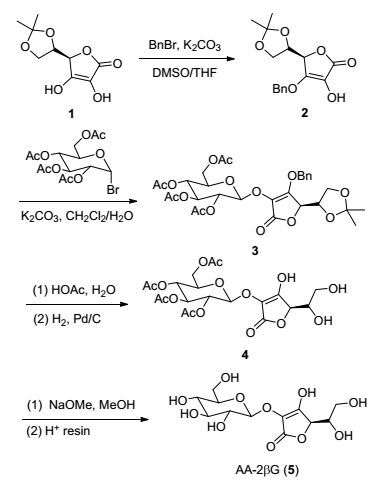
 图式 1
AA-2βG的合成路线
Scheme1.
Synthetic route to AA-2βG
图式 1
AA-2βG的合成路线
Scheme1.
Synthetic route to AA-2βG
引用本文:
马济美, 谢凌云, 王龙文, 陈杭炜, 曾贞, 陈浩, 江洪. 三种抗坏血酸糖苷的合成及其α-糖苷酶抑制活性[J]. 有机化学,
2017, 37(6): 1426-1432.
doi:
10.6023/cjoc201612037

Citation: Ma Jimei, Xie Lingyun, Wang Longwen, Chen Hangwei, Zeng Zhen, Chen Hao, Jiang Hong. Synthesis and α-Glycosidase Inhibitory Activity of Three Ascorbic Acid Glycosides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1426-1432. doi: 10.6023/cjoc201612037
Citation: Ma Jimei, Xie Lingyun, Wang Longwen, Chen Hangwei, Zeng Zhen, Chen Hao, Jiang Hong. Synthesis and α-Glycosidase Inhibitory Activity of Three Ascorbic Acid Glycosides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1426-1432. doi: 10.6023/cjoc201612037
三种抗坏血酸糖苷的合成及其α-糖苷酶抑制活性
English
Synthesis and α-Glycosidase Inhibitory Activity of Three Ascorbic Acid Glycosides
Abstract:
2-O-(β-D-Glucopyranosyl)-L-ascorbic acid, a natural β-glycoside with ascorbic acid moiety present in Lycium barbarum, and its two analogues were chemically synthesized. Their inhibitory effect on yeast α-glucosidase and α-amylase were investigated with commercially available acarbose as positive control. The biological assays demonstrated the distinct inhibitory effect of these compounds on α-glucosidase activity and weak inhibitory activity against pancreatic α-amylase. The kinetic study indicated that these compounds were competitive inhibitors. These results imply that these three β-glycosides have the potential to be developed as α-glucosidase inhibitors.
-
Key words:
- 2-O-(β-D-glucopyranosyl)-L-ascorbic acid
- / analogue
- / synthesis
- / α-glucosidase
- / α-amylase
- / inhibitor
-
糖尿病是一种代谢紊乱疾病, 已成为三大严重威胁人类健康的慢性疾病之一[1].根据2014年国际糖尿病联合会的统计, 全世界糖尿病患者超过4.5亿人, 其中Ⅱ型糖尿病(非胰岛素依耐性糖尿病)占比超过90%[2].在治疗Ⅱ型糖尿病的药物中, α-葡萄糖苷酶抑制剂如阿卡波糖等疗效非常显著, 但是阿卡波糖也常常会引起腹鸣、腹胀、腹痛等副作用[3], 因此寻求更高效安全低副作用的抗糖尿病药物一直是药物研究的重要目标[4].
枸杞果实是一种传统的中药, 在中国和一些亚洲国家已经当作保健品食用了两千多年.研究发现, 枸杞具有丰富的药理活性, 如抗氧化、降血压、降血糖等效果[5]. 2-O-β-D-葡萄糖基-L-抗坏血酸(AA-2βG)是一种天然的抗坏血酸衍生物, 最早由日本学者Toyada-ono从宁夏枸杞中分离得到[6], AA-2βG在体内只能被小肠中的β-糖苷酶水解, 具有缓释抗坏血酸的作用, 能达到持续清除自由基和抗氧化的效果[7], 还能抑制Hela细胞增殖生长[8].根据AA-2βG分子的结构特点, 我们推测AA-2βG有可能会与食物中糖类化合物在结合α-糖苷酶时产生竞争, 起到抑制血糖急剧升高的作用.为了证实这个推测, 我们合成了AA-2βG及其两种类似物2-O-β-D-半乳糖基-L-抗坏血酸(AA-2βGal)和3-O-β-D-葡萄糖基-L-抗坏血酸(AA-3βG), 并研究了它们在体外对α-糖苷酶的影响.
1 结果与讨论
AA-2βG的合成路线如Scheme 1所示.由于抗坏血酸的2位羟基容易与羰基形成分子内氢键, 导致2位羟基反应活性比其他羟基低, 因此制备AA-2βG时需先保护抗坏血酸的3、5、6位羟基[9].以商品化的5, 6-O-异丙叉基-L-抗坏血酸1为原料, 先后尝试了使用叔丁基二甲基硅基(TBS)、苄氧羰基(Cbz)和苄基(Bn)保护3位羟基.以Cbz为保护基时, 在碳酸氢钠/THF/H2O、碳酸钾/ THF/H2O、碳酸钾/DMSO/THF等条件下, 3-O-苄氧羰基-5, 6-O-异丙叉基-L-抗坏血酸产率均较低; 控制试剂的用量, 在碳酸钾和N, N-二甲基甲酰胺(DMF)中, TBS能选择性地保护化合物1的3位羟基, 但遗憾的是在随后以溴代糖为糖基供体, 碳酸钾反应条件中不稳定, TBS基团会发生脱除导致糖苷化反应发生在抗坏血酸的3位羟基(经核磁氢谱确认); 最后我们使用酸碱耐受性较高的苄基保护基, 控制保护基试剂的用量和反应时间, 以较高的产率(81%)得到化合物2.
图式 1
AA-2βG的合成路线
Scheme1.
Synthetic route to AA-2βG
在糖苷化反应中, 我们首先尝试了Schmidt给体法, 即以三氯乙酰亚胺酯为糖基供体, 但此类反应在Lewis酸催化条件(TMSOTf或三氟化硼乙醚)下进行, 得到的反应产物复杂, 难以纯化.后改用溴代糖作为糖基供体, 在碱性条件下发生糖苷化, 以碳酸钾为碱、DMF为溶剂的常规条件下, 但反应产率依然非常低, 分析原因可能是碳酸钾在DMF中溶解度较低, 而糖基受体2位羟基本身活性偏低所致.最后通过探索条件, 发现使用二氯甲烷和水作为溶剂, 同时在相转移催化剂四丁基溴化铵作用下, 反应效率大为提高, 以80%的产率得到了化合物3.
化合物3水解条件也必须注意, 为避免糖苷键在强酸性条件下断裂, 选择用醋酸水溶液加热条件下脱除异丙叉保护基, 然后氢化脱除苄基得到化合物4, 最后用甲醇钠脱除乙酰基得到AA-2βG (Scheme 1).与文献合成路线方法[6]相比(13%), 本路线大大提高了反应总收率(53%).以1-溴-2, 3, 4, 6-四乙酰基半乳糖为原料, 用相同的方法可以制备AA-2βGal(总产率51%, Scheme 2); AA-3βG则通过5, 6-O-异丙叉基-L-抗坏血酸直接与1-溴-2, 3, 4, 6-四乙酰基葡萄糖反应后, 依次脱除保护基得到(总产率51%, Scheme 3).
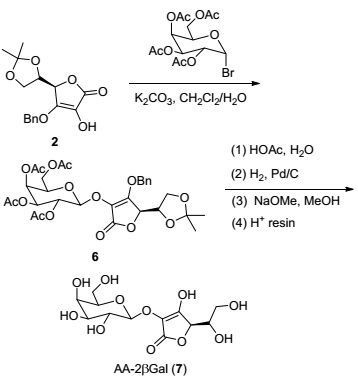 图式 2
AA-2βGal的合成路线
Scheme2.
Synthetic route to AA-2βGal
图式 2
AA-2βGal的合成路线
Scheme2.
Synthetic route to AA-2βGal
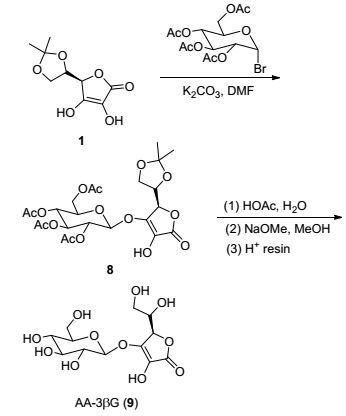 图式 3
AA-3βG的合成路线
Scheme3.
Synthetic route to AA-3βG
图式 3
AA-3βG的合成路线
Scheme3.
Synthetic route to AA-3βG
所得化合物经过NMR和HRMS确认结构, 然后分别进行了对酿酒酵母α-葡萄糖苷酶和α-猪胰腺淀粉酶活性的测试.结果显示, 三种化合物均对α-葡萄糖苷酶有明显的抑制活性, 抑制率与浓度成正相关性, 通过计算得出AA-2βG、AA-2βGal和AA-3βG的IC50值分别为0.98, 1.15, 63.67 mmol/L(表 1).其中, AA-2βG和AA-2βGal的IC50值小于阿卡波糖阳性对照组(4.54 mmol/L), 说明AA-2βG和AA-2βGal对酿酒酵母α-葡萄糖苷酶的抑制能力高于阿卡波糖.
 表 1
测试化合物对α-葡萄糖苷酶的半抑制浓度
Table 1.
IC50 value of the tested compounds against α-gluco-sidase
表 1
测试化合物对α-葡萄糖苷酶的半抑制浓度
Table 1.
IC50 value of the tested compounds against α-gluco-sidase

我们通过动力学研究了三种抗坏血酸糖苷化合物对α-葡萄糖苷酶的抑制类型.根据不同浓度下的三种化合物水解pNPG的速率, 作出Linewaever-Burk图如图 1所示, 随着浓度的增加, 米氏常数Km变大, 最大速率保持不变, 说明三种化合物为竞争性抑制, 通过Dixon方程计算得到AA-2βG、AA-2βGal和AA-3βG的抑制常数Ki分别为1.98, 7.25和19.56 mmol/L.
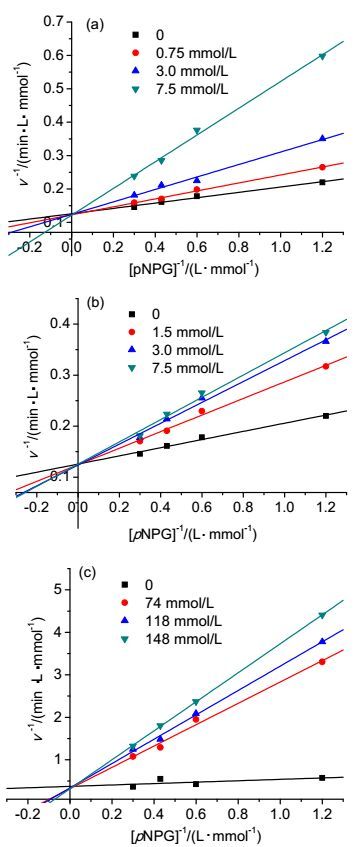 图 1
AA-2βG (a)、AA-2βGal (b)和AA-3βG (c)对α-葡萄糖苷酶的抑制类型曲线
Figure Figure1.
Determination of the inhibitory type of AA-2βG (a), AA-2βGal (b) and AA-3βG (c) on α-glucosidase
图 1
AA-2βG (a)、AA-2βGal (b)和AA-3βG (c)对α-葡萄糖苷酶的抑制类型曲线
Figure Figure1.
Determination of the inhibitory type of AA-2βG (a), AA-2βGal (b) and AA-3βG (c) on α-glucosidase
AA-2βG、AA-2βGal和AA-3βG对猪胰腺α-淀粉酶的抑制活性结果如图 2所示.作为阳性对照的阿卡波糖, 在浓度为0.015 mmol/L时, 对α-淀粉酶的抑制率为42.7%, 而当浓度增加到0.077 mmol/L时, 抑制率达到了90.6%.而AA-2βG、AA-2βGal和AA-3βG对α-淀粉酶的影响则较小, 在浓度为0.015 mmol/L时抑制率分别为5.9%、19.4%和4.6%, 浓度增加到0.77 mmol/L时也仅为12.9%、25.3%和5.9%.
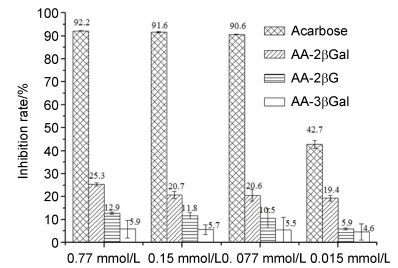 图 2
测试物对α-淀粉酶的抑制活性
Figure Figure2.
Inhibitory activity of the tested compounds on α-amy-lase
图 2
测试物对α-淀粉酶的抑制活性
Figure Figure2.
Inhibitory activity of the tested compounds on α-amy-lase
阿卡波糖作为α-糖苷酶抑制剂, 是一种有效的糖尿病治疗药物, 然而, 服用阿卡波糖常常伴有胃肠胀气和肠鸣音及腹痛、腹胀、腹泻等副作用, 这可能是由于阿卡波糖对α-淀粉酶的强抑制作用, 导致未消化的淀粉在肠道内聚集, 被肠道菌群发酵释放出气体和低分子量物质引起的[10].相比之下, AA-2βG、AA-2βGal和AA-3βG对猪胰腺α-淀粉酶的抑制效果远不如阿卡波糖, 如果作为α-糖苷酶抑制剂药物开发, 这三种糖苷可能不会引起以上不良反应.
2 结论
通过化学方法合成了AA-2βG、AA-2βGal和AA-3βG, 并发现三种化合物对α-葡萄糖苷酶具备明显的抑制能力, 抑制类型为竞争性抑制, 而对α-淀粉酶影响很小, 说明这类抗坏血酸糖苷化合物可以作为α-葡萄糖苷酶抑制剂.尤其是AA-2βG, 本身是一种天然安全的保健品成分, 具有作为低副作用抗糖尿药物开发的潜质, 同时本研究内容也为开发传统保健品枸杞降血糖功能提供了新的线索.
3 实验部分
3.1 仪器与试剂
核磁共振仪(AV400, AV600, USA, TMS为内标); 质谱仪(Agilent 6100 ESI MS, USA); 高分辨质谱仪(Agilent 6540 ESI MS, USA; Bruker MicroTof Q Ⅱ, USA); 葡萄糖苷酶(EC 3.2.1.20, Sigma), 淀粉酶(EC 3.2.1.1, Sigma), 其余试剂为市售化学纯或分析纯产品.
3.2 化合物的合成
3.3 生物活性测试
3.2.1 3-O-苄基-5, 6-异丙叉基-L-抗坏血酸(2)的合成
5, 6-O-异丙叉基-L-抗坏血酸1 (1.5 g, 6.93 mmol)溶于四氢呋喃/二甲亚砜(24 mL, V/V=1:1) 中, 加入碳酸钾(0.957 g, 6.93 mmol), 然后逐滴滴加苄溴(4.05 mL, 34.65 mmol), 滴加完毕, 室温(25 ℃)条件下反应0.5 h, 反应进程薄层色谱(TLC)检测跟踪.反应结束后减压蒸除溶剂四氢呋喃, 加入二氯甲烷(20 mL), 用柠檬酸水溶液洗涤数次, 至体系呈中性, 有机相用MgSO4干燥, 过滤, 滤液浓缩, 硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:4]分离得到1.72 g白色固体2[9a], 产率81%. m.p. 200~201 ℃(文献值[11]202.7~203.5 ℃); 1H NMR (CDCl3, 600 MHz) δ: 7.42~7.34 (m, 5H), 5.53 (d, J=11.8 Hz, 1H), 5.49 (d, J=11.8 Hz, 1H), 4.57 (d, J=3.7 Hz, 1H), 4.26 (dt, J=6.7, 3.8 Hz, 1H), 4.10 (dd, J=8.5, 6.9 Hz, 1H), 4.02 (dd, J=8.5, 6.8 Hz, 1H), 1.38 (s, 3H), 1.35 (s, 3H).
3.2.4 2-O-β-D-葡萄糖基-L-抗坏血酸(5)的合成
将化合物4 (0.51 g, 1 mmol)溶于30 mL甲醇中, 加入CH3ONa (21.6 mg, 0.4 mmol), 室温下反应3 h.反应结束加入5 g的732阳离子交换树脂, 室温搅拌30 min, 反应体系成弱酸性.抽滤收集溶液, 旋干得到0.33 g白色发泡状固体5, 产率98%. m.p. 62~63 ℃; [α]D20+29.30 (c 1.0, H2O); 1H NMR (D2O, 400 MHz) δ: 4.72 (d, J=7.8 Hz, 1H), 4.56 (d, J=1.8 Hz, 1H), 4.04~4.00 (m, 1H), 3.88~3.84 (m, 1H), 3.80~3.70 (m, 3H), 3.55~3.40 (m, 4H); 13C NMR (D2O, 100 MHz) δ: 175.4, 173.9, 115.8, 102.9, 77.8, 76.2, 75.4, 72.8, 69.3, 69.3, 62.2, 60.4; HRMS (ESI) calcd for C12H19O11[M+H]+339.0922, found 339.0917.
3.2.2 2-O-(2, 3, 4, 6-四乙酰基-β-D-葡萄糖基)-3-O-苄基-5, 6-O-异丙叉基-L-抗坏血酸(3)的合成
将化合物2 (1.72 g, 5.6 mmol)加入到二氯甲烷/水(34 mL, V:V=1:1) 的混合溶液中, 室温下搅拌均匀后加入溴代五乙酰葡萄糖(2.77 g, 6.72 mmol), 随之加入碳酸钾(0.93 g, 6.72 mmol)和四丁基溴化铵(2.16 g, 6.72 mmol), 25 ℃下搅拌反应5 h.反应停止, 将水相与有机相分离, 有机相用柠檬酸水溶液洗涤2次, 用MgSO4干燥, 过滤, 滤液浓缩, 残余物经硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:3]分离得到2.86 g白色固体3, 产率80%. m.p. 138~140 ℃; [α]D20-4.73 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 7.41~7.34 (m, 5H), 5.55 (d, J=11.6 Hz, 1H), 5.41 (d, J=11.6 Hz, 1H), 5.39 (d, J=8.0 Hz, 1H), 5.28 (t, J=9.5 Hz, 1H), 5.17~5.08 (m, 2H), 4.56 (d, J=3.3 Hz, 1H), 4.30~4.24 (m, 2H), 4.14 (dd, J=12.4, 2.3 Hz), 4.08 (dd, J=8.5, 6.8 Hz, 1H), 3.99 (dd, J=8.5, 6.8 Hz, 1H), 3.76 (ddd, J=10.0, 4.4, 2.3 Hz, 1H), 2.11 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 1.97 (s, 3H), 1.37 (s, 3H), 1.34 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.9 (2C), 169.5, 167.8, 158.5, 135.2, 128.9, 128.7, 128.2, 118.1, 110.4, 98.3, 74.9, 74.5, 73.9, 72.4, 72.1, 71.0, 68.1, 65.2, 61.4, 25.8, 25.6, 20.8, 20.6 (2C); HRMS (ESI) calcd for C30H36O15Na[M+Na]+ 659.1946, found 659.1959.
3.2.6 2-O-β-D-半乳糖基-L-抗坏血酸(7)的合成
化合物6 (1.91 g, 3 mmol)溶于50 mL 80% CH3COOH水溶液, 80 ℃下加热反应1 h.反应停止, 反应物加入50 mL乙酸乙酯稀释, 依次用水、饱和NaHCO3溶液洗涤, 中和多余的CH3COOH, 有机相用MgSO4干燥, 过滤, 旋干, 柱层析[V(乙酸乙酯):V(石油醚)=2:1]分离得到白色发泡状固体化合物.向该化合物(1.61 g, 2.70 mmol)中加入0.32 g质量分数为10%的Pd/C湿剂.将体系抽成真空状态, 加入甲醇, 通入氢气, 室温下反应2.5 h.反应物抽滤, 滤液旋干, 柱层析(乙酸乙酯)得到1.30 g白色发泡状固体2-O-(2, 3, 4, 6-四乙酰基-β-D-半乳糖基)-L-抗坏血酸, 产率85%. m.p. 109~110 ℃; [α]D20-11.03 (c 1.0, CH3OH); 1H NMR (DMSO-d6, 400 MHz) δ: 5.28~5.21 (m, 3H), 5.11~5.07 (m, 1H), 4.80~4.79 (d, J=8.8 Hz, 1H), 4.24 (t, J=6.6 Hz, 1H), 4.04 (ddd, J=25.4, 11.2, 6.6 Hz, 2H), 3.79 (t, J=7.0 Hz, 1H), 3.46~3.38 (m, 2H), 2.12 (s, 3H), 2.04 (s, 3H), 1.96 (s, 3H), 1.92 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ: 170.0, 169.9, 169.5, 169.4, 168.6, 161.7, 117.3, 99.3, 79.2, 74.8, 70.2, 70.1, 68.7, 68.5, 67.2, 61.8, 60.8, 20.6, 20.5 (2C), 20.4; HRMS (ESI) calcd for C20H26O15Na [M+Na]+ 529.1164, found 529.1170.
2-O-(2, 3, 4, 6-四乙酰基-β-D-半乳糖基)-L-抗坏血酸(0.50 g, 1 mmol)溶于30 mL甲醇中, 加入CH3ONa (21.6 mg, 0.4 mmol), 室温反应3 h.反应结束加入5 g的732阳离子交换树脂室温搅拌30 min.抽滤, 滤液旋干得到0.33 g白色发泡状固体7, 产率98%. m.p. 90~91 ℃; [α]D20+36.67 (c 1.0, H2O); 1H NMR (D2O, 600 MHz) δ: 4.64 (d, J=7.1 Hz, 1H), 4.53 (d, J=1.9 Hz, 1H), 4.01~3.99 (m, 1H), 3.99~3.87 (m, 1H), 3.78~3.69 (m, 4H), 3.67~3.62 (m, 3H); 13C NMR (D2O, 150 MHz) δ: 177.7, 176.5, 115.1, 103.8, 78.3, 75.5, 72.5, 70.7, 69.5, 68.6, 62.4, 61.0; HRMS (ESI) calcd for C12H18O11Na[M+Na]+ 361.0741, found 361.0748.
3.2.3 2-O-(2, 3, 4, 6-四乙酰基-β-D-葡萄糖基)-L-抗坏血酸(4)的合成
将化合物3 (2.00 g, 3.14 mmol)溶于50 mL体积分数为80%的CH3COOH水溶液, 80 ℃下加热反应1 h.冷却, 体系中加入20 mL乙酸乙酯, 用饱和NaHCO3溶液洗涤分液, 中和多余的CH3COOH, 有机相用MgSO4干燥, 过滤, 旋干, 柱层析[V(乙酸乙酯):V(石油醚)=2:1]分离得到1.80 g白色发泡状固体化合物, 向该化合物(1.80 g, 2.83 mmol)中加入其质量0.02倍质量分数为10%的Pd/C湿剂.将体系抽成真空状态, 加入甲醇, 通入氢气, 室温下反应2.5 h, 抽滤, 滤液旋干, 柱层析(乙酸乙酯)得到1.36 g白色固体4, 产率85%. m.p. 163~165 ℃; [α]D20-3.4 (c 1.0, CH3OH); 1H NMR (DMSO-d6, 400 MHz) δ: 5.38~5.29 (m, 2H), 4.96~4.90 (m, 2H), 4.74 (s, 1H), 4.23 (dd, J=12.3, 4.8 Hz, 1H), 4.05~4.00 (m, 1H), 3.95 (dd, J=12.3, 2.1 Hz, 1H), 3.78~3.74 (m, 1H), 3.46~3.41 (m, 2H), 2.02 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ: 170.1, 169.6, 169.4, 169.3, 168.6, 162.0, 117.1, 98.6, 74.8, 71.9, 71.0, 70.8, 68.6, 68.0, 61.8, 61.5, 20.5 (2C), 20.4, 20.3; HRMS (ESI) calcd for C20H26O15Na [M+Na]+ 529.1164, found 529.1163.
3.2.8 3-O-β-D-葡萄糖基-L-抗坏血酸(9)的合成
3-O-β-D-四乙酰葡萄糖基-5, 6-O-异丙叉基-L-抗坏血酸(1.05 g, 1.92 mmol)溶于65 mL 80% CH3COOH溶液中, 80 ℃下反应2.5 h.反应停止, 加入50 mL乙酸乙酯稀释, 水洗分液, 水相用乙酸乙酯萃取1次, 合并有机相, 依次用水和饱和NaHCO3溶液洗涤至中性, 有机相用MgSO4干燥, 过滤, 旋干, 柱层析分离得到0.79 g白色发泡状产物3-O-β-D-四乙酰葡萄糖基-L-抗坏血酸, 产率80%.
将3-O-β-D-四乙酰葡萄糖基-L-抗坏血酸(0.60 g, 1.16 mmol)溶于15 mL甲醇中, 随后加入甲醇钠(6.28 mg, 0.46 mmol)室温下搅拌反应3 h.停止反应, 反应体系中加入阳离子交换树脂, 搅拌约30 min.过滤, 滤液旋干, 得到0.38 g白色发泡状固体9, 产率98%. m.p. 71~72 ℃; [α]D20-2.67 (c 1.0, H2O); 1H NMR (D2O, 600 MHz) δ: 5.13 (d, J=7.9 Hz, 1H), 4.95 (d, J=3.1 Hz, 1H), 4.08~4.05 (m, 1H), 3.86 (dd, J=12.5, 2.0 Hz, 1H), 3.74~3.67 (m, 3H), 3.54~3.48 (m, 2H), 3.45~3.41 (m, 2H); 13C NMR (D2O, 150 MHz) δ: 177.2, 171.1, 142.5, 133.1, 101.5, 76.3, 76.0, 75.4, 73.1, 69.2, 69.0, 62.4, 60.4; HRMS (ESI) calcd for C12H18O11Na[M+Na]+ 361.0748, found 361.0741.
3.2.7 3-O-(2, 3, 4, 6-四乙酰基-β-D-葡萄糖基)-5, 6-O-异丙叉基-L-抗坏血酸(8)的合成
将5, 6-O-异丙叉基-L-抗坏血酸(2.00 g, 9.26 mmol)溶于60 mL DMF溶剂中, 随后加入K2CO3 (1.92 g, 14 mmol)和四乙酰基溴代葡萄糖(5.71 g, 14 mmol), 室温下搅拌反应10 h.停止反应, 反应体系用乙酸乙酯稀释, 水洗分液, 水相用乙酸乙酯萃取1次, 合并有机相, 用柠檬酸溶液洗涤两次, 再用饱和食盐水洗涤一次, 有机相用无水硫酸镁干燥, 过滤, 滤液旋干, 柱层析分离得到3.31 g白色发泡状固体8, 产率65%. m.p. 68~69 ℃; [α]D20+8.70 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 5.56 (d, J=8.0 Hz, 1H), 5.30 (t, J=9.4 Hz, 1H), 5.20~5.11 (m, 2H), 4.61 (d, J=2.4 Hz, 1H), 4.27 (dd, J=12.5, 5.1 Hz, 1H), 4.22~4.17 (m, 2H), 4.12 (dd, J=8.4, 6.9 Hz, 1H), 4.04 (dd, J=8.4, 6.6 Hz, 1H), 3.91 (ddd, J=10.1, 5.0, 2.4 Hz, 1H), 2.09 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.35 (s, 3H), 1.33 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 170.7, 170.1, 169.8, 169.5, 169.4, 144.8, 120.8, 110.4, 98.8, 74.8, 73.2, 72.5, 72.2, 70.7, 67.9, 65.2, 61.5, 25.7, 25.6, 20.6, 20.5 (3C); HRMS (ESI) calcd for C23H30O15Na [M+Na]+ 569.1477, found 569.1458.
3.2.5 2-O-(2, 3, 4, 6-四乙酰基-β-D-半乳糖基)-3-O-苄基-5, 6-O-异丙叉基-L-抗坏血酸(6)的合成
将化合物2 (2.45 g, 8 mmol)加入到二氯甲烷/水(50 mL, V:V=1:1) 的混合溶液中, 室温下搅拌均匀后加入溴代四乙酰半乳糖(3.95 g, 9.6 mmol), 随之加入K2CO3 (1.33 g, 9.6 mmol)和四丁基溴化铵(3.1 g, 9.6 mmol), 室温25 ℃下搅拌反应5 h.反应停止, 将水相与有机相分离, 有机相用柠檬酸水溶液洗涤数次, MgSO4干燥, 过滤, 滤液旋干, 柱层析[V(乙酸乙酯):V(石油醚)=1:3]分离得到3.87 g白色发泡状固体6, 产率76%. m.p. 96~98 ℃; [α]D20+26.73 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ: 7.40~7.3 (m, 5H), 5.58 (d, J=11.7 Hz, 1H), 5.47~5.43 (m, 2H), 5.37~5.30 (m, 2H), 5.09 (dd, J=9.8, 3.5 Hz, 1H), 4.56 (d, J=3.3 Hz, 1H), 4.27 (td, J=6.6, 3.3 Hz, 1H), 4.19 (dd, J=11.2, 6.6 Hz, 1H), 4.13~4.07 (m, 2H), 4.02~3.96 (m, 2H), 2.16 (s, 6H), 2.03 (s, 3H), 1.99 (s, 3H), 1.38 (s, 3H), 1.35 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 170.3, 170.1 (2C), 169.9, 167.8, 158.4, 135.3, 128.9, 128.7, 128.3, 118.3, 110.4, 99.0, 74.9, 74.5, 73.9, 71.4, 70.5, 68.5, 66.8, 65.3, 60.8, 25.8, 25.6, 20.9, 20.6, 20.5; HRMS (ESI) calcd for C30H36O15Na[M+Na]+ 659.1946, found 659.1936.
3.3.3 α-淀粉酶活性测试
参考文献方法[14], 取0.25 mL猪胰腺α-淀粉酶溶液(3.54 U/mL), 加入0.25 mL待测样品溶液(0.015、0.077、0.15、0.77 mmol/L), 在37 ℃的温度下预热10 min, 然后加入0.5 mL的淀粉溶液在37 ℃水浴加热3 min.完毕后加入0.5 mL DNS显色溶液, 混合均匀, 置于80 ℃沸水浴中反应8 min.反应完毕将体系冷却至室温, 加入5 mL蒸馏水进行稀释.显色溶液在540 nm波长下进行吸光值检测.空白对照:用PBS (pH=6.9) 代替待测样品进行上述反应.样品对猪胰腺α-淀粉酶抑制率的计算公式为:抑制率(%)=(D空白-D样品)/D空白×100%, D样品表示样品的吸光度值(样品无背景吸收), D空白表示空白的吸光度值, 每组实验测三个平行, 抑制率取平均值.
辅助材料(Supporting Information) 化合物3~9的核磁共振氢谱和碳谱谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
3.3.2 α-葡萄糖苷酶抑制类型测定
参考文献方法[13], 固定酿酒酵母源α-葡萄糖苷酶的浓度为0.45 U/mg, 配置AA-2βG浓度分别为0.75、3、7.5 mmol/L; AA-2βGal浓度分别为1.5、3、7.5 mmol/L; AA-3βG浓度分别为74、118、148 mmol/L, 分别在这些浓度下, 改变底物pNPG浓度分别为0.3、0.43、0.6、1.2 mmol/L.反应每隔1 min记录吸光度值随时间推移而增长的曲线.计算反应的初速度及其倒数与底物的pNPG浓度倒数进行作图, 得到Lineweaver-Burk曲线.
3.3.1 α-葡萄糖苷酶活性测试
采用96孔板法[12], 每孔取50 μL的待测溶液(AA-2βG、AA-2βGal和阿卡波糖浓度为0.75, 1.5, 3, 7.5, 15, 30 mmol/L; AA-3βG浓度为30, 44, 59, 74, 118, 148 mmol/L), 加入50 μL磷酸二氢钠缓冲溶液(PBS, pH 6.5) 及10 μL的酿酒酵母源α-葡萄糖苷酶液(0.45 U/mL), 37 ℃恒温预热15 min, 加入50 μL的pNPG溶液(0.83 mmol/L), 在37 ℃恒温条件下继续反应15 min.加入100 μL 1 mol/L的碳酸钠溶液终止反应.然后用酶标仪测定405 nm下吸光度D值.实验以PBS代替待测样品为空白对照组, 阿卡波糖作为阳性对照组.阴性对照组: PBS代替对硝基苯基α-D-葡萄糖苷(pNPG)对照组, 在405 nm处无明显吸收.
样品对α-葡萄糖苷酶抑制率的计算公式为:抑制率(%)=(D空白-D样品)/D空白×100%, D样品表示样品的吸光度值(排除样品自身的干扰), D空白表示空白的吸光度值, 每组实验重复三组平行, 抑制率取平均值.以浓度为横坐标, 抑制率为纵坐标作图得到各样品的IC50值.
-
-
[1]
(a) Zhu, J.; Liu, W.; Yu, J.; Zou, S.; Wang, J.; Yao, W.; Gao, X. Carbohydr. Polym. 2013, 98, 8.
(b) Zhou, Z.; Yan, J.; Tang, X.; Zhang, W.; Zhang, Y.; Chen, X.; Su, X.; Yang, D. Chin. J. Org. Chem. 2010, 30, 582 (in Chinese).
(周祖文, 晏菊芳, 唐雪梅, 张蔚瑜, 张映霞, 陈欣, 苏小燕, 杨大成, 有机化学, 2010, 30, 582.) -
[2]
Ruan, C.-T.; Lam, S.-H.; Lee, S.-S.; Su, M.-J. Phytomedicine 2013, 20, 667. doi: 10.1016/j.phymed.2013.02.009
-
[3]
(a) Aoki, K.; Muraoka, T.; Ito, Y.; Togashi, Y.; Terauchi, Y. Intern. Med. 2010, 49, 1085.
(b) Derosa, G.; Maffioli, P. Clin. Ther. 2012, 34, 1221.
(c) Hollander, P. Drugs 2012, 44, 47. -
[4]
(a) Escandon-Rivera, S.; Gonzalez-Andrade, M.; Bye, R.; Linares, E.; Navarrete, A.; Mata, R. J. Nat. Prod. 2012, 75, 968.
(b) Huang, Y.; Fu, W.; Li, Y.; Liu, Yu.; Li, Z.; Liu, Y.; Liu, S.; Sun, J.; Li, N.; Wang, B.; Gao, X.; Zhang, D. J. Evidence-Based Complementary Altern. 2014, 2014, 9.
(c) Kim, Y.-M.; Wang, M.-H.; Rhee, H.-I. Carbohydr. Res. 2004, 339, 715.
(d) Alarcon-Aguilara, F. J.; Roman-Ramos, R.; Perez-Gutierrez, S.; Aguilar-Contreras, A.; Contreras-Weber, C. C.; Flores-Saenz, J. L. J. Ethnopharmacol. 1998, 61, 101. -
[5]
(a) Cheng, J.; Zhou, Z.-W.; Sheng, H.-P.; He, L.-J.; Fan, X.-W.; He, Z.-X.; Sun, T.; Zhang, X.; Zhao, R. J.; Gu, L.; Cao, C.; Zhou, S.-F. Drug Des., Dev. Ther. 2015, 9, 33.
(b) Cai, H.; Liu, F.; Zuo, P.; Huang, G.; Song, Z.; Wang, T.; Lu, H.; Guo, F.; Han, C.; Sun, G. Med. Chem. 2015, 11, 383. -
[6]
Toyoda-Ono, Y.; Maeda, M.; Nakao, M.; Yoshimura, M.; Sugiura-Tomimori, N.; Fukami, H. J. Agric. Food Chem. 2004, 52, 2092. doi: 10.1021/jf035445w
-
[7]
(a) Takebayashi, J.; Yagi, Y.; Ishii, R.; Abe, S.; Yamada, K.; Tai, A. Biosci. Biotechnol. Biochem. 2008, 72, 1558.
(b) Zhang, Z.; Liu, X.; Zhang, X.; Liu, J.; Hao, Y.; Yang, X.; Wang, Y. Arch. Pharm. Res. 2011, 34, 801.
(c) Zhang, Z.; Zhang, L.; Hao, Y. J. Practic. Med. 2011, 27, 11 (in Chinese).
(张自萍, 张立平, 郝艳芳, 实用医学杂志, 2011, 27, 11.)
(d) Li, H.; Zhang, Z.; Liao, G. Chin. J. New Drug 2007, 16, 212 (in Chinese).
(李弘武, 张自萍, 廖国玲, 中国新药杂志, 2007, 16, 212.)
(e) Xie, R.; Zhang, Z.; Qiu, J. Bull. Sci. Technol. 2010, 26, 362 (in Chinese).
(谢若男, 张正波, 裘娟萍, 科技通报, 2010, 26, 362.) -
[8]
Zhang, Z.; Liu, X.; Wu, T.; Liu, J.; Zhang, X.; Yang, X.; Goodheart, M. J.; Engelhardt, J. F.; Wang, Y. Cell Biol. Toxicol. 2011, 27, 107. doi: 10.1007/s10565-010-9174-2
-
[9]
(a) Olabisi, A. O.; Wimalasena, K. J. Org. Chem. 2004, 69, 7026.
(b) Zheng, H. Pharmaceutical Chemistry, People's Medical Publishing House, Bejing, 2007, p. 426 (in Chinese).
(郑虎, 药物化学, 人民卫生出版社, 北京, 2007, p. 426.) -
[10]
Cho, B.-H.; Kim, J.-H.; Jeon, H.-B.; Kim, K.-W. Tetrahedron 2005, 61, 4341. doi: 10.1016/j.tet.2005.03.001
-
[11]
Kim, G.-N.; Kwon, Y.-I.; Jang, H.-D. J. Med. Food 2011, 14, 712. doi: 10.1089/jmf.2010.1368
-
[12]
(a) Balan, K.; Perumal, P.; Sundar abaalaji, N.; Palvannan, T. J. Mol. Struct. 2015, 1081, 62.
(b) Sheng, Z.; Dai, H.; Pan, S.; Wang, H.; Hu, Y.; Ma, W. Molecules 2014, 19, 10563. -
[13]
Brindis, F.; Rodríguez, R.; Bye, R.; Gonzalez-Andrade, M.; Mata, R. J. Nat. Prod. 2011, 74, 314. doi: 10.1021/np100447a
-
[14]
Thongprajukaew, K.; Choodum, A.; Sa-E, B.; Hayee, U. Food Chem. 2014, 163, 87. doi: 10.1016/j.foodchem.2014.04.080
-
[1]
-

图 1 AA-2βG (a)、AA-2βGal (b)和AA-3βG (c)对α-葡萄糖苷酶的抑制类型曲线
Figure 1 Determination of the inhibitory type of AA-2βG (a), AA-2βGal (b) and AA-3βG (c) on α-glucosidase
表 1 测试化合物对α-葡萄糖苷酶的半抑制浓度
Table 1. IC50 value of the tested compounds against α-gluco-sidase
 下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 26
- 文章访问数: 2294
- HTML全文浏览量: 357
 下载:
下载:
