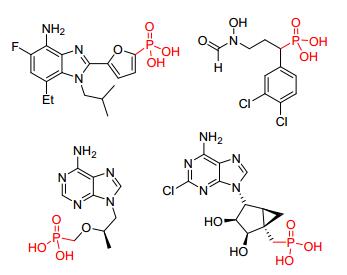
 图 1
含磷碳键的药物
Figure 1.
Drugs containing P—C bonds
图 1
含磷碳键的药物
Figure 1.
Drugs containing P—C bonds
引用本文:
邵长伟, 徐炜刚, 李亮, 张兴华. 过渡金属催化P-C键偶联反应的研究进展[J]. 有机化学,
2017, 37(2): 335-348.
doi:
10.6023/cjoc201608030

Citation: Shao Changwei, Xu Weigang, Li Liang, Zhang Xinghua. Recent Advances of Transition Metal-Catalyzed P-C Coupling Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 335-348. doi: 10.6023/cjoc201608030
Citation: Shao Changwei, Xu Weigang, Li Liang, Zhang Xinghua. Recent Advances of Transition Metal-Catalyzed P-C Coupling Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 335-348. doi: 10.6023/cjoc201608030
过渡金属催化P-C键偶联反应的研究进展
English
Recent Advances of Transition Metal-Catalyzed P-C Coupling Reactions
Abstract:
Organophosphorus compounds which contain P-C bonds have been widely used in photoelectric materials, retardant materials and medicinal chemistry. It is an important method for the synthesis of functional organophosphorus compounds from P (O)-H reagents using transition metal-catalyzed cross coupling reaction to form P-C bond. The recent development in this area is summarized on the basis of different types of carbon atom.
-
Key words:
- transition metal-catalyzed
- / P-C bond
- / cross-coupling
-
含有磷碳 (P—C) 键的有机膦化合物在有机合成、催化、功能材料以及药物化学等领域具有广泛的应用[1~5], 并展现出许多优异性能.由于P—C键特殊的化学性质, 含有膦酸基团[R—PO (OH)2]结构的药物被广泛用于糖尿病、心力衰竭、疟疾、艾滋病等[6~10]的治疗 (图 1).天然产物中也存在很多含有P—C键的有机膦化合物[11], 如Bialaphos和Fosfomycin就已经商业化, 并作为除草剂和抗生素被广泛使用.
图 1
含磷碳键的药物
Figure 1.
Drugs containing P—C bonds
功能材料方面, 目前研究比较广泛的含磷有机阻燃剂, 因其无毒无害且具有良好的阻燃效果, 已经得到了世界范围的公认.分子中所含有的P—C键使之具有较高的热稳定性, 而P=O键的存在也使得五价氧膦化合物 (Phosphine Oxide) 的抗氧化性能明显高于三价有机膦化合物 (Phosphine).
由此可见, 含有P—C键的有机膦化合物在有机合成以及新材料等的研发中具有广泛应用.如何通过简单、高效、低毒的方法构建这一类化合物, 尤其是构建具有高度官能化的膦酸酯以及氧膦类化合物, 已成为近年来有机磷化学家研究的热点.
早期构建P—C键的方法主要是通过Arbuzov或类Arbuzov反应来实现的 (Scheme 1).该方法使用三配位磷化合物与烷基卤代物反应, 反应的原子经济性较差 (需消除一分子的卤代烷烃), 并且反应底物比较单一, 尤其是在构建具有特殊结构的有机膦化合物时, 此类方法不能满足实际的需要.采用格氏试剂或者锂试剂与氯代磷 (膦) 酸酯反应的方法也可构建P—C键, 但该方法所使用的氯代磷试剂不稳定, 并且有机金属试剂对水和氧气比较敏感, 不利于广泛推广.
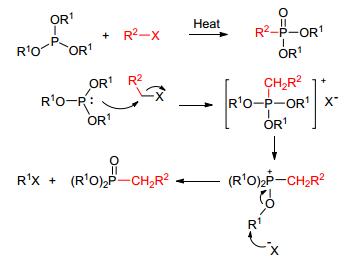 图 图式1
Michaelis-Arbuzov反应及机理
Figure 图式1.
A
plausible mechanism for the reaction of Michaelis-Arbuzov
图 图式1
Michaelis-Arbuzov反应及机理
Figure 图式1.
A
plausible mechanism for the reaction of Michaelis-Arbuzov
采用过渡金属催化的方法进行P—C键的构建[12~14], 因其具有高效、高选择性等特点而成为有机磷化学研究比较活跃的领域, 目前主要集中在过渡金属Pd、Cu、Ni的催化研究.
1 过渡金属催化构建P—C (sp3) 键
如前所述, 传统构建P—C (sp3) 键的方法主要依赖Michaelis-Arbuzov、Michaelis-Becker或类Arbuzov反应, 但高温及强碱条件大大限制了此类反应的进一步应用.近期对Michaelis-Arbuzov反应的改进取得了很好的进展, 2011年Mohanakrishnan等[15a]首次报道了室温条件下Lewis酸催化的Michaelis-Arbuzov反应, 从三配位磷出发, 以苄卤、苄醇、杂环苄卤、杂环苄醇为原料, 在ZnBr2催化下制备了相应的膦酸酯 (Eq. 1). 2011年, Wiemer等[15b]对此类反应底物进行拓展, 对于含不同取代基的苄醇或烯丙醇, 在Lewis酸ZnI2的催化下均可与亚磷酸三乙酯反应生成苄基或烯丙基膦酸酯, 收率15%~85% (Eq. 2).
近年来, 从P (O)—H化合物出发构建P—C键的方法也受到广泛关注, 并得到不断深入的研究. 2003年, Salvatore等[16]发展了一种便捷合成膦酸酯的方法, 室温条件下以Cs2CO3作碱, 即可实现从P (O)—H化合物到P—C (sp3) 键转变, 但反应底物大多适用于1°卤代烷烃以及苄基卤代化合物, 具有一定的局限性 (Eq. 3). 2009年, Stawinski等[17]报道了醋酸钯催化下苄卤与氢亚磷酸酯构建P—C (sp3) 键的方法 (Eq. 4), 同样存在底物适用性不强等问题 (仅适用于苄基卤代物).
过渡金属催化P (O)—H化合物的交叉脱氢偶联反应 (Cross-Dehydrogenative-Coupling, CDC) 也是构建P—C (sp3) 键的主要方法. 2009年, 李朝军等[18]以CuBr盐为催化剂, 氧气作为氧化剂实现了P (O)—H化合物与C (sp3)—H键 (邻位为氮原子) 的脱氢氧化偶联反应, 该方法在较温和的条件下即可合成具有生物活性的α-氨基膦酸酯 (Eq. 5).同样, Ofial等[19]也报道了一种二价铁盐催化氢亚磷酸酯经脱氢氧化制备P—C (sp3) 键化合物的新方法, 该方法以过氧化叔丁醇为氧化剂, 10 mol%氯化亚铁催化下高效制备了α-氨基膦酸酯类化合物 (Eq. 6), 反应的副产物为水.
由此可见, 金属Cu、Fe催化P (O)—H键的脱氢氧化偶联反应实现了P—C (sp3) 键化合物的绿色构建, 也拓展了合成α-氨基膦酸酯类化合物的新方法.在此基础上杨尚东等[20]以Ag2CO3为氧化剂, 在手性质子酸诱导下进一步实现了α-氨基膦酸酯类化合物的不对称合成 (Eq. 7), 该反应可适用于含有不同取代基团的烯丙胺及P—H化合物, 拓展了氧化脱氢偶联反应的底物适用范围, 并且取得了ee值高达92%的手性选择.
2015年, 赵玉芬等[21]采用相同的氧化脱氢偶联策略, 以CuSO4•5H2O为催化剂, 过氧化叔丁醇 (TBHP) 为氧化剂, 通过脱羧氧化偶联实现了由炔基羧酸到β-羰基膦氧化合物的转化.该反应可在较温和条件下快速、高效地制备β-酮类含磷衍生物, 这也为氧化脱羧偶联构建P—C (sp3) 键提供了新的途径 (Eq. 8).
可见光作为新型绿色能源, 在催化构建P—C (sp3) 键方面也得到了很好的利用. 2011年, Koenigs等[22a]首次报道了光照条件下, 利用Ru (Ⅱ) 或Ir (Ⅲ) 作催化剂, 通过氧化脱氢高效制备α-氨基膦酸酯, 开辟了可见光催化构建P—C (sp3) 键的新领域 (Eq. 9).通过对催化剂的筛选, 此类反应得到进一步深入的研究, 曙红Y (eosinY)、Au (Ⅲ) 络合物、氟硼荧 (BODIPY) 染料等[22b, 22c, 22d]均可在相应光源照射下实现P—C (sp3) 键的转化 (Eqs. 10, 11, 12).此外, Rueping等[22e]也报道了金属氧化物同样可以在光照条件下高产率合成α-氨基膦酸酯 (Eq. 13), 并且, 金属氧化物具有可循环利用、价格低廉等优点, 具有很好的应用前景.
近年来, 金属锰盐在实现P—C (sp3) 键的转化中也有突出表现. 2015年, 赵玉芬课题组[23a]报道了系列锰盐催化构建P—C (sp3) 键的新方法.在2.5 equiv.二水合醋酸锰的促进下, 以廉价的端烯、TMSN3、亚磷酸酯为原料, 温和条件下即可合成β-叠氮膦酸酯, 产率28%~88% (Eq. 14).同样以锰盐为催化剂[23b], 以不饱和烯酸为原料, 通过与P (O)—H试剂偶联的同时也生成了五、六元环内酯含磷衍生物, 产率中等以上.该反应具有官能团耐受性好以及反应条件温和等特点 (Eq. 15).
2016年, 赵玉芬课题组[24]实现了Cu (OTf)2催化P (O)—H试剂与端烯类化合物的环化反应, 并成功合成了异喹啉二酮含磷衍生物.实验表明, Cu (Ⅱ)/TBHP协同作用下所产生的磷自由基是促进反应的关键中间体, 通过改变P (O)—H试剂的种类, 可以在温和条件下制备不同类型的含磷异喹啉二酮, 并得到中等以上的收率, 这也为P—C (sp3) 键构建提供了新的途径 (Eq. 16).
2 过渡金属催化构建P—C (sp2) 键
近年来, 随着过渡金属催化构建C—X (X=C, N, S, Si, B) 键的研究深入, 金属催化领域所取得的突破性研究成果显示了其与传统合成方法相比所具有的优势, 因此, 采用简单金属催化P—C (sp2) 键的合成方法, 因其高效、高选择性等特点越来越引起人们的关注[25~27].其中, 过渡金属Pd、Cu、Ni对催化实现P—C (sp2) 的构建具有广泛的应用前景 (Scheme 2).
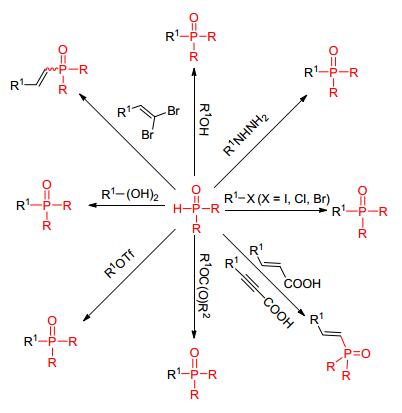 图 图式2
过渡金属催化构建P—C (sp2) 键
Figure 图式2.
Transition
metal-catalyzed coupling of P—C (sp2) bond
图 图式2
过渡金属催化构建P—C (sp2) 键
Figure 图式2.
Transition
metal-catalyzed coupling of P—C (sp2) bond
2.1 Pd催化
相对于其它过渡金属, 钯催化的反应具有反应底物适用性强、反应条件温和等特点.目前, 绝大多数的偶联反应都采用Pd盐作为催化剂, 尤其是在构建P—C (sp2) 键化合物领域, Pd催化P (O)—H键与卤代芳香化合物之间的偶联反应是研究得较为深入的一个方向. 1980年, Hirao等[28~30]首先采用Pd (PPh3)4催化体系成功实现了卤代芳烃与P (O)—H键化合物之间的偶联 (Eq. 17).
在此基础之上, 2007年, Stawinski等[31]以Pd (OAc)2作为催化剂, 通过醋酸根离子的协同作用, 实现了芳基卤代化合物与P (O)—H键的偶联反应 (Scheme 3).在对反应机理的研究中发现, 该反应经历了Pd (0) 的氧化加成以及Pd (Ⅱ) 的还原消除历程, 从而促进了整个偶联反应的循环进行.
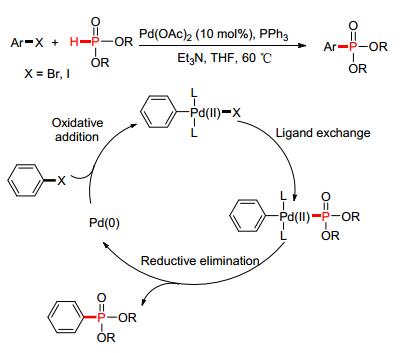 图 图式3
Pd催化的P-芳基化反应
Figure 图式3.
Pd-catalyzed P-arylation
图 图式3
Pd催化的P-芳基化反应
Figure 图式3.
Pd-catalyzed P-arylation
在Hirao等的研究工作发表之后, Pd催化卤代芳烃的P芳基化反应受到广泛关注, 主要研究工作集中在反应底物及催化体系的拓展, 如反应的亲核磷试剂由亚磷酸二烷基酯扩展到亚膦酸酯、次磷酸酯等各种P (O)—H试剂, 包括各种钯盐催化体系及反应条件也被系统考察[32~60].
值得注意的是, 长期以来与P (O)—H化合物偶联的卤代芳烃仅局限于溴、碘代物, 而对于C (sp2)—Cl键的活化一直未能实现. 2011年, Montchamp等[61]报道了金属钯催化卤代芳香体系与亚磷 (膦) 酸酯的偶联反应, 在膦配体的辅助作用下, 首次实现了芳基氯代物与磷原子的交叉偶联, 产率为47%~86% (Eq. 18).
随着对反应机理的进一步认识, 关于金属钯催化P—C (sp2) 键的反应得到了更深入的研究, 通过对反应底物的拓展, 已有实验表明除卤代芳烃之外, 芳基硼酸类底物也适用于此类交叉偶联反应. 2009年, Larhed等[62]以芳基硼酸化合物替代卤代芳烃与P (O)—H键反应, 微波辅助钯催化30 min即可实现P芳基化反应, 极大地缩短了反应时间 (Eq. 19).
在通过缩短反应时间提高反应效率的同时, 原子经济性的提高也成为构建P—C (sp2) 键偶联反应的主要目标.随着钯催化脱氢交叉偶联反应的研究深入, 2012年, 李福伟等[63]报道了醋酸钯催化咪唑类化合物与亚磷酸酯的氧化偶联反应.在脯氨酸等配体的协同作用下, 通过对C (sp2)—H键的活化, 从而实现了P—C (sp2) 键的高效构建 (Eq. 20).此方法具有较高的区域选择性, 并且反应过程中无需碱的加入, 与传统的反应底物卤代芳烃相比, 此类反应的副产物为H2O, 这为合成芳基膦酸酯类化合物提供了更为经济、有效的方法.
2013年, 吴养洁课题组[64]报道了氯化钯催化香豆素与亚膦酸酯脱氢交叉偶联反应 (Scheme 4), 合成了多种天然产物香豆素的磷酰化衍生物.通过对反应机理的推测, P (O)—H亲核试剂首先进攻Pd (Ⅱ) 催化体系形成中间体B, 香豆素C-3位氢原子在K2S2O8的协同作用下被氧化的同时形成中间体D, 在经历了还原消除得到目标化合物的同时, 二价钯催化体系继续进入下一轮催化循环.
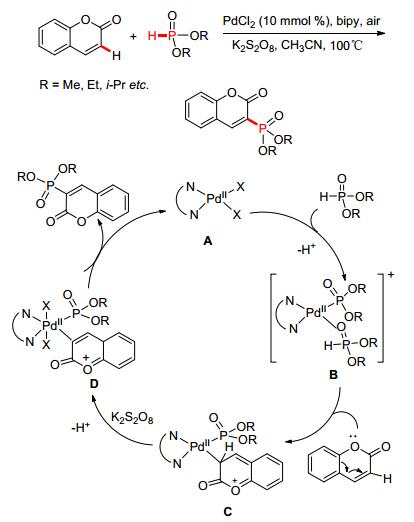 图 图式4
Pd催化脱氢偶联构建P—C (sp2) 键
Figure 图式4.
Pd-catalyzed P—C (sp2) bond coupling via CDC
图 图式4
Pd催化脱氢偶联构建P—C (sp2) 键
Figure 图式4.
Pd-catalyzed P—C (sp2) bond coupling via CDC
同年, 余金权等[65]也以钯催化C—H键活化的策略实现了P—C (sp2) 键的构建 (Scheme 5), 通过与芳环相连吡啶基团的导向作用, 使之与催化活性中心Pd (Ⅱ) 形成五元环过渡态, 在活化C—H键的同时, 通过脱氢氧化构建目标化合物3a并释放出Pd (0), 在Ag (Ⅰ) 的氧化作用下产生的Pd (Ⅱ) 继续进入下一个催化循环.
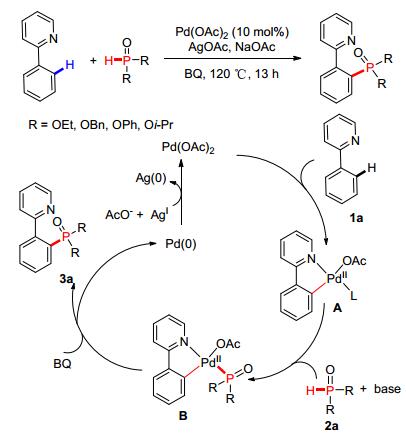 图 图式5
Pd催化C—H活化构建P—C (sp2) 键
Figure 图式5.
Pd-catalyzed
P—C (sp2)
bond coupling via C-H activation
图 图式5
Pd催化C—H活化构建P—C (sp2) 键
Figure 图式5.
Pd-catalyzed
P—C (sp2)
bond coupling via C-H activation
2014年, 赵玉芬课题组[66]发展了合成P—C (sp2) 键的新方法, 通过钯催化C—N键的断裂, 实现P—C (sp2) 键的氧化偶联反应 (Scheme 6).这也是首次报道利用金属钯催化实现芳肼与P (O)—H键偶联形成P—C (sp2) 键的反应, 进一步丰富了Pd催化构建P—C (sp2) 键反应的适用范围.值得注意的是, 该方法采用环境友好性的O2作为氧化剂, 反应的副产物为H2O和N2, 在实现高产率目标产物转化的同时, 也使反应具有良好的官能团适应性.通过对反应机理的推测, Pd (Ⅱ) 催化体系对C (sp2)—N键的活化是整个偶联反应的决速步.
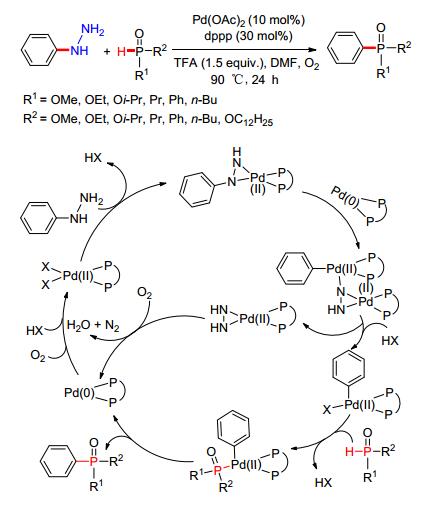 图 图式6
Pd催化C—N活化构建P—C (sp2) 键
Figure 图式6.
Pd-catalyzed
P—C (sp2)
bond formation via C—N activation
图 图式6
Pd催化C—N活化构建P—C (sp2) 键
Figure 图式6.
Pd-catalyzed
P—C (sp2)
bond formation via C—N activation
由此可见, 钯催化构建P—C (sp2) 键的偶联反应具有条件温和、反应简单以及底物适应性强等特点, 因此在其被发现之后的几十年里研究力度日益增加, 然而, 经过多年对钯试剂催化反应的研究与应用, 人们也逐渐认识到了钯试剂催化反应的内在缺陷, 如金属毒性较大、价格昂贵、对不稳定且剧毒的有机膦配体具有一定的依赖性.
2.2 Cu催化
铜作为催化剂, 具有价格低廉、毒性低等优点, 并且铜的化学性质比较温和, 配体简单, 正因为如此, 应用铜盐进行反应催化是目前有机化学非常热门的一个研究方向.
2003年, Venkataraman等[67]以CuI为催化剂 (N, N-二甲基乙二胺为配体), 发展了一种芳基或烯基卤代化合物与含P (O)—H键有机磷化合物的偶联方法, 拓展了此类反应的催化体系, 取得了较好的收率 (Eq. 21). 2006年, 付华等[68]对CuI催化的P芳基化反应进行改进, 采用价廉易得的脯氨酸和2-哌啶甲酸为配体, 在很大程度上改善了卤代芳烃与含P (O)—H键有机磷化合物的偶联反应 (Eq. 22).
2011年, 赵玉芬等[69]以苯硼酸为底物替代卤代芳烃, 并在一价铜盐的催化下成功地实现了P芳基化反应.该反应操作简便, 室温条件下, 无需惰性气体保护即可实现高产率转化, 以含氮双齿配体1, 10-菲罗啉替代含膦配体的使用, 也极大地促进了反应的进行 (Eq. 23).
2011年, Evano等[70]以CuI为催化剂, 以N, N-二甲基乙二胺为配体、K3PO4为碱, 甲苯为溶剂, 成功地实现了1, 1-二溴-1-烯烃与二烷基亚磷酸酯的偶联, 得到了高立体选择性的 (E)-1-烯基膦酸酯, 进一步拓展了此类反应的底物适用范围 (Eq. 24).
与偶联反应过程中脱除一分子的HX相比, Cu催化脱羧偶联反应副产物仅为CO2气体, 此类方法具有后处理简单, 反应过程无需碱的加入等优点, 在烯基膦酸酯类化合物的合成中得到很好的应用. 2011年, 杨尚东等[71]通过Cu2O/Ag2O催化脱羧反应实现了P—C (sp2) 键的交叉偶联 (Scheme 7).该方法可适应于不同的芳基氧膦类化合物, 进一步拓展了P (O)—H试剂的适用范围.研究发现, 反应底物首先在Ag (Ⅰ) 催化下发生脱羧反应, 原位生成的“烯基银”进攻二苯基氧膦参与活化的Cu催化中心, 形成P—C (sp2) 键的同时释放出Cu (Ⅰ) 络合物进入下一个催化循环.在构建P—C (sp2) 键的过程中, 此方法有很好的区域选择性和化学选择性.
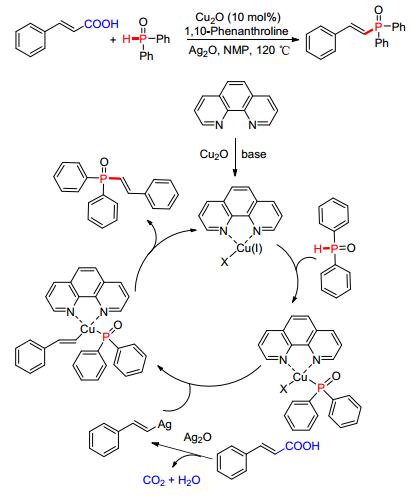 图 图式7
Cu催化脱羧偶联构建P—C (sp2) 键
Figure 图式7.
Cu-catalyzed
P—C (sp2)
bond formation via decarboxylation-coupling
图 图式7
Cu催化脱羧偶联构建P—C (sp2) 键
Figure 图式7.
Cu-catalyzed
P—C (sp2)
bond formation via decarboxylation-coupling
2014年, 赵玉芬课题组[72]报道了以CuCl为催化剂催化炔基羧酸与P (O)—H化合物的P—C (sp2) 交叉偶联反应.该反应条件温和, 在氮气保护下反应6 h即可高产率实现P—C (sp2) 键的转化.在无任何配体、助剂以及碱参与的条件下, 一价铜盐参与脱羧、偶联过程, 大大简化了此类偶联反应的操作 (Eq. 25), 具有良好的应用前景.
除此之外, 采用“一锅法”多组分反应策略也是高效构建P—C (sp2) 键的又一研究方向. 2013年, 张礼和课题组[73]报道了CuCl催化端炔、叠氮化合物以及亚磷酸酯类化合物的三组分偶联反应, 在碱促进下, 室温条件高产率地实现了1, 2, 3-三唑基-5-膦酸酯类化合物的合成, 该方法具有良好的立体选择性和基团适应性 (Eq. 26).
2016年, 韩立彪等[74]发展了以铜催化炔丙基醋酸酯与P (O)—H化合物在低温条件下的偶联反应 (Eq. 27).该反应以四甲基乙二胺 (TMEDA) 为配体, N, N-二异丙基乙胺 (DIPEA) 为缚酸剂, 在乙醇溶液中0 ℃的条件下制备了联烯类膦酸酯, 进一步丰富了铜盐催化P—C (sp2) 键的反应类型.
2.3 Ni催化
综上所述, 从P (O)—H键化合物出发构建P—C (sp2) 键的方法主要集中在钯、铜催化, 而近几年来, 过渡金属镍参与的P芳基化反应正逐渐成为有机磷化学研究的热点.其主要原因是因为相对于Pd等贵金属而言, 金属Ni价廉易得, 不仅可以催化一些钯盐不能催化的偶联反应, 而且在官能团选择性以及区域选择性等方面表现优异.
2004年, 韩立彪等[75]首次报道了室温下镍催化P (O)—H试剂与端炔反应制备磷烯类化合物的方法, 通过研究发现, 改变镍催化剂以及添加有机膦配体, 可以实现P—C (sp2) 键的区域选择构建 (Scheme 8).
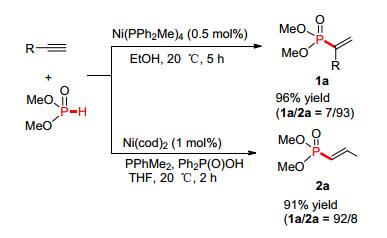 图 图式8
Ni催化P (O)—H化合物与炔烃选择性构建P—C (sp2) 键
Figure 图式8.
Selective
nickel-catalyzed addition of P (O)—H bonds to alkynes
图 图式8
Ni催化P (O)—H化合物与炔烃选择性构建P—C (sp2) 键
Figure 图式8.
Selective
nickel-catalyzed addition of P (O)—H bonds to alkynes
2011年, 赵玉芬等[76]报道了一种镍催化卤代芳烃与二苯基膦氧偶联制备三芳基氧膦的新方法 (Eq. 28).该方法以价廉易得的镍盐和锌粉作为催化体系, 芳基卤与二苯基膦氧在水中即可实现还原偶联合成三芳基膦氧化合物.该方法反应条件温和、操作简单、用水替代了普遍使用的有机溶剂, 是目前合成三芳基膦氧类化合物的绿色、经济方法.
在此基础上, 赵玉芬等[77]对该反应体系进一步拓展, 以NiBr2为催化剂, Mg为还原剂, Bpy为配体, 在碱的促进下可实现1, 1-二溴-1-烯烃与P—H化合物的偶联反应.该方法具有很好的区域选择性以及较高的收率, 也是首次利用Ni-Mg体系催化P—C (sp2) 键的合成 (Scheme 9).
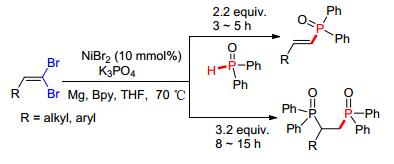 图 图式9
Ni催化还原偶联反应构建P-C键
Figure 图式9.
Ni-catalyzed P-C bond formation via reductive
coupling reaction
图 图式9
Ni催化还原偶联反应构建P-C键
Figure 图式9.
Ni-catalyzed P-C bond formation via reductive
coupling reaction
将反应底物扩展到芳基硼酸类化合物, 也是对金属镍催化体系的进一步完善, 2013年, 赵玉芬等[78]报道了Ni催化芳基硼酸与P (O)—H化合物的偶联反应, 该反应以NiBr2为催化剂、K2CO3为缚酸剂、吡啶为配体实现了多种P (O)—H键与芳基硼酸化合物的高效交叉偶联, 最高收率达到99% (Eq. 29).
通过不同配体的选择, 在改变催化活性中心活性的同时, 也实现了镍催化反应的多样性. 2014年, 赵玉芬等[79]以有机膦配体dppf代替含氮配体继续对镍催化体系进行拓展, 在Ag (Ⅰ) 的辅助下, 成功地实现了Ni催化脱羧交叉偶联构建P—C (sp2) 键.该方法适用于多种不同的P (O)—H试剂, 并且具有很好的区域选择性.通过密度泛函理论 (DFT) 计算发现, 由于膦配体具有更强的亲和性和更大的立体空间效应, 对于此类反应, 相比氮配体而言有机膦配体的参与使之具有更好的催化性能 (Eq. 30).
近年来, 通过C (sp2)—O键活化构建P—C (sp2) 键, 也是镍催化酚及其衍生物与磷试剂偶联反应的热点. 2011年, 张万斌等[80]报道了镍催化亚磷酸三乙酯与芳基三氟甲磺酸酯的类阿尔布佐夫反应.该反应体系以4 equiv. KBr为活化试剂可以实现较高收率的芳基膦酸酯的合成 (Eq. 31).
2012年, 韩福社等[81]报道了用Ni催化酚类化合物与P (O)—H键偶联构建P—C (sp2) 键的反应.通过“一锅法”策略, 当量三吡咯烷基溴化膦六氟磷酸盐 (PyBroP) 的加入, 使得原位活化的酚类底物在NiCl2(dppp) 催化下与多种的P (O)—H试剂交叉偶联, 并得到了较高的产率 (Eq. 32).
2015年, 韩立彪等[82]发展了Ni催化C—O键活化构建P—C (sp2) 键的新方法.该方法从萘酚羧酸酯类衍生物出发, 以Ni (COD)2为催化剂, K2CO3为缚酸剂在较温和条件下实现了其与P (O)—H键的高效偶联.反应以Ni (0) 为催化活性中心, 通过氧化加成, Ni (Ⅱ) 插入到C (sp2)—O键之间形成活化中间体, 经历配体交换以及还原消除反应之后, 释放出新的零价镍进入下一个催化循环的同时P—C (sp2) 键得以构建 (Scheme 10).
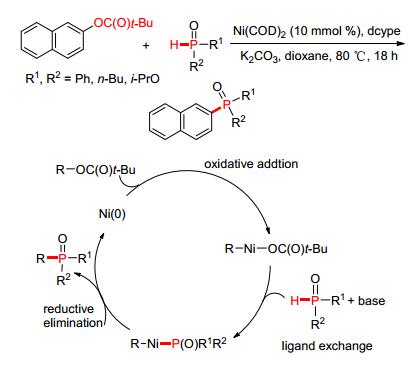 图 图式10
Ni催化C—O活化构建P—C (sp2) 键
Figure 图式10.
Ni-catalyzed
P—C (sp2)
coupling via C—O
activation
图 图式10
Ni催化C—O活化构建P—C (sp2) 键
Figure 图式10.
Ni-catalyzed
P—C (sp2)
coupling via C—O
activation
2.4 其他金属催化
随着脱氢氧化偶联反应的深入研究, 2012年, 黄志真等[83]报道了银催化杂环化合物与亚磷酸酯的氧化偶联反应 (Eqs. 33, 34).该反应以Ag+/S2O82-为催化体系, 通过C (sp2)—H与P (O)—H键的活化, 从而达到氧化脱氢以及P—C (sp2) 键的构建.此类方法适用于包括呋喃、噻吩、吡咯以及吡啶衍生物在内多种杂环化合物的磷酰化反应, 并且具有底物适用广泛、反应条件温和、区域选择性好等特点.
2015年, 谭泽等[84]报道了一种Ag催化端烯与亚磷酸酯的脱氢偶联反应 (Scheme 11).通过自由基捕获剂2, 2, 6, 6-四甲基哌啶氧化物 (TEMPO) 的加入, 很好地实现了烯烃端位磷酰化的区域选择.反应过程中Ag+/ S2O82-的协同作用, 保证了反应体系中足量的Ag (Ⅰ) 与P (O)—H化合物作用生成磷自由基B, B进攻端烯所形成的自由基中间体C被TEMPO捕获后形成化合物D, D经过消除反应最终得到具有良好区域选择性的目标化合物.
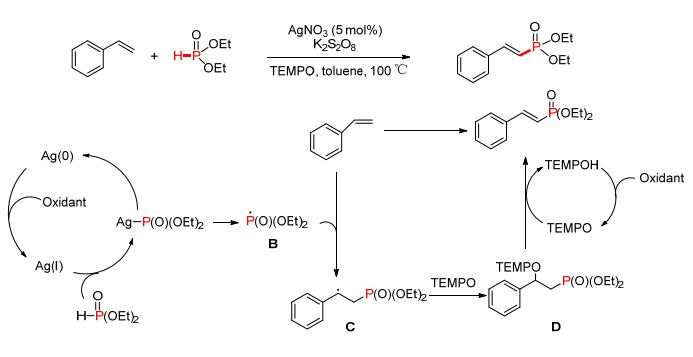 图 图式11
Ag催化脱氢偶联反应构建P—C (sp2)
Figure 图式11.
Ag-catalyzed P—C (sp2) coupling via CDC
图 图式11
Ag催化脱氢偶联反应构建P—C (sp2)
Figure 图式11.
Ag-catalyzed P—C (sp2) coupling via CDC
2015年, 黄志真等[85]利用同样的催化体系, 对反应底物进一步拓展, 在C—H活化方向又实现新的突破 (Eq. 35).他们利用芳香醛或酮与亚膦酸酯在Ag+/ S2O82-催化体系下的脱氢偶联反应, 高产率地合成了芳醛或芳酮的对位磷酰化产物.这种新型的交叉脱氢偶联 (CDC) 反应具有选择性高、产率优良、官能团耐受性好等特点.
2016年, 赵玉芬课题组[86]报道了Ag (Ⅰ) 盐催化合成吲哚类含磷衍生物的新方法 (Eq. 36), 在AgOAc (3 equiv.) 催化下, 芳基膦氧类P (O)—H试剂所产生的膦自由基进攻取代炔烃的同时, 也实现了碳氮键的构建, 但该反应需在较高温度下进行, 产率28%~74%.
Mn (Ⅲ) 盐作为新型的催化试剂在C—H键活化、P—C键偶联方面具有很重要的应用. 2010年, 张伟、邹建平等[87]利用三价锰实现了单取代或多取代芳烃的邻、对位磷酰化反应 (Eq. 37).在此基础之上, 他们也报道了利用相同催化体系对α, β-不饱和化合物的α-H的选择性活化, 制备了具有区域选择性的芳基烯烃磷酰化合产物 (Eq. 38).此方法具有很好的底物适用性, 对于含有酮基、酰胺基、硝基或酯基的底物均取得了较高的收率[88].
3 过渡金属催化构建P—C (sp) 键
2000年, Hayes研究小组[89]报道了以醋酸钯、dppf、环氧丙烷为催化体系, 以1, 1-二溴-1-烯烃、氢亚磷酸酯为原料构建P—C (sp) 键的方法 (Eq. 39), 成功制备了炔基磷酸酯类化合物.
2011年, Gaumont等[90]报道了在使用CuI作催化剂的碱性条件下催化合成炔基磷硼烷的反应 (Eq. 40), 并且得到了50%~74%的产率.
2009年, 赵玉芬等[91]采用脱氢偶联的方法, 在空气氧化下实现了由简单稳定的氢亚磷酸酯与端炔偶联获得1-炔基膦酸酯, 并且氢亚磷酸酯可以定量转化 (Eq. 41).该方法反应条件温和、操作简单、绿色经济 (副产物为水)、底物适用性广 (对含卤代基、羟基、氰基、酯基、羰基、烯键、酰胺和磺酰胺等底物都适合), 是目前合成1-炔基膦酸酯的最简单、经济有效的方法, 有着广阔的应用前景.
2011年, 王磊等[92]对此类反应进行了改进, 使用固定相铜盐作为催化剂合成炔基磷酸酯, 室温条件下P (O)—H试剂即可与端炔化合物发生脱氢偶联 (Eq. 42), 反应以空气中的氧气作为氧化剂, 并且避免了反应体系中碱的使用, 大大简化了反应的操作.值得注意的是, 固定相铜催化剂连续使用六次以后仍具有很好的催化活性.
2012年, 赵玉芬课题组[93]改变了铜盐的种类, 以价廉易得的CuSO4•5H2O为催化剂, 在较温和的条件下就可以实现端炔与亚膦酸酯的偶联反应.经过对反应条件的优化筛选, 确定以三乙胺 (TEA) 为碱, 二甲亚砜 (DMSO) 为溶剂在55 ℃的条件下合成出产率优良的炔基膦酸酯类化合物 (Eq. 43).但是, 该方法仅适用于芳基炔丙醚类底物, 具有一定的局限性.
2014年, 赵玉芬课题组[94]继续对铜催化下端炔与P (O)—H键的反应机理研究 (Scheme 12).通过实验进一步证明了空气中氧气的参与是构建磷炔[P—C (sp)]键的关键, 氧气的存在促进了端炔中C (sp)—H与P (O)—H键之间的脱氢氧化, 但若以惰性气体氮气替代氧气, 则相同条件下生成了完全不同的磷烯[P—C (sp2)]键, 属于完全不同类型的加成反应.
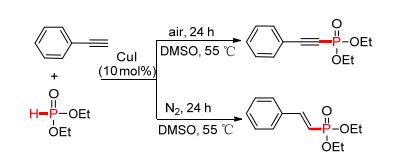 图 图式12
Cu催化端炔磷酰化反应的机理研究
Figure 图式12.
Theoretical
and experimental study on Cu-catalyzed phosphorylation of terminal alkynes
图 图式12
Cu催化端炔磷酰化反应的机理研究
Figure 图式12.
Theoretical
and experimental study on Cu-catalyzed phosphorylation of terminal alkynes
综上所述, Cu盐催化磷炔键的P—C (sp) 偶联反应在反应条件及机理方面的研究已经比较完善, 但此类反应的P (O)—H试剂仅适用于含有烷氧基团的亚磷酸酯类化合物, 对于芳 (烷) 基氧膦类底物适用性不强.鉴于此, 2015年, 韩立彪等[95]发展了一种钯催化端炔与二苯基氧膦的脱氢偶联反应 (Scheme 13).在温和的条件下, 以醋酸钯为催化剂, 通过添加过量的AgBF4, 从而实现了P—C (sp) 键的转化, 产率高达99%.在反应过程中, Ag (Ⅰ) 对还原消除过程中产生的Pd (0) 进行氧化, 保证了反应体系中Pd (Ⅱ) 的催化循环, 这种方法可以将不同的P (O)—H试剂与端炔进行偶联, 弥补了铜催化此类反应的不足.
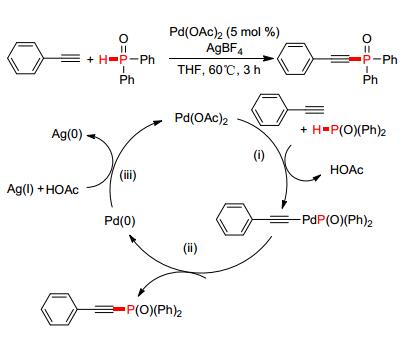 图 图式13
Pd催化脱氢偶联构建P—C (sp) 键
Figure 图式13.
Pd-catalyzed P—C (sp) coupling via CDC
图 图式13
Pd催化脱氢偶联构建P—C (sp) 键
Figure 图式13.
Pd-catalyzed P—C (sp) coupling via CDC
同年, 雷爱文课题组[96]也报道了一种新颖的银促进端炔与P (O)—H试剂的氧化偶联反应 (Scheme 14).反应中过量的Ag (Ⅰ) 选择性氧化C—H、P—H键形成C—Ag、P—Ag键, 经历中间体A, 并通过单电子氧化释放Ag (0) 的同时生成炔基磷酸酯.
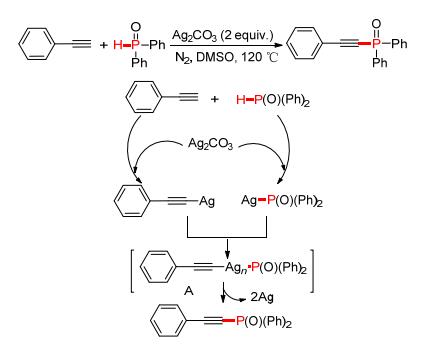 图 图式14
Ag催化脱氢偶联构建P—C (sp) 键
Figure 图式14.
Ag-catalyzed P—C (sp) coupling via CDC
图 图式14
Ag催化脱氢偶联构建P—C (sp) 键
Figure 图式14.
Ag-catalyzed P—C (sp) coupling via CDC
吴养洁等[97]继续对反应底物拓展, 采用2-甲基-3-丁炔-2-醇类化合物为炔源与P (O)—H试剂偶联, 通过脱除丙酮进一步得到含有P—C (sp) 键的炔基磷酸酯 (Scheme 15).反应以Pd (PPh3) Cl2为催化剂, 在缚酸剂K3PO4的作用下脱除丙酮形成炔钯中间体, 经过配体交换, 形成P—C (sp) 键的同时释放Pd (0), Ag (Ⅰ) 将其氧化成Pd (Ⅱ) 继续进入到下一催化循环.
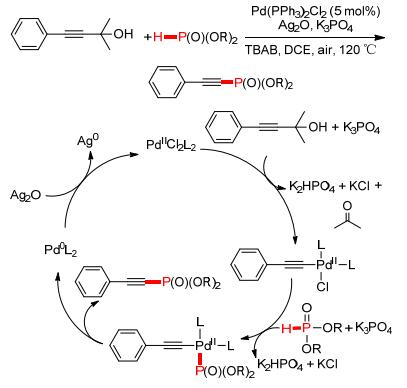 图 图式15
Pd/Ag催化脱丙酮反应构建P—C (sp) 键
Figure 图式15.
Pd/Ag catalyzed P—C (sp) coupling via oxidative deacetonative
图 图式15
Pd/Ag催化脱丙酮反应构建P—C (sp) 键
Figure 图式15.
Pd/Ag catalyzed P—C (sp) coupling via oxidative deacetonative
过渡金属催化脱羧偶联反应同样也可应用于P—C (sp) 的构建. 2014年, 吴玉生等[98]报道了以Cu盐为催化剂催化芳基丙炔酸与亚磷酸酯脱羧偶联合成炔基磷酸酯的反应 (Eq. 44).该反应以Cu (OAC)2•H2O为催化剂, 1, 10-菲罗啉为配体, 异丙醇为添加剂, 以K3PO4为缚酸剂, 在水溶液中就可以进行.该反应具有条件温和、绿色环保、官能团耐受性好等特点, 并给出了中等以上的产率.
4 总结
综上所述, 过渡金属催化P—C键的构建在近几年得到了快速发展, 从传统的高温、对水和氧敏感等苛刻反应发展到简单、高效的绿色合成, 实现了巨大的飞跃, 也为有机膦化合物在光电、阻燃材料以及药物化学等研究领域的广泛应用奠定了良好基础.目前, 对过渡金属催化构建P—C键的研究已相对成熟, 以光催化实现C—H键的活化来构建P—C键已经得到逐步发展, 因其反应条件温和, 以及较高的原子经济性等特点符合绿色化学的发展要求, 具有广阔的发展空间.
-
-
[1]
Baumgartner, T.; Réau, R. Chem. Rev. 2006, 106, 4681. doi: 10.1021/cr040179m
-
[2]
White, A. K.; Metcalf, W. W. Annu. Rev. Microbiol. 2007, 61, 379. doi: 10.1146/annurev.micro.61.080706.093357
-
[3]
Metcalf, W. W.; van der Donk, W. A. Annu. Rev. Biochem. 2009, 78, 65. doi: 10.1146/annurev.biochem.78.091707.100215
-
[4]
De Clercq, E. Med. Res. Rev. 2010, 30, 667. http://europepmc.org/abstract/med/19626594
-
[5]
De Clercq, E. Med. Res. Rev. 2011, 31, 118. doi: 10.1002/med.v31.1
-
[6]
Dang, Q.; Kasibhatla, S. R.; Xiao, W.; Liu, Y.; DaRe, J.; Taplin, F.; Reddy, K. R.; Scarlato, G. R.; Gibson, T.; van Poelje, P. D.; Potter, S. C.; Erion, M. D. J. Med. Chem. 2010, 53, 441. doi: 10.1021/jm901420x
-
[7]
Maryanoff, B. E. J. Med. Chem. 2004, 47, 769. doi: 10.1021/jm030493t
-
[8]
Kumar, T. S.; Zhou, S.-Y.; Joshi, B. V.; Balasubramanian, R.; Yang, T.; Liang, B.-T.; Jacobson, K. A. J. Med. Chem. 2010, 53, 2562. doi: 10.1021/jm9018542
-
[9]
Kang, S.-U.; Shi, Z.-D.; Worthy, K. M.; Bindu, L. K.; Dhar-mawardana, P. G.; Choyke, S. J.; Bottaro, D. P.; Fisher, R. J.; Burke, T. R., Jr. J. Med. Chem. 2005, 48, 3945. doi: 10.1021/jm050059m
-
[10]
Haemers, T.; Wiesner, J.; Van Poecke, S.; Goeman, J.; Henschker, D.; Beck, E.; Jomaa, H.; Van Calenbergh, S. Bioorg. Med. Chem. Lett. 2006, 16, 1888. doi: 10.1016/j.bmcl.2005.12.082
-
[11]
Seto, H.; Kuzuyama, T. Nat. Prod. Rep. 1999, 16, 589. doi: 10.1039/a809398i
-
[12]
Schwan, A. L. Chem. Soc. Rev. 2004, 33, 218. doi: 10.1039/B307538A
-
[13]
Glueck, D. S. Top. Organomet. Chem. 2010, 31, 65. doi: 10.1007/978-3-642-12073-2_4?no-access=true
-
[14]
Tappe, F. M. J.; Trepohl, V. T.; Oestreich, M. Synthesis 2010, 3037. https://www.researchgate.net/publication/259139618_Transition-Metal-Catalyzed_C-P_Cross-Coupling_Reactions
-
[15]
(a) Rajeshwaran, G. G.; Nandakumar, M.; Sureshbabu, R.; Mohanakrishnan, A. K. Org. Lett. 2011, 13, 1270.
(b) Barney, R. J.; Richardson, R. M.; Wiemer, D. F. J. Org. Chem. 2011, 76, 2875. -
[16]
Cohen, R. J.; Fox, D. L.; Eubank, J. F.; Salvatore, R. N. Tetrahedron Lett. 2003, 44, 8617. doi: 10.1016/j.tetlet.2003.09.045
-
[17]
Lavén, G.; Stawinski, J. Synlett 2009, 225. doi: 10.1002/chin.200922173/pdf
-
[18]
Baslé, O.; Li, C.-J. Chem. Commun. 2009, 4124 https://www.ncbi.nlm.nih.gov/pubmed/19568654
-
[19]
Han, W.; Ofial, A. R. Chem. Commun. 2009, 6023. http://europepmc.org/abstract/med/19809631
-
[20]
Cheng, M.-X.; Ma, R.-S.; Yang, Q.; Yang, S.-D. Org. Lett. 2016, 18, 3262. doi: 10.1021/acs.orglett.6b01514
-
[21]
Zhang, P.-B.; Zhang, L.-L.; Gao, Y.-Z.; Xu, J.; Fang, H.; Tang, G.; Zhao, Y.-F. Chem. Commun, 2015, 51, 7839. doi: 10.1039/C5CC01904D
-
[22]
(a) Rueping, M.; Zhu, S.-Q; Koenigs, R. M. Chem. Commun. 2011, 47, 8679.
(b) Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852.
(c) Xue, Q.-C.; Xie, J.; Jin, H.-M.; Cheng, Y.-X.; Zhu, C.-J. Org. Biomol. Chem. 2013, 11, 1606.
(d) Wang, X.-Z.; Meng, Q.-Y.; Zhong, J.-J.; Gao, X.-W.; Lei, T.; Zhao, L.-M.; Li, Z.-J.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Chem. Commun. 2015, 51, 11256.
(e) Rueping, M.; Zoller, J.; Fabry, D. C.; Poscharny, K.; Koenigs, R. M.; Weirich, T. E.; Mayer, J. Chem. Eur. J. 2012, 18, 3478. -
[23]
(a) Xu, J.; Li, X.-Q.; Gao, Y.-Z.; Zhang, L.-L; Chen, W.-Z.; Fang, H.; Tang, G.; Zhao, Y.-F. Chem. Commun. 2015, 51, 11240.
(b) Gao, Y.-Z.; Li, X.-Q.; Xu, J.; Wu, Y.-L.; Chen, W.-Z.; Tang, G.; Zhao, Y.-F. Chem. Commun. 2015, 51, 1605. -
[24]
Wu, J.; Gao, Y.-Z.; Zhao, X.; Zhang, L.-L; Chen, W.-Z.; Tang, G.; Zhao, Y.-F. RSC Adv. 2016, 6, 303. doi: 10.1039/C5RA22570A
-
[25]
(a) Tappe, F. M. J.; Trepohl, V. T.; Oestreich, M. Synthesis 2010, 3037.
(b) Han, L.-B.; Tanaka, M. Chem. Commun. 1999, 395.
(c) Coundray, L.; Montchamp, J. Eur. J. Org. Chem. 2008, 3601.
(d) Prim, D.; Campagne, J.; Joseph, D.; Andrioletti, B. Tetrahedron 2002, 58, 2041.
(e) Schwan, A. L. Chem. Soc. Rev. 2004, 33, 218.
(f) Glueck, D. S. Synlett 2007, 2627. -
[26]
Seto, H.; Kuzuyama, T. Nat. Prod. Rep. 1999, 16, 589. doi: 10.1039/a809398i
-
[27]
(a) Glueck, D. S. Top. Organomet. Chem. 2010, 31, 65.
(b) Niu, M.; Fu, H.; Jiang, Y.; Zhao, Y. Chem. Commun. 2007, 272.
(c) Han, L.-B.; Tanaka, M. J. Am. Chem. Soc. 1996, 118, 1571.
(d) Han, L.-B.; Ono, Y.; Shimada, S. J. Am. Chem. Soc. 2008, 130, 2752. -
[28]
Hirao T.; Masunaga T.; Ohshiro Y.; Agawa. T. Tetrahedron Lett. 1980, 21, 3595. doi: 10.1016/0040-4039(80)80245-0
-
[29]
Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Synthesis 1981, 56. https://www.researchgate.net/publication/273798969_A_Novel_Synthesis_of_Dialkyl_Arenephosphonates
-
[30]
Hirao T.; Masunaga T.; Yamada N, Ohshiro, Y.; Agawa, T. Bull. Chem. Soc. Jpn. 1982, 55, 909. doi: 10.1246/bcsj.55.909
-
[31]
Kalek, M.; Stawinski, J. Organometallics 2007, 26, 5840. doi: 10.1021/om700797k
-
[32]
Xu, Y.-Y.; Zhang, J. Synthesis 1984, 778. https://www.researchgate.net/publication/244564020_Palladium-Catalysed_Synthesis_of_Functionalised_Alkyl_Alkylarylphosphinates
-
[33]
Bulot, J. J.; Aboujaoude, E. E.; Collignon N.; Savignac, P. Phosphorus Sulfur Relat. Elem. 1984, 21, 197. doi: 10.1080/03086648408077657
-
[34]
Xu, Y.-Y.; Li, Z.; Xia, J.-Z.; Guo, H.; Huang, Y.-Z. Synthesis 1984, 781. https://www.researchgate.net/publication/244564021_Palladium-Catalysed_Synthesis_of_Alkylarylphenylphosphine_Oxides
-
[35]
Xu, Y.-Y.; Zhang, J. J. Chem. Soc., Chem. Commun. 1986, 1606. http://pubs.rsc.org/en/content/articlepdf/1986/c3/c39860001606
-
[36]
Lu, X.-Y.; Zhu, J.-Y. Synthesis 1987, 726
-
[37]
Zhang, J.; Xu, Y.-Y.; Huang, G.-H.; Guo, H. Tetrahedron Lett. 1988, 29, 1955. doi: 10.1016/S0040-4039(00)82088-2
-
[38]
Petrakis, K. S.; Nagabhushan, T. L. J. Am. Chem. Soc. 1987, 109, 2831. doi: 10.1021/ja00243a050
-
[39]
Holt, D.A.; Erb, J. M. Tetrahedron Lett. 1989, 30, 5393. doi: 10.1016/S0040-4039(01)80576-1
-
[40]
Xu, Y.-Y.; Wei, H.-X.; Zhang, J.; Huang, G.-H. Tetrahedron Lett. 1989, 30, 949. doi: 10.1016/S0040-4039(00)95287-0
-
[41]
Bennett, S. N. L.; Hall, R. G. J. Chem. Soc., Perkin Trans. 1 1995, 1145. doi: 10.1002/chin.199536207/pdf
-
[42]
Trost, B. M.; Radinov, R. J. Am. Chem. Soc. 1997, 119, 5962. doi: 10.1021/ja9706864
-
[43]
Machnitzki, P.; Nickel, T.; Stelzer, O.; Landgrafe, C. J. Inorg. Chem. 1998, 1029. doi: 10.1002/(SICI)1099-0682(199807)1998:7<1029::AID-EJIC1029>3.0.CO;2-7/full
-
[44]
Kazankova, M. A.; Trostyanskaya, I. G.; Lutsenko, S. V.; Beletskaya, I. P. Tetrahedron Lett 1999, 40, 569. doi: 10.1016/S0040-4039(98)02358-2
-
[45]
Hall, R. G.; Riebli, P. Synlett 1999, 1633. doi: 10.1002/chin.200003167/full
-
[46]
Zhong, P.; Xiong, Z.-X.; Huang, X. Synth. Commun. 2000, 30, 273. doi: 10.1080/00397910008087318
-
[47]
Schuman, M.; Lopez, X.; Karplus, M.; Gouvemeur, V. Tetrahedron. 2001, 57, 10299. doi: 10.1016/S0040-4020(01)01048-1
-
[48]
Kant, M.; Bischoff, S.; Siefken, R.; Gründemann, E.; Köckritz, A. J. Org. Chem. 2001, 477. doi: 10.1002/1099-0690(200102)2001:3<477::AID-EJOC477>3.0.CO;2-3/pdf
-
[49]
Kobayashi, Y.; William, A.; Tokoro, Y. J. Org. Chem. 2001, 66, 7903. doi: 10.1021/jo010701u
-
[50]
Kim, Y.-C.; Brown, S. G.; Harden, T. K.; Boyer, J. L.; Dubyak, G.; King, B. F.; Burnstock, G.; Jacobson, K. A. J. Med. Chem. 2001, 44, 340. doi: 10.1021/jm9904203
-
[51]
Cristau, H.-J.; Hervé, A.; Loiseau, F.; Virieux, D. Synthesis 2003, 2216.
-
[52]
Xie, J.-H.; Wang, L.-X.; Fu, Y.; Zhu, S.-F.; Fan, B.-M.; Duan, H.-F.; Zhou, Q.-L. J. Am. Chem. Soc. 2003, 125, 4404. doi: 10.1021/ja029907i
-
[53]
Muthukumaran, K.; Loewe, R. S.; Ambroise, A.; Tamaru, S. I.; Li, Q.; Mathur, G.; Bocian, D. F.; Misra, V. J. Org. Chem. 2004, 69, 1444. doi: 10.1021/jo034945l
-
[54]
Kobayashi, Y.; William, A. D. Adv. Synth. Catal. 2004, 346, 1749. doi: 10.1002/adsc.200404191
-
[55]
Ziessel, R. F.; Charbonnière, L. J.; Mameri, S.; Camerel, F. J. Org. Chem. 2005, 70, 9835. doi: 10.1021/jo051586g
-
[56]
Chauhan, S. S.; Varshney, A.; Verma, B.; Pennington, M. W. Tetrahedron 2007, 48, 4051. doi: 10.1016/j.tetlet.2007.04.027
-
[57]
Bennett, J. A.; Hope, E. G.; Singh, K.; Stuart, A. M. J. Fluorine Chem. 2009, 130, 615. doi: 10.1016/j.jfluchem.2009.04.008
-
[58]
Kotek, J.; Hermann, P. L. V.; Muller, R. N.; Peters, J. A. J. Mater. Chem. 2009, 19, 1494. doi: 10.1039/b817065g
-
[59]
Ghalib, M.; Niaz, B.; Jones, P. G.; Heinicke, J. W. Tetrahedron Lett. 2012, 53, 5012. doi: 10.1016/j.tetlet.2012.07.037
-
[60]
Rankic, D. A.; Parvez, M.; Keay, B. A. Tetrahedron: Asymmetry 2012, 23, 754. doi: 10.1016/j.tetasy.2012.04.022
-
[61]
Deal, Er. L.; Petit, C.; Montchamp, J. L. Org. Lett. 2011, 13, 3270. doi: 10.1021/ol201222n
-
[62]
Andaloussi, M.; Lindh, J.; Sävmarker, J.; Sjöberj, P. J. R.; Larhed, M. Chem. Eur. J. 2009, 15, 13069. doi: 10.1002/chem.v15:47
-
[63]
Hou, C.-D.; Ren, Y.-L.; Lang, R.; Hu, X.-X.; Xia, C.-G.; Li, F.-W. Chem. Commun. 2012, 48, 5181. doi: 10.1039/c2cc30429e
-
[64]
Mi, X.; Huang, M.-M.; Zhang, J.-Y.; Wang, C.-Y.; Wu, Y.-J. Org. Lett. 2013, 15, 6266. doi: 10.1021/ol4031167
-
[65]
Feng, C.-G.; Ye, M.-C.; Xiao, K.-J.; Li, S.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 9322. doi: 10.1021/ja404526x
-
[66]
Xu, W.-T.; Hu, G.-B.; Xu, P.-X.; Gao, Y.-X.; Yin, Y.-W.; Zhao, Y.-Z. Adv. Synth. Catal. 2014, 356, 2948. doi: 10.1002/adsc.201400155
-
[67]
Van Allen, D.; Venkataraman, D. J. Org. Chem. 2003, 68, 4590. doi: 10.1021/jo0343376
-
[68]
Huang, C.; Tang, X.; Fu, H.; Jiang, Y.-Y.; Zhao, Y.-F. J. Org. Chem. 2006, 71, 5020. doi: 10.1021/jo060492j
-
[69]
Zhuang, R.-Q.; Xu, J.; Cai, Z.-S.; Tang, G.; Fang, M.-J.; Zhao, Y.-F. Org. Lett. 2011, 13, 2110. doi: 10.1021/ol200465z
-
[70]
Evano, G.; Tadiparthi, K.; Couty, F. Chem. Commun. 2011, 47, 179. doi: 10.1039/C0CC01617A
-
[71]
Hu, J.; Zhao, N.; Yang, B.; Wang, G.; Guo, L.-N.; Liang, Y.-M.; Yang, S.-D. Chem. Eur. J. 2011, 17, 5516. doi: 10.1002/chem.v17.20
-
[72]
Hu, G.-B.; Gao, Y.-X.; Zhao, Y.-F. Org. Lett. 2014, 16, 4464. doi: 10.1021/ol502009b
-
[73]
Li, L.-J.; Hao, G.-L.; Zhu, A.-L.; Fan, X.-C.; Zhang, G.-S.; Zhang, L.-H. Chem. Eur. J. 2013, 19, 14403. doi: 10.1002/chem.v19.43
-
[74]
Shen, R.-W.; Luo, B.; Yang, J.-L.; Zhang, L.-X.; Han, L.-B. Chem. Commun. 2016, 52, 6451. doi: 10.1039/C6CC02563C
-
[75]
Han, L.-B.; Zhang, C.; Yazawa, H.; Shimada, S. J. Am. Chem. Soc. 2004, 126, 5080. doi: 10.1021/ja0494297
-
[76]
Zhang, X.-H.; Liu, H.-Z.; Hu, X.-M.; Tang, G.; Zhu, J.; Zhao, Y.-F. Org. Lett. 2011, 13, 3478. doi: 10.1021/ol201141m
-
[77]
Liu, L.; Wang, Y.-L.; Zeng, Z.-P.; Xu, P.-X.; Gao, Y.-X.; Yin, Y.-W.; Zhao, Y.-F. Adv. Synth. Catal. 2013, 355, 659. doi: 10.1002/adsc.201200853
-
[78]
Hu, G.-B.; Chen, W.-Z.; Fu, T.-T.; Peng, Z.-M.; Qiao, H.-W.; Gao, Y.-X.; Zhao, Y.-F. Org. Lett. 2013, 15, 5362. doi: 10.1021/ol402672e
-
[79]
Wu, Y.-L.; Liu, L.; Yan, K.-L.; Xu, P.-X.; Gao, Y.-X.; Zhao, Y.-F. J. Org. Chem. 2014, 79, 8118. doi: 10.1021/jo501321m
-
[80]
Yang, G.-Q.; Shen, C.-R.; Zhang, L.; Zhang, W.-B. Tetrahedron Lett. 2011, 52, 5032. doi: 10.1016/j.tetlet.2011.07.077
-
[81]
Zhao, Y.-L.; Wu, G.-J.; Han, F.-S. Chem. Commun. 2012, 48, 5868. doi: 10.1039/c2cc31718d
-
[82]
Yang, J.; Chen, T.-Q.; Han, L.-B. J. Am. Chem. Soc. 2015, 137, 1782. doi: 10.1021/ja512498u
-
[83]
Xiang, C.-B.; Bian, Y.-J.; Mao, X.-R.; Huang, Z.-Z. J. Org. Chem. 2012, 77, 7706. doi: 10.1021/jo301108g
-
[84]
Gui, Q.-W.; Hu, L.; Chen, X.; Liu, J.-D.; Tan, Z. Chem. Commun. 2015, 51, 13922. doi: 10.1039/C5CC04826E
-
[85]
Huang, X.-F.; Wu, Q.-L.; He, J.-S.; Huang, Z.-Z. Org. Biomol. Chem. 2015, 13, 4466. doi: 10.1039/C5OB00161G
-
[86]
Gao, Y.-Z.; Lu, G.-Z.; Zhang, P.-B.; Zhang, L.-L.; Tang, G.; Zhao, Y.-F. Org. Lett. 2016, 18, 1242. doi: 10.1021/acs.orglett.6b00056
-
[87]
Xu, W.; Zou, J.-P.; Zhang, W. Tetrahedron Lett. 2010, 51, 2639. doi: 10.1016/j.tetlet.2010.03.029
-
[88]
Pan, X.-Q.; Zou, J.-P.; Zhang, G.-L.; Zhang, W. Chem. Commun. 2010, 46, 1721. doi: 10.1039/b925951a
-
[89]
Lera, M.; Hayes, C. J. Org. Lett. 2000, 2, 3873. doi: 10.1021/ol0066173
-
[90]
Bernoud, E.; Alayrac, A.; Delacroix, O.; Gaumont, A. C. Chem. Commun. 2011, 47, 3239. doi: 10.1039/c0cc04645k
-
[91]
Gao, Y.-X.; Wang, G.; Chen, L.; Xu, P.-X; Zhao, Y.-F.; Zhou, Y.-B.; Han, L.-B. J. Am. Chem. Soc. 2009, 131, 7956. doi: 10.1021/ja9023397
-
[92]
Liu, P.; Yang, J.; Li, P.-H.; Wang, L. Appl. Organometal. Chem. 2011, 25, 830. doi: 10.1002/aoc.v25.11
-
[93]
Qu, Z.-B.; Chen, X.-L.; Yuan, J.-W.; Qu, L.-B.; Wang, F.-J.; Ding, X.-L.; Zhao, Y.-F. Can. J. Chem. 2012, 90, 747. doi: 10.1139/v2012-029
-
[94]
Liu, L.; Wu, Y.-L.; Wang, Z.-S.; Zhu, J.; Zhao, Y.-F. J. Org. Chem. 2014, 79, 6816. doi: 10.1021/jo5007174
-
[95]
Yang, J.; Chen, T.-Q.; Zhou, Y.-B.; Yin, S.-F.; Han, L.-B. Chem. Commun. 2015, 51, 3549. doi: 10.1039/C4CC09567G
-
[96]
Wang, T.; Chen, S.-T.; Shao, A.-L.; Gao, M.; Huang, Y.-F.; Lei, A.-W. Org. Lett. 2015, 17, 118. doi: 10.1021/ol503341t
-
[97]
Li, X.; Sun, S.-Y.; Yang, F.; Kang, J.-X.; Wu, Y.-S.; Wu, Y.-J. Org. Biomol. Chem. 2015, 13, 2432. doi: 10.1039/C4OB02410A
-
[98]
Li, X.; Yang, F.; Wu, Y.-J.; Wu, Y.-S. Org. Lett. 2014, 16, 992. doi: 10.1021/ol4037242
-
[1]
-

图式1 Michaelis-Arbuzov反应及机理
Scheme 1 A plausible mechanism for the reaction of Michaelis-Arbuzov

图式2 过渡金属催化构建P—C (sp2) 键
Scheme 2 Transition metal-catalyzed coupling of P—C (sp2) bond

图式5 Pd催化C—H活化构建P—C (sp2) 键
Scheme 5 Pd-catalyzed P—C (sp2) bond coupling via C-H activation

图式6 Pd催化C—N活化构建P—C (sp2) 键
Scheme 6 Pd-catalyzed P—C (sp2) bond formation via C—N activation

图式7 Cu催化脱羧偶联构建P—C (sp2) 键
Scheme 7 Cu-catalyzed P—C (sp2) bond formation via decarboxylation-coupling

图式8 Ni催化P (O)—H化合物与炔烃选择性构建P—C (sp2) 键
Scheme 8 Selective nickel-catalyzed addition of P (O)—H bonds to alkynes

图式9 Ni催化还原偶联反应构建P-C键
Scheme 9 Ni-catalyzed P-C bond formation via reductive coupling reaction

图式10 Ni催化C—O活化构建P—C (sp2) 键
Scheme 10 Ni-catalyzed P—C (sp2) coupling via C—O activation

图式12 Cu催化端炔磷酰化反应的机理研究
Scheme 12 Theoretical and experimental study on Cu-catalyzed phosphorylation of terminal alkynes
-


 下载:
下载:















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 142
- 文章访问数: 6655
- HTML全文浏览量: 2509
 下载:
下载:
