修饰提供简洁高效的策略.
同年, Singh小组[59]首次实现了铜催化芳基硼酸与α-烯醇二硫代酯112 的Chan-Evans-Lam交叉偶联反应得到不对称的α-氧代乙烯S, S-缩醛.该反应在室温下, 中性条件并且不需要任何的碱与配体就可以进行.反应条件温和, 操作简单, 反应时间短(5 min).在该反应条件下, 可以选择性地得到S-芳基化产物, 且反应底物适用性范围广, 产率达到优秀(Eq. 49).
4 C—C键形成的偶联反应
4.1 TMSCF3参与的C—C键偶联反应
由于碳原子作为亲核试剂, 亲核性不够, 通过Chan-Lam反应构建C—C键会受到很大限制, 研究得相对较少. 2012年Fu课题组[60]首次报道了铜催化的一级、二级烷基硼酸衍生物的三氟甲基化来构建C—C键.底物的官能团兼容性比较好, 包括酯、酰胺、醚、胺和羰基等都能发生反应, 环状和链状烷烃都可以通过这种方法成功地实现三氟甲基化(Eq. 50).
4.2 丙二酸酯衍生物参与的C—C键偶联反应
2016年, Lundgren小组[61]报道了铜催化芳基硼试剂与sp3-C亲核试剂的氧化偶联反应.在温和的条件下, 三氟甲磺酸铜选择性地催化芳基硼试剂与丙二酸酯类衍生物的sp3-C亲核试剂芳基化反应, 叔丙基丙二酸酯和酰胺酯可用作产生季碳中心的底物.与传统的交叉偶联反应相比, 这种方法在卤素亲电体(包括芳基溴化物和碘化物)的存在下是具有化学选择性的.反应底物范围比较广, 带有酰胺、磺酰基和膦酰基的底物都可以进行反应(Eq. 51).
同年, Lundgren小组[62]在进一步研究sp3-C亲核试剂的氧化偶联反应时发现, 丙二酸单酯衍生物在进行Chan-Evans-Lam反应时, 可以发生脱羧, 然后再发生偶联反应, 从而构建新的C—C键(Eq. 52).芳基硼酸的底物范围适用性很广, 包括含有给电子和拉电子基团的芳基硼酸及杂环硼酸, 能以中等到良好的收率得到产物.
5 Chan-Evans-Lam偶联反应在天然产物合成中的应用
恶性疟原虫是发展中国家非常普遍引起疟疾的一种致病性寄生物. 2014年, Bogyo小组鉴定了一类可以选择性抑制这类疾病的帽型多肽123.这类帽型多肽具有很好的生物活性(IC50=35 nmol/L).为了进一步研究这类多肽的生物活性, Bogyo小组以Boc保护的3-溴苯丙氨酸120 为起始原料通过9步合成了该多肽.其中在关键成环步骤化合物121 到化合物122 合成中, 就使用Chan-Evans-Lam反应进行关环生成二苯醚骨架, 脱除Boc保护基得到目标多肽123 (Scheme 11).
 图 图式11
Chan-Evans-Lam 反应合成多肽中的应用
Figure 图式11.
Chan-Evans-Lam reaction in total synthesis of peptides
图 图式11
Chan-Evans-Lam 反应合成多肽中的应用
Figure 图式11.
Chan-Evans-Lam reaction in total synthesis of peptides
Baran小组[64]在2015年进行生物碱Verruculogen和Fumitremorgin A的全合成时, 其中最关键的一步就是Ir催化的C—H键硼化和Chan-Lam串联一锅法的策略在吲哚的C(6) 位高效地引入甲氧基团, 值得一提的是, 这一步串联反应可以实现克级量制备, 在C(6) 位和C(5) 位引进甲氧基的比例为8:1.从而以11步和12步分别成功地实现了生物碱Verruculogen和Fumitremorgin A全合成(Scheme 12).
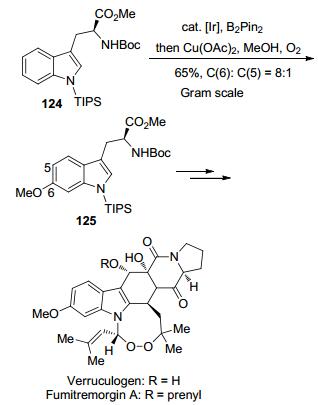 图 图式12
Chan-Evans-Lam 反应在生物碱合成中的应用
Figure 图式12.
Chan-Evans-Lam reaction in total synthesis of alkaloids
图 图式12
Chan-Evans-Lam 反应在生物碱合成中的应用
Figure 图式12.
Chan-Evans-Lam reaction in total synthesis of alkaloids
2016年, Lundgren小组[62]利用发展以sp3-C为亲核试剂进行的Chan-Evans-Lam反应, 结合钯催化的Suzuki偶联反应, 经过两步反应以较好的产率合成天然产物Nicergoline (Eq. 53).
6 结论与展望
从上面所举的例子以及其它许多成功例子来看, 铜催化的Chan-Evans-Lam偶联反应自从20世纪90年代发现以来, 已经取得了长足发展.目前, 这一反应在C—N, C—O, C—S, C—C键的构建上得到了成功的应用, 尤其在天然产物全合成、制药及材料科学领域, 已经有了广泛的应用, 但在机理认识上还相对滞后, 铜在反应中的具体中间体并不是特别清楚.
尽管Chan-Evans-Lam反应已经取得了很大的进展, 是一个充满活力的前沿研究领域.但仍然还有很多问题值得进行深入地研究.特别是对于含有多个亲核位点的底物, 选择性的Chan-Evans-Lam反应目前研究得并不多, 需要发展新的策略解决这一反应的选择性.同时其它过渡金属参与的Chan-Evans-Lam仍然有待开发. Chan-Evans-Lam反应的研究工作不仅具有重要的理论价值, 同时也有潜在的应用前景.我们相信, 经过科研工作者的不断努力, 这一反应在不久的将来会变得更加完善.
-
-
[1]
See reviews: (a) Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780.
(b) Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215.
(c) Mousseau, J. J.; Charette, A. B. Acc. Chem. Res. 2013, 46, 412.
(d) Liu, C.; Liu, D.; Lei, A. Acc. Chem. Res. 2014, 47, 3459. -
[2]
(a) Tan, M. ; Gu, Y. ; Luo, X. ; Zhang, P. Chin. J. Org. Chem. 2015, 35, 781 (in Chinese).
(谭明雄, 顾运琼, 罗旭健, 张培, 有机化学, 2015, 35, 781. )
(b) Zhang, Y. ; Nie, B. ; Zhang, J. Chin. J. Org. Chem. 2015, 35, 2067 (in Chinese).
(张英俊, 聂飚, 张霁, 有机化学, 2015, 35, 2067. )
(c) Lu, Q. ; Yi, H. ; Lei, A. Acta Chim. Sinica 2015, 73, 1245 (in Chinese).
(陆庆全, 易红, 雷爱文, 化学学报, 2015, 73, 1245. )
(d) Zhang, J. ; Lu, Q. ; Liu, C. ; Lei, A. Chin. J. Org. Chem. 2015, 35, 743 (in Chinese).
(张剑, 陆庆全, 刘超, 雷爱文, 有机化学, 2015, 35, 743. ) -
[3]
(a) Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winters, M. P. Tetrahedron Lett. 1998, 39, 2933.
(b) Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937.
(c) Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941. -
[4]
(a) Ley, S. V.; Thomas, A. W. Angew. Chen., Int. Ed. 2003, 42, 5400.
(b) Thomas, A. W.; Ley, S. V. In Modern Arylation Methods, Ed.: Ackermann, L., Wiley-VCH, Weinherim, 2008, p. 121. -
[5]
(a) Chan, D. M. T.; Lam, P. Y. S. In Boronic Acids, Ed.: Hall, D. G., Wiley-VCH, Weinheim, 2005, Chapter 5, p. 205.
(b) Li, J. J. In Name Recations: A Collection of Detailed Reaction Mechanisms and Synthetic Applications, 4th ed., Springer, New York, 2009, 102. -
[6]
(a) King, A. E.; Brunold, T. C.; Stahl, S. S. J. Am. Chem. Soc. 2009, 131, 5044.
(b) King, A. E.; Ryland, B. L.; Brunold, T. C.; Stahl, S. S. Organometallics 2012, 31, 7948. -
[7]
Larsson, P. F.; Wallentin, C. J.; Norrby, P. O. ChemCatChem 2014, 6, 1277.
-
[8]
Kumar, K. A.; Kannaboina, P.; Jaladanki, C. K.; Bharatam, P. V.; Das, P. ChemistrySelect 2016, 3, 601.
-
[9]
Qiao, J. X.; Lam, P. Y. S. Synlett 2011, 829.
-
[10]
Rao, K. S.; Wu, T. S. Tetrahedron 2012, 68, 7735. doi: 10.1016/j.tet.2012.06.015
-
[11]
Bruneau, A.; Brion, J. D.; Alami, M.; Messaoudi, S. Chem. Commun. 2013, 49, 8359. doi: 10.1039/c3cc44780d
-
[12]
Zhu, X.; Zhang, Q.; Su, W. RSC Adv. 2014, 4, 22775. doi: 10.1039/c4ra02952f
-
[13]
Gogoi, A.; Sarmah, G.; Dewan, A.; Bora, U. Tetrahedron Lett. 2014, 55, 31. doi: 10.1016/j.tetlet.2013.10.084
-
[14]
Rasheed, S.; Rao, D. N.; Reddy, K. R.; Aravinda, S.; Vishwakarma, R. A.; Das, P. RSC Adv. 2014, 4, 4960. doi: 10.1039/c3ra44903c
-
[15]
Yoo, W. J.; Tsukamoto, T.; Kobayashi, S. Angew. Chem., Int. Ed. 2015, 54, 6587. doi: 10.1002/anie.201500074
-
[16]
Rasheed, S.; Rao, D. N.; Das, P. J. Org. Chem. 2015, 80, 9321. doi: 10.1021/acs.joc.5b01396
-
[17]
Chen, J.; Natte, K.; Man, N. Y. T.; Stewart, S. G.; Wu, X. F. Tetrahedron Lett. 2015, 56, 4843. doi: 10.1016/j.tetlet.2015.06.092
-
[18]
Rizwan, K.; Karakaya, I.; Heitz, D.; Zubair, M.; Rasool, N.; Molander, G. A. Tetrahedron Lett. 2015, 56, 6839. doi: 10.1016/j.tetlet.2015.10.080
-
[19]
Mallia, C. J.; Burton, P. M.; Smith, A. M. R.; Walter, G. C.; Baxendale, L. R. Beilstein J. Org. Chem. 2016, 12, 1598. doi: 10.3762/bjoc.12.156
-
[20]
Roy, S.; Sarma, M. J.; Kashyap, B.; Phukan, P. Chem. Commun. 2016, 52, 1170. doi: 10.1039/C5CC04619J
-
[21]
Reddy, A. S.; Reddy, K. R.; Rao, D. N.; Jaladanki, C. K.; Bharatam, P. V.; Lam. P. Y. S.; Das, P. Org. Biomol. Chem. 2017, 15, 801. doi: 10.1039/C6OB02444K
-
[22]
Vantourout, J. C.; Law, R. P.; Isidro-Llobet, A.; Atkinson, S. J.; Watson, A. J. B. J. Org. Chem. 2016, 81, 3942. doi: 10.1021/acs.joc.6b00466
-
[23]
Krasavin, M. Eur. J. Med. Chem. 2015, 97, 525. doi: 10.1016/j.ejmech.2014.11.028
-
[24]
Sarnpitak, P.; Mujumdar, P.; Morisseau, C.; Hwang, C. H.; Hammock, B.; Iurchenko, V.; Zozulya, S.; Gavalas, A.; Geronikaki, A.; Ivanenkov, Y.; Krasavin, M. Eur. J. Med. Chem. 2014, 84, 160. doi: 10.1016/j.ejmech.2014.07.023
-
[25]
Darin, D.; Krasavin, M. J. Org. Chem. 2016, 81, 12514. doi: 10.1021/acs.joc.6b02404
-
[26]
Onaka, T.; Umemoto, H.; Miki, Y.; Nakamura, A.; Maegawa, T. J. Org. Chem. 2014, 79, 6703. doi: 10.1021/jo500862t
-
[27]
Boykin, D. W.; Farehat, A. A. Synth. Commun. 2015, 45, 245. doi: 10.1080/00397911.2014.961196
-
[28]
Mandal, P. S.; Kumar, A. V. Synlett 2016, 27, 1408. doi: 10.1055/s-00000083
-
[29]
Garnier, T.; Sakly, R.; Danel, M.; Chassaing, S.; Pale, P. Synthesis 2017, 49, 1223.
-
[30]
Rao, D. N.; Rasheed, S.; Vishwakarma, R. A.; Das, P. Chem. Commun. 2014, 50, 12911. doi: 10.1039/C4CC05628K
-
[31]
Beyer, A.; Castanheiro, T.; Busca, P.; Prestat, G. ChemCatChem 2015, 7, 2433. doi: 10.1002/cctc.201500510
-
[32]
Rao, D. N.; Rasheed, S.; Kumar, K. A.; Reddy, A. S.; Das, P. Adv. Synth. Catal. 2016, 358, 2126. doi: 10.1002/adsc.v358.13
-
[33]
Kumar, K. A.; Kannaboina, P.; Rao, D. N.; Das, P. Org. Biomol. Chem. 2016, 14, 8989. doi: 10.1039/C6OB01307D
-
[34]
Nandi, G. C.; Kota, S. R.; Govender, T.; Kruger, H. G.; Arvidsson, P. I. Tetrahedron 2014, 70, 5428. doi: 10.1016/j.tet.2014.06.122
-
[35]
Moon, S. Y.; Nam, J.; Rathwell, K.; Kim, W. S. Org. Lett. 2014, 16, 338. doi: 10.1021/ol403717f
-
[36]
You, C.; Yao, F.; Yan, T.; Cai, M. RSC Adv. 2016, 6, 43605. doi: 10.1039/C6RA04298H
-
[37]
Mo, D.-L.; Anderson, L. L. Angew. Chem., Int. Ed. 2013, 52, 6722. doi: 10.1002/anie.v52.26
-
[38]
Mo, D.-L.; Wink, D. A.; Anderson, L. L. Org. Lett. 2012, 14, 5180. doi: 10.1021/ol3022885
-
[39]
Kontokosta, D.; Mueller, D. S.; Mo, D.-L.; Pace, W. H.; Simpson, R. A.; Anderson, L. L. Beilstein J. Org. Chem. 2015, 11, 2097. doi: 10.3762/bjoc.11.226
-
[40]
Ohata, J.; Minus, M. B.; Abernathy, M. E.; Ball, Z. T. J. Am. Chem. Soc. 2016, 138, 7472. doi: 10.1021/jacs.6b03390
-
[41]
Rossi, S. A.; Shimkin, K. W.; Xu, Q.; Mori-Quiroz, L. M.; Watson, D. A. Org. Lett. 2013, 15, 2314. doi: 10.1021/ol401004r
-
[42]
Racine, E.; Monnier, F.; Vors, J.-P.; Taillefer, M. Chem. Commun. 2013, 49, 7412. doi: 10.1039/c3cc42575d
-
[43]
(a) Tabolin, A. A.; Ioffe, S. L. Chem. Rev. 2014, 114, 5426.
(b) Shi, W.-M.; Ma, X.-P.; Su, G.-F.; Mo, D.-L. Org. Chem. Front. 2016, 3, 116. -
[44]
Mondal, M.; Sarmah, G.; Gogoi, K.; Bora, U. Tetrahedron Lett. 2012, 53, 6219. doi: 10.1016/j.tetlet.2012.09.003
-
[45]
Mulla, S. A. R.; Chavan, S. S.; Inamdar, S. M.; Pathan, M. Y.; Shaikh, T. M. Y. Tetrahedron Lett. 2014, 55, 5327. doi: 10.1016/j.tetlet.2014.07.056
-
[46]
Medda, A.; Pal, G.; Singha, R.; Hossain, T.; Saha, A.; Das, A, R. Synth. Commun. 2013, 43, 169. doi: 10.1080/00397911.2011.594544
-
[47]
Čermak, J. K.; Církva, V. Tetrahedron Lett. 2014, 55, 4185.
-
[48]
Wang, R.; Wang, L.; Zhang, K.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Tetrahedron Lett. 2015, 56, 4815. doi: 10.1016/j.tetlet.2015.06.066
-
[49]
Khatib, M. E.; Molander, G. A. Org. Lett. 2014, 16, 4944. doi: 10.1021/ol5024689
-
[50]
Marcum, J. S.; McGarry, K. A.; Ferber, C. J.; Clark, T. B. J. Org. Chem. 2016, 81, 7963. doi: 10.1021/acs.joc.6b01254
-
[51]
Huang, F.; Quach, T. D.; Batey, R. A. Org. Lett. 2013, 15, 3150. doi: 10.1021/ol4013712
-
[52]
Patil, A. S.; Mo, D.-L.; Wang, H. Y.; Mueller, D. S.; Anderson, L. L. Angew. Chem., Int. Ed. 2012, 51, 7799. doi: 10.1002/anie.201202704
-
[53]
Wang, H. Y.; Anderson, L. L. Org. Lett. 2013, 15, 3362. doi: 10.1021/ol401416r
-
[54]
Kontokosta, D.; Mueller, D. S.; Wang, H. Y.; Anderson, L. L. Org. Lett. 2013, 15, 4830. doi: 10.1021/ol402237w
-
[55]
Xu, H. J.; Zhao, Y. Q.; Feng, T.; Feng, Y. S. J. Org. Chem. 2012, 77, 2878. doi: 10.1021/jo300100x
-
[56]
Lin, Y.; Cai, M.; Fang, Z.; Zhao, H. Tetrahedron 2016, 72, 3335. doi: 10.1016/j.tet.2016.04.063
-
[57]
Qian, Z.; Ge, N.; Jiang, X. Chem. Commun. 2015, 51, 10295. doi: 10.1039/C5CC03038B
-
[58]
Xiao, X.; Feng, M.; Jiang, X. Angew. Chem., Int. Ed. 2016, 55, 14121. doi: 10.1002/anie.v55.45
-
[59]
Koley, S.; Chowdhury, S.; Chanda, T.; Ramulu, B. J.; Anand, N.; Singh, M. S. Eur. J. Org. Chem. 2015, 409.
-
[60]
Xu, J.; Xiao, B.; Xie, C. Q.; Luo, D. F.; Liu, L.; Fu, Y. Angew. Chem., Int. Ed. 2012, 51, 12551. doi: 10.1002/anie.201206681
-
[61]
Moon, P. J.; Halperin, H. M.; Lundgren, R. J. Angew. Chem., Int. Ed. 2016, 55, 1894. doi: 10.1002/anie.201510558
-
[62]
Moon, P. J.; Yin, S. K.; Lundgren, R. J. J. Am. Chem. Soc. 2016, 138, 13826. doi: 10.1021/jacs.6b08906
-
[63]
Li, H.; Tsu, C.; Blackburn, C.; Li, G.; Hales, P.; Dick, L.; Bogyo, M. J. Am. Chem. Soc. 2014, 136, 13562. doi: 10.1021/ja507692y
-
[64]
Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L. R.; Baran. P. S. J. Am. Chem. Soc. 2015, 137, 10160. doi: 10.1021/jacs.5b07154
-
[1]
-

图式3 Cu-Salen 络合物催化的 C—N 偶联反应
Scheme 3 Cu-Salen complex-catalyzed C—N coupling reaction

图式4 Chan-Evans-Lam 反应合成咔唑化合物
Scheme 4 Synthesis of carbazoles via the Chan-Evans-Lam reaction

图式5 光介导的铜催化芳胺与芳基硼酸偶联反应机理
Scheme 5 Proposed mechanism for Visible-Light-Mediated copper-catalyzed coupling of arylamines and aryl boronic acids

图式8 2-氨基苯并咪唑的选择性 N-芳基化反应
Scheme 8 Selective N-arylation of 2-aminobenzimidazole compounds

图式9 磺酰叠氮与芳基硼酸的偶联反应机理
Scheme 9 Proposed mechanism for coupling of sulfonyl azides and aryl boronic acids

图式11 Chan-Evans-Lam 反应合成多肽中的应用
Scheme 11 Chan-Evans-Lam reaction in total synthesis of peptides
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 838
- 文章访问数: 53671
- HTML全文浏览量: 8600
 下载:
下载:
