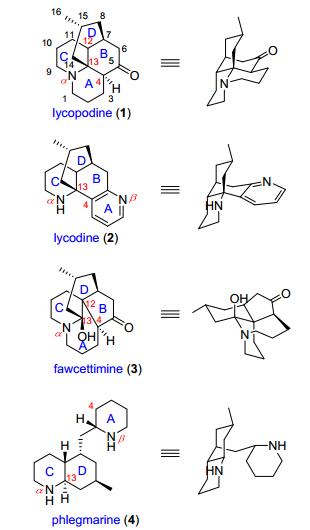
 图 1
四大类石松生物碱的代表化合物
Figure 1.
Representative compounds of the four major classes of Lycopodium alkaloids.
图 1
四大类石松生物碱的代表化合物
Figure 1.
Representative compounds of the four major classes of Lycopodium alkaloids.
引用本文:
肖春霞, 曹林, 王佳, 苗银龙, 范华芳. 石松生物碱集成合成的研究进展[J]. 有机化学,
2017, 37(4): 810-823.
doi:
10.6023/cjoc201611032

Citation: Xiao Chunxia, Cao Lin, Wang Jia, Miao Yinlong, Fan Huafang. Advances in the Collective Synthesis of Lycopodium Alkaloids[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 810-823. doi: 10.6023/cjoc201611032
Citation: Xiao Chunxia, Cao Lin, Wang Jia, Miao Yinlong, Fan Huafang. Advances in the Collective Synthesis of Lycopodium Alkaloids[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 810-823. doi: 10.6023/cjoc201611032
石松生物碱集成合成的研究进展
English
Advances in the Collective Synthesis of Lycopodium Alkaloids
Abstract:
In view of the relevance between chemical structures and biosynthetic pathways of compounds that belong to the same family of natural products, MacMillan et al. (2011) proposed a strategy for the rapid and efficient synthesis of natural product within a family from a common intermediate or a group of similar intermediates, which is termed as collective synthesis. In recent years, this strategy has been applied for the synthesis of multiple family of natural products. Lycopodium alkaloids are a class of structurally diverse alkaloids, and many of them exhibiting good biological activity. In this review, the progress in the collective synthesis of Lycopodium alkaloids is summarized.
-
Key words:
- Lycopodium alkaloids
- / natural products
- / total synthesis
- / collective synthesis
-
石松生物碱 (Lycopodium alkaloids) 是存在于石松科 (Lycopodiaceae) 蕨类植物石松和其近缘石杉科 (Huperziaceae) 植物中的一类生物碱.石松类植物如玉柏、石松、千层塔等自古以来就是传统中药和民族药的重要组成部分, 主要功效有活血镇痛、散瘀止血、清热除湿、舒筋通络等, 常用于治疗风湿痹痛、肢体麻木、皮肤瘙痒、跌打损伤等病症[1].此外, 研究还发现石松生物碱具有抗乙酰胆碱酯酶[2]、抗肿瘤[3]、抗艾滋病毒[4]、神经营养[5]、杀虫[6]等生物活性. 1881年德国化学家Bödeker从L. complanatum中分离得到了首个石松生物碱——石松碱 (lycopodine), 至今文献报道的天然石松生物碱已接近300个[7].其中, 1986年刘嘉森等[8]从传统中药千层塔 (蛇足石杉, Huperzia serrata) 中分离得到的石杉碱甲 (huperzine A) 是一种强效、高选择性、可逆的乙酰胆碱酯酶抑制剂, 能有效地提高记忆效率, 临床试验显示在治疗阿尔兹海默病 (Alzheimer's disease, AD) 方面具有显著疗效.石杉碱已成为药学界的研究热点之一, 同时也大大推动了石松生物碱的研究.
石松生物碱的基本骨架主要是C16N或C16N2组成的三环或四环化合物, 也有少量是C27N3或少于C16的骨架.按结构特点被分为四个类型 (图 1), 即 (ⅰ) lycopodine型, (ⅱ) lycodine型, (ⅲ) fawcettimine型和 (ⅳ) 其他类型, 代表化合物分别是lycopodine (1), lycodine (2), fawcettimine (3) 和phlegmarine (4)][7a].
图 1
四大类石松生物碱的代表化合物
Figure 1.
Representative compounds of the four major classes of Lycopodium alkaloids.
石松生物碱结构复杂多样、手性中心密集等特点, 使得该类化合物极具合成挑战性.同时, 它们显著的生物活性和潜在的成药性也吸引了有机化学家的研究兴趣, 近年来有大量关于石松生物碱全合成研究被报道.
天然产物生物活性多样和生物相容性高等特点使它们长期以来一直是发现和发展新医药和新农药的重要源泉[9].同时天然产物作为小分子探针也被用于疾病相关的重要生物学靶点的发现.然而, 天然产物化学结构复杂、自然界含量微少一直是制约人类开发利用天然产物的“瓶颈”.发展天然化合物和类天然产物的高效合成方法, 特别是针对家族化合物的合成方法, 建立化合物库, 供生物学家进行系统生物活性和构效关系研究, 已经成为当代合成化学家面临的挑战性任务[10].
具有相似核心骨架的同一家族天然产物在生物体内一般是由相同的生源前体化合物经不同的生物代谢途径产生.利用仿生合成的思路, 2011年MacMillan等[10a]提出针对天然产物家族化合物的集成合成 (collective synthesis) 策略, 即通过汇聚合成方式快速地、规模合成积累目标家族化合物的共同关键中间体, 然后从关键中间体出发, 以发散合成方式高效地完成家族成员化合物的合成.本文将简要综述石松生物碱的集成合成研究进展.
1 Lycopodine型石松生物碱的集成合成
Lycopodine型生物碱发现最早, 数量最多.一般是含有一个喹诺里西啶 (quinolizidine) 环的四个叠六元环结构 (C-4和C-13相连), 喹诺里西啶环一般为顺式, 大部分含有C-5位羰基, 代表化合物有10-hydroxylycopodine, deacetylpaniculine和paniculine.
2008年, Carter小组[11]完成 (—)-lycopodine的首次对映选择性合成之后, 最近他们以集成合成方式完成了lycopodine型石松生物碱10-hydroxylycopodine, deacetylpaniculine和paniculine的合成 (Scheme 1)[12].如图所示, 手性化合物5经双羟基保护、酯缩合和关环复分解 (RCM) 反应给出Michael加成产物前体化合物6.在手性小分子催化剂7的催化下经分子内Michael加成反应得到含有D环的化合物8.转化化合物8未端羟基成为叠氮基后, 保护其仲羟基并选择性地转化其甲基酮成为相应的烯醇硅醚得化合物9.它经Aza-Wittig反应、分子内Mannich反应和还原脱砜基反应构建出含B/C/D三环的中间体11.化合物11按照文献方法引入A环得到关键中间体12.化合物12经脱硅保护基、羰基还原和乙酰化等官能团转化分别得到目标分子10-hydroxylycopodine, deacetylpaniculine和paniculine.
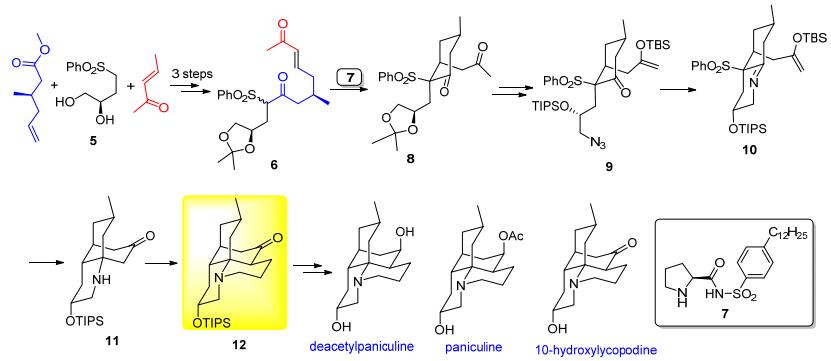 图 图式1
Carter小组对lycopodine型石松生物碱的集成合成
Figure 图式1.
Collective synthesis of lycopodine type Lycopodium alkaloids from Carter's lab
图 图式1
Carter小组对lycopodine型石松生物碱的集成合成
Figure 图式1.
Collective synthesis of lycopodine type Lycopodium alkaloids from Carter's lab
2 Lycodine型石松生物碱的集成合成
目前为止, 所发现的具有乙酰胆碱酯酶 (acetylcholine esterase, AChE) 抑制活性的石松生物碱大都属该类化合物, 如著名的石杉碱甲 (huperzine A).这类化合物一般也是四环结构, 只是由喹诺里西啶环 (A环并C环) 形式转化成分开的吡啶或者吡啶酮环A环和六氢吡啶C环, 该类化合物与lycopodine型的B、C、D环的构型是一致的, 骨架变化一般发生在C环, 如石杉碱甲 (huperzine A), 可以看作是C环断开, 并且脱去了C-9的结果, 是少见的C15N2骨架的石松生物碱.此外, huperzines B和U、α-obscurine、N-desmethyl-α-obscurine、β-obscurine和N-desmethyl-β-obscurine均为lycodine型石松生物碱.
林国强等[13]从商品化手性试剂 (R)-长叶薄荷酮 (pulegone) 出发经关键合成中间体18完成了它们的集成合成 (Scheme 2). (R)-长叶薄荷酮经三氟甲磺酰化/臭氧化得烯醇磺酸酯13.它经Buchwald-Hartwig偶联反应形成关键的C—N键得到合成砌块14.化合物14经羰基的α位不对称烷基化引入含有吡啶环的片段得反式构型产物15.化合物15经还原得1:1的烯醇混合物16.化合物16经分子内Heck偶联反应构建出石杉碱甲的核心骨架得17.它经Ley-Griffith氧化得关键中间体18.化合物18发生格氏试剂加成/消除反应引入环外双键得19, 接下来用氢溴酸处理19, 经脱甲基/脱Boc/双键迁移即可得目标产物石杉碱甲 (huperzine A).
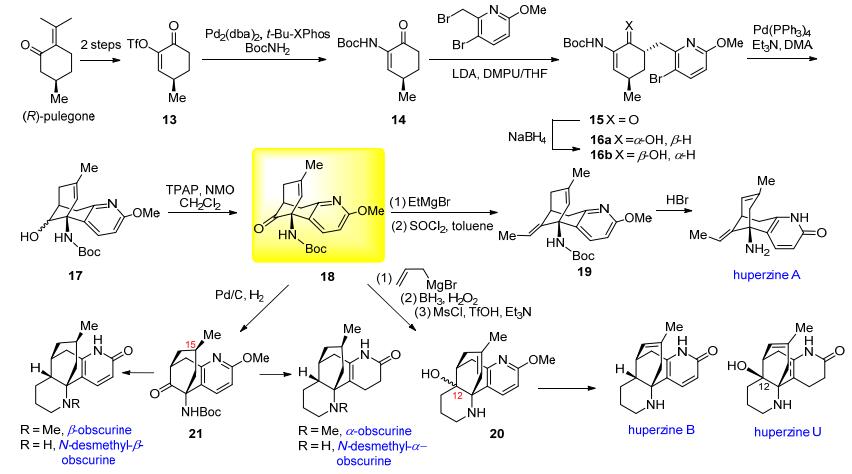 图 图式2
林国强、孙炳峰组对lycodine型石松生物碱的集成合成
Figure 图式2.
Collective synthesis of lycodine type Lycopodium alkaloids from Lin & Sun's lab
图 图式2
林国强、孙炳峰组对lycodine型石松生物碱的集成合成
Figure 图式2.
Collective synthesis of lycodine type Lycopodium alkaloids from Lin & Sun's lab
随后, 该小组[14]又利用关键中间体18完成了其他6个lycodine类石松生物碱的合成.化合物18经烯丙基格氏试剂加成, 硼氢化/氧化, 分子内环化等反应构建哌啶环得四环中间体20.化合物20经双键迁移、脱甲基、还原吡啶环等步骤得huperzines B和U.化合物18经立体选择性加氢反应构建C-15手性甲基得21, 化合物21经类似前面的方法可得4个石松生物碱 (α-ob-scurine, N-desmethyl-α-obscurine, β-obscurine和N-des-methyl-β-obscurine).
3 Fawcettimine型石松生物碱的集成合成
Fawcettimine型石松生物碱是骨架变化最为复杂的一类石松生物碱, 生源上可看作是lycopodine型的C (4)—C (13) 键断开形成C (4)—C (12) 键的结果.化合物fawcettimine已被证实是醇胺式 (carbinal-amine form, 22a) 和酮胺式 (keto-amine form, 22b) 的平衡体 (Eq. 1), 由此演化成醇胺式和酮胺式两部分, 并衍生出其他的亚类.
根据C (12)—C (13) 键的断开与否, 醇胺式又分为fawcettidine和phlegmariurine A两种亚型, 前者大都是6/6/5/7四环体系, 代表化合物有 (图 2): 8α-OH-fawcettimine (A1), fawcettidine (A2), alopecuridine (A3), lycoposerramine C (A4), lycoposerramine Q (A5) 和lycopoclavamine A (A6). Lycojapodine A (A7) 是C (4)—C (5) 键氧化断裂, C (5) 与13-OH构成内酯结构的产物. Huperzine Q (A8), N-oxyhuperzine Q (A9), lycopladines B, C (A10, A11), lycopoclavamine B (A12) 和lycopladine D (A13) 是C (15), C (16) 被氧化的产物, 其中lycopladine D (A13) 是五环笼状分子结构的石松生物碱, 该化合物具有独特的缩醛胺内酯结构, 其中C (4), C (5), C (15) 三个手性中心的构型与大多数fawcettimine型石松生物碱相反. Phlegmariurine A (A14) 是断开C (12)—C (13) 键, 只有三个环的醇胺式结构.
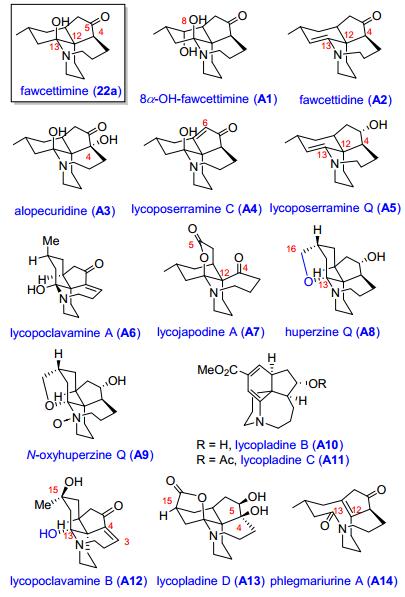 图 2
醇胺式fawcettimine型石松生物碱的代表化合物
Figure 2.
Representative compounds with carbinol-amine form of fawcettimine type Lycopodium alkaloids
图 2
醇胺式fawcettimine型石松生物碱的代表化合物
Figure 2.
Representative compounds with carbinol-amine form of fawcettimine type Lycopodium alkaloids
酮胺式的生物碱没有C (13)—N键, 根据C (4)—N或C (3)—C (10) 键的存在与否, 也可以分为三环和四环骨架两个亚型 (图 3). lycoposerramine B (B1) 是含有酮肟的三环化合物形式, lycoposerramine T (B2) 是C (15) 被氧化的三环结构. magellanine (B3), magellaninone (B4) 和paniculatine (B5) 是具有C (3)—C (10) 键的四环石松生物碱. sieboldine A (B6) 是C (1) 和C (4) 通过一个氧原子成环的四环生物碱. cernupalhine A (B7), lycoflexine (B8) 和lycoposerramine U (B9) 是罕见的C17N生物碱, 其中cernupalhine A (B7) 分子中含有新颖的羟基二氢呋喃酮片段. serratine (B10), serratanidine (B11), serratinine (B12), 8-deoxyserratinine (B13), lycojaponicumin C (B14) 和serratezomine A (B15) 是C (4) 与N原子相连的酮胺式四环结构的代表化合物.
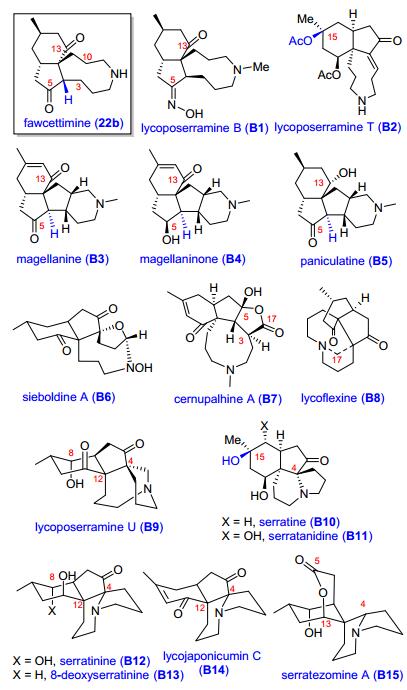 图 3
酮胺式fawcettimine型石松生物碱的代表化合物
Figure 3.
Representative compounds with keto-amine form of fawcettimine type Lycopodium alkaloids
图 3
酮胺式fawcettimine型石松生物碱的代表化合物
Figure 3.
Representative compounds with keto-amine form of fawcettimine type Lycopodium alkaloids
1986年Heathcock小组[15]报道了一条fawcettimine的高效合成路线, 合成包括立体选择性Hosomi-Sakurai烯丙基化/分子内Michael加成等关键步骤 (Scheme 3). Heathcock小组的研究还揭示了C (4) 位的立体构型在fawcettimine分子的互变异构现象中的作用, 作者首先合成了C (4) 位是α-构型的6/5/9三环中间体23a, 1H NMR显示有少量5-半胺缩醛的异构体24出现, 但是不存在4-epi-fawcettimine (23b); 碱性条件下C (4) 位发生差相异构化生成热力学稳定的β-构型产物fawcettimine (22b), 进一步异构化得22a, 整个合成过程共16步, 总收率16.6%.
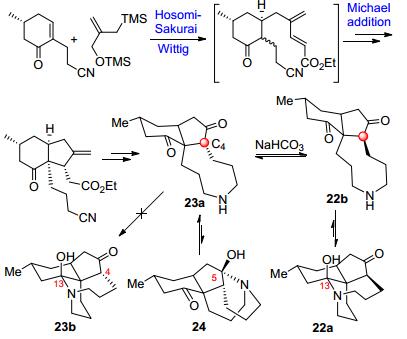 图 图式3
Heathcock小组对fawcettimine的早期合成研究
Figure 图式3.
Heathcock's originally total synthesis of fawcettimine
图 图式3
Heathcock小组对fawcettimine的早期合成研究
Figure 图式3.
Heathcock's originally total synthesis of fawcettimine
Heathcock小组的工作对fawcettimine型生物碱的后续合成产生了巨大影响, 时至今日, 通过6/5/9三环中间体来合成该类生物碱仍然是最佳策略, 被用于多个fawcettimine型生物碱的合成中.合成化学家通过各种不同方法来构建Heathcock三环中间体 (图 4), 关键步骤涉及6/5并环体系和C (12) 位手性季碳的构建. 2014年Murphy和Sarpong等[16]按反应类型对Heathcock三环中间体的构建方法进行了总结 (利用Pauson-Khand反应的综述, 另见文献[17]).
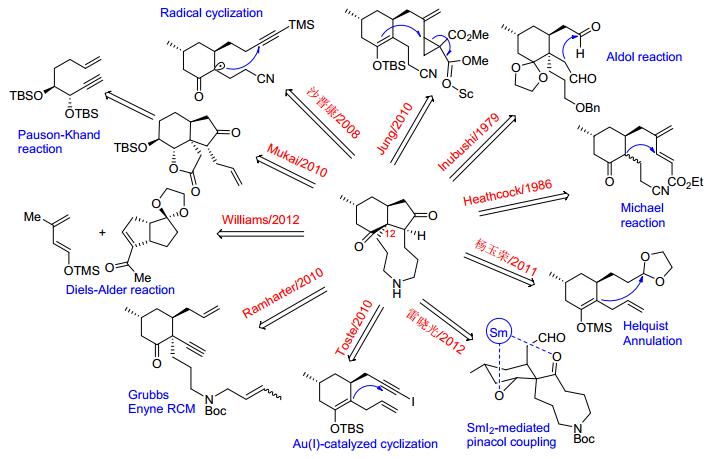 图 4
Heathcock三环中间体合成策略汇总
Figure 4.
Summary of synthetic strategies on Heathcock-type tricycle intermediates
图 4
Heathcock三环中间体合成策略汇总
Figure 4.
Summary of synthetic strategies on Heathcock-type tricycle intermediates
Mukai小组[18]从酒石酸二乙酯衍生出的烯炔25出发, 以Pauson-Khand反应为关键反应构筑6/5并环体系得到共同中间体27, 最后经引入C (15)-手性甲基等步骤完成了fawcettimine (22), lycoposerramine B (B1), magellanine (B3), magellaninone (B4) 和paniculatine (B5) 等5种石松生物碱的多样性合成, 其中在合成magel-lanine类化合物时, 作者两次应用立体选择性Pauson-Khand反应来合成目标分子的关键骨架28. 2013年, 该小组[18c]又发表了第二代fawcettimine型石松生物碱的多样性合成工作, 与前面最后引入甲基的策略不同, Pauson-Khand反应是从含有支链亚甲基的烯炔29出发, 双环产物30经立体选择性氢化反应高效快速地引入C (15)-手性甲基从而大大减少了合成步骤, 最后经heathcock中间体32完成了fawcettimine (22), fawcettidine (A2), lycoflexine (B8), lycoposerramine Q (A5) 等4种石松生物碱的集成合成.继Mukai小组之后, Takayama小组[17]也利用Pauson-Khand反应经关键中间体35完成了lycoposerramine C (A4), phlegmariurine A (A14), huperzine Q (A8), fawcettimine (22) 和fawcettidine (A2) 等5种石松生物碱的集成合成, 合成之亮点在于利用乙烯基Claisen重排反应引入C3片段构筑C (12) 手性中心 (Scheme 4).
 图 图式4
以Pauson-Khand反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式4.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the PKR
图 图式4
以Pauson-Khand反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式4.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the PKR
2012年Williams小组[19]首次以Diels-Alder反应为关键步骤构建fawcettimine型石松生物碱的6/5并环骨架, 底物37作为惰性亲双烯体使得该策略极具挑战性, 最后经共同中间体39实现了fawcettimine (22), fawcettidine (A2), lycoflexine (B8), lycoposerramine B (B1) 等4种石松生物碱的集成合成.随后, Taniguchi小组[20]也发表了Diels-Alder (D-A) 应用于石松生物碱多样性合成的工作, 作者提出氧化还原多样性合成 (redox divergent synthesis) 策略, D-A反应产物42经多步反应转化为不同氧化态的三环中间体43a ~ 43d, 最终完成了fawcettimine (22), 8-deoxyserratinine (B13), lycopoclavamine A (A6), serratine (B10), lycopoclavamine B (A12), serratanidine (B11) 和lycoposerramine T (B2) 等7种fawcettimine石松生物碱的集成合成 (Scheme 5).
 图 图式5
以Diels-Alder反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式5.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the Diels-Alder reaction
图 图式5
以Diels-Alder反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式5.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the Diels-Alder reaction
2010年起杨玉荣课题组[21]陆续发表了多篇关于石松生物碱全合成的工作 (Scheme 6), 他们从手性环己烯酮化合物44出发, 利用Helquist拼环策略立体选择性地构筑6/5并环体系得关键中间体46, 化合物46经三环化合物47得到fawcettimine (22), lycoflexine (B8), 8-deoxyserratinine (B13), cernupalhine A (B7) 等4种fawcettimine型石松生物碱.此外, 经化合物47还可合成三环石松生物碱lycopladine A[22]. 2014年该小组[23]又发表了针对diquinane型四环石松生物碱的集成合成研究, 手性化合物48与碘代物49发生烷基化反应, 产物经氧化脱硫得环己烯酮50, 中间体50立体选择性引入烯丙基, 后经选择性双羟化, 活化羟基关闭B环得含有A、B、D环的三环中间体52, 化合物52通过分子内选择性aldol反应关C环得关键中间体53, 最终实现了diquinane型四环石松碱magellanine (B3), magellaninone (B4), paniculatine (B5) 和两个paniculatine非天然类似物的不对称全合成.
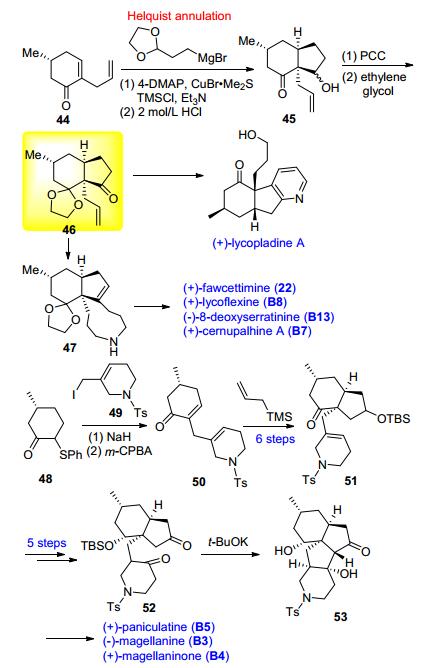 图 图式6
杨玉荣组对fawcettimine型石松生物碱的集成合成
Figure 图式6.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Yang's lab
图 图式6
杨玉荣组对fawcettimine型石松生物碱的集成合成
Figure 图式6.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Yang's lab
涂永强课题组[24]从 (R)-长叶薄荷酮衍生出的溴代物54和八元环化合物55出发, 以中环的Semipinacol重排和SmI2介导的自由基偶联反应为关键步骤, 完成了 (+)-alopecuridine (A3) 的首次全合成, 并通过仿生氧化反应实现了从 (+)-alopecuridine (A3) 到 (+)-sieboldine A (B6) 和 (-)-lycojapodine A (A7) 的仿生合成, 该工作发展了以Semipinacol重排为关键反应构筑fawcettimine类石松生物碱的全新策略 (Scheme 7).
 图 图式7
以自由基偶联反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式7.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the radical coupling
图 图式7
以自由基偶联反应为关键步骤合成fawcettimine型石松生物碱
Figure 图式7.
Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the radical coupling
雷晓光课题组[25]围绕fawcettimine型石松生物碱的集成合成做了大量工作, 取得了系列研究成果. 2012年该小组[25a~25c]以手性甲基环己烯酮60为原料, 经共轭加成串联羟醛缩合、分子内烷基化等步骤合成出关键中间体61, 螺环化合物61经与涂永强组类似的自由基偶联反应, 成功地完成了 (+)-fawcettimine (22), (+)-fawcettidine (A2), (—)-8-deoxyserratinine (B13), (—)-alopecuridine (A3) 和 (—)-lycojapodine A (A7) 等5个石松生物碱家族天然产物的集成合成, 特别是在最有挑战性的 (—)-lycojapodine A (A7) 的全合成过程中, 作者提出了通过利用互变异构体构型锁定的策略 (Tautomer Locking Strategy) 实现最终的串联反应, 从而一步高效构造了天然产物的多环体系 (Scheme 7).
2014年雷晓光课题组[25d]在多样化导向合成 (DOS) 基础上提出一种全新的“官能团配对特征识别”(FGPPR) 的合成策略, 旨在通过系统地发现和利用天然产物中不同的连接形式 (C—C, C—N, C—O键等) 结合在一起的官能团单元.该策略可以系统地指引合成化学家发现并拆解在复杂天然产物结构中存在的配对官能团, 从而得到一个含有多种独立官能团的简单前体, 而后利用Build/Couple可以快速合成该前体67, 最后通过不同类型的官能团配对反应 (Pair), 就可以并行地合成出多种不同骨架的天然和非天然有机化合物.采用该策略, 雷晓光课题组高效地完成了4种石松生物碱[(+)-serratezomine A (B15), (-)-serratinine (B12), (+)-8α-hydroxyfawcettimine (A1) 和 (-)-lycoposerramine U (B9)]以及6种非天然石松生物碱类似物68~73, 共10种不同骨架类型复杂分子的构建.同年, 该课题组[25e]在前期工作的基础上, 又完成了 (-)-Huperzine Q (A8), (+)-lycopladines B, C (A10, 11) 的高效集成合成.螺环中间体76经区域选择性羰基-烯烃复分解反应 (carbonylolefin metathesis) 完成了关键的6/5/9三环骨架的构建, 三环中间体77中的环戊烯经硼氢化/氧化一锅法转化为环戊酮得关键中间体78, 接下来作者运用新颖的烯胺溴代官能团化策略 (enamine bromofunctionalization strategy), 完成了分子结构中非常具有挑战的aminal和dienamine官能团的合成, 分别经中间体79→81, 80→82得到三个目标化合物.文章中首次报道了罕见的epoxyamine中间体83的X-ray单晶结构, 并首次将该中间体运用于复杂天然产物的全合成中 (Scheme 8).
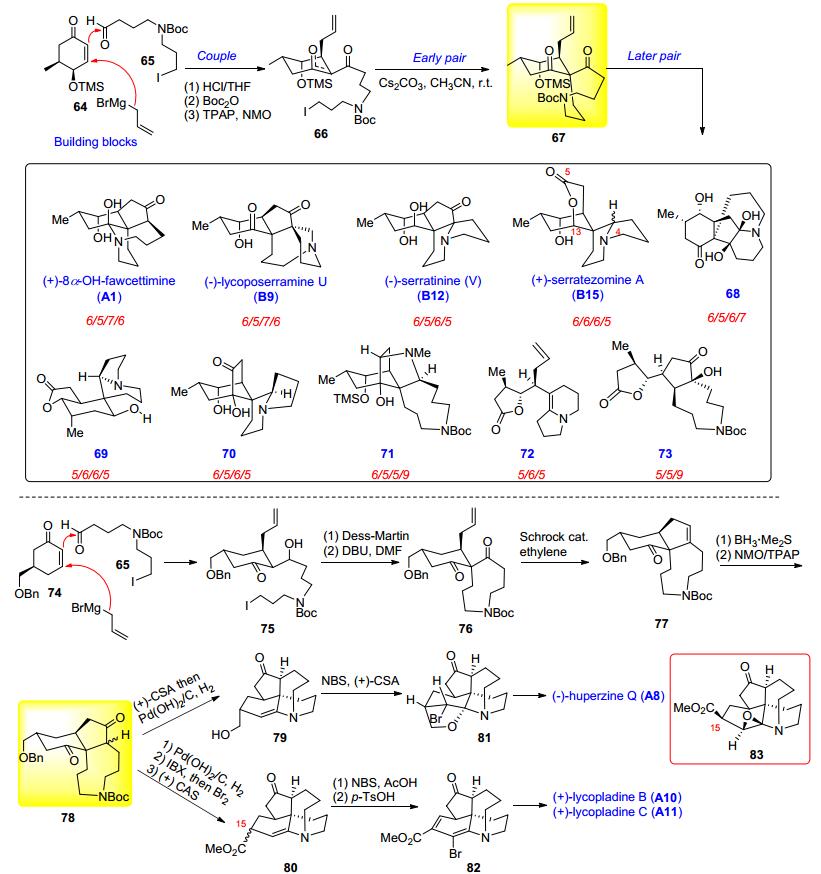 图 图式8
雷晓光课题组对fawcettimine型石松生物碱的集成合成
Figure 图式8.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Lei's lab
图 图式8
雷晓光课题组对fawcettimine型石松生物碱的集成合成
Figure 图式8.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Lei's lab
赵刚课题组[26]从简单易得的手性Hajos-Parrish酮84出发开展对fawcettimine型石松生物碱的多样性合成研究.如Scheme 9, 化合物84经5步反应得到关键中间体85, 化合物85经硼氢化/氧化, 关闭九元环, 引入C (15) 手性甲基得Heathcock三环中间体86.化合物86分别经87和88可实现 (+)-fawcettimine (22), (+)-fawcettidine (A2), (+)-lycoflexine (B8), (+)-lycoposerramine Q (A5), (—)-huperzine Q (A8) 和 (+)-N-oxyhuperzine Q (A9) 等6种石松生物碱的多样性合成.由于lycopladine D (A13) 分子中C (4), C (5), C (15) 三个手性中心的构型与大多数fawcettimine型石松生物碱相反, 为了构建合适构型的目标分子, 作者改变合成策略, 先关闭九元环, 接下来对环内双键进行硼氢化/氧化, 由于九元环的空间位阻效应, 得到的是与86分子中C (4), C (5) 构型完全相反的产物89, 接下来利用金属钯催化的插羰反应引入C (15)-酯基得Heathcock三环中间体90, 最后脱除硅保护基/半胺缩醛化/内酯化得目标分子lycopladine D (A13).该合成工作充分展示出Hajos-Parrish酮在fawcettimine型石松生物碱合成中的独特优势.
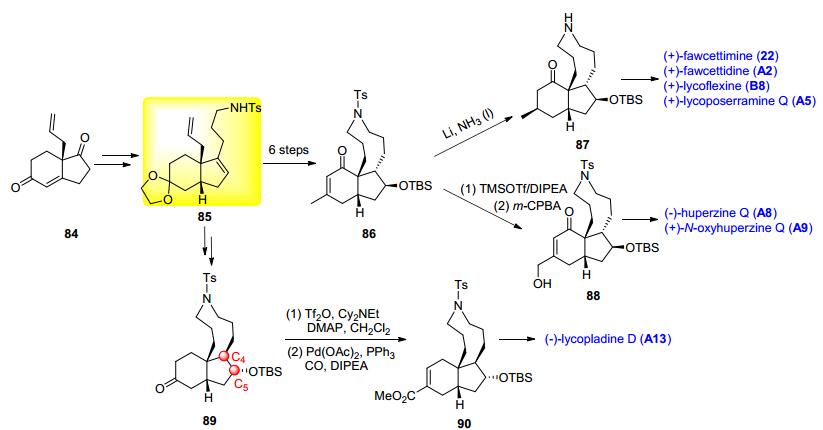 图 图式9
Hajos-Parrish酮在fawcettimine型石松生物碱集成合成中的应用
Figure 图式9.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Hajos-Parrish-like diketone
图 图式9
Hajos-Parrish酮在fawcettimine型石松生物碱集成合成中的应用
Figure 图式9.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Hajos-Parrish-like diketone
除了通过经典的Heathcock三环中间体来合成fawcettimine型石松生物碱, 其他一些巧妙的合成切断思路也有报道. 2013年, 涂永强小组[27]报道了利用新颖的6/5/5三环共同中间体95对石松生物碱 (—)-lycojaponicumin C (B14), (—)-8-deoxyserratinine (B13), (+)-fawcettimine (22) 和 (+)-fawcettidine (A2) 多样性合成的研究, 其关键反应是利用分子内卡宾环化反应来构建化合物94的6/5并环体系 (Scheme 10).
 图 图式10
涂永强课题组对fawcettimine型石松生物碱的集成合成
Figure 图式10.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Tu's lab
图 图式10
涂永强课题组对fawcettimine型石松生物碱的集成合成
Figure 图式10.
Collective synthesis of fawcettimine type Lycopodium alkaloids from Tu's lab
4 其他类型石松生物碱的集成合成
该类生物碱是所有不能归于前三类的石松生物碱的总和, 所以没有统一的骨架类型, 数量相对较多的是phlegmarine型. 2010年Comins小组[28]发表了对phlegmarine类石松生物碱的集成合成研究, 合成中两次利用该小组发展的对手性多取代吡啶盐的格氏加成反应来引入分子中的两个手性哌啶环.含手性辅基的多取代吡啶盐97经格氏加成得二氢哌啶酮98.化合物98经臭氧化、分子内Aldol环合、Michael加成等反应得双环中间体99, 碘代物99经锂化/金属交换得混合格氏试剂100, 对另一分子77发生第二次加成反应得到具有全部所需手性中心的三环中间体101, 最终共同中间体102经一系列官能团转化即完成5个Phlegmarine类化合物的合成, 其中4个为天然产物 (Scheme 11).
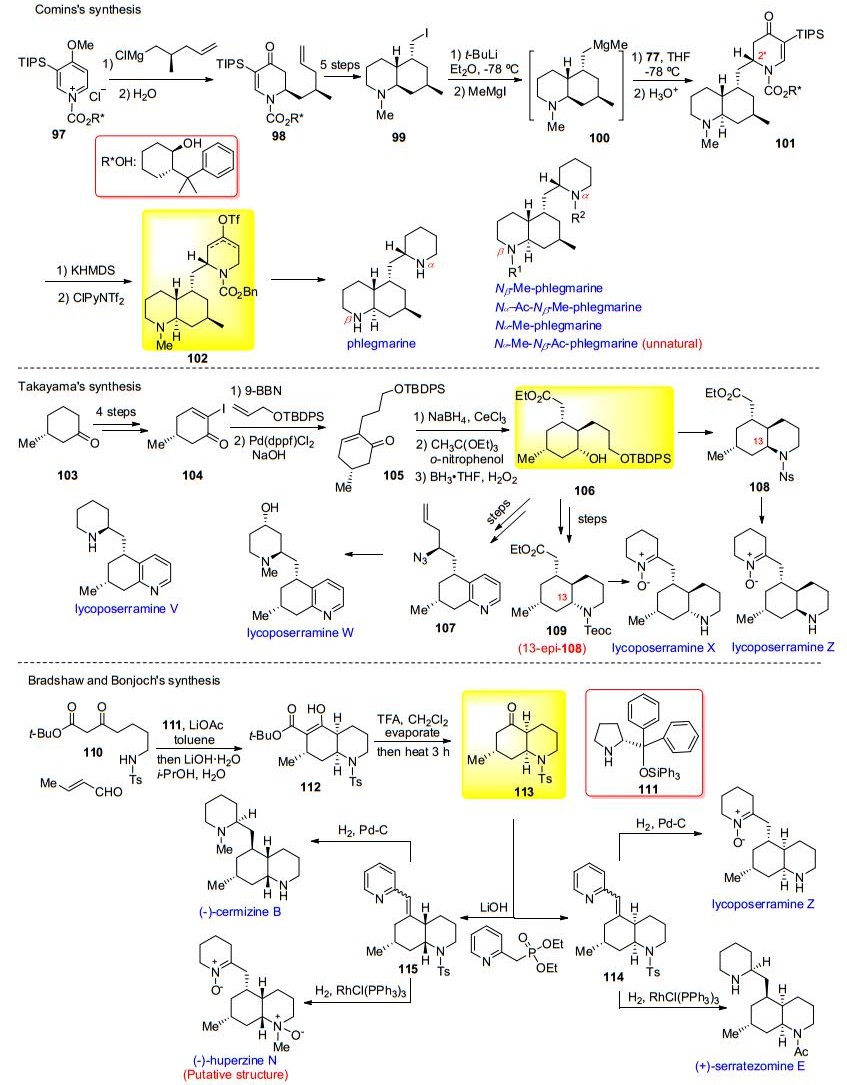 图 图式11
Phlegmarine类石松生物碱的集成合成研究
Figure 图式11.
Collective synthesis of phlegmarine type Lycopodium alkaloids
图 图式11
Phlegmarine类石松生物碱的集成合成研究
Figure 图式11.
Collective synthesis of phlegmarine type Lycopodium alkaloids
2007年Takayama小组[29]发表了对两个新phlegmarine类石松生物碱分离合成工作 (Scheme 11).合成从商品试剂手性甲基环己酮103出发, 经氧化消除, 碘代得甲基环己烯酮碘代物104.碘代物104经SuzukiMiyaura偶联引入羟丙基片段得105, 化合物105经Luche还原/Johnson-Claisen重排/硼氢化-氧化等3步立体/区域选择性反应构筑关键的手性多取代环己烷中间体106.化合物106经Knoevenagel反应引入吡啶环, 利用Brown不对称烯丙基化反应构建C (5) 手性中心, 最终引入哌啶环分别完成了两个目标分子lycoposerramines V, W的全合成, 并确定了天然产物的绝对构型. 2009年, 该小组[30]采用类似策略经共同中间体106完成了对另外一对含硝酮基phlegmarine类石松生物碱lycoposerramines X和Z的全合成工作, 其关键步骤为多次使用Mitsunobu反应构建C (13) 的不同立体构型.
Bradshaw、Bonjoch等[31]陆续发表了对4个不同立体构型的phlegmarine类石松生物碱的多样性合成 (Scheme 11).β-酮酸酯110与丁烯醛发生小分子催化的不对称Michael加成串联Robinson环化/分子内azaMichael加成, 一锅法合成顺式十氢喹啉中间体112.化合物112经酸催化脱羧得关键中间体113.接下来作者利用立体多样性策略, 由关键中间体113经多步区域/立体选择性反应分别完成了lycoposerramine Z, (—)-cermizine B, (+)-serratezomine E和 (—)-huperzine N原推测结构的全合成, 并通过合成给出了 (—)-huperzine N的正确结构.
Cernuane类 (cernuine, cermizine D) 和喹诺里西啶类石松生物碱 (cermizine C, senepodine G) 含有共同的6/6并环结构. 2009年Takayama小组[32]发表了对这两类石松生物碱的多样性合成工作, 作者从天然手性原料 (+)-香茅醛出发, 经缩醛化, 切断双键, 小分子催化的α-氨基化反应引入手性氨基, 脱羰基保护/环化得噁唑烷酮116.化合物116经Hosomi-Sakura烯丙基化, 水解噁唑烷酮, 烯丙酰化得117.化合物117经关环复分解 (RCM) 反应关环, 还原双键得具有喹诺里西啶环的关键中间体118, 化合物118经还原羰基、脱羟基即得 (+)-cermizine C, 进一步按文献方法转化为 (—)-senepodine G.随后作者又利用118进行另外两个石松生物碱 (—)-cernuine和 (+)-cerminzine D的全合成 (Scheme 12).
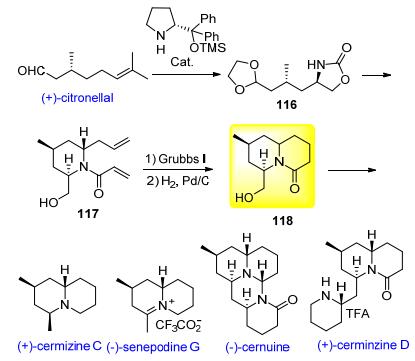 图 图式12
Cernuane类和喹诺里西啶类石松生物碱的集成合成
Figure 图式12.
Collective synthesis of cernuane-type and quinolizidine-type Lycopodium alkaloids
图 图式12
Cernuane类和喹诺里西啶类石松生物碱的集成合成
Figure 图式12.
Collective synthesis of cernuane-type and quinolizidine-type Lycopodium alkaloids
2010年Shair小组[33]完成了对 (+)-fastigiatine的首次全合成, 随后在2014年该课题组[34]又发表了对其他5个含七元环石松生物碱的集成合成 (Scheme 13).由环氧氯丙烷衍生得到的环丙烷化合物119与铜锂试剂120发生片段偶联反应 (fragment coupling), 区域选择性地开环得关键中间体121. β-羧基酰亚胺121经烷基化反应引入C3片段, 随后乙酸叔丁脂的烯醇锂盐进攻C (5)-羰基得酮酸酯化合物122, 通过与不同的烷基化试剂反应得到结构多样化的122.接下来β-酮酸酯122经立体选择性7-endo-trig分子内1, 4-加成构建C (6)—C (7) 键, C (7), C (12) 手性中心; 同时发生两次缩合反应引入Nα— C (5) 键和Nβ—C (12) 键, 一锅法合成具有目标分子七元环骨架的关键中间体123.化合物123经还原C (13) 亚胺/氧化胺化可合成lyconadins A和B, 化合物123发生分子内跨环Mannich反应构筑C (4)—C (13) 键可合成 (—)-fastigiatine, (—)-himeradine A, (—)-lycopecurine, (—)-dehydrolycopecurine. Shair小组完成的6个目标分子中, (—)-lycopecurine, (—)-dehydrolycopecurine属于lycopodine型, (—)-fastigiatine, (—)-himeradine A属于lycodine型, 而lyconadins A和B为其他类型, 这是首例针对不同大类石松生物碱的集成合成.
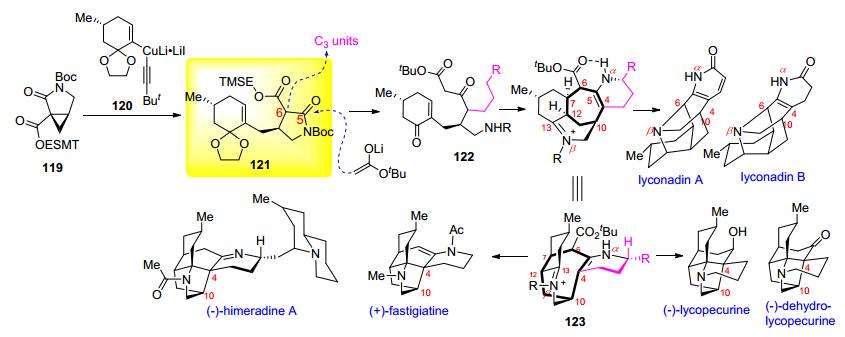 图 图式13
含七元环石松生物碱的集成合成
Figure 图式13.
Collective synthesis of 7-membered-ring-containing Lycopodium alkaloids from Shair's lab
图 图式13
含七元环石松生物碱的集成合成
Figure 图式13.
Collective synthesis of 7-membered-ring-containing Lycopodium alkaloids from Shair's lab
纵观石松生物碱合成文献报道, 早期多侧重于石松生物碱家族成员的合成策略或方法的研究, 有机合成之目的重在展示人类合成能力和合成艺术.近年来为了满足化学生物学、药物化学对天然产物分子的需求, 合成化学家们开始追求更高效的合成.在此背景下, 集成合成策略作为合成家族天然产物的一种有效策略, 引起了有机化学家们的广泛关注.本文仅简单总结了石松生物碱的集成合成研究概况, 相信未来该策略将在其他天然产物的全合成和骨架多样化合成中得到更广泛地应用, 为药物筛选以及化学生物学研究提供足够多的结构新颖的化合物, 从而推动相关学科的发展.
-
-
[1]
赵国平, 戴慎, 陈仁寿, 中药大辞典, 第二版, 上海科学技术出版社, 上海, 2009, pp. 284~285, 774~775, 1590~1591.(a) Zhao, G.; Dai, S.; Chen, R. Dictionary of Traditional Chinese Medicine, 2 ed., Shanghai Scienfitic & Technical Publishers, Shanghai, 2009, pp. 284~285, 774~775, 1590~1591 (in Chinese).
-
[2]
Choo, C. Y.; Hirasawa, Y.; Karimata, C.; Koyama, K.; Sekiguchi, M.; Kobayashi, J. I.; Morita, H. Bioorg. Med. Chem. 2007, 15, 1703. doi: 10.1016/j.bmc.2006.12.005
-
[3]
(a) Mandal, S. K.; Biswas, R.; Bhattacharyya, S. S.; Paul, S.; Dutta, S.; Pathak, S.; Khuda-Bukhsh, A. R. Eur. J. Pharmacol. 2010, 626, 115.
(b) Ham, Y.-M.; Yoon, W.-J.; Park, S.-Y.; Jung, Y.-H.; Kim, D.; Jeon, Y.-J.; Wijesinghe, W.; Kang, S.-M.; Kim, K.-N. Food Chem. Toxicol. 2012, 50, 2629. -
[4]
He, J.; Chen, X.-Q.; Li, M.-M.; Zhao, Y.; Xu, G.; Cheng, X.; Peng, L.-Y.; Xie, M.-J.; Zheng, Y.-T.; Wang, Y.-P. Org. Lett. 2009, 11, 1397. doi: 10.1021/ol900079t
-
[5]
Yuan, C.; Chang, C.-T.; Axelrod, A.; Siegel, D. J. Am. Chem. Soc. 2010, 132, 5924. doi: 10.1021/ja101956x
-
[6]
Feng, X.; Jiang, H.; Zhang, Y.; He, W.; Zhang, L. J. Med. Plants Res. 2012, 6, 1263.
-
[7]
(a) Ayer, W. A. Nat. Prod. Rep. 1991, 8, 455.
(b) Maclean, D. B. In The Alkaloids: Chemistry and Pharmacology, Vol. 26, Ed. : Arnold, B. , Academic Press, New York, 1985, Chapter 5.
(c) Luan, X. ; Xu, Z. Acta Pharm. Sin. 1986, 21, 310 (in Chinese). (栾新慧, 徐择邻, 药学学报, 1986, 21, 310. )
(d) Yuan, S. ; Luan, X. Bull. Acad. Mil. Med. Sci. 1999, 23, 58 (in Chinese). (袁珊琴, 栾新慧, 军事医学科学院院刊, 1999, 23, 58. )
(e) Tan, C. ; Zhu, D. Chin. J. Nat. Med. 2003, 1, 1 (in Chinese). (谭昌恒; 朱大元, 中国天然药物, 2003, 1, 1. )
(f) Chen, Y. ; Liu, Y. ; Jiang, J. ; Liu, B. J. Yunnan Normal Univ. (Nat. Sci. Ed. ) 2010, 30, 12 (in Chinese). (陈业高; 刘怡君; 蒋金和; 刘波, 云南师范大学学报 (自然科学版), 2010, 30, 12. -
[8]
刘嘉森; 俞超美; 周有作, 化学学报, 1986, 44, 1035. doi: 10.3321/j.issn:0251-0790.1986.11.021Liu, J.; Yu, C.; Zhou, Y.; Han, Y.; Qi, B.; Zhu, Y. Acta Chim. Sinica 1986, 44, 1035 (in Chinese). doi: 10.3321/j.issn:0251-0790.1986.11.021
-
[9]
(a) Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2012, 75, 311.
(b) Li, J. W.-H.; Vederas, J. C. Science 2009, 325, 161. -
[10]
(a) Jones, S. B.; Simmons, B.; Mastracchio, A.; MacMillan, D. W. C. Nature 2011, 475, 183.
(b) Schreiber, S. L. Science 2000, 287, 1964. -
[11]
(a) Yang, H.; Carter, R. G.; Zakharov, L. N. J. Am. Chem. Soc. 2008, 130, 9238.
(b) Yang, H.; Carter, R. G. J. Org. Chem. 2010, 75, 4929. -
[12]
(a) Saha, M.; Carter, R. G. Org. Lett. 2013, 15, 736.
(b) Saha, M.; Li, X.; Collett, N. D.; Carter, R. G. J. Org. Chem. 2016, 81, 5963. -
[13]
Ding, R.; Sun, B.-F.; Lin, G.-Q. Org. Lett. 2012, 14, 4446. doi: 10.1021/ol301951r
-
[14]
(a) Ding, R.; Fu, J.-G.; Xu, G.-Q.; Sun, B.-F.; Lin, G.-Q. J. Org. Chem. 2014, 79, 240.
(b) Fu, J.-G.; Xu, G.-Q.; Ding, R.; Lin, G.-Q.; Sun, B.-F. Org. Chem. Front. 2016, 3, 62. -
[15]
(a) Heathcock, C. H.; Smith, K. M.; Blumenkopf, T. A. J. Am. Chem. Soc. 1986, 108, 5022.
(b) Heathcock, C. H.; Blumenkopf, T. A.; Smith, K. M. J. Org. Chem. 1989, 54, 1548. -
[16]
Murphy, R. A.; Sarpong, R. Chem.-Eur. J. 2014, 20, 42. doi: 10.1002/chem.201303975
-
[17]
Nakayama, A.; Kitajima, M.; Takayama, H. Synlett 2012, 23, 2014. doi: 10.1055/s-0032-1316680
-
[18]
(a) Otsuka, Y.; Inagaki, F.; Mukai, C. J. Org. Chem. 2010, 75, 3420.
(b) Kozaka, T.; Miyakoshi, N.; Mukai, C. J. Org. Chem. 2007, 72, 10147.
(c) Itoh, N.; Iwata, T.; Sugihara, H.; Inagaki, F.; Mukai, C. Chem.-Eur. J. 2013, 19, 8665. -
[19]
Pan, G.; Williams, R. M. J. Org. Chem. 2012, 77, 4801. doi: 10.1021/jo3006045
-
[20]
(a) Zaimoku, H.; Taniguchi, T. Chem.-Eur. J. 2014, 20, 9613.
(b) Zaimoku, H.; Nishide, H.; Nishibata, A.; Goto, N.; Taniguchi, T.; Ishibashi, H. Org. Lett. 2013, 15, 2140. -
[21]
(a) Yang, Y.-R.; Lai, Z.-W.; Shen, L.; Huang, J.-Z.; Wu, X.-D.; Yin, J.-L.; Wei, K. Org. Lett. 2010, 12, 3430.
(b) Yang, Y.-R.; Shen, L.; Wei, K.; Zhao, Q.-S. J. Org. Chem. 2010, 75, 1317.
(c) Yang, Y.-R.; Shen, L.; Huang, J.-Z.; Xu, T.; Wei, K. J. Org. Chem. 2011, 76, 3684.
(d) Dong, L.-B.; Wu, Y.-N.; Jiang, S.-Z.; Wu, X.-D.; He, J.; Yang, Y.-R.; Zhao, Q.-S. Org. Lett. 2014, 16, 2700. -
[22]
Xu, T.; Luo, X.-L.; Yang, Y.-R. Tetrahedron Lett. 2013, 54, 2858. doi: 10.1016/j.tetlet.2013.03.097
-
[23]
Jiang, S.-Z.; Lei, T.; Wei, K.; Yang, Y.-R. Org. Lett. 2014, 16, 5612. doi: 10.1021/ol502679v
-
[24]
(a) Zhang, X.-M.; Tu, Y.-Q.; Zhang, F.-M.; Shao, H.; Meng, X. Angew. Chem., Int. Ed. 2011, 50, 3916.
(b) Zhang, X.-M.; Shao, H.; Tu, Y.-Q.; Zhang, F.-M.; Wang, S.-H. J. Org. Chem. 2012, 77, 8174. -
[25]
(a) Li, H.; Wang, X.; Lei, X. Angew Chem., Int. Ed. 2012, 51, 491.
(b) Li, H.; Wang, X.; Hong, B.; Lei, X. J. Org. Chem. 2013, 78, 800.
(c) Wang, X.; Li, H.; Lei, X. Synlett 2013, 24, 1032.
(d) Zhang, J.; Wu, J.; Hong, B.; Ai, W.; Wang, X.; Li, H.; Lei, X. Nat. Commun. 2014, 5, 4614.
(e) Hong, B.; Li, H.; Wu, J.; Zhang, J.; Lei, X. Angew. Chem., Int. Edit. 2015, 54, 1011. -
[26]
(a) Zeng, C.; Zheng, C.; Zhao, J.; Zhao, G. Org. Lett. 2013, 15, 5846.
(b) Zeng, C.; Zhao, J.; Zhao, G. Tetrahedron 2015, 71, 64. -
[27]
Hou, S.-H.; Tu, Y.-Q.; Liu, L.; Zhang, F.-M.; Wang, S.-H.; Zhang, X.-M. Angew. Chem., Int. Ed. 2013, 52, 11373. doi: 10.1002/anie.v52.43
-
[28]
Wolfe, B. H.; Libby, A. H.; Al-Awar, R. S.; Foti, C. J.; Comins, D. L. J. Org. Chem. 2010, 75, 8564. doi: 10.1021/jo1019688
-
[29]
Shigeyama, T.; Katakawa, K.; Kogure, N.; Kitajima, M.; Takayama, H. Org. Lett. 2007, 9, 4069. doi: 10.1021/ol701871v
-
[30]
Tanaka, T.; Kogure, N.; Kitajima, M.; Takayama, H. J. Org. Chem. 2009, 74, 8675. doi: 10.1021/jo9018182
-
[31]
(a) Bradshaw, B.; Luque-Corredera, C.; Bonjoch, J. Org. Lett. 2013, 15, 326.
(b) Bradshaw, B.; Luque-Corredera, C.; Bonjoch, J. Chem. Commun. 2014, 50, 7099.
(c) Bosch, C.; Bradshaw, B.; Bonjoch, J.; Fiser, B.; Gomez-Bengoa, E. Org. Lett. 2015, 17, 5084. -
[32]
Nishikawa, Y.; Kitajima, M.; Kogure, N.; Takayama, H. Tetrahedron 2009, 65, 1608. doi: 10.1016/j.tet.2008.12.067
-
[33]
Liau, B. B.; Shair, M. D. J. Am. Chem. Soc. 2010, 132, 9594. doi: 10.1021/ja104575h
-
[34]
Lee, A. S.; Liau, B. B.; Shair, M. D. J. Am. Chem. Soc. 2014, 136, 13442. doi: 10.1021/ja507740u
-
[1]
-

图 1 四大类石松生物碱的代表化合物
Figure 1 Representative compounds of the four major classes of Lycopodium alkaloids.

图式1 Carter小组对lycopodine型石松生物碱的集成合成
Scheme 1 Collective synthesis of lycopodine type Lycopodium alkaloids from Carter's lab

图式2 林国强、孙炳峰组对lycodine型石松生物碱的集成合成
Scheme 2 Collective synthesis of lycodine type Lycopodium alkaloids from Lin & Sun's lab

图 2 醇胺式fawcettimine型石松生物碱的代表化合物
Figure 2 Representative compounds with carbinol-amine form of fawcettimine type Lycopodium alkaloids

图 3 酮胺式fawcettimine型石松生物碱的代表化合物
Figure 3 Representative compounds with keto-amine form of fawcettimine type Lycopodium alkaloids

图式3 Heathcock小组对fawcettimine的早期合成研究
Scheme 3 Heathcock's originally total synthesis of fawcettimine

图 4 Heathcock三环中间体合成策略汇总
Figure 4 Summary of synthetic strategies on Heathcock-type tricycle intermediates

图式4 以Pauson-Khand反应为关键步骤合成fawcettimine型石松生物碱
Scheme 4 Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the PKR

图式5 以Diels-Alder反应为关键步骤合成fawcettimine型石松生物碱
Scheme 5 Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the Diels-Alder reaction

图式6 杨玉荣组对fawcettimine型石松生物碱的集成合成
Scheme 6 Collective synthesis of fawcettimine type Lycopodium alkaloids from Yang's lab

图式7 以自由基偶联反应为关键步骤合成fawcettimine型石松生物碱
Scheme 7 Collective synthesis of fawcettimine type Lycopodium alkaloids utilizing the radical coupling

图式8 雷晓光课题组对fawcettimine型石松生物碱的集成合成
Scheme 8 Collective synthesis of fawcettimine type Lycopodium alkaloids from Lei's lab

图式9 Hajos-Parrish酮在fawcettimine型石松生物碱集成合成中的应用
Scheme 9 Collective synthesis of fawcettimine type Lycopodium alkaloids from Hajos-Parrish-like diketone

图式10 涂永强课题组对fawcettimine型石松生物碱的集成合成
Scheme 10 Collective synthesis of fawcettimine type Lycopodium alkaloids from Tu's lab

图式11 Phlegmarine类石松生物碱的集成合成研究
Scheme 11 Collective synthesis of phlegmarine type Lycopodium alkaloids

图式12 Cernuane类和喹诺里西啶类石松生物碱的集成合成
Scheme 12 Collective synthesis of cernuane-type and quinolizidine-type Lycopodium alkaloids
-


 下载:
下载:
















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 67
- 文章访问数: 6099
- HTML全文浏览量: 862
 下载:
下载:
