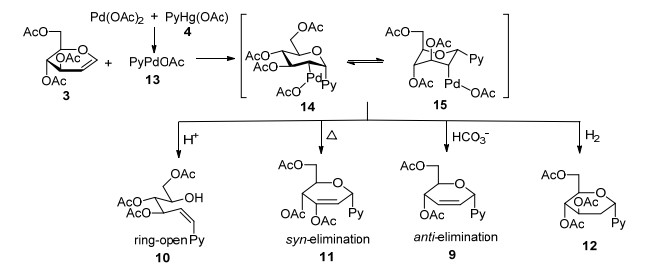
 图式1
Pd催化的糖烯与嘧啶汞盐的偶联反应机理
Scheme1.
Proposed mechanism of C-glycoside formation by coupling of glycals and oragnomercury compounds catalyzed by Pd
图式1
Pd催化的糖烯与嘧啶汞盐的偶联反应机理
Scheme1.
Proposed mechanism of C-glycoside formation by coupling of glycals and oragnomercury compounds catalyzed by Pd
引用本文:
廖进喜, 刘慧, 孙建松. 过渡金属催化的偶联反应在合成C-糖苷中的应用[J]. 有机化学,
2017, 37(6): 1382-1391.
doi:
10.6023/cjoc201612031

Citation: Liao Jinxi, Liu Hui, Sun Jiansong. Applications of Transition Metal Catalyzed Coupling Reactions in the Synthesis of C-Glycosides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1382-1391. doi: 10.6023/cjoc201612031
Citation: Liao Jinxi, Liu Hui, Sun Jiansong. Applications of Transition Metal Catalyzed Coupling Reactions in the Synthesis of C-Glycosides[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1382-1391. doi: 10.6023/cjoc201612031
过渡金属催化的偶联反应在合成C-糖苷中的应用
English
Applications of Transition Metal Catalyzed Coupling Reactions in the Synthesis of C-Glycosides
Abstract:
C-Glycosides in which the interglycosidic oxygen atom have been replaced by carbon atom are widely found in natural products and drug molecules. They have better enzymatic and hydrolytic stablility compared to their corresponding O-glycosides and N-glycosides. The syntheses of them have received considerable attention because of their unique chemical structure and extensive application value. Rencent advances in the synthesis of C-glycosides through coupling reactions catalyzed by transition metal are summarized in this review. A summary of the advantages and disadavatages of different synthetic methods will be beneficial to the develepment of new synthetic method for C-glycosides.
-
Key words:
- C-glycosides
- / synthesis
- / transition metal catalyzed
- / coupling reactions
-
糖, 又称碳水化合物, 与蛋白质、核酸和脂质一起并称为生命活动过程中的四类重要的生物大分子.它广泛分布于自然界中, 不仅以葡萄糖、果糖、核糖、蔗糖、麦芽糖、几丁质、淀粉和纤维素等还原糖的形式存在, 还与其它非糖分子通过共价键连接形成糖缀合物.
所谓C-糖苷是指糖苷键的环外氧原子被碳所取代的一类化合物的总称.这类化合物的合成在近年来受到了越来越多的研究人员的重视, 究其原因, 一方面是因为C-糖苷与其具有类似结构的O-糖苷和N-糖苷相比, 具有更好的酶稳定性[1]及耐水解性能[2]; 而另一方面则是因为越来越多的含C-糖苷键的天然产物的发现[3], 例如海葵毒素[4]等, 使人们迫切地需要寻找到更高效的合成C-糖苷键的方法.
迄今为止, 已有许多关于C-糖苷的合成方法的报道[5], 主要包括过渡金属催化的偶联反应、亲核取代、重排反应和自由基反应等.但是鉴于过渡金属催化的C—C键的偶联反应已经成为有机合成中构建C—C键的强有力的工具[6], 但是在糖化学中的应用却还不是很普遍, 因此本文主要针对过渡金属催化的偶联反应合成C-糖苷的研究方法进行综述, 试图根据不同的过渡金属参与的C-糖苷的合成研究现状来归纳总结出各类过渡金属催化剂的特点以及不足, 为过渡金属催化的偶联反应制备C-糖苷的进一步研究提供帮助.
1 Pd催化
1.1 Pd催化的糖烯参与的偶联反应制备C-苷
1976年, Bergstrom等[7]发现嘧啶的汞盐与烯烃能够在Pd(Ⅱ)的催化下发生偶联反应, Daves等[8]据此推测, 与烯烃具有相似化学性质的糖烯应该也能够在这一条件下与嘧啶汞盐发生偶联反应生成碳核苷类化合物.经过试验, 他们发现3, 4-二氢吡喃1与嘧啶的醋酸汞盐4在1 equiv.醋酸钯和2 equiv.氯化锂的存在下, 得到双键转移的偶联产物5 (66%)和6 (24%).当采用3, 4-二-O-乙酰基-D-阿拉伯糖烯时, 却只以20%的收率得到3-脱乙酰基的偶联产物7 (20%), 另外还得到了部分开环产物8 (32%), 而当采用3, 4, 6-三-O-乙酰基葡萄糖烯时, 同样也得到了少量的偶联产物9 (20%)和大部分的开环产物10 (73%) (Eq. 1).
作者通过对反应机理的研究, 发现造成各偶联产物不同的原因在于Pd加成产物的还原脱除步骤[9](Scheme 1).嘧啶的醋酸汞盐先与醋酸钯发生金属转移反应, 接着该嘧啶金属试剂再与糖烯3的双键发生加成反应.加成会发生在空间位阻相对更小的α面得到加成产物14(或15), 接下来该加成产物不同的还原脱除路径便会导致不同的偶联产物的生成.
图式1
Pd催化的糖烯与嘧啶汞盐的偶联反应机理
Scheme1.
Proposed mechanism of C-glycoside formation by coupling of glycals and oragnomercury compounds catalyzed by Pd
上述产物中各组分的比例除了底物本身的影响外, 与反应体系的组成特别是反应中所添加的盐的性质密切相关.而反应的立体选择性则取决于分子中各个羟基保护基的大小和化学性质, 这一特性在呋喃型糖烯与嘧啶汞盐在Pd催化下进行偶联时表现相当明显[10](Eq. 2).例如当α, β面空间位阻差不多时(如16a), 得到的是α, β混合物(17a, 18a), 而当β面有大位阻的TBDPS保护时(16b), 只得到了α型产物17b.
2001年, Maddaford等[11]发现全乙酰化的葡萄糖烯也能够与芳基硼酸在Pd的催化作用下发生偶联反应, 生成2, 3-二脱氧的芳基碳苷, 但是反应产物并不是他们所预期的β-H消除的产物, 而是类Ferrier重排类型的产物(Eq. 3).作者推测此反应发生的主要原因是由于在这一条件下, 反式消除要优先于顺式的Heck-β-H消除[12].他们也曾试图通过改变反应体系的酸碱性及反应温度, 以期能够得到正常的β-H消除的产物, 但是都没有成功.
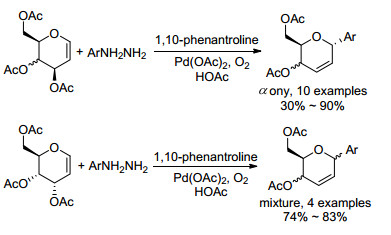
2009年, Ye等[13]发现糖烯与芳基硼酸在氧气和醋酸铜的存在下, 通过醋酸钯催化, 能生成对应的β-H消除的Heck反应产物19.更有意思的是, 研究人员发现在使用不同的氧化剂进行氧化时, 得到的最终产物也各不相同.例如采用醋酸铜和氧气氧化时, 得到的是β-H消除的2, 3-糖烯碳苷19, 采用苯醌(BQ)作氧化剂时, 得到的是3-羰基碳苷21, 而采用2, 3-二氯-5, 6-二氰基苯醌(DDQ)作氧化剂时, 得到的是烯酮类碳苷20 (Scheme 2).作者推测反应发生的主要原因是因为在使用DDQ或者BQ作氧化剂时, 引起了3-OH上硅基保护基的水解.作者提出反应的立体选择性取决于3位取代基的立体构型[14], 因为大位阻的活性Pd物种对糖烯双键进行加成时, 更倾向于从空间位阻更小的3位取代基的另一面进行加成.
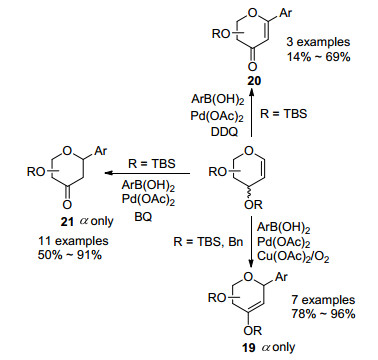 图式2
Pd催化的不同氧化剂存在下的糖烯与芳基硼酸的偶联反应
Scheme2.
Reactions of glycals and arylboronic acids catalyzed by Pd with different oxidants
图式2
Pd催化的不同氧化剂存在下的糖烯与芳基硼酸的偶联反应
Scheme2.
Reactions of glycals and arylboronic acids catalyzed by Pd with different oxidants
然而, 当Mukherjee等[15]在使用TEMPO作为氧化剂时, 却能发生Heck-Suzuki类双偶联反应, 得到cis-1, 2-二芳基化的碳苷产物22 (Eq. 4).研究人员分析反应发生的主要原因可能是因为四甲基六氢吡啶氧化物(TEMPO)本身所具有的自由基氧化特性, 能够使Pd配位上去后连续发生两次氧化加成反应(Scheme 3).
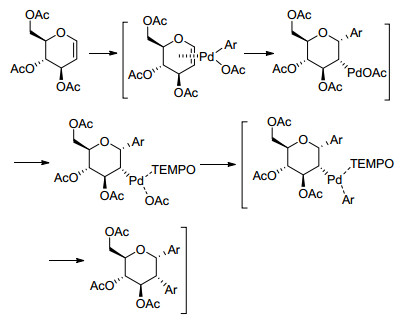 图式3
Pd催化的以TEMPO为氧化剂的糖烯与芳基硼酸的偶联反应机理
Scheme3.
Plausible mechanism for the reactions of glycals and arylboronic acids catalyzed by Pd with TEMPO as the oxidant
图式3
Pd催化的以TEMPO为氧化剂的糖烯与芳基硼酸的偶联反应机理
Scheme3.
Plausible mechanism for the reactions of glycals and arylboronic acids catalyzed by Pd with TEMPO as the oxidant
除了芳基硼酸以外, 芳基碘代物[14]、芳基溴代物[16]和烯醇磺酸酯[17]同样也能在Pd催化下与糖烯发生Heck偶联反应(Scheme 4).
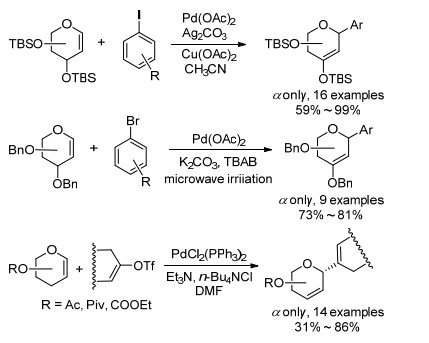 图式4
Pd催化的糖烯与其他芳基试剂的偶联反应
Scheme4.
Reactions of glycals and other aryl reagents catalyzed by Pd
图式4
Pd催化的糖烯与其他芳基试剂的偶联反应
Scheme4.
Reactions of glycals and other aryl reagents catalyzed by Pd
Dunkerton等[18]发现除了1, 2-不饱和糖烯化合物外, 2, 3-不饱和糖烯化合物在Pd(0) 的催化下能够形成Pd-π-烯丙基的复合物, 而亲核试剂在该条件下能够直接与该复合物发生亲核加成反应生成对应的C-糖苷.不久, Sinou等[19]还发现, 通过控制参加反应的配体的种类和配比, 可以完全得到单一构型的C-苷产物(Scheme 5).
 图式5
Pd催化的2, 3-不饱和糖烯与亲核试剂的偶联反应
Scheme5.
Reactions of 2, 3-unsaturated glycals and nucleophiles catalyzed by Pd
图式5
Pd催化的2, 3-不饱和糖烯与亲核试剂的偶联反应
Scheme5.
Reactions of 2, 3-unsaturated glycals and nucleophiles catalyzed by Pd
1.2 Pd催化的1-取代糖烯参与的偶联反应制备C-苷
早在1990年, Friesen等[20]就发现糖烯的1-锡基取代化合物与芳基溴化物能够在Pd催化下发生偶联反应, 生成对应的芳基糖烯碳苷.芳基溴的对位或邻位有吸电子取代基更有利于此反应的进行, 同时反应的副产物主要是糖烯的自身偶联产物(Eq. 5).
此外, 糖烯的金属衍生物, 如糖烯铟化合物[21](Eq. 6) 和糖烯锌化合物[22](Scheme 6), 也能够与芳基卤化物在Pd的催化下合成糖烯碳苷. Augé等[23]还将此方法用于克级C-糖肽的合成中(Eq. 7).
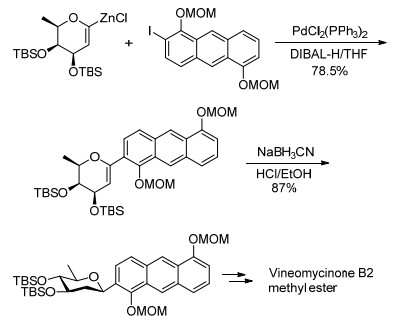 图式6
Pd催化的糖烯锌化合物与芳基碘试剂的偶联反应
Scheme6.
Reaction of zincated glycal and aryl iodide catalyzed by Pd
图式6
Pd催化的糖烯锌化合物与芳基碘试剂的偶联反应
Scheme6.
Reaction of zincated glycal and aryl iodide catalyzed by Pd
除了芳基卤化物外, 2010年, Werz等[24]利用1-碘代糖烯或1-三氟甲磺酸酯糖烯(23)与糖基炔化物24在Pd催化下, 合成了一系列1→6炔键连接的二糖碳苷25.接着利用Raney-Ni可以选择性地将25中的连接键炔键进行还原, 生成对应的糖烯碳苷26.并且在将糖烯的不饱和双键进行氧化还原时, 除了可以使用传统的硼氢化氧化得到1, 2反式的β-C-苷27外[25], 研究人员还发现, 使用氧化剂过氧丙酮(DMDO)可以立体专一性地生成α面的1, 2-环氧糖(>20:1), 接着使用与环氧的氧原子具有配位效用的还原剂二异丁基氢化铝(DIBAL)则可以选择性地得到β-C-苷27, 而使用位阻更小的超氢(LiBH-Et3)则可以得到具有相反构型的α-C-苷28 (Scheme 7).
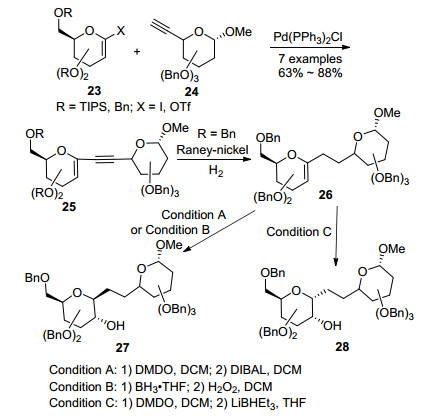 图式7
Pd催化的1→6连接碳苷二糖的制备
Scheme7.
Synthesis of (1→6)-linked C-glycosides catalyzed by Pd
图式7
Pd催化的1→6连接碳苷二糖的制备
Scheme7.
Synthesis of (1→6)-linked C-glycosides catalyzed by Pd
随后, Werz等[26]又利用糖基锡试剂与环外的溴代烯烃在Pd的催化下, 发生Still偶联得到连二烯型的碳苷30, 通过这一反应可以快速地制备出各种(1→2), (1→3) 和(1→4) C—C键连接的二糖.这种连二烯的结构可以进一步还原生成2-脱氧碳苷31, 也可以通过硼氢化氧化制备2-OH碳苷32 (Scheme 8).
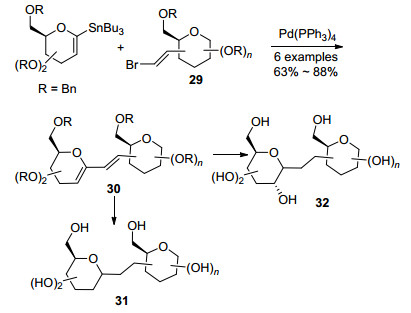 图式8
Pd催化的(1→2), (1→3) 和(1→4) 连接的碳苷二糖的制备
Scheme8.
Synthesis of (1→2), (1→3) and (1→4)-linked C-glycosides catalyzed by Pd
图式8
Pd催化的(1→2), (1→3) 和(1→4) 连接的碳苷二糖的制备
Scheme8.
Synthesis of (1→2), (1→3) and (1→4)-linked C-glycosides catalyzed by Pd
1.3 Pd催化的1-取代饱和糖基参与的偶联反应制备C-苷
除了1-取代糖烯外, 饱和的1-锡基取代的糖基化合物也能够与芳基卤化物发生偶联反应. 1995年, Falck等[27]利用糖基1-锡基取代物33与酰氯在Pd/Cu共催化作用下, 制备出了α-羰基碳苷34.其中化合物33中糖基2位的甲氧基甲基(MOM)保护基非常重要, 因为MOM保护基可以有效地稳定反应过程中生成的有机铜中间体, 有利于异头位C—C键的构建, 而改用简单的醚键保护时, 例如Me或者Bn保护时, 则不会有此效果, 原料在这种条件下会发生降解(Eq. 8).
2016年, Walczak等[28]也发现1-锡基取代的糖基化合物与芳基卤化物在Pd催化下能发生偶联反应, 生成芳基碳苷.这一反应最大的一个特色也就是可以通过控制底物糖基锡的立体构型来控制生成的产物芳基碳苷的立体构型, 产物异头位的立体构型与原料糖基锡的立体构型保持一致(Eq. 9).
1.4 Pd催化的1-取代糖烯参与的CO插入反应制备C-苷
1997年, Vogel等[29]发现在CO存在下, 1-碘代或者1-三丁基锡代糖烯能够与对应的烯烃锡化物或者卤化物发生CO插入的偶联反应, 生成对应的α-羰基糖烯碳苷(Eq. 10).
随后, Tan研究小组[30]在此基础上也发现1-碘代糖烯与原位生成的末端烯烃的硼酸酯也同样能够在Pd催化下生成糖烯碳苷.并且该反应在CO存在下, 也能发生CO插入反应, 生成α-羰基糖烯碳苷(Scheme 9).
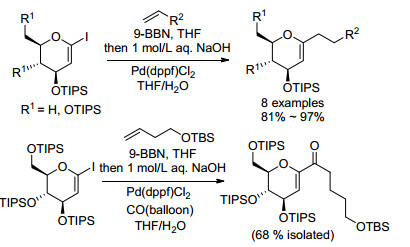 图式9
Pd催化的C(1)-烷基或羰基碳苷的合成
Scheme9.
Synthesis of C(1)-alkyl-and acyl-glycals catalyzed by Pd
图式9
Pd催化的C(1)-烷基或羰基碳苷的合成
Scheme9.
Synthesis of C(1)-alkyl-and acyl-glycals catalyzed by Pd
1.5 Pd催化的C—H活化制备C-苷
过渡金属催化的导向基团诱导的C—H键直接官能团化在过去的几十年里已经成为构建C—C键的重要方法之一[31]. 2016年, Ye等[32]发展了Pd催化的芳基C—H键活化直接与碘代糖烯进行偶联制备芳基碳苷的方法.该方法采用N-喹啉基作为反应的导向基团, 可以选择性地活化苯甲酸类化合物的酰基邻位C(sp2)—H键.反应的副产物主要是二糖苷化产物, 但是通过调节参与反应的氨基酸配体, 可以主要得到单糖苷化产物(Eq. 11).
1.6 Pd催化的脱羧偶联反应制备C-苷
自2002年, Myers等[33]报道首例Pd催化的脱羧偶联反应以来, 过渡金属催化的交叉脱羧偶联反应就开始广泛应用于有机合成领域[34]. 2011年, Liu等[35]首次发现芳基甲酸在Pd催化下也能够与糖烯发生脱羧偶联反应, 生成立体专一性的α-芳基碳苷.与前述的Heck偶联反应相比, 该类型反应在底物糖烯羟基上的保护基可选择范围更广一些, 除了酯基、苄基、硅基外还包括叔丁氧羰基和苄叉等保护基(Eq. 12).
随后, 该研究小组[36]又发现糖环上的3-位酯基也能够在Pd催化下, 发生分子内的脱羧重排反应生成C-苷.与分子间的脱羧偶联反应及传统的Pd催化的Heck偶联产物不同的是, 该类反应产物的立体构型主要为β构型(Eq. 13).通过利用这一高立体选择性的C-糖苷键构建方法, 该小组[37]快速地完成了抗肿瘤活性小分子Asper-gillide A的合成.
除了芳基羧酸可以与糖烯发生脱羧偶联外, 芳基磺酸钠[38]和芳基磺酰氯[39]同样也能够在Pd催化下与糖烯发生脱磺酸偶联反应生成芳基碳苷(Scheme 10).
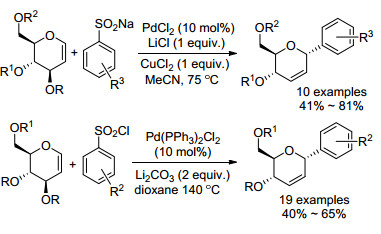 图式10
Pd催化的利用芳基磺酸化合物制备碳苷
Scheme10.
Synthesis of C-glycosides from glycals and arylsulfonyl sodium (or chloride)
图式10
Pd催化的利用芳基磺酸化合物制备碳苷
Scheme10.
Synthesis of C-glycosides from glycals and arylsulfonyl sodium (or chloride)
除脱羧外, 芳基肼与糖烯在Pd催化下也能够发生C—N键的断裂, 生成芳基C-苷[40].但是研究人员发现, 当糖烯3位为R构型时, 能够得到立体转移性的α-构型产物; 而3位为S构型时, 得到的却是α, β的混合物(Scheme 11).作者由此认为反应的立体选择性并不如Ye等[14]预测的一样仅仅取决于3位取代基的立体构型.
 图式11
Pd催化的糖烯与芳基肼反应
Scheme11.
Reactions of glycals and aryl hydrazines catalyzed by Pd
图式11
Pd催化的糖烯与芳基肼反应
Scheme11.
Reactions of glycals and aryl hydrazines catalyzed by Pd
1.7 Pd参与的共催化反应制备C-苷
自从Trost等[41]利用钯与硼共催化实现了醇对环氧化合物的高效开环反应以来, 共催化在有机合成中也得到了非常广泛的应用, 它实现了传统催化反应不能实现的新的化学转化和高效不对称催化等.例如Nicolaou等[42]利用Pd/Cu共催化1-锡基取代的糖烯化合物与烯醇磺酸酯发生偶联, 生成C-苷(Eq. 14).
2014年, Liu等[43]也利用共催化的方法实现了糖烯和2-吡啶甲醛的偶联反应, 生成α-羰基碳苷.他们采用过渡金属Pd和有机小分子3, 4-二甲基-5-(2-羟乙基)碘代噻唑(NHC))作为共催化剂, 其中, Pd与糖烯形成亲电性的Pd-π-烯丙基复合物, 而NHC活化2-醛基吡啶形成亲核性的Breslow中间体, 随后两种中间体发生加成反应生成糖烯碳苷(Scheme 12).
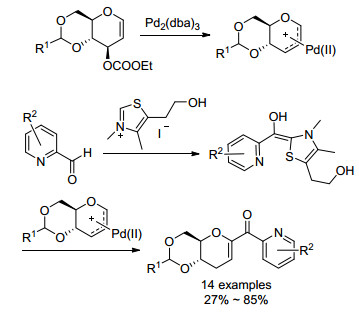 图式12
Pd/NHC共催化的糖烯与芳基醛反应
Scheme12.
Reactions of glycals and (o-azaaryl) carboxaldehyde co-catalyzed by Pd and NHC
图式12
Pd/NHC共催化的糖烯与芳基醛反应
Scheme12.
Reactions of glycals and (o-azaaryl) carboxaldehyde co-catalyzed by Pd and NHC
2 Ni催化的偶联反应制备C-苷
虽然金属Pd催化的偶联反应在糖烯类型的C-苷合成中较为多见, 但是对于C(1)-卤素取代的糖苷来说, 由于反应中生成的C(1)—Pd键非常容易发生β-H消除, 因此很难通过Pd催化的偶联反应来构建1, 2饱和的C-糖苷键. 2000年, Marsden等[44]发现糖基溴苷与缺电子的末端烯烃在Ni催化下, 可以发生偶联反应生成C-苷.作者推测反应发生的可能历程是糖基溴苷在过渡金属Ni(Ⅱ)和Mn共催化作用下形成了糖基自由基[45], 随后该糖基自由基与双键进行加成后得到α构型占主要的烷基C-苷.其中当R1为酯基取代时, 反应具有非常好的α构型立体选择性(36a, 36b), 但是R1为氰基取代时, 却还得到了少量β构型的产物(36c).而当R1为苯基取代时, 没有任何目标C-苷的生成(36d) (Eq. 15).
2007年, Gagne等[46]又发现糖基溴苷或者氯苷与烷基锌试剂在NiCl2的催化下, 室温下就能发生Negishi偶联, 生成烷基碳苷产物.该类型反应具有较好的底物普适性, 且反应产物在甘露糖底物上具有非常好的α型立体选择性, 但是遗憾的是对于葡萄糖型的底物, 立体选择性并不是太好, 反应的副产物主要是糖烯(Eq. 16).
紧接着, 在进一步的研究过程中他们发现当使用Ni(COD)2作为催化剂时, 通过筛选合适的配体t-Bu-Terp, 除了烷基锌试剂外, 芳基锌试剂同样也能发生偶联反应, 并且该类型反应在葡萄糖型底物上具有非常好的β型立体选择性[47](Eq. 17).
除了烷基和芳基锌试剂外, 缺电子烯烃也能够与葡萄糖溴苷在锌粉的还原下, 通过Ni(COD)2催化发生偶联反应得到α型C-苷产物[48](Eq. 18).
2014年, Gong等[49]又利用糖基溴苷和酸通过Ni催化下的还原偶联反应得到了α-羰基碳苷.在产物的立体选择性方面, 对于甘露糖底物得到了完全α构型的产物, 而对于葡萄糖和半乳糖型的底物, 也是以α构型为主(Eq. 19).
除了α-羰基碳苷外, 他们还利用糖基溴苷与氯甲酸酯在Ni的催化下进行还原偶联反应, 制备得到了α-酯基碳苷[50](Eq. 20).
而Ye等[51]在尝试使用糖烯与芳基硼酸酯或芳基三氟硼酸钾在Ni催化下进行偶联反应时, 只得到了开链的C-苷产物37.但是他们在进一步的研究过程中发现, 该产物在酸等关环反应的条件下, 能够分子内关环生成所需要的碳苷产物, 并且当采用不同的关环条件时, 可以得到结构完全不同的关环碳苷产物.例如在使用路易斯酸Sc(OTf)3关环时, 主要得到β构型的2, 3-不饱和C-苷, 而采用质子酸PhPHBr关环时, 却主要得到α构型的2, 3-不饱和C-苷.在采用PhSeCl/Bu3SnH时得到2-脱氧的β-C-苷(40a, 40b), 而采用NBS或NIS/ Bu3SnH时得到的却是2-脱氧的α-C-苷42 (Scheme 13).
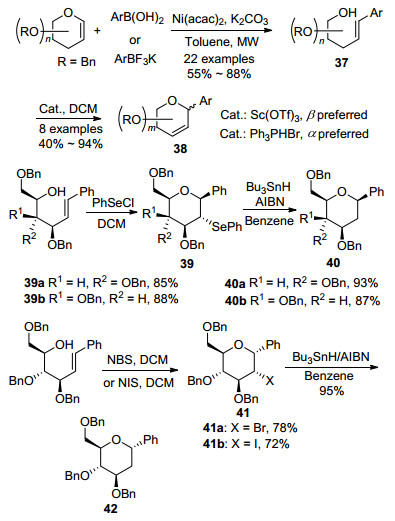 图式13
Ni催化的糖烯与芳基硼酸反应制备C-苷
Scheme13.
Synthesis of C-glycosides from glycals and aryl boronic acid catalyzed by Ni
图式13
Ni催化的糖烯与芳基硼酸反应制备C-苷
Scheme13.
Synthesis of C-glycosides from glycals and aryl boronic acid catalyzed by Ni
3 其他金属催化的偶联反应制备C-苷
2012年, Cossy等[52]发现在金属Co催化下, 芳基格氏试剂或烯基格氏试剂能够与糖基溴苷发生亲核取代反应, 生成芳基碳苷(Eq. 21).由于Co不易发生β-H消除, 因此反应几乎不会生成糖烯的副产物.同时该类型反应在全乙酰化的α-溴代甘露糖和半乳糖底物上具有非常好的α选择性, 但是在葡萄糖底物上的立体选择性较为一般.
2007年, Somsák等[53]发现利用糖基溴苷与缺电子的末端烯烃在Cr的催化下, 能够发生加成反应生成C-苷.但是只有在底物为葡萄糖时, 产物不仅具有较好的α-选择性, 产率也较高(46a~46c).当底物为半乳糖和木糖时, 虽然反应仍然具有很好的立体选择性, 但是产率都不是太高(47a, 48b); 但是当底物为阿拉伯糖时, 得到的却是α, β的混合物(50a, 50b, α:β=1:1) (Scheme 14).
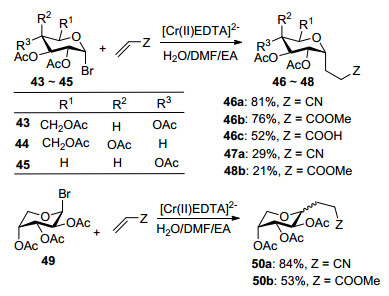 图式14
Cr催化的糖基溴苷与烯烃反应制备C-苷
Scheme14.
Synthesis of C-glycosides from glycosyl bromides and alkenes catalyzed by Cr
图式14
Cr催化的糖基溴苷与烯烃反应制备C-苷
Scheme14.
Synthesis of C-glycosides from glycosyl bromides and alkenes catalyzed by Cr
2010年, Gagné等[54]发现糖基溴苷能够与末端烯烃在金属Ru催化下, 发生偶联反应生成α-C-苷.研究表明反应中Hantzsch ester的使用对反应产率的提高非常重要, 可以在很大程度上抑制糖二聚产物的生成(Eq. 22).
研究人员推测该反应是通过可见光诱导下的自由基反应历程进行的, 同时反应具有很好的立体选择性.后续研究表明Ru(dmb)32+比Ru(bpy)32+具有更好的催化活性[55], 作者还利用这一方法完成了C-糖肽等的合成[56](Scheme 15).
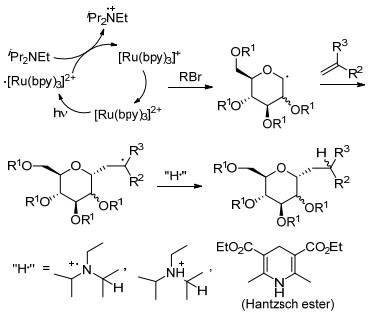 图式15
Ru催化的糖基溴苷与烯烃反应制备C-苷的机理
Scheme15.
A plausible mechanism for the synthesis of C-gly-cosides from glycosyl bromides and alkenes catalyzed by Ru
图式15
Ru催化的糖基溴苷与烯烃反应制备C-苷的机理
Scheme15.
A plausible mechanism for the synthesis of C-gly-cosides from glycosyl bromides and alkenes catalyzed by Ru
2001年, Maddaford等[57]利用金属Rh催化的芳基硼酸和糖烯酮反应得到了立体专一性α-C-苷, 且当芳基上有给电子取代基时会更有利于反应的发生(Eq. 23).
反应发生的可能机理如Scheme 16所示:芳基硼酸先与Rh催化剂发生转金属化, 接着芳基金属化合物对糖烯酮发生α-面的加成反应, 接着加成产物在水的存在下进行水解即得到了α-C-苷.研究表明反应中水的存在对Rh—O键的解离至关重要, 若在无水条件下进行反应, 几乎得不到目标产物(<5%).
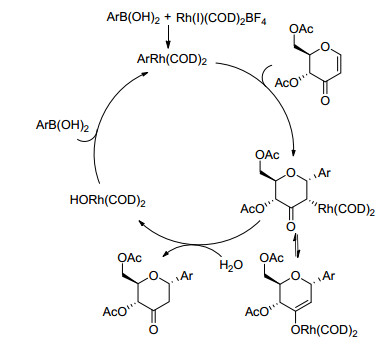 图式16
Rh催化的糖烯与芳基硼酸反应制备C-苷的机理
Scheme16.
A plausible mechanism for the synthesis of C-glycosides from glycals and arylboronic acids catalyzed by Rh
图式16
Rh催化的糖烯与芳基硼酸反应制备C-苷的机理
Scheme16.
A plausible mechanism for the synthesis of C-glycosides from glycals and arylboronic acids catalyzed by Rh
4 总结与展望
与传统的糖苷化反应相比, 过渡金属催化的偶联反应制备C-苷具有立体选择性好, 产率高且反应多样化的特点, 同时它与传统糖苷化完全不同的催化体系也大大丰富了寡糖合成中糖苷化正交策略的选择范围.其中在各类不同类型的异头位C-苷的合成方面, 芳香C-苷和烯基C-苷的研究相对来说比较多, 而α-羰基C-苷和烷基C-苷的合成研究还不是很多.在反应的立体选择性方面, 目标产物立体构型的影响因素主要包括哪些, 以及如何更好地控制目标产物的立体构型还需要更进一步的研究结果来证实.在各类不同金属的催化活性方面, 金属Pd催化的偶联反应在制备C-苷反应中已经得到了较为广泛的应用, 接着是Ni催化, 但是其他过渡金属催化的偶联反应制备C-苷还是不多见.总体来说, 过渡金属催化的偶联反应制备C-糖苷的合成研究与现有的偶联反应生成C—C键的反应相比, 还大有发展空间.如何更加充分地利用过渡金属催化的偶联反应来制备C-糖苷是糖化学工作者们接下来需要重点关注的一个研究方向.
-
-
[1]
Zou, W. Curr. Top. Med. Chem. 2005, 5, 1363. doi: 10.2174/156802605774642999
-
[2]
Hultin, P. G. Curr. Top. Med. Chem. 2005, 5, 1299. doi: 10.2174/156802605774643015
-
[3]
(a) Cao, X.; Tian, Y.; Zhang, T.; Li, X.; Ito, Y. J. Chromatogr. A 1999, 855, 709.
(b) Nomura, S.; Sakamaki, S.; Hongu, M.; Kawanishi, E.; Koga, Y.; Sakamoto, T.; Yamamoto, Y.; Ueta, K.; Kimata, H.; Nakayama, K.; Tsuda-Tsukimoto, M. J. Med. Chem. 2010, 53, 6355. -
[4]
Moore, R. E.; Scheuer, P. J. Science 1971, 172, 495. doi: 10.1126/science.172.3982.495
-
[5]
(a) De Clercq, E. J. Med. Chem. 2016, 59, 2301.
(b) Lalitha, K.; Muthusamy, K.; Prasad, Y. S.; Vemula, P. K.; Nagarajan, S. Carbohydr. Res. 2015, 402, 158.
(c) Yuan, X. J.; Linhardt, R. J. Curr. Top. Med. Chem. 2005, 5, 1393.
(d) Du, Y.; Linhardt, R. J.; Vlahov, I. R. Tetrahedron 1998, 54, 9913.
(e) Lee, D. Y. W.; He, M. S. Curr. Top. Med. Chem. 2005, 5, 1333.
(f) Wellington, K. W.; Benner, S. A. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1309.
(g) Li, X.; Zhu, J. Eur. J. Org. Chem. 2016, 2016, 4724. -
[6]
(a) Yang, D. -C. ; Zhang, Q. -H. ; Xiong, W. -W. ; Yuan, L. -G. ; Cai, Q. -S. ; Yang, M. -M. ; Li, X. ; Jiang, Y. -J. ; Liu, Y. ; Li, P. ; Xu, Z. -S. ; Sun, P. -P. ; Geng, H. -L. Chin. J. Org. Chem. 2015, 35, 961 (in Chinese).
(袁定重, 张庆华, 廖世军, 熊文文, 元利刚, 蔡奇胜, 杨梦梅, 李雄, 蒋烨佳, 刘妍, 李萍, 徐贞帅, 孙盼盼, 耿会玲, 有机化学, 2015, 35, 961. )
(b) Zhang, J. ; Lu, Q. -Q. ; Liu, C. ; Lei, A. -W. Chin. J. Org. Chem. 2015, 35, 743 (in Chinese).
(张剑, 陆庆全, 刘超, 雷爱文, 有机化学, 2015, 35, 743. )
(c) Liu, J. ; Zhu, Q. -R. ; Du, J. -Z. ; Xiu, L. Chin. J. Org. Chem. 2015, 35, 15 (in Chinese).
(刘杰; 朱庆仁; 杜娟张; 袖丽, 有机化学, 2015, 35, 15. )
(d) Zhang, W. -M. ; Dai, J. -J. ; Xu, H. -J. Chin. J. Org. Chem. 2015, 35, 1820 (in Chinese).
(张文曼, 戴建军, 许华建, 有机化学, 2015, 35, 1820. ) -
[7]
Bergstrom, D. E.; Ruth, J. L. J. Am. Chem. Soc. 1976, 98, 1587. doi: 10.1021/ja00422a056
-
[8]
Arai, I.; Daves, G. D. J. Am. Chem. Soc. 1978, 100, 287. doi: 10.1021/ja00469a051
-
[9]
Daves, G. D. Acc. Chem. Res. 1990, 23, 201. doi: 10.1021/ar00174a006
-
[10]
Cheng, J. C. Y.; Daves, G. D. J. Org. Chem. 1987, 52, 3083. doi: 10.1021/jo00390a022
-
[11]
Ramnauth, J.; Poulin, O.; Rakhit, S.; Maddaford, S. P. Org. Lett. 2001, 3, 2013. doi: 10.1021/ol010070q
-
[12]
Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009. doi: 10.1021/cr9903048
-
[13]
Xiong, D. C.; Zhang, L. H.; Ye, X. S. Org. Lett. 2009, 11, 1709. doi: 10.1021/ol900273d
-
[14]
Li, H. H.; Ye, X. S. Org. Biomol. Chem. 2009, 7, 3855. doi: 10.1039/b909248j
-
[15]
Kusunuru, A. K.; Jaladanki, C. K.; Tatina, M. B.; Bharatam, P. V.; Mukherjee, D. Org. Lett. 2015, 17, 3742. doi: 10.1021/acs.orglett.5b01722
-
[16]
Lei, M.; Gao, L.; Yang, J.-S. Tetrahedron Lett. 2009, 50, 5135. doi: 10.1016/j.tetlet.2009.06.116
-
[17]
Bai, Y.; Leow, M.; Zeng, J.; Liu, X. W. Org. Lett. 2011, 13, 5648. doi: 10.1021/ol202368n
-
[18]
Dunkerton, L. V.; Euske, J. M.; Serino, A. J. Carbohydr. Res. 1987, 171, 89. doi: 10.1016/S0008-6215(00)90881-4
-
[19]
Brakta, M.; Lhoste, P.; Sinou, D. J. Org. Chem. 1989, 54, 1890. doi: 10.1021/jo00269a026
-
[20]
Friesen, R. W.; Sturino, C. F. J. Org. Chem. 1990, 55, 2572. doi: 10.1021/jo00296a005
-
[21]
Lehmann, U.; Awasthi, S.; Minehan, T. Org. Lett. 2003, 5, 2405. doi: 10.1021/ol0345428
-
[22]
Tius, M. A.; Gomez-Galeno, J.; Gu, X. Q.; Zaidi, J. H. J. Am. Chem. Soc. 1991, 113, 5775. doi: 10.1021/ja00015a035
-
[23]
Ousmer, M.; Boucard, V.; Lubin-Germain, N.; Uziel, J.; Augé, J. Eur. J. Org. Chem. 2006, 1216.
-
[24]
Koester, D. C.; Leibeling, M.; Neufeld, R.; Werz, D. B. Org. Lett. 2010, 12, 3934. doi: 10.1021/ol101625p
-
[25]
Schmidt, R. R.; Preuss, R.; Betz, R. Tetrahedron Lett. 1987, 28, 6591. doi: 10.1016/S0040-4039(00)96921-1
-
[26]
Koester, D. C.; Kriemen, E.; Werz, D. B. Angew. Chem., Int. Ed. 2013, 52, 2985. doi: 10.1002/anie.201209697
-
[27]
Belosludtsev, Y. Y.; Bhatt, R. K.; Falck, J. R. Tetrahedron Lett. 1995, 36, 5881. doi: 10.1016/00404-0399(50)1183I-
-
[28]
Zhu, F.; Rourke, M. J.; Yang, T. Y.; Rodriguez, J.; Walczak, M. A. J. Am. Chem. Soc. 2016, 138, 12049. doi: 10.1021/jacs.6b07891
-
[29]
Jeanneret, V.; Meerpoel, L.; Vogel, P. Tetrahedron Lett. 1997, 38, 543. doi: 10.1016/S0040-4039(96)02367-2
-
[30]
Potuzak, J. S.; Tan, D. S. Tetrahedron Lett. 2004, 45, 1797. doi: 10.1016/j.tetlet.2003.12.006
-
[31]
(a) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(b) Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780. -
[32]
Liu, M.; Niu, Y.; Wu, Y. F.; Ye, X. S. Org. Lett. 2016, 18, 1836. doi: 10.1021/acs.orglett.6b00566
-
[33]
Myers, A. G.; Tanaka, D.; Mannion, M. R. J. Am. Chem. Soc. 2002, 124, 11250. doi: 10.1021/ja027523m
-
[34]
Rodriguez, N.; Goossen, L. J. Chem. Soc. Rev. 2011, 40, 5030. doi: 10.1039/c1cs15093f
-
[35]
Xiang, S.; Cai, S.; Zeng, J.; Liu, X. W. Org. Lett. 2011, 13, 4608. doi: 10.1021/ol201820m
-
[36]
Zeng, J.; Ma, J.; Xiang, S.; Cai, S.; Liu, X. W. Angew. Chem., Int. Ed. 2013, 52, 5134. doi: 10.1002/anie.201210266
-
[37]
Kito, K.; Ookura, R.; Yoshida, S.; Namikoshi, M.; Ooi, T.; Kusumi, T. Org. Lett. 2008, 10, 225. doi: 10.1021/ol702598q
-
[38]
Ma, J.; Xiang, S.; Jiang, H.; Liu, X.-W. Eur. J. Org. Chem. 2015, 2015, 949. doi: 10.1002/ejoc.v2015.5
-
[39]
Kusunuru, A. K.; Yousuf, S. K.; Tatina, M.; Mukherjee, D. Eur. J. Org. Chem. 2015, 2015, 459. doi: 10.1002/ejoc.201403195
-
[40]
Bai, Y.; Kim, L. M. H.; Liao, H.; Liu, X.-W. J. Org. Chem. 2013, 78, 8821. doi: 10.1021/jo401032r
-
[41]
Trost, B. M.; McEachern, E. J.; Toste, F. D. J. Am. Chem. Soc. 1998, 120, 12702. doi: 10.1021/ja983238k
-
[42]
Nicolaou, K. C.; Sato, M.; Miller, N. D.; Gunzner, J. L.; Renaud, J.; Untersteller, E. Angew. Chem., Int. Ed. 1996, 35, 889. doi: 10.1002/(ISSN)1521-3773
-
[43]
Bai, Y.; Leng, W. L.; Li, Y.; Liu, X. W. Chem. Commun. (Camb.) 2014, 50, 13391. doi: 10.1039/C4CC06111J
-
[44]
Readman, S. K.; Marsden, S. P.; Hodgson, A. Synlett 2000, 1628.
-
[45]
(a) Adlington, R. M.; Baldwin, J. E.; Basak, A.; Kozyrod, R. P. J. Chem. Soc., Chem. Commun. 1983, 944.
(b) Dunach, E.; Esteves, A. P.; Freitas, A. M.; Medeiros, M. J.; Olivero, S. Tetrahedron Lett. 1999, 40, 8693. -
[46]
Gong, H. G.; Sinisi, R.; Gagne, M. R. J. Am. Chem. Soc. 2007, 129, 1908. doi: 10.1021/ja068950t
-
[47]
Gong, H. G.; Gagne, M. R. J. Am. Chem. Soc. 2008, 130, 12177. doi: 10.1021/ja8041564
-
[48]
Gong, H. G.; Andrews, R. S.; Zuccarello, J. L.; Lee, S. J.; Gagne, M. R. Org. Lett. 2009, 11, 879. doi: 10.1021/ol8028737
-
[49]
(a) Zhao, C.; Jia, X.; Wang, X.; Gong, H. J. Am. Chem. Soc. 2014, 136, 17645.
(b) Jia, X.; Zhang, X.; Qian, Q.; Gong, H. Chem. Commun. (Camb.) 2015, 51, 10302. -
[50]
Zheng, M.; Xue, W.; Xue, T.; Gong, H. Org. Lett. 2016, 18, 6152. doi: 10.1021/acs.orglett.6b03158
-
[51]
Liu, C. F.; Xiong, D. C.; Ye, X. S. J. Org. Chem. 2014, 79, 4676. doi: 10.1021/jo500730y
-
[52]
Nicolas, L.; Angibaud, P.; Stansfield, I.; Bonnet, P.; Meerpoel, L.; Reymond, S.; Cossy, J. Angew. Chem. 2012, 124, 11263. doi: 10.1002/ange.201204786
-
[53]
Juhász, Z.; Micskei, K.; Gál, E.; Somsák, L. Tetrahedron Lett. 2007, 48, 7351. doi: 10.1016/j.tetlet.2007.08.019
-
[54]
Andrews, R. S.; Becker, J. J.; Gagné, M. R. Angew. Chem. 2010, 122, 7432. doi: 10.1002/ange.v122:40
-
[55]
Andrews, R. S.; Becker, J. J.; Gagne, M. R. Org. Lett. 2011, 13, 2406. doi: 10.1021/ol200644w
-
[56]
Andrews, R. S.; Becker, J. J.; Gagne, M. R. Angew. Chem., Int. Ed. 2012, 51, 4140. doi: 10.1002/anie.201200593
-
[57]
Ramnauth, J.; Poulin, O.; Bratovanov, S. S.; Rakhit, S.; Maddaford, S. P. Org. Lett. 2001, 3, 2571. doi: 10.1021/ol016245d
-
[1]
-

图式1 Pd催化的糖烯与嘧啶汞盐的偶联反应机理
Scheme 1 Proposed mechanism of C-glycoside formation by coupling of glycals and oragnomercury compounds catalyzed by Pd

图式2 Pd催化的不同氧化剂存在下的糖烯与芳基硼酸的偶联反应
Scheme 2 Reactions of glycals and arylboronic acids catalyzed by Pd with different oxidants

图式3 Pd催化的以TEMPO为氧化剂的糖烯与芳基硼酸的偶联反应机理
Scheme 3 Plausible mechanism for the reactions of glycals and arylboronic acids catalyzed by Pd with TEMPO as the oxidant

图式4 Pd催化的糖烯与其他芳基试剂的偶联反应
Scheme 4 Reactions of glycals and other aryl reagents catalyzed by Pd

图式5 Pd催化的2, 3-不饱和糖烯与亲核试剂的偶联反应
Scheme 5 Reactions of 2, 3-unsaturated glycals and nucleophiles catalyzed by Pd

图式6 Pd催化的糖烯锌化合物与芳基碘试剂的偶联反应
Scheme 6 Reaction of zincated glycal and aryl iodide catalyzed by Pd

图式8 Pd催化的(1→2), (1→3) 和(1→4) 连接的碳苷二糖的制备
Scheme 8 Synthesis of (1→2), (1→3) and (1→4)-linked C-glycosides catalyzed by Pd

图式9 Pd催化的C(1)-烷基或羰基碳苷的合成
Scheme 9 Synthesis of C(1)-alkyl-and acyl-glycals catalyzed by Pd

图式10 Pd催化的利用芳基磺酸化合物制备碳苷
Scheme 10 Synthesis of C-glycosides from glycals and arylsulfonyl sodium (or chloride)

图式12 Pd/NHC共催化的糖烯与芳基醛反应
Scheme 12 Reactions of glycals and (o-azaaryl) carboxaldehyde co-catalyzed by Pd and NHC

图式13 Ni催化的糖烯与芳基硼酸反应制备C-苷
Scheme 13 Synthesis of C-glycosides from glycals and aryl boronic acid catalyzed by Ni

图式14 Cr催化的糖基溴苷与烯烃反应制备C-苷
Scheme 14 Synthesis of C-glycosides from glycosyl bromides and alkenes catalyzed by Cr

图式15 Ru催化的糖基溴苷与烯烃反应制备C-苷的机理
Scheme 15 A plausible mechanism for the synthesis of C-gly-cosides from glycosyl bromides and alkenes catalyzed by Ru
-


 下载:
下载:















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 16
- 文章访问数: 2963
- HTML全文浏览量: 525
 下载:
下载:
