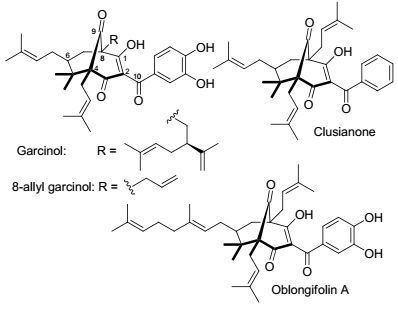
 图 1
山竹醇、8-烯丙基山竹醇、Clusianone、Oblongifolin A的化学结构
Figure 1.
Chemical structure of garcinol, 8-allyl garcinol, clusianone and oblongifolin A
图 1
山竹醇、8-烯丙基山竹醇、Clusianone、Oblongifolin A的化学结构
Figure 1.
Chemical structure of garcinol, 8-allyl garcinol, clusianone and oblongifolin A
引用本文:
曹菁, 韩超明, 张桂莲, 周新莹, 李舒雯, 杜银端, 赵帅, 张辛燕, 陈新. 8-烯丙基山竹醇的合成及其抗癌活性研究[J]. 有机化学,
2017, 37(8): 2086-2093.
doi:
10.6023/cjoc201701017

Citation: Cao Jing, Han Chaoming, Zhang Guilian, Zhou Xinying, Li Shuwen, Du Yinduan, Zhao Shuai, Zhang Xinyan, Chen Xin. Synthesis and Anticancer Activity of 8-Allyl Garcinol[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 2086-2093. doi: 10.6023/cjoc201701017
Citation: Cao Jing, Han Chaoming, Zhang Guilian, Zhou Xinying, Li Shuwen, Du Yinduan, Zhao Shuai, Zhang Xinyan, Chen Xin. Synthesis and Anticancer Activity of 8-Allyl Garcinol[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 2086-2093. doi: 10.6023/cjoc201701017
8-烯丙基山竹醇的合成及其抗癌活性研究
摘要:
山竹醇来源于印度藤黄,具有抗癌和抗炎活性.用烯丙基取代山竹醇结构中C(8)位置上的过大支链5-甲基-2-(1-甲基乙烯基)-4-己烯基,因为这个过大支链可能会影响山竹醇的生物活性和吸收度.以1,3-环己二酮为起始原料,经过Effenburger环化等一系列步骤得到关键桥环中间体(±)-1-羟基-5,5-二甲基-4,6-双(3-甲基-丁-2-烯基)-二环[3.3.1]壬-2-烯-3,9-二酮(8),随后通过五步反应得到8-烯丙基山竹醇,并用噻唑蓝(MTT)法对其进行生物活性测试.研究结果表明,8-烯丙基garcinol对口腔鳞癌细胞具有显著的抑制增殖作用.
-
关键词:
- 山竹醇
- / Effenburger环化
- / 合成
- / 结构改造
English
Synthesis and Anticancer Activity of 8-Allyl Garcinol
Abstract:
Garcinol, isolated from the fruit rind of Garcinia indica, shows anti-carcinogenic and anti-inflammatory activities. However, the C(8) side chain of garcinol is so large that it may influence the bioactivity of the compound. 8-Allyl garcinol, in which the bulky side chain at C(8) position of garcinol is replaced with much smaller allyl group, was synthesized through a 12-step procedure. Starting from 1, 3-cyclohexandione, the key intermediate (±)-1-hydroxy-5, 5-dimethyl-4, 6-bis(3-methylbut-2-en-1-yl)bicyclo[3.3.1]non-2-ene-3, 9-dione (8) was obtained via Effenburger cyclization. Through additional 5-steps, 8-allyl garcinol was prepared. The thiazolyl blue tetrazolium bromide (MTT) results indicated that 8-allyl garcinol shows strong inhibitory activity on cell proliferation of oral cancer cell lines.
-
Key words:
- garcinol
- / Effenburger cyclization
- / synthesis
- / structural modification
-
山竹醇, 英文名Garcinol, 为多聚异戊二烯基苯甲酮[1], 是从印度藤黄的干果皮中提取出来的一种黄色晶体化合物[2].在印度和东南亚地区, 印度藤黄的干果皮用来作为一种香料, 常用于一些民间医学, 印度藤黄的干果皮在治疗糖尿病、肥胖、溃疡和其它疾病有着重要作用[3~5].山竹醇的化学结构与含有酚羟基和β-二酮的姜黄素类似, 都具有很强的抗氧化活性[6, 7].山竹醇还具有抗炎, 抗癌活性[8~10], 同时是组蛋白乙酰转化酶p300抑制剂[11].此外, 在细胞培养研究中, 山竹醇还表现出抗增殖作用, 诱导细胞凋亡作用, 也是环氧化酶-2抑制剂, 5-脂氧化酶(5-LOX)抑制剂[3, 9, 12].
我们曾报道, 山竹醇通过抑制5-LOX, 对7, 12-二甲基苯并蒽(DMBA)引导的急性炎症和仓鼠口腔颊囊的口腔鳞状细胞癌有化学预防作用[13].计算机建模预测, 山竹醇与5-LOX的活性位点相结合从而抑制5-LOX, 并且, 山竹醇的13和14位的两个羟基对抑制5-LOX起着至关重要的作用[13].我们近期研究表明, 这两个羟基也是山竹醇抗癌活性的必需官能团[14].计算机建模还表明, 山竹醇结构中C(8) 位置上的过大支链可能会阻碍山竹醇与5-LOX的活性位点相结合, 进而不能有效抑制5-LOX.这个过大的支链还可能会导致山竹醇的抗癌活性降低, 膜渗透性变差[13].因此, 我们计划对山竹醇的C(8) 位置进行结构改造, 用烯丙基代替过大的5-甲基-2-(1-甲基乙烯基)-4-己烯基支链, 合成8-烯丙基山竹醇(8-allyl garcinol) (图 1).
图 1
山竹醇、8-烯丙基山竹醇、Clusianone、Oblongifolin A的化学结构
Figure 1.
Chemical structure of garcinol, 8-allyl garcinol, clusianone and oblongifolin A
目前, 8-烯丙基山竹醇还未见报道, 合成山竹醇及其衍生物的文献也不多. Socolsky等[15]以乙酰丙酮作为起始原料, 经过aldol缩合、Tsuji-Trost烷基化、Claisen环化等多步反应合成得到山竹醇. Biber等[16]从乙酰丙酮开始, 通过α-亚甲基化、Michael加成、Knoevenagel缩合、烷基化、Dieckmann缩合等反应, 合成了与8-烯丙基山竹醇结构相似的Oblongifolin A[15] (图 1). Rodeschini等[17]和Nuhant等[18]合成了与山竹醇结构相似的Clusianone (图 1).
1 结果与讨论
8-烯丙基山竹醇的合成路线如Scheme 1所示.参考Nuhant等[18]合成Clusianone的方法, 我们以1, 3-环己二酮作为原料, 以N, N-二异丙基乙胺(DIEA)作为碱, 与3, 3-二甲基烯丙基溴发生烷基化反应生成3-羟基-2-(3-甲基-2-丁烯基)环己-2-烯酮(2). 2与2-碘丙烷进行烷基化, 得到3-异丙氧基-2-(3-甲基-2-丁烯基)环己-2-烯酮(3), 两步产率为60%.在二异丙基氨基锂(LDA)作用下, 3再与3, 3-二甲基烯丙基溴发生烷基化, 得到3-异丙氧基-2, 6-双(3-甲基-2-丁烯基)环己-2-烯酮(4), 产率为71%.化合物4与甲基锂反应并酸化后, 以70%产率生成3-甲基-2, 4-双(3-甲基-2-丁烯基)环己-2-烯酮(5).接着, 5与甲基铜锂进行Michael加成, 得到双甲基化产物3, 3-二甲基-2, 4-双(3-甲基丁-2-烯基)环己酮(6).经过Me2SO4作用, 环己酮6被转化为甲醚化合物7.该中间体无需纯化, 直接与丙二酰氯发生环化反应, 得到关键的三环中间体8, 两步产率为27%.在对甲苯磺酸催化下, 8在甲醇中与原甲酸三甲酯反应, 将其中的羟基保护为甲醚9, 以便在下一步中, 在C(8) 位引入烯丙基生成10.
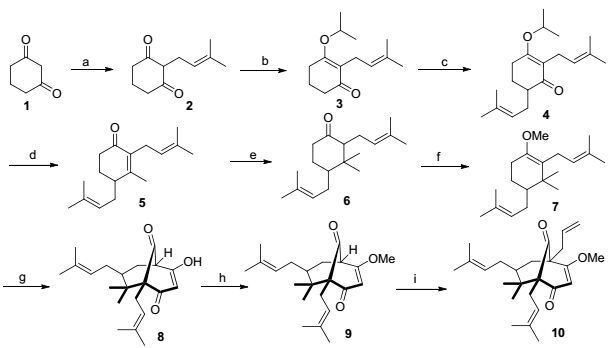 图式 1
三环中间体10的合成
图式 1.
Synthetic route for tricyclic intermediate 10
图式 1
三环中间体10的合成
图式 1.
Synthetic route for tricyclic intermediate 10
Rodeschini等[17]利用三环化合物1-甲氧基-5, 5-二甲基-4, 6, 8-三(3-甲基-丁-2-烯基)二环[3.3.1]壬-2-烯-3, 9-二酮(A1)与苯甲酰氯在强碱2, 2, 6, 6-四甲基哌啶锂(LTMP)作用下进行酰基化反应, 所得产物2-苯甲酰基-1-甲氧基-5, 5-二甲基-4, 6, 8-三(3-甲基-丁-2-烯基)二环[3.3.1]壬-2-烯-3, 9-二酮(A2)再经过LiOH水解而得到Clusianone (Scheme 2).然而, 在此条件下, 我们尝试了10与3, 4-二甲氧基苯甲酰氯反应, 却无法得到预期的酰基化产物11.我们将10在LiOH作用下进行水解, 以60%产率得到12 (Scheme 3).此反应看似简单, 却易导致很低的产率, 关键是要控制好反应温度和反应时间, 因为10不太容易被水解, 但温度过高或者反应时间过长, 却可能使三环中间体分解.用3, 4-二甲氧基苯甲酰氰[18]取代3, 4-二甲氧基苯甲酰氯后, 在三环化合物12 C2位上可以顺利进行酰化.对于3, 4-二甲氧基苯甲酰氰和相应酰氯同三环中间体(10和12)亲电反应的差异, 我们推测其可能原因是“氰基效应”[19], 也就是酰氰化合物的氰基能与烯醇化合物形成氢键, 而且在有机溶剂中呈现强碱性质, 因此, 酰氰比酰氯更有利于亲电取代反应.
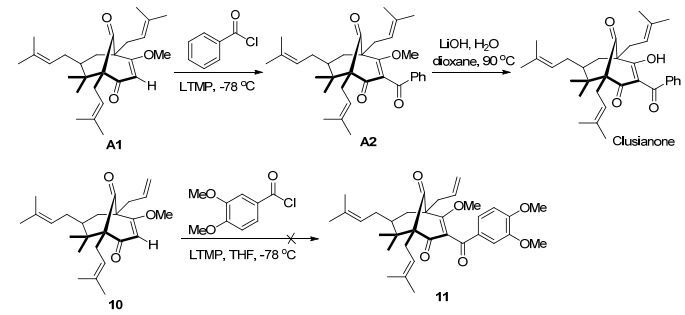 图 2
三环中间体与苯甲酰氯的酰基化反应
图 2.
Acylation of the tricyclic intermediates with benzoyl chlorides
图 2
三环中间体与苯甲酰氯的酰基化反应
图 2.
Acylation of the tricyclic intermediates with benzoyl chlorides
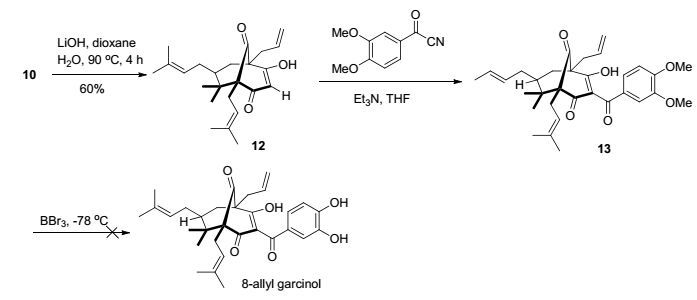 图式3
三环中间体与苯甲酰氰的酰基化反应
图式3.
Acylation of the tricyclic intermediates with benzoyl cyanides
图式3
三环中间体与苯甲酰氰的酰基化反应
图式3.
Acylation of the tricyclic intermediates with benzoyl cyanides
不过, 所得产物13在进行去甲基化反应时, 并未得到目标产物(Scheme 3).可能的原因是, 13在低温(-78 ℃)下反应不完全, 单羟基和双羟基化产物混在一起, 不易有效分离; 但若提高反应温度, 三溴化硼则会破坏三环骨架.
最终, 我们改变了酚羟基的保护基, 用3, 4-二乙酰氧基苯甲酰氰代替3, 4-二甲氧基苯甲酰氰, 再与化合物12进行酰基化反应, 以73%产率得到了酰基化物14. 最后, 14在碳酸钾和甲醇体系中进行水解, 以64%产率顺利得到目标产物8-烯丙基山竹醇(Scheme 4).
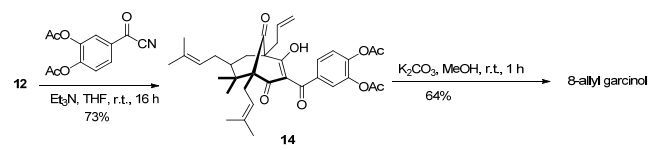 图式4
8-烯丙基山竹醇的合成
图式4.
Synthesis of 8-allyl garcinol
图式4
8-烯丙基山竹醇的合成
图式4.
Synthesis of 8-allyl garcinol
1.2 山竹醇和8-烯丙基山竹醇的生物活性比较
我们利用噻唑蓝(MTT)法, 比较了山竹醇和8-烯丙基山竹醇对口腔鳞癌细胞SCC-15和CAL-27细胞增殖的抑制活性.与阴性对照组相比, 山竹醇和8-烯丙基山竹醇均能够显著抑制口腔鳞癌细胞SCC-15和CAL-27细胞的增殖, 差异具有统计学意义(P<0.01).随着山竹醇和8-烯丙基山竹醇浓度的增高和作用时间延长, 对SCC-15和CAL-27细胞的抑制作用逐渐增强, 呈一定浓度和时间依赖关系(图 1和图 2).本研究发现, 在药物低至中浓度(5~20 μmol/L)时, 8-烯丙基山竹醇的抑制效果优于山竹醇(P<0.01).
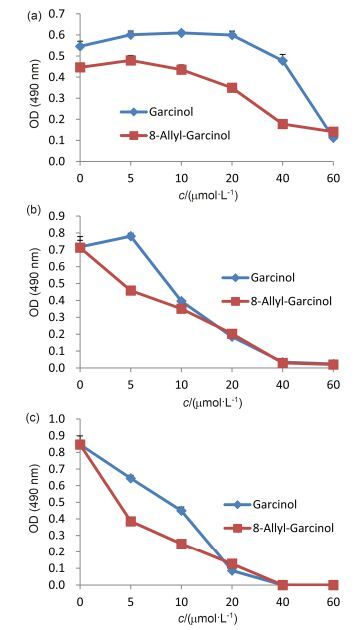 图 2
不同浓度gacinol和8-烯丙基garcinol处理SCC-15细胞24 (a)、48 (b)和72 (c) h的OD值
Figure 2.
OD values of SCC-15 cells treated with different concentrations of garcinol and 8-allyl garcinol at 24 (a), 48 (b) and 72 (c) h
图 2
不同浓度gacinol和8-烯丙基garcinol处理SCC-15细胞24 (a)、48 (b)和72 (c) h的OD值
Figure 2.
OD values of SCC-15 cells treated with different concentrations of garcinol and 8-allyl garcinol at 24 (a), 48 (b) and 72 (c) h
2 结论
以1, 3-环己二酮为起始原料, 采用了Effenburger环化为关键步骤, 经过12步反应得到了8-烯丙基山竹醇, 总产率为1.2%.该方法避免了使用贵重试剂和有毒试剂, 操作简单, 为山竹醇新衍生物的合成提供了参考, 具有潜在的应用价值.并且, 利用MTT法测得8-烯丙基山竹醇对人口腔鳞癌细胞具有显著的抑制增殖作用.
3 实验部分
3.1 仪器与试剂
熔点使用上海申光公司的显微熔点仪, 型号为SGW X-4B; NMR用Bruker (300和400 MHz)型核磁共振仪测定(CDCl3, DMSO-d6作溶剂, TMS为内标); HRMS用Thermo Orbitrap Elite测定; 昆山市超声仪器有限公司SQ-5200型超声波清洗器; 160~200目硅胶(青岛海洋化工厂生产); 薄层色谱使HSGF-254(购自烟台江友硅胶开发有限公司).试剂均为分析纯.
3.2 实验方法
3.2.1 3-羟基-2-(3-甲基-2-丁烯基)环己-2-烯酮(2)的合成
取1, 3-环己二酮(50 g, 446.2 mmol)溶于水中(100 mL), 冷却至0 ℃, 向其中缓慢加入N, N-二异丙基乙胺(57.53 g, 446.2 mmol), 之后再缓慢加入3, 3-二甲基烯丙基溴(73.08 g, 491 mmol), 在室温搅拌过夜(TLC监测反应进程).反应毕, 向反应液加入2 mol/L NaOH溶液, 调节pH≈11, 分液, 水相用乙酸乙酯萃取(300 mL), 之后用冰醋酸酸化, 直到白色固体不再析出, 此时pH≈3.抽滤, 并放在真空干燥箱干燥, 得到白色固体2 (48.16 g, 产率60%). m.p. 141~145 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 5.00 (t, J=7.1 Hz, 1H), 2.81 (d, J=7.0 Hz, 2H), 2.31~2.28 (m, 4H), 1.85~1.78 (m, 2H), 1.63 (s, 3H), 1.59 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ:129.74, 124.23, 114.70, 33.01, 26.06, 21.25, 21.13, 18.18; HRMS calcd for C11H17O2[M+H]+ 181.1223; found 181.1229..
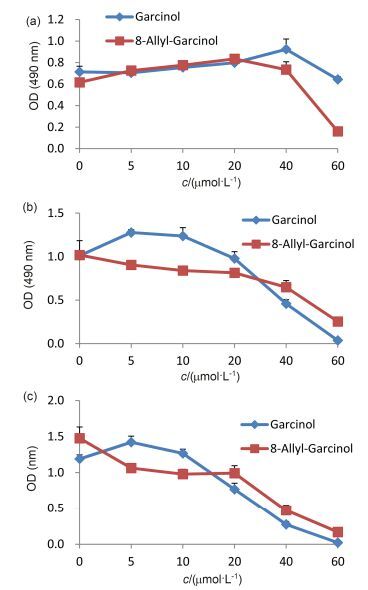 图 图 1
不同浓度gacinol和8-烯丙基garcinol处理CAL-27细胞24 (a)、48 (b)和72 (c) h的OD值
Figure 图 1.
OD values of CAL-27 cells treated with different concentrations of garcinol and 8-allyl garcinol at 24 (a), 48 (b) and 72 (c) h
图 图 1
不同浓度gacinol和8-烯丙基garcinol处理CAL-27细胞24 (a)、48 (b)和72 (c) h的OD值
Figure 图 1.
OD values of CAL-27 cells treated with different concentrations of garcinol and 8-allyl garcinol at 24 (a), 48 (b) and 72 (c) h
3.2.2 3-异丙氧基-2-(3-甲基-2-丁烯基)环己-2-烯酮(3)的合成
取化合物2 (35.27 g, 196 mmol)溶于丙酮(300 mL)中, 加入异丙基碘(116.62 g, 686 mmol)和碳酸钾(64.04 g, 784 mmol), 在60 ℃下反应4 h, TLC检测.反应结束后温度降至室温, 浓缩得到粗产物, 向粗产物中加入水(400 mL)和二氯甲烷(400 mL)溶解粗产物.溶液分液, 水相用二氯甲烷萃取(300 mL×3), 合并有机相, 有机相用饱和碳酸钠溶液洗(200 mL), 水洗(200 mL), 饱和食盐水洗(200 mL), 无水硫酸钠干燥, 浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=10:1], 得到黄色液体3 (40.4 g, 产率93%). 1H NMR (300 MHz, CDCl3) δ: 5.06~5.02 (m, 1H), 4.57~4.51 (m, 1H), 2.94 (d, J=7.2 Hz, 2H), 2.50 (t, J=6.2 Hz, 2H), 2.33 (t, J=6.2, 2H), 2.03~1.91 (m, 2H), 1.69 (s, 3H), 1.62 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 197.24, 169.64, 129.46, 121.93, 119.38, 69.15, 35.63, 24.71, 24.65, 22.00, 20, 19, 20.51, 16.80; HRMS calcd for C14H23O2[M+H]+ 223.1693; found 223.1699.
3.2.3 3-异丙氧基-2, 6-双(3-甲基-2-丁烯基)环己-2-烯酮(4)的合成
量取THF (100 mL)加入烧瓶中, 在氮气保护下, 加入二异丙胺(10.39 g, 103 mmol), 温度降至-78 ℃, 之后缓慢滴加正丁基锂(2.5 mol/L in Et2O, 41.1 mL, 103 mmol), 搅拌30 min, 再缓慢加入溶于THF化合物3 (20.74 g, 93 mmol).在-78 ℃下搅拌1 h后缓慢加入3, 3-二甲基烯丙基溴(16.7 g, 112 mmol), 在此温度下继续搅拌1 h后缓慢升至室温, 搅拌过夜, TLC监测.反应结束后, 用饱和氯化铵溶液淬灭反应, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取(300 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥.减压浓缩, 粗产品通过柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=50:1]得到黄色液体4 (18.9 g, 产率71%). 1H NMR (300 MHz, CDCl3) δ: 5.10~5.04 (m, 1H), 5.06~5.02 (m, 1H), 4.52~4.48 (m, 1H), 2.95 (d, J=4.3 Hz, 2H), 2.60~2.54 (m, 2H), 2.48~2.40 (m, 1H), 2.18~1.12 (m, 1H), 2.08~2.00 (m, 2H), 1.70 (s, 3H), 1.69 (s, 3H), 1.63 (s, 3H), 1.60 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 199.78, 169.66, 133.09, 130.43, 122.41, 122.16, 119.91, 45.33, 28.34, 25.87, 25.75, 25.67, 24.77, 24.50, 21.89, 17.72; HRMS calcd for C19H31O2[M+H]+291.2319, found 291.2321.
3.2.4 3-甲基-2, 4-双(3-甲基-2-丁烯基)环己-2-烯酮(5)的合成
取化合物4 (31 g, 106.8 mmol)加入到烧瓶中, 在氮气保护下加入无水THF (250 mL), 在0 ℃缓满滴加甲基锂(1.3 mol/L in Et2O, 124 mL, 160.2 mmol)后, 直接在室温中反应30 min, 随后再次将反应液冷却至0 ℃并缓慢滴加稀盐酸, 调节至弱酸性并搅拌30 min, TLC监测.反应毕, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取(200 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩并柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=150:1], 得到黄色液体5 (23.52 g, 产率71%). 1H NMR (400 MHz, CDCl3) δ: 5.11~5.06 (m, 1H), 4.89~4.82 (m, 1H), 3.03~2.92 (m, 2H), 2.41 (ddd, J=17.3, 12.2, 5.2 Hz, 1H), 2.29 (dt, J=17.2, 4.9 Hz, 1H), 2.25~2.09 (m, 2H), 2.14~2.11 (m, 1H), 1.98~1.90 (m, 1H), 1.94 (s, 3H), 1.85~1.80 (m, 1H), 1.83 (s, 3H), 1.82 (s, 3H), 1.81 (s, 3H), 1.80 (s, 3H); 13C NMR (100 MHz, CDCl3)δ: 198.91, 159.53, 135.64, 134.27, 132.09, 123.08, 122.83, 42.35, 34.45, 30.40, 26.50, 26.37, 26.21, 25.15, 20.68, 18.51; HRMS calcd for C17H27O [M+H]+ 247.2056; found 247.2061.
3.2.5 3, 3-二甲基-2, 4-双(3-甲基丁-2-烯基)环己酮(6)的合成
取溴化亚铜二甲硫醚(0.512 g, 2.5 mmol)于反应瓶中, 在氮气保护下加入无水THF (250 mL), -78 ℃滴加甲基溴化镁(3 mol/L in Et2O, 250 mmol)和六甲基磷酰三胺(17.92 g, 100 mmol), 搅拌20 min后, 通过注射器滴加溶于THF的5 (12.26 g, 50 mmol)和三甲基氯硅烷(10.86 g, 100 mmol), -78 ℃下搅拌2 h后, 滴加稀盐酸至酸性, 搅拌30 min, 升至室温搅拌10 min, TLC监测.反应毕分液, 减压浓缩有机相, 水相用乙酸乙酯萃取(300 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥.减压浓缩, 粗产物通过柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=400:1], 得到黄色液体6 (11.35 g, 产率86%). 1H NMR (300 MHz, CDCl3) δ: 5.05~5.01 (m, 1H), 4.95~4.90 (m, 1H), 2.36~2.32 (m, 1H), 2.26~2.20 (m, 2H), 2.23~2.09 (m, 1H), 2.17 (dd, J=9.5, 2.0 Hz, 1H), 2.08~1.99 (m, 2H), 1.71 (s, 3H), 1.69~1.66 (m, 1H), 1.63 (s, 6H), 1.64 (s, 3H), 1.55~1.48 (m, 1H), 1.41~1.35 (m, 1H), 1.18 (s, 3H), 0.56 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 211.32, 131.35, 130.29, 123.23, 122.69, 60.96, 47.03, 42.26, 41.29, 28.55, 27.85, 27.43, 25.70, 25.60, 20.75, 16.78, 16.45, 14.38; HRMS calcd for C18H31O [M+H]+ 263.2369; found 263.2368.
3.2.6 甲醚化合物7的合成
取化合物6 (8 g, 30.5 mmol)溶于干燥的DMSO (70 mL), 并向其中加入叔丁醇钾(5.13 g, 45.8 mmol), 在室温下搅拌30min, 随后缓慢滴加Me2SO4 (5.78 g, 45.8 mmol), 搅拌1 h, 之后向其中加入石油醚(200 mL), 分液, DMSO层用石油醚萃取(100 mL×2), 石油醚相用水洗, 饱和食盐水洗, 无水硫酸钠干燥.浓缩直接投下一步.
3.2.7 (±)-4-羟基-8, 8-二甲基-1, 7-双(3-甲基-丁-2-烯基)-二环[3.3.1]壬-3-烯-2, 9-二酮(8)的合成
取混合物7 (5.94 g, 21.6 mmol)溶于乙醚(12 mL)中, 在氮气保护下缓慢加入丙二酰氯(2.1 mL, 21.6 mmol), -20 ℃搅拌过夜后, 再加入氢氧化钾水溶液(4.84 g, 86.4 mmol)和苄基三乙基氯化铵(329 mg, 1.1 mmol), 之后在室温下搅拌5 h.反应毕, 向反应液加入水(200 mL)和石油醚(200 mL), 并滴加氢氧化钠溶液调节至强碱性, 分液, 水相用石油醚萃取(250 mL×3), 合并有机相, 用无水硫酸钠干燥, 减压浓缩, 得到化合物6.水相冰浴, 用稀盐酸酸化至强碱性, 随后用二氯甲烷萃取(400 mL×3), 合并有机相, 无水硫酸镁干燥, 减压浓缩, 粗产物柱层析分离纯化[洗脱剂: V(二氯甲烷):V(甲醇)=200:1], 得到黄色固体8 (1.94 g). m.p. 166~170 ℃; 1H NMR (300 MHz, CDCl3) δ: 8.73 (s, 1H, OH), 5.85 (s, 1H), 4.96~4.92 (m, 1H), 4.64~4.60 (m, 1H), 3.71~3.68 (m, 1H), 3.15~3.11 (m, 1H), 2.47~2.44 (m, 2H), 2.05~2.02 (m, 2H), 1.82~1.70 (m, 3H), 1.65 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H), 1.55 (s, 3H), 1.04 (s, 3H), 0.73 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 207.21, 191.08, 183.32, 132.74, 132.25, 121.43, 118.81, 108.34, 67.01, 55.15, 44.73, 40.04, 31.87, 27.15, 24.91, 24.81, 22.73, 22.16, 17.08, 16.87, 14.98; HRMS calcd for C21H31O3[M+H]+331.2268, found 331.2272.
3.2.8 (±)-1-甲氧基-5, 5-二甲基-4, 6-二-(3-甲基-丁-2-烯基)-二环[3.3.1]壬-2-烯-3, 9-二酮(9)的合成
称取化合物8 (500 mg, 1.51 mmol)加入到反应瓶中, 溶于无水甲醇(20 mL)中, 在氮气保护下加入对甲苯磺酸(23 mg, 0.134 mmol)和原甲酸三甲酯(4.3 mL, 39 mmol), 在50 ℃回流, TLC监测.反应毕, 滴加两滴三乙胺, 减压浓缩, 粗产物通过柱层析分离纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=30:1], 得到化合物微黄固体9 (420 mg, 产率81%). m.p. 65~70 ℃; 1H NMR (300 MHz, CDCl3) δ: 5.67 (s, 1H), 4.87~4.85 (m, 1H), 4.62~4.57 (m, 1H), 3.69 (s, 3H), 3.12 (s, 1H), 2.53 (dd, J=14.0, 6.8 Hz, 1H), 2.35 (dd, J=14.0, 6.5 Hz, 1H), 2.09~2.05 (m, 1H), 2.03~2.02 (m, 1H), 1.97~1.96 (m, 3H), 1.94 (2s, 6H), 1.59 (s, 3H), 1.52 (s, 3H), 1.02 (s, 3H), 0.71 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 208.99, 197.33, 175.04, 134.12, 133.74, 123.27, 120.80, 107.28, 71.45, 57.31, 53.86, 46.92, 41.66, 33.35, 28.55, 26.57, 26.49, 24.71, 23.29, 18.76, 18.54, 16.65; HRMS calcd for C22H33O3 [M+H]+ 345.2430, found 345.2424.
3.2.9 (±)-1-甲氧基-8-烯丙基-5, 5-二甲基-4, 6-二-(3-甲基-丁-2-烯基)-二环[3.3.1]壬-2-烯-3, 9-二酮(10)的合成
取二异丙胺(0.8 mL, 5.56 mmol)于反应瓶中, 用无水THF (16 mL), 在-78 ℃下缓慢加入正丁基锂(1.6 mol/L in Et2O, 3.5 mL, 5.56 mmol), 加毕反应30 min后, 再缓慢加入溶于THF的化合物9 (372 mg, 1.11 mmol), 搅拌40 min后, 加入3-溴丙烯(0.5 mL, 5.56 mmol).反应30 min后用饱和氯化铵溶液淬灭反应, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取, 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产品通过柱层析分离纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=30:1], 得到化合物黄色液体10 (307 mg, 产率72%). 1H NMR (300 MHz, CDCl3) δ: 5.82~5.68 (m, 2H), 5.07~4.90 (m, 2H), 4.60 (td, J=5.6, 1.3 Hz, 1H), 3.71 (s, 1H), 2.61~2.54 (m, 2H), 2.42~2.39 (m, 2H), 2.08~2.05 (m, 1H), 1.88~1.82 (m, 1H), 1.68~1.65 (m, 1H), 1.65 (s, 6H), 1.62~1.58 (m, 1H), 1.55 (s, 3H), 1.52 (s, 3H), 1.39~1.32 (m, 1H), 1.03 (s, 3H), 0.70 (s, 3H); 13C NMR (75 MHz, CDCl3)δ: 208.17, 195.95, 175.66, 134.22, 133.18, 133.07, 122.71, 120.41, 117.74, 107.64, 71.03, 56.61, 55.91, 46.16, 41.97, 39.65, 35.29, 27.91, 25.96, 25.88, 24.46, 22.77, 18.14, 17.94, 15.88; HRMS calcd for C25H37O3 [M+H]+ 385.2737, found 385.2740.
3.2.10 (±)-1-羟基-8-烯丙基-5, 5-二甲基-4, 6-二-(3-甲基-丁-2-烯基)-二环[3.3.1]壬-2-烯-3, 9-二酮(12)的合成
取化合物10 (260 mg, 0.68 mmol)于反应瓶中, 加入二氧六环溶解, 再加入氢氧化锂(142 mg, 3.38 mmol)和水, 在氮气保护下90 ℃回流, 反应4 h.反应毕, 将反应液冰浴, 滴加稀盐酸至酸性, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取, 合并有机相, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物经过柱层析分离纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=7:1], 得到白色固体12 (127 mg, 60%产率). m.p. 115~118 ℃; 1H NMR (300 MHz, DMSO-d6) δ: 5.71 (s, 1H), 5.68~5.60 (m, 1H), 5.03~4.94 (m, 3H), 4.66~4.63 (m, 1H), 2.45 (d, J=7.0, 2H), 2.36 (d, J=6.7, 2H), 2.06~2.02 (m, 1H), 1.71~1.68 (m, 2H), 1.65 (s, 3H), 1.62 (s, 3H), 1.55 (s, 3H), 1.53 (s, 3H), 1.35~1.25 (m, 2H), 1.04 (s, 3H), 0.67 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ: 208.30, 134.73, 132.40, 131.65, 123.06, 121.05, 117.71, 108.92, 44.60, 41.65, 34.79, 27.92, 25.94, 25.71, 23.81, 23.14, 18.09, 17.76, 15.67; HRMS calcd for C24H33O3 [M+H]- 369.2435, found 369.2467.
3.2.11 中间体14的合成
取化合物12 (77 mg, 0.21mmol)溶于无水THF中, 在氮气保护下缓慢加入三乙胺(87 μL, 0.63 mmol)和溶于THF的3, 4-二乙酰氧基苯甲酰氰(233 mg, 0.95 mmol), 搅拌过夜.反应毕, 加入饱和的氯化铵溶液, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取, 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物经过柱层析分离纯化[洗脱剂: V(二氯甲烷):V(甲醇)=400:1], 得到淡黄色固体14 (76 mg, 产率73%). m.p. 89~91 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.52~7.41 (m, 2H), 7.22~7.18 (m, 1H), 5.94~5.81 (m, 1H), 5.21~5.10 (m, 2H), 4.99~4.66 (m, 2H), 2.78~2.65 (m, 2H), 2.62~2.44 (m, 2H), 2.35~2.32 (m, 1H), 2.30 (s, 3H), 2.29 (s, 3H), 2.17~1.99 (m, 2H), 1.72 (m, 1H), 1.69 (s, 3H), 1.68~1.65 (m, 1H), 1.61 (d, J=4.4 Hz, 3H), 1.57~1.54 (m, 1H), 1.53 (s, 3H), 1.40~1.37 (m, 1H), 1.24~1.22 (m, 3H), 1.05~1.01 (m, 1H), 0.70~0.66 (m, 3H); 13C NMR (CDCl3, 100 MHz) δ: 207.15, 195.54, 192.33, 167.84, 167.53, 145.66, 141.36, 135.38, 134.87, 134.64, 133.53, 133.21, 127.94, 124.85, 122.75, 122.07, 120.31, 118.80, 115.95, 71.18, 59.22, 48.62, 47.77, 42.46, 41.67, 29.70, 26.08, 24.84, 23.75, 22.62, 20.77, 20.61, 18.15, 17.92, 16.19; HRMS calcd for C35H43O8Na[M+Na]+ 613.6772, found 613.6773.
3.2.12 (±)-8-烯丙基山竹醇的合成
取化合物14 (76 mg, 0.13 mmol)用无水甲醇(3 mL)溶解后, 在氮气保护下加入碳酸钾(71 mg, 0.51 mmol), 室温搅拌10 min.反应完全后, 加入饱和的氯化铵溶液, 用乙酸乙酯萃取三次, 合并有机相, 有机相用饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物柱层析分离纯化[洗脱剂: V(二氯甲烷):V(甲醇)=80:1], 得到产物 (23 mg, 产率64%). 1H NMR (400 MHz, CDCl3) δ: 7.06~7.01 (m, 2H), 6.61 (d, J=8.2 Hz, 1H), 5.92~5.86 (m, 1H), 5.20~5.07 (m, 2H), 4.96~4.78 (m, 2H), 2.73~2.70 (m, 2H), 2.65~2.60 (m, 2H), 2.11~1.99 (m, 2H), 1.70 (s, 3H), 1.66 (s, 3H), 1.63 (s, 3H), 1.54 (s, 3H), 1.28~1.23 (m, 5H), 1.13~1.10 (m, 1H), 0.79~0.77 (m, 3H); 13C NMR (CDCl3, 100 MHz) δ: 195.88, 149.37, 143.54, 134.94, 133.64, 129.93, 128.75, 123.91, 122.10, 118.99, 116.51, 114.50, 42.84, 35.94, 31.92, 29.22, 27.23, 26.03, 25.29, 22.70, 18.17, 17.94, 16.29, 14.14; HRMS calcd for C31H37O6[M+H]- 505.2596, found 505.2591.
3.2.13 细胞增殖分析
MTT法检测细胞增殖:将对数生长期的SCC-15和CAL-27细胞悬液调整浓度至1×104个/mL后, 接种到96孔培养板, 每孔200 μL, 细胞贴壁后, 根据预实验结果, 分别加入不同浓度的Garcinol和8-烯丙基Garcinol (5、10、20、40、60 μmol/L).每个浓度设置6个复孔, 同时设置不加细胞的调零孔和不加药物的阴性对照孔.在37 ℃ 5% CO2分别孵育24、48、72 h, 每孔加入20 μL MTT溶液(5 mg/mL), 继续培养4 h后终止培养, 吸去孔内培养液, 每孔加入150 μL二甲基亚砜, 置摇床上低速振荡10 min, 使结晶物充分溶解.在酶联免疫检测仪波长OD 490 nm处测量各孔的吸光值.应用SPSS 17.0 for windows统计软件进行统计分析.采用ANOVA检验, 以α=0.05作为检验水准.
辅助材料(Supporting Information) 目标化合物的1H NMR, 13C NMR, 和MS数据.这些材料可以免费从本刊网站(http://siocjournal.cn/)上下载.
-
-
[1]
Aggarwal, S.; Das, S. N. Tumor. Biol. 2016, 37, 7175. doi: 10.1007/s13277-015-4583-8
-
[2]
Saadat, N.; Gupta, S. V. J. Oncol. 2012, 84, 647206.
-
[3]
Koeberle, A.; Northoff, H.; Werz, O. Biochem. Pharmacol. 2009, 77, 1513. doi: 10.1016/j.bcp.2009.02.005
-
[4]
Li, F.; Shanmugam, M. K.; Siveen, K. S. Oncotarget 2015, 6, 5147. doi: 10.18632/oncotarget.v6i7
-
[5]
Wu, A. T.; Lee, W. H.; Huang, C. C.; Lin, C. M.; Huang, Y. J.; Wei, L.; Ting, L. L.; Kuo, C. C.; Hsu, C.; Chiou, J. Biotechnol. Appl. Biochem. 2017, 64, 165. doi: 10.1002/bab.2017.64.issue-2
-
[6]
Liao, C. H.; Sang, S.; Liang, Y. C.; Ho, C. T.; Lin, J. K. Mol. Carcinog. 2004, 41, 140. doi: 10.1002/(ISSN)1098-2744
-
[7]
Padhye, S.; Ahmad, A.; Oswal, N.; Sarkar, F. C. J. Hematol. Oncol. 2009, 2, 38. doi: 10.1186/1756-8722-2-38
-
[8]
Tanaka, T.; Kohno, H.; Shimada, R.; Kagami, S.; Yamaguchi, F.; Kataoka, S.; Ariga, T.; Murakami, A.; Koshimizu, K.; Ohigashi, H. Carcinogenesis 2000, 21, 1183. doi: 10.1093/carcin/21.6.1183
-
[9]
Hong, J.; Sang, S.; Park, H. J.; Kwon, S. J.; Suh, N.; Huang, M. T.; Ho, C. T.; Yang, C. S. Carcinogenesis 2006, 27, 278. doi: 10.1093/carcin/bgi208
-
[10]
Ahmad, A.; Wang, Z.; Ali, R.; Maitah, M. Y.; Kong, D.; Banerjee, S.; Padhye, S.; Sarkar, F. H. J. Cell. Biochem. 2010, 109, 1134.
-
[11]
Balasubramanyam, K.; Altaf, M.; Varier, R. A.; Swaminathan, V. Ravindran, A.; Sadhale, P. P.; Kundu, T. K. J. Biol Chem. 2004, 279, 33716. doi: 10.1074/jbc.M402839200
-
[12]
Ahmad, A.; Sarkar, S. H.; Aboukameel A.; Ali, S.; Biersack, B.; Seibt, S.; Li, Y. W.; Bao, B.; Kong, D.; Banerjee, S.; Schobert, R.; Padhye, S. B.; Sarkar, F. H. Carcinogenesis 2012, 33, 2450. doi: 10.1093/carcin/bgs290
-
[13]
Chen, X.; Zhang, X. Y.; Lu, Y.; Shim, J. Y.; Sang, S.; Sun, Z.; Chen, X. X. Nutr. Cancer. 2012, 64, 1211. doi: 10.1080/01635581.2012.718032
-
[14]
Han, C. M.; Zhou, X. Y.; Cao, J.; Zhang, X. Y.; Chen, X. Bioorg. Chem. 2015, 60, 123. doi: 10.1016/j.bioorg.2015.04.010
-
[15]
Socolsky, C.; Plietker, B. Chem.-Eur. J. 2015, 21, 3053. doi: 10.1002/chem.201406077
-
[16]
Biber, N.; Plietker, B.; Mows, K. Nat. Chem. 2011, 3, 938. doi: 10.1038/nchem.1170
-
[17]
Rodeschini, V.; Ahmad, N. M.; Simpkins, N. S. Org. Lett. 2006, 8, 5283. doi: 10.1021/ol0620592
-
[18]
Nuhant, P.; David, M.; Pouplin, T.; Delpech, B.; Marazano, C. Org. Lett. 2007, 9, 287. doi: 10.1021/ol062736s
-
[19]
Peng, P.; Linseis, L.; Winter, F. R.; Schmidt, R. R. J. Am. Chem. Soc. 2016, 138, 6002. doi: 10.1021/jacs.6b02454
-
[1]
-

图 1 山竹醇、8-烯丙基山竹醇、Clusianone、Oblongifolin A的化学结构
Figure 1 Chemical structure of garcinol, 8-allyl garcinol, clusianone and oblongifolin A

图式 1 三环中间体10的合成
Scheme 1 Synthetic route for tricyclic intermediate 10
Reagents and conditions: (a) 3, 3-dimethylallyl bromide, DIEA, H2O, r.t., 12 h, 60%; (b) i-PrI, K2CO3, 60 ℃, 4 h, 93%; (c) LDA, 3, 3-dimethylallyl bromide, -78 ℃, 1 h, 71%; (d) MeLi, THF, 0 ℃~r.t., 1 h; 1 mol/L HCl, r.t., 30 min, 70%; (e) CuBr.Me2S, MeMgBr, HMPA, TMSCl, THF, -78 ℃, 2 h, 86%; (f) Me2SO4, t-BuOK, DMSO, r.t., 1 h; (g) malonyl dichloride, benzyltriethylammonium chloride, ether, -20 ℃, 24 h; (h) trimethyl orthoformate, TsOH, MeOH, 50 ℃, 16 h, 81%; (i) allyl bromide, LDA, THF, -78 ℃, 1 h, 72%.

图 2 三环中间体与苯甲酰氯的酰基化反应
Figure 2 Acylation of the tricyclic intermediates with benzoyl chlorides

图式3 三环中间体与苯甲酰氰的酰基化反应
Scheme 3 Acylation of the tricyclic intermediates with benzoyl cyanides

图 2 不同浓度gacinol和8-烯丙基garcinol处理SCC-15细胞24 (a)、48 (b)和72 (c) h的OD值
Figure 2 OD values of SCC-15 cells treated with different concentrations of garcinol and 8-allyl garcinol at 24 (a), 48 (b) and 72 (c) h
-


 下载:
下载:






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 8
- 文章访问数: 3013
- HTML全文浏览量: 762
 下载:
下载:
