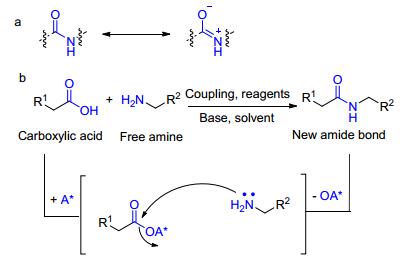
 图 1
酰胺的化学结构和合成酰胺键的传统化学方法
Figure 1.
Chemical
structure of amides and the conventional chemical method for amide bond
synthesis
图 1
酰胺的化学结构和合成酰胺键的传统化学方法
Figure 1.
Chemical
structure of amides and the conventional chemical method for amide bond
synthesis
引用本文:
董浩, 侯梅芳. 酰胺类化合物合成的最新研究进展[J]. 有机化学,
2017, 37(2): 267-283.
doi:
10.6023/cjoc201608014

Citation: Dong Hao, Hou Meifang. Recent Progress in Synthesis of Amides[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 267-283. doi: 10.6023/cjoc201608014
Citation: Dong Hao, Hou Meifang. Recent Progress in Synthesis of Amides[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 267-283. doi: 10.6023/cjoc201608014
酰胺类化合物合成的最新研究进展
English
Recent Progress in Synthesis of Amides
Abstract:
The formation of amide bond is one of the most key reactions in organic chemistry. How to generate amide bond economically is often overlooked as a contemporary challenge as a result of the widespread occurrence of amide polymers, natural products marketed drugs as well as synthetic intermediates. There are inherent drawbacks in the methods. Concerns about their waste and expense are becoming sharper. Thus, Novel chemical protocols to amide formation are being raised. Among a large amount of ways of preparing a amide bond, there has been a growing attention in the use of catalyzed methods for preparing such important functional group. In this review, the catalyst for the formation of an amide bond has become the highlight of some the latest literature in key areas. Alcohols, aldehydes, ketones, unsaturated hydrocarbons, etc., react with organic amines or inorganic amines under a variety of conditions to give the corresponding amide. The review of a new generation of amide-forming reactions may be beneficial to solving these problems.
-
Key words:
- amides
- / catalyst
- / organic amine
- / ammonia
-
酰胺键不仅是蛋白质类中的关键化学键, 同时也是合成很多高分子的基础单元[1].它们形成的化学反应大多是在有机化学中完成的. 图 1(a)为酰胺基的互变异构; 图 1(b)酰胺键的传统合成方法, 包括活化基团 (A*) 对羧酸的活化, 接着在耦合试剂、碱和溶剂存在下通过自由胺进行亲核置换得到新的酰胺键[2].酰胺的功能性是普遍存在的, 尤其是在肽类和蛋白质类中, 有时候会使人们产生错觉, 认为这类物质的合成没有挑战性.但事实上很多简单的酰胺很难合成, 我们需要从国外购买昂贵的试剂用于这些酰胺的合成.此外, 酰胺良好的性质, 如大的极性、稳定性和结构的多样性使得它成为有机化学所有分支中最普遍和最值得依赖的官能团之一.合成酰胺官能团方法的改进, 无论是催化、无污染的, 又或是具有化学选择性用于片段耦合均势在必行.
图 1
酰胺的化学结构和合成酰胺键的传统化学方法
Figure 1.
Chemical
structure of amides and the conventional chemical method for amide bond
synthesis
目前合成酰胺的方法大多比较普遍, 而且成本较高, 工艺比较粗糙. 2007年美国化学学会绿色化学研究所投票决定“高效、经济地合成酰胺”作为有机化学中的顶级挑战[3].此外, 合成具有空间位阻的酰胺时常常不能用最优的化学当量的试剂, 试剂的昂贵和浪费问题经常出现在合成中.
为了能够帮助解决现有的合成方法中存在的弊端, 本文在综合已发表的文献基础上进行简要的总结和评述.
1 含氮有机物作为胺源
1.1 以醇作底物合成酰胺
2012年Bantreil等[4]用氧化铜作催化剂, 叔丁基过氧化氢 (TBHP) 作氧化剂, 醇和胺的盐酸盐在80 ℃下反应4 h, 得到酰胺 (Eq. 1).该反应中会产生副产物酸, 加入过量的醇可避免因副产物的生成而减少酰胺的产率, 且副产物酸在后处理时容易除去.含手性原料参与反应, 生成酰胺的选择性也相当好.
反应机理如Scheme 1: A分子内β-氢脱掉生成苯甲醛和叔丁醇同时生成有活性的二价铜, 生成的苯甲醛和铜离子络合物和自由胺、碳酸钙反应生成中间体B, 在过氧化物的存在下B进化成C, 接着β-氢离去生成酰胺 D, 并且再次生成二价铜催化剂.实验表明第一次催化循环即氧化成醛是比第二次催化循环即醛的酰胺化要慢.
 图 图式1
铜催化机理
Figure 图式1.
Copper-catalyzed mechanism
图 图式1
铜催化机理
Figure 图式1.
Copper-catalyzed mechanism
2013年Ghosh小组[5]提出用硝酸铁作催化剂, 在空气中合成酰胺的方法 (Scheme 2).该反应分两步进行, 首先铁盐和2, 2, 6, 6-四甲基哌啶-氮-氧化物 (TEMPO) 联合作催化剂, 空气中的氧作为氧化剂, 氧化醇到醛; 第二步醛和胺的盐酸盐在叔丁基过氧化氢 (TBHP) 作氧化剂, 碳酸钙作碱的条件下反应得到酰胺.用纯氧作氧化剂存在一定的安全隐患, 改用大气中的氧气, 对反应亦没有影响.但同时出现新的问题, 由于是开放系统, 反应液中的溶剂处在暴露中, 就会不断向大气中挥发, 对环境和人身都会造成一定伤害, 急需改善.
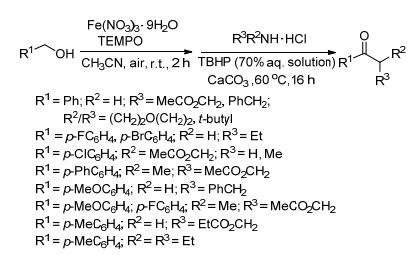 图 图式2
铁催化生成酰胺
Figure 图式2.
Amide formation by iron
catalysts
图 图式2
铁催化生成酰胺
Figure 图式2.
Amide formation by iron
catalysts
机理如Scheme 3: Fe-TEMPO介导氧化醇生成醛, 然后和胺在原位发生反应形成半缩醛中间体, 进一步通过游离基机理被Fe-TBHP催化氧化.
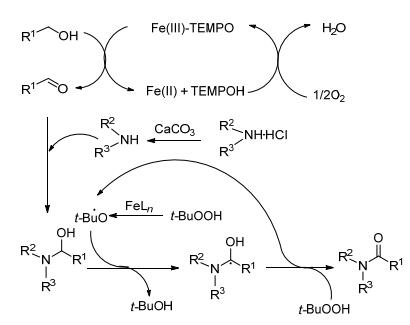 图 图式3
(Fe-TEMPO) 催化氧化氨化机理
Figure 图式3.
A mechanism for (Fe-TEMPO)-catalysed tandem oxidative
amidation
图 图式3
(Fe-TEMPO) 催化氧化氨化机理
Figure 图式3.
A mechanism for (Fe-TEMPO)-catalysed tandem oxidative
amidation
2014年Islam等[6]提出用聚合物锚定二价钌作为制备二级酰胺的催化剂 (Eq. 2).醇和胺在该催化剂作用下, 碳酸铯作碱, 3-甲基-2-丁酮作氢的接受体, 正丁醇作溶剂回流18 h得到酰胺.醇中甲氧基的引入会使得反应收率明显提高 (87%~96%), 且几乎没有副产物.催化剂可以重复使用六次而不失活性, 在工业上的应用有着光明的前景. Hong等[7]报道N杂环卡宾钌配合物被用于催化醇和胺在无碱的条件下合成酰胺, 反应受所带基团的电子效应的影响明显.反应条件对于仲胺参与反应不是很适用, 需要加入催化量的碱, 促进醇和胺的反应.
2015年Rokade等[8]提出用高氯酸铜作催化剂, 催化氧化α, β-不饱和烯醇和叠氮三甲基硅烷 (TMSN3) 反应生成α, β-不饱和酰胺 (Eq. 3).叠氮三甲基硅烷作为氮的来源, 2, 3-二氯-5, 6-二氰基-1, 4-苯醌 (DDQ) 作氧化剂, 醋酸和水作溶剂, 在室温下和醇反应1 h得到酰胺.该工艺是专门为α, β-不饱和烯醇向不饱和酰胺转化所设计, 收率很好.但反应中使用的TMSN3是有毒的危险品, 对环境和人身都有危害, 不适宜在放大工艺中使用.
2016年, Hull等[9]用金属铑作催化剂, 醇和胺一锅法得到酰胺.同年Hong等[10]提出一种高效、无金属催化的直接由醇合成酰胺的方法 (Eq. 4).偶氮化合物作为氮的来源和醇反应, 叔丁基过氧化氢作氧化剂, 乙腈作溶剂, 在120 ℃下反应24 h得到相应的酰胺, 电子效应对该反应并没有明显的影响.苯系醇和脂肪族甚至萘系醇参与反应都有不错的收率.
反应机理如Scheme 4:苯甲醇先被过氧化物氧化成醛, 再转变成自由基, 偶氮苯和自由基反应生成中间体, 再吸引叔丁基过氧化氢上的氢原子得到另一中间体, 最后水解得到酰胺.
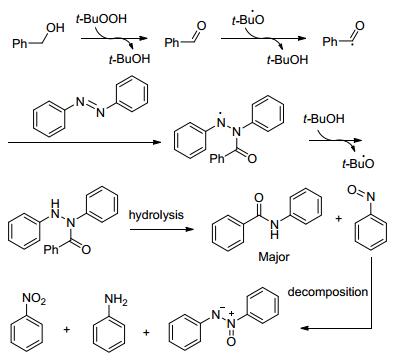 图 图式4
反应机理
Figure 图式4.
Proposed mechanism
图 图式4
反应机理
Figure 图式4.
Proposed mechanism
1.2 以醛酮为底物合成酰胺
2006年Yoo等[11]用醛和有机胺的盐酸盐在碘化亚铜作催化剂, 碳酸钙提供碱性, 乙腈作溶剂, TBHP作氧化剂条件下合成酰胺 (Eq. 5).反应中加入少量的碘酸银可提高反应收率, 且连有空间位阻较大基团的胺盐酸盐参与反应会使得收率大大降低; 脂肪醛参与反应效果不理想且收率很低.当带有旋光性的胺盐参与反应, 收率同样很高, 且不会发生外消旋化, ee值达到99%.
2007年Bode小组[12]用N杂环卡宾和1, 8-二氮杂二环[5.4.0]十一碳-7-烯 (DBU) 联合作催化剂, α, β-不饱和醛、苯胺在碱性条件下反应15 h得到酰胺.该方法对于底物范围的扩展有着很大的局限性, α, β-不饱和醛作为原料是很难制备的, 也就限制其大范围的使用, 且在反应中产物易形成亚胺, 使得实验方案变得相对复杂.该小组[13]为了解决底物应用的局限性, 用α′-羟基烯酮代替α, β-不饱和醛与伯胺在两种催化剂和三氮唑作用下反应生成酰胺 (Eq. 6). α′-羟基烯酮很容易通过一步法反应得到, 所以原料来源广泛.优化后的方案, 不仅使得底物范围扩大, 同时反应时间大大缩短, 反应效率明显提高.
2012年Leighey等[14]用醛和溴代硝基甲烷在含手性铜配合物的催化下经Henry加成[15]生成含手性化合物, 再和含手性胺反应形成酰胺.整体反应能具有如此高度选择性需归功于手性邻碘苯甲酸二噁唑啉配体铜盐催化剂的使用, 产物ee值达到99%.同年Woog小组[16]发现了一种用于合成酰胺的高效催化剂四氯金酸钾.该组除了探索一些结构简单的带有不同基团的芳香醛和脂肪醛, 还研究结构较复杂的含多羟基寡糖醛和二级胺在该催化剂作用下生成酰胺的反应情况 (Eq. 7).反应收率高, 且有很高的选择性, 但因金价格昂贵, 使得它不能大数量级使用.
2014年Ambreen等[17]使用流体化学对酰胺的合成工艺进行研究.该反应必须在高温、高压下进行, 醛和羟胺盐酸盐, 在250 ℃, 20 MPa下反应48 h, 得到酰胺 (Eq. 8).
一个高压液相泵连接到一个金属流动反应器, 该反应器放在恒温器中.反压调节器使得反应混合物保留液体形态, 维持反应器内的温度保持不变 (Scheme 5).
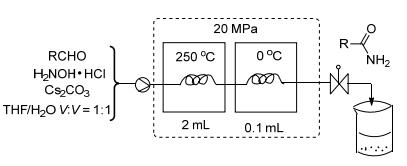 图 图式5
高温高压反应流程示意图
Figure 图式5.
Schematic flow setup for high-temperature and high-pressure
reactions
图 图式5
高温高压反应流程示意图
Figure 图式5.
Schematic flow setup for high-temperature and high-pressure
reactions
流体化学[18]在实验室的应用是在最近几年才开始的.流动化学技术促使流动化学反应器逐渐取代烧瓶间歇反应器和微波反应器.微波反应器不能够工业化, 其致命缺点注定其在化学研究手段短命, 能够进行微波反应器不可能完成的危险反应, 克服了微波反应器不能放大的缺陷.中试流动反应釜可以轻易将合成放大到吨级反应.
2015年Ding等[19]用一价铜作催化剂, TBHP作氧化剂, 醛和胺在室温、大气中、无溶剂条件下反应12 h得到酰胺. 2016年Lu等[20]同样也用一价铜作催化剂, 叔丁基过氧化氢作氧化剂, 水作反应媒介, 脂肪醛、芳香醛和各种胺反应得到酰胺 (Eq. 9).很多文章也报道过用铜作催化剂合成酰胺, 但用脂肪醛和脂肪胺作为偶联伙伴, 很多文章也报道过用铜作催化剂合成酰胺, 但据文献[20]报道用脂肪醛和脂肪胺作为偶联伙伴合成酰胺实属首例.
1.3 以羧酸为底物合成酰胺
2003年Grzyb小组[21]用羧酸和胺甲酰咪唑盐在弱碱性下室温反应16 h得到叔酰胺.咪唑盐是由仲胺和N-N'-羰基二咪唑 (CDI) 反应, 接着用碘甲烷甲基化得到 (Scheme 6).咪唑盐在通常情况下是固体结晶, 比氨基甲酰氯稳定得多, 可以长时间的储存, 如此就更加方便地用于酰胺的合成.
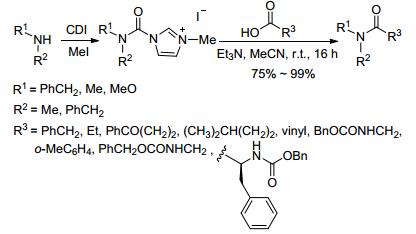 图 图式6
羧酸合成叔酰胺
Figure 图式6.
The synthesis of tertiary
amides from carboxylic acid
图 图式6
羧酸合成叔酰胺
Figure 图式6.
The synthesis of tertiary
amides from carboxylic acid
反应机理如Scheme 7:胺甲酰咪唑盐和羧酸阴离子反应释放出N-甲基咪唑, 生成酸酐, 接着N-甲基咪唑进攻酸酐, 释放出二氧化碳和游离胺的同时, 生成烷基酰咪唑, 再和游离胺的反应生成最后产物酰胺.
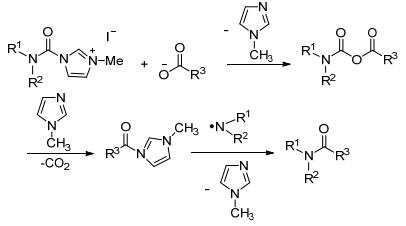 图 图式7
叔酰胺的合成机理
Figure 图式7.
Synthesis mechanism of tertiary amide
图 图式7
叔酰胺的合成机理
Figure 图式7.
Synthesis mechanism of tertiary amide
2011年Liebeskind等[22]发现硫代羧酸直接与胺反应活性很低, 得到的产物收率也低.于是该组在反应中加入了N, N-二 (三甲基硅基) 乙酰胺 (BSA), 提高反应效率.硫代羧酸和BSA快速 (4 min) 反应成O-硅烷基硫羰酯, 这是一种在原位上被活化的酯, 接着和胺发生高效率反应生成酰胺 (Eq. 10).反应机理如Scheme 8.
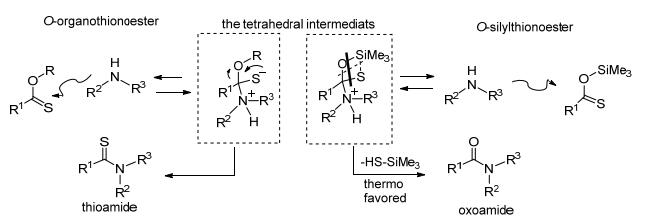 图 图式8
硫到氧转换酰胺化机理
Figure 图式8.
S-to-O switch amidation mechanism
图 图式8
硫到氧转换酰胺化机理
Figure 图式8.
S-to-O switch amidation mechanism
2012年Townsend等[23]用α, β-不饱和羧酸和异腈在微波的辅助下发生双组份偶联生成FCMA (formimidate-carboxylate mixed anhydride) 的中间体, 再通过1, 3-O-N的迁移得到N-甲酰基酰胺 (Scheme 9).产物是含有双键的酰胺, 因氮上连有甲酰基, 使得该类物质活性增强, 可以作为亲双烯体发生DA反应[28].
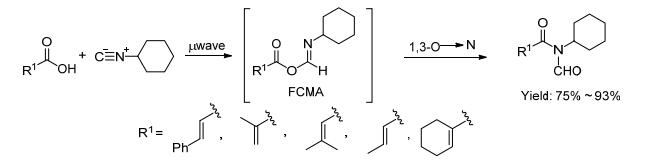 图 图式9
N-甲酰基酰胺的合成
Figure 图式9.
Synthesis of N-formyl
amide
图 图式9
N-甲酰基酰胺的合成
Figure 图式9.
Synthesis of N-formyl
amide
2013年Vogel等[24]用羧酸和亚磺酸三甲基硅烷在20 ℃下快速反应生成羧酸三甲硅烷盐, 得到的产物不做处理, 反应液直接升温到70 ℃再和亚硫酰氟反应生成羧酸和亚磺酸的混合酸酐, 再和胺、四氟硼酸银在20 ℃下反应生成最终产物酰胺 (Scheme 10).该反应是通过一锅法实现, 反应过程中释放出易挥发的二氧化硫、异丁烯和三甲基氟硅烷.
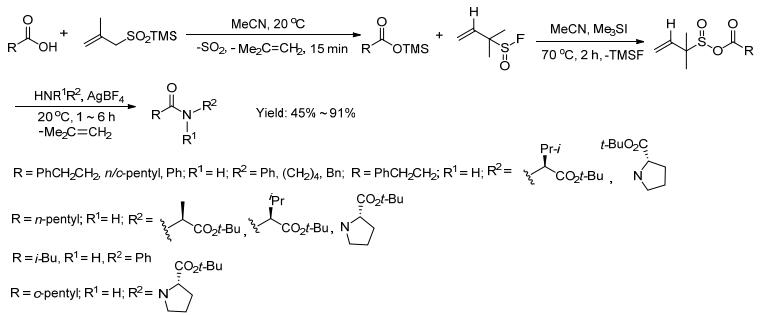 图 图式10
混合酸酐法合成酰胺
Figure 图式10.
Amide formation by mixed anhydrides
图 图式10
混合酸酐法合成酰胺
Figure 图式10.
Amide formation by mixed anhydrides
最后一步反应中如果不加入四氟硼酸银, 那么胺就有机会和酸反应生成亚硫酰胺而不是预期的酰胺.为了改变这种状况, 用非亲核的四氟硼酸阴离子代替亲核的碘离子.用四氟硼酸银中银离子, 沉降碘离子, 如此就可以让反应朝着预设的方向进行 (Scheme 11).
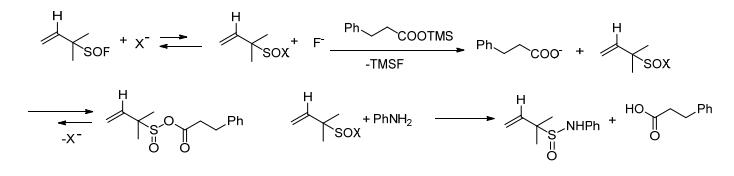 图 图式11
形成混合酸酐的机理
Figure 图式11.
Mechanism for formation of mixed anhydride
图 图式11
形成混合酸酐的机理
Figure 图式11.
Mechanism for formation of mixed anhydride
2015年Lipshutz等[25]描述一种在水性胶束介质中形成酰胺/肽键且对环境友好的方法.用 (2-肟基-氰基乙酸乙酯)-N, N-二甲基-吗啉基脲六氟磷酸酯 (COMU) 作偶联试剂, 2, 6-二甲基吡啶和TPGS-750-M提供温和的条件, 羧酸和胺的盐酸盐在室温下反应生成酰胺 (Eq. 11).水系反应介质是可回收的, 减少了对环境的危害.
2016年Thiemann等[26]用PPh3/CBrCl3联合试剂参与不饱和酸和胺偶合生成酰胺的反应, 避免了处理酰氯的麻烦, 收率60%~84% (Eq. 12).此方法同样适用于合成酯、酸酐和O-酰基肟.
1.4 以酯为底物合成酰胺
2002年Kawahata等[27]用芳酯或内酯经水解, 脱水生成酰胺 (Eq. 13).酯在四烷基羟胺下水解, 水解后加入对甲苯磺酸处理掉多余的羟胺, 再加入胺和碳二亚胺和1-羟基苯并三唑 (HOBt), 脱水生成酰胺.反应液中再加对甲苯磺酸处理过量的胺, 用大孔聚合物碳酸酯树脂去萃取HOBt和残留的酸, 最后过滤, 浓缩得到酰胺.
2005年Hosmane小组[28]用异咪唑插烯酯和过量的胍反应生成酰胺.碳酸钾催化酯和卤代烃发生烷基化反应, 得到产物和过量胍在无水乙醇中回流15 h, 得到醛基被保护的酰胺, 再在水中回流8 h, 去保护得到含甲酰基的酰胺 (Scheme 12).因底物中含有活泼性好的醛基, 和一般的胺反应, 醛基也会参与反应, 而改用过量的胍作为胺的来源, 即解决了上述的问题, 也可以起到对基团的保护作用, 对于应用到其它反应中也是一种很好的办法, 且去保护也比较简单, 不会给整个方案带来额外复杂步骤.
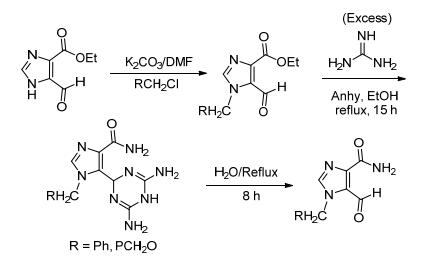 图 图式12
胍进行选择性官能团转换
Figure 图式12.
Selective
functional group transformation using guanidine
图 图式12
胍进行选择性官能团转换
Figure 图式12.
Selective
functional group transformation using guanidine
2014年An小组[29]用二异丁基 (吗啉) 铝和酯反应制备酰胺.用吗啉和三烷基铝在0 ℃下反应3 h得到中间体, 再和酯反应10 min, 即得到吗啉酰胺 (Scheme 13).
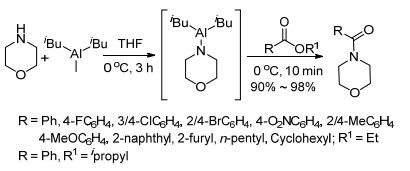 图 图式13
吗啉酰胺的合成新方法
Figure 图式13.
New
synthetic method of morpholine amides
图 图式13
吗啉酰胺的合成新方法
Figure 图式13.
New
synthetic method of morpholine amides
2015年Mecinovic等[30]和Wang等[31]分别提出用锆的二茂铁和1-甲基-3-(3-磺丙基) 咪唑磷钨酸盐催化饱和酯和胺反应制备酰胺.反应操作简便, 收率高, 且底物应用范围广, 催化剂可回收使用, 实际应用前景光明.除用饱和酯作为底物制备酰胺, Du等[32]又以不饱和酯为原料, N-杂环卡宾为催化剂和胺在60 ℃下反应得到酰胺 (Eq. 14).反应机理如Scheme 14.
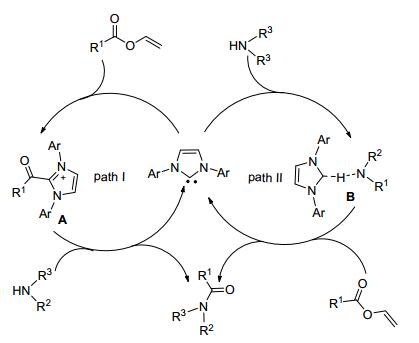 图 图式14
反应机理
Figure 图式14.
Proposed mechanism
图 图式14
反应机理
Figure 图式14.
Proposed mechanism
1.5 以腈为底物合成酰胺
2004年Mizuno小组[33]用氧化铝负载钌的氢氧化物作催化剂, 腈在140 ℃下水解成酰胺 (Eq. 15).用芳香腈、脂肪腈和不饱和脂肪腈对反应进一步验证, 数据得出, 芳香腈反应6 h原料就全部转化完, 而脂肪腈和α, β-不饱和腈反应时间就大大延长, 脂肪族链越长, 反应时间也相应延长.
2009年Varma等[34]同样使用钌金属作为催化剂用于合成酰胺, 与Mizuno小组[33]不同的是用纳米铁氧体负载钌的氢氧化物作为催化剂, 在微波辅助下, 含多种官能团的腈水解成相应的酰胺 (Eq. 16).该方案较Mizuno小组的方案, 反应时间短, 收率高.
2011年Kim等[35]用银纳米粒作催化剂, 在150 ℃下催化腈水解成酰胺 (Eq. 17).但因银是贵金属, 价格昂贵, 不能大量使用在工业上.
2012年Shimizu等[36]对Kim组[35]的方案做了改善, 用二氧化硅负载银纳米粒催化腈在水中回流水解成酰胺 (Eq. 18).金的部分表面会覆盖着氧原子, 增加了催化剂活性, 提高整体的反应收率.
催化剂SiO2/Ag参与反应机理如Scheme 15, 腈和催化剂表面形成相互吸附的复合物, 氧原子表面区域作为Br nsted碱吸引水中的质子, 使得氧原子表面形成Hδ+, 在银的表面形成OHδ-, 接着OHδ-亲核加成到腈上的碳原子, 通过带负电荷过渡态实现酰胺的生成.
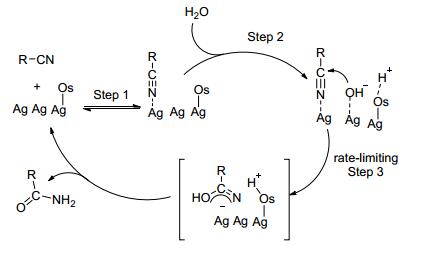 图 图式15
Ag/SiO2催化腈水解机理
Figure 图式15.
Mechanism
of Ag/SiO2-catalyzed hydration of nitriles
图 图式15
Ag/SiO2催化腈水解机理
Figure 图式15.
Mechanism
of Ag/SiO2-catalyzed hydration of nitriles
2013年Tyler等[37]用钌的配合物催化α-羟基腈快速水解成α-羟基酰胺 (Eq. 19).该催化剂在pH=3.5和低温下仍具有活性, 在此条件下α-羟基腈会产生很少能让催化剂中毒的氰化物.该催化剂可以让乙醇腈和乳腈的水解速度超过使用其它任何现有催化剂的水解速度.
2015年Qu等[38]用硫桥双核铁复合物帮助腈水解成酰胺 (Scheme 16). 2016年Jin等[39]用双氧水, 在室温、碱性条件下帮助高分子腈水解成酰胺高分子 (Eq. 20).
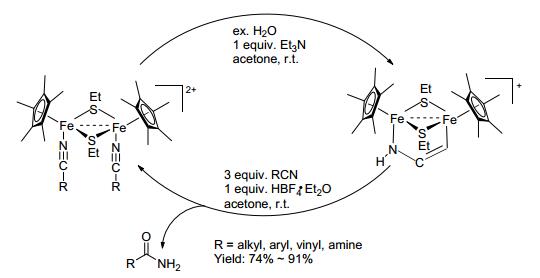 图 图式16
硫桥双核铁复合物促进腈水解成酰胺
Figure 图式16.
Hydration of nitriles to amides by thiolate-bridged
diiron complexes
图 图式16
硫桥双核铁复合物促进腈水解成酰胺
Figure 图式16.
Hydration of nitriles to amides by thiolate-bridged
diiron complexes
1.6 以卤代烃为底物合成酰胺
2000年Alper等[40]用1, 4-双 (二苯膦) 丁烷 (DPPB) 作配体的醋酸钯络合物作催化剂, 邻碘苯胺和对氯苯异氰酸酯、一氧化碳在80 ℃下反应12 h得到环内酰胺 (Eq. 21).
2003年Inoue等[41]发现PNNP型双核钯络合物可以有效地催化卤代烃和胺发生双羰基化反应.磷酸钾作碱, 1, 4-二氧六环作溶剂, 碘代烃和一氧化碳在催化剂的作用下反应15 h, 得到两种酰胺 (Eq. 22).碘苯做原料选择性可达到96%, 2-碘代呋喃参与反应选择性可以达到100%.卤代烃的双羰基化机理如Scheme 17.
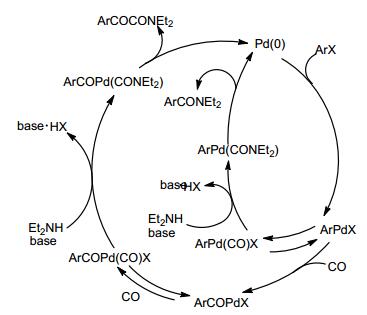 图 图式17
反应机理
Figure 图式17.
Mechanism for the reaction
图 图式17
反应机理
Figure 图式17.
Mechanism for the reaction
1.7 以肟为底物合成酰胺
2006年Ding等[42]用硅硫酸盐高效, 可循环, 高转换, 高选择性, 无污染地催化肟转变成酰胺 (Eq. 23).用微波代替传统加热回流, 提高了收率, 大大缩短反应时间.
2010年Crabtree等[43]用二价钌金属的配合物作催化剂, 促使肟发生贝克曼重排[44]得到酰胺 (Eq. 24).
反应机理如Scheme 18, 第一步催化剂和肟氧化加成, 再亲核加成, 接着发生β-消除, 最后还原消除得到酰胺.
 图 图式18
钌催化肟重排反应机理
Figure 图式18.
Proposed mechanism for
the Ru (Ⅱ) catalyzed rearrangement of oximes
图 图式18
钌催化肟重排反应机理
Figure 图式18.
Proposed mechanism for
the Ru (Ⅱ) catalyzed rearrangement of oximes
2012年Deng等[45]用纳米级Solicalite-1作催化剂, 在400 ℃的氮气流下活化0.5 h, 然后将催化剂置于设定好的反应温度下, 加入溶于乙醇的环己酮肟, 在氮气流下, 发生贝克曼重排生成环己内酰胺 (Eq. 25).
Maronna等[46]在NbOx/SiO2催化下研究环己酮肟的气相贝克曼重排生成酰胺的反应, 反应中加入醇对于催化剂的高活性和反应的选择性是至关重要的.当方案应用到工业上时, 可采用正环己醇, 虽然价格上比乙醇要贵, 但反应可在高压下操作且不失高的选择性.
2013年Lee等[47]用苯酚作为原料一锅法合成己内酰胺, 钯/三氟甲烷磺酸钪/离子液体1-丁基-3-甲基咪唑六氟磷酸盐作为多功能催化剂催化苯酚经肟中间体转变成酰胺 (Scheme 19).苯酚先由钯/三氟甲烷磺酸钪在氢气的存在下共同催化氢化生成环己酮, 再由三氟甲基磺酸钪/离子液体1-丁基-3-甲基咪唑六氟磷酸盐共同催化得到的环己酮肟发生贝克曼重排得到己内酰胺.整个催化体系作为一种多功能催化剂, 将会有着更加广泛的使用性.
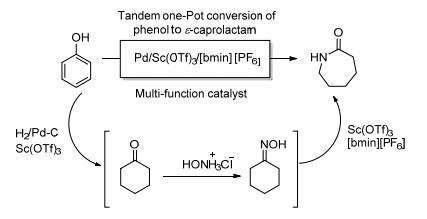 图 图式19
苯酚作原料一锅法合成己内酰胺
Figure 图式19.
A one-pot conversion of phenol to ε-caprolactam
图 图式19
苯酚作原料一锅法合成己内酰胺
Figure 图式19.
A one-pot conversion of phenol to ε-caprolactam
2016年Mhaske等[48]用过硫酸铵 (APS) 和二甲基亚砜 (DMSO) 联合催化肟在100 ℃下重排生成酰胺 (Eq. 26).
反应机理如Scheme 20:过硫酸铵和二甲基亚砜在加热条件下生成甲基硫自由基, 再和肟A反应经过B、C、D自由基的转变得到酰胺.
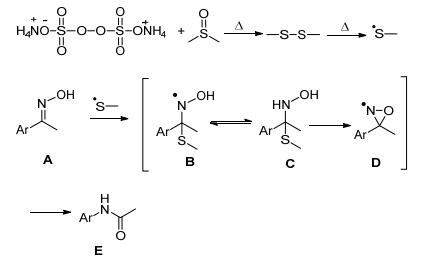 图 图式20
反应机理
Figure 图式20.
Reaction mechanism
图 图式20
反应机理
Figure 图式20.
Reaction mechanism
1.8 以叠氮为底物合成酰胺
2014年Zhang等[49]和Zhu等[50]在三氟硼酸乙醚存在下, 用叠氮化合物和亚胺、醇反应生成酰胺 (Eqs. 27, 28).
机理如Scheme 21.烯烃叠氮化合物亲核进攻碳亲电子体, 形成中间体亚氨基重氮离子A, 接着释放出氮气, 生成腈离子中间体B, 最后水解得到酰胺.但这些过程中是需要过量的三氟硼酸乙醚参与反应的, 烯烃基叠氮化合物的缓慢加成, 防止叠氮化合物的分解.
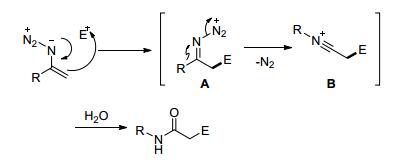 图 图式21
叠氮化合物合成酰胺
Figure 图式21.
Synthesis amide from azide compound
图 图式21
叠氮化合物合成酰胺
Figure 图式21.
Synthesis amide from azide compound
因此Zhang等[51]提出一条新的更温和且简单的反应路线. Tf2NH作催化剂, 醇作碳正离子的来源和叠氮反应生成酰胺 (Eq. 29).
2015年Howell等[52]以叠氮化合物为原料, 经钯碳还原成酯, 再和对硝基酚酯类反应生成α-N-糖基类酰胺 (Eq. 30).
2016年Lüdtke等[53]用硒化学一锅法合成糖基酰胺 (Scheme 22), 可避免处理含硒化合物, 简化处理反应步骤.硒先经三乙基硼氢化锂还原成Se2-, 再和具有活性的羧酸衍生物反应, 在原位上生成硒羧酸盐, 再加入含糖基的叠氮化合物, 发生环加成反应, 生成中间体硒三唑, 最后分解生成酰胺, 并释放出氮气和硒.
 图 图式22
硒化学合成羰基酰胺
Figure 图式22.
Synthesis of glycosyl
amides using selenocarboxylates
图 图式22
硒化学合成羰基酰胺
Figure 图式22.
Synthesis of glycosyl
amides using selenocarboxylates
1.9 以其他物质为底物合成酰胺
1.9.1 以酰氯为底物
2005年Babu等[54]用三甲基氯硅烷和锌粉、酰氯、胺反应可以得到高收率的酰胺 (Eq. 31).胺和三甲基氯硅烷反应生成N-硅烷胺, 接着和酰氯发生偶联反应生成酰胺.
2011年Li等[55]研究用镝金属作还原剂, 促使酰氯和胺发生偶合作用生成酰胺 (Eq. 32).反应中该金属无需活化和预处理, 就可以展现出较高的催化活性, 得到较高收率的酰胺.
2015年Liguori等[56]用醋酸银催化含手性的酰氯和胺反应生成酰胺 (Eq. 33).条件温和, 反应时间短, 收率高, 且不会发生外消旋化反应.
1.9.2 以不饱和烃为底物
2005年Goo en等[57]用钌金属作催化剂, 端位炔和胺反应可以得到区域选择和立体选择性很好的酰胺 (Eq. 34).
2006年Chang等[58]用Pd/Cu作催化剂, 一系列共轭烯烃和酰胺反应得到 (Z)-酰胺 (Eq. 35), 反应同样有很好的立体选择性.反应中加入亚甲基二磷酸四乙酯 (TMEDP), 可以抑制环化的烯酰胺副产物的生成.
2007年Takai等[59]提出一条铼催化的具有区域和立体选择性的氢胺化端位炔制备烯酰胺的方案 (Eq. 36), 端位炔和环酰胺偶联生成高选择性酰胺 (E:Z>99:1).
2013年Beller等[60]开发出一种基于钯的催化体系, 用于烯烃分别和含胺基和硝基的化合物有选择性地氨基羰基化生成烷基酰胺 (Scheme 23).胺参与反应机理如Scheme 24.
 图 图式23
不饱和烃合成酰胺
Figure 图式23.
Synthesis of amide from unsaturated
hydrocarbons
图 图式23
不饱和烃合成酰胺
Figure 图式23.
Synthesis of amide from unsaturated
hydrocarbons
 图 图式 24
催化循环反应
Figure 图式 24.
Proposed catalytic cycle for this reaction
图 图式 24
催化循环反应
Figure 图式 24.
Proposed catalytic cycle for this reaction
1.9.3 以酰胺类为底物合成酰胺
2009年Stephenson等[61]利用高效经济的锆和铪氨基复合物实现叔酰胺与仲胺的相互转换生成新的酰胺 (Scheme 25).在甲苯做溶剂下, 大部分底物在空气环境下就可以发生互换反应, 脂肪族酰胺的活性要高于芳香族酰胺, 脂肪族在室温下即可完成反应, 芳香族需要50 ℃的温度下才能发生转变.该反应是个可逆反应, 在相同条件下, 反应物与产物是可以互相转换的.
 图 图式25
催化叔酰胺间的互相转换
Figure 图式25.
Catalytic
transamidation reactions with tertiary amide
图 图式25
催化叔酰胺间的互相转换
Figure 图式25.
Catalytic
transamidation reactions with tertiary amide
2015年Srinivas等[62]提出一条高效, 对环境友好, 反应条件温和的制备酰胺的方案.在过硫酸钾存在的条件下, 酰胺和胺在水中, 普通加热或微波加热, 生成新的酰胺 (Eq. 37).
具体的反应历程如Scheme 26.酰胺和过硫酸钾混合下, 会促使过硫酸钾的过氧键发生断裂, 生成中间体1, 胺对1加成后得到中间体2, 最后释放放出氨气和硫酸氢钾得到新的酰胺.
 图 图式26
酰胺和胺转换机理
Figure 图式26.
Mechanism
for the formation of transamidated product from amide with aniline
图 图式26
酰胺和胺转换机理
Figure 图式26.
Mechanism
for the formation of transamidated product from amide with aniline
炔酰胺是一种典型的缺电子酰胺, 关于它的描述要追述到1972年Viehe所做的先驱工作[63]. 2012年Jin等[64]利用端位炔和酰胺在氢氧化铜催化剂作用下, 相互耦合得到炔酰胺, 再在锡和钨的混合催化氧化下, 水解得到酰亚胺. 2015年Xu等[65]用炔酰胺和乙酸在100 ℃高温下, 用水和丙酮做混合溶剂发生水解反应得到N-酰基磺酰胺化合物, 产物经过滤就可分离出来. 2016年Huang等[66]用三氟乙酸做介导炔酰胺发生水解得到新的酰胺 (Scheme 27).
 图 图式27
炔酰胺的水解
Figure 图式27.
Hydration of ynamides
图 图式27
炔酰胺的水解
Figure 图式27.
Hydration of ynamides
2 以含氮无机物作为胺源
2.1 以醇为底物合成酰胺
2010年Togo等[67]用碘单质作催化剂, 双氧水作氧化剂, 醇和氨水一锅法合成相应的酰胺 (Eq. 38).带有吸电子基团的醇参与反应得到的酰胺收率偏低, 是因为生成活性较低的芳香腈, 抑制了过氧化氢对其氧化.
Scheme 28描述了整个反应的历程:该方法分两步, 第一步是醇和氨水在碘的催化下生成相应的腈; 第二步腈在碱性条件用过氧化氢氧化水解得到酰胺.
 图 图式28
反应历程
Figure 图式28.
Reaction pathway
图 图式28
反应历程
Figure 图式28.
Reaction pathway
2012年Mizuno等[68]利用KMn8O16(OMS-2) 催化醇和氧气、氨水在无有机溶剂条件下反应生成酰胺 (Eq. 39).从各个类型产物的收率分析, 脂肪醇为原料参与反应收率偏低, 其他都在87%以上.该催化剂可循环使用8次, 在目前金属催化剂中, 锰系催化剂相对价格要便宜.副产物只有水, 对环境友好, 催化剂与产物易分离; 反应时间适中, 但反应温度稍高, 且用到氧气, 存有潜在安全问题.
2.2 以醛酮为底物合成酰胺
2009年Wu等[69]用酮和氨水在碘的作用下反应得到酰胺 (Eq. 40).该反应用水作溶剂, 较之有机溶剂有更多的优势.所使用的原料来源于傅克烷基化产物, 该反应简单, 可放大.当底物中是带有氧或硫的杂环, 对反应的整体有效性影响并不大.但是当R是4-吡啶基时, 得不到预期的产物, 可能是发生N的碘代导致2-位上的碳发生亲核进攻, 产生了副反应.随着反应的进行会看到沉淀生成, 且反应液的颜色也由黑色转变成黄色, 这表明反应中生成了碘仿而沉积下来.反应机理如Scheme 29.
 图 图式29
甲基酮转变成一级酰胺的反应机理
Figure 图式29.
Mechanism
for the transformation of methyl ketone to primary amide
图 图式29
甲基酮转变成一级酰胺的反应机理
Figure 图式29.
Mechanism
for the transformation of methyl ketone to primary amide
2.3 以羧酸为底物合成酰胺
2002年Peng小组[70]用羧酸和碳酸氢氨在甲酰胺中反应得到相应的酰胺 (Eq. 41).微波辅助代替传统搅拌加热, 减少反应时间, 收率高, 用无机盐碳酸氢氨作为氨源, 甲酰胺即作为溶剂又作脱水剂.当羧基上连有的苯基团上带有羟基时, 是观察不到产物的生成.肉桂酸参与反应时, 因在高温下大量升华, 故很难生成酰胺.
2.4 以卤代烃为底物合成酰胺
2015年Ahmed等[71]用二茂铁作配体的钯配合物作为卤代烃酰胺化反应的催化剂.在氯化铵作为胺的来源, 八羰基二钴作为一氧化碳的来源, N, N-二异丙基乙胺 (DIPEA) 作为碱性试剂的条件下, 卤代烃发生酰胺化得到酰胺 (Eq. 42).咪唑作为适当的添加物可有效地增加产物的收率.
2.5 以甲基芳烃为底物合成酰胺
Mizuno等[72]用甲基芳烃和尿素在505 kPa O2, 150 ℃下反应生成酰胺.该反应是用甲基芳烃代替醇或醛合成酰胺的一个重大突破, 但反应条件较为苛刻.
Wang等[73]用甲基芳烃和有机胺在TBHP和TBAI作为氧化剂, 三氯化铁作为催化剂的条件下反应生成相应的酰胺.为了能够使得该反应能够具有更加广泛的应用, 2015年Wang等[74]用氨水作为氮的来源和甲基芳烃在无溶剂, 相同的氧化和催化体系下反应, 得到酰胺 (Eq. 43).该反应通过对芳香烃类的C (sp3)—H直接功能化转变实现了便宜易得化学试剂向高价值产物的转变.
2.6 以酯为底物合成酰胺
2016年Powles等[75]以甘油三油酸酯作底物在南极假丝酵母脂肪酶 (CALB) 催化作用下和氨水发生氨解反应得到油酰胺 (Eq. 44). CALB在氨水中具有很好的催化活性且对甘油酯类氨解表现出链长的选择性.天然酶因不溶于氨水无法催化该类酯发生氨解反应.酯的氨解不是同时发生, 是分阶段进行的, 随着残留的羧基中烃基链长的增加, 氨解的速率也随之在下降.
2.7 以端位炔烃为底物合成酰胺
2016年Batra等[76]用可见光引发碘催化氧化端位炔的碳碳叁键的断裂和氨水反应生成酰胺 (Eq. 45).反应机理如Scheme 30.炔被碘活化生成碘正离子A, 接着氢胺化生成中间体B并释放出碘化铵, 中间体B进攻碘单质, 得到α, α-二碘中间体C, 中间体C发生水解得到D, D的C—H断裂, 消去CH2I2得到酰胺. CH2I2在可见光作用下形成中间体 (CH2OO), 并释放出碘单质.双氧水和碘化铵反应得到氢氧化铵和氢碘酸, 氢碘酸提供碘正离子.
 图 图式30
端位炔转化成伯酰胺的机理
Figure 图式30.
Mechanism for the transformation terminal
alkyne to primary amide
图 图式30
端位炔转化成伯酰胺的机理
Figure 图式30.
Mechanism for the transformation terminal
alkyne to primary amide
3 总结与展望
目前, 用于酰胺合成的催化类方法, 对于合成含酰胺键的化合物有着直接的影响.这些反应不仅对环境友好, 也可以将成本降到最低.这些方法最初都是应用在合成小分子上, 但是原理和机理都可以扩展应用到合成肽类结构上, 尤其适用于那些含有非天然的氨基酸残基的肽类结构的合成.催化方法的高效以及固有的选择性都会给对映选择和化学选择的反应提供新的机会, 当然也包括一些活性基团的酰胺化.
当我们研究出更好的方法来合成复杂的、高性能的酰胺类化合物, 且这些方法中无需腐蚀性的试剂和繁琐的保护基团, 酰胺的合成就会提供新一代带有如粘合、细胞生长调节和生物矿化等高性能功能材料.直到现在, 合成方法仍没有向如此有前途的材料看齐.化学家们应该注意挑战和机遇存在于酰胺的合成中且设计的方案应该具有化学选择性、催化性和合理性.
-
-
[1]
(a) Qin, W. W.; Liu, B.; Ma, J. Chin. J. Org. Chem. 2012, 32, 896 (in Chinese).(秦伟伟, 刘波, 马静, 有机化学, 2012, 32, 896.)
(b) Burwell, D. A.; Thompson, M. E. Chem. Mater. 1991, 3, 730.
(c) Barrett, A. G. M.; Patel, B. H. J. Org. Chem. 2012, 77, 11296.
(d) Yin, L. A.; Brewitz, L.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2014, 136, 17958. -
[2]
Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 22. doi: 10.1038/480022a
-
[3]
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R. Green Chem. 2007, 9, 411. doi: 10.1039/B703488C
-
[4]
Bantreil, X.; Fleith, C.; Martinez, J.; Lamaty, F. ChemCatChem 2012, 4, 1922. doi: 10.1002/cctc.201200441
-
[5]
Ghosh, S. C.; Ngiam, J. S. Y.; Seayad, A. M.; Tuan, D. T.; Johannes, C. W.; Chen, A. Tetrahedron Lett. 2013, 54, 4922. doi: 10.1016/j.tetlet.2013.07.005
-
[6]
Islam, S. M.; Molla, R. A.; Roy, S.; Ghosh, K. Appl. Organomet. Chem. 2014, 28, 900. doi: 10.1002/aoc.v28.12
-
[7]
Hong, S. H.; Kim, K.; Kang, B. Tetrahedron 2015, 71, 4565. doi: 10.1016/j.tet.2015.02.016
-
[8]
Rokade, B. V.; Gadde, K.; Prabhu, K. R. Eur. J. Org. Chem. 2015, (12), 2706. doi: 10.1002/chin.201533108/pdf
-
[9]
Wu, Z.; Hull, K. L. Chem. Sci. 2016, 7, 969. doi: 10.1039/C5SC03103F
-
[10]
Hong, G.; Wu, S. Y.; Zhu, X. Y.; Mao, D.; Wang, L. M. Tetrahedron 2016, 72, 436. doi: 10.1016/j.tet.2015.11.063
-
[11]
Yoo, W. J.; Li, C. J. J. Am. Chem. Soc. 2006, 128, 13064. doi: 10.1021/ja064315b
-
[12]
Bode, J. W.; Sohn, S. S. J. Am. Chem. Soc. 2007, 129, 13798. doi: 10.1021/ja0768136
-
[13]
Chiang, P. C.; Kim, Y. J.; Bode, J. W. Chem. Comm. 2009, 30, 4566. http://www.ncbi.nlm.nih.gov/pubmed/19617985
-
[14]
Leighty, M. W.; Shen, B.; Johnston, J. N. J. Am. Chem. Soc. 2012, 134, 15233. doi: 10.1021/ja306225u
-
[15]
Luzzio, F. A. Tetrahedron 2001, 57, 915. doi: 10.1016/S0040-4020(00)00965-0
-
[16]
Woog, M. K.; Li, G. L.; Kung, K. K. Chem. Commun. 2012, 48, 4112. doi: 10.1039/c2cc17689k
-
[17]
Ambreen, N.; Wirth, T. Eur. J. Org. Chem. 2014, 34, 7590.
-
[18]
(a) Heffner, J. E.; Brown, L. K.; Deleo, J. M. Am. J. Respir. Crit. Care Med. 1995, 151, 1700.
(b) Sarkar, G.; Siddiqua, S. Eng. Geol. 2016, 202, 153. -
[19]
Ding, Y. Z.; Zhang, D. Y.; Zhang, X.; Chen, Y. T.; Wu, Z. B.; Wang, P. Y.; Xue, W.; Song, B. A.; Yang, S. Tetrahedron Lett. 2015, 56, 831. doi: 10.1016/j.tetlet.2014.12.113
-
[20]
Lu, S. Y.; Badsara, S. S.; Wu, Y. C.; Reddy, D. M.; Lee, R. C. Tetrahedron Lett. 2016, 57, 633. doi: 10.1016/j.tetlet.2015.12.060
-
[21]
Grzyb, J. B.; Batey, R. A. Tetrahedron Lett. 2003, 44, 7485. doi: 10.1016/j.tetlet.2003.08.026
-
[22]
Liebeskind, L. S.; Wu, W. T.; Zhang, Z. H. J. Am. Chem. Soc. 2011, 133, 14256. doi: 10.1021/ja2065158
-
[23]
Townsend, S. D.; Wu, X. Y.; Danishefsky, S. J. J. Am. Chem. Soc. 2012, 134, 10659. doi: 10.1021/ja303876e
-
[24]
Vogel, P.; Zambron, B. K.; Dubbaka, S. R.; Markovic, D. Org. Lett. 2013, 15, 2550. doi: 10.1021/ol401053y
-
[25]
Lipshutz, B. H.; Gabriel, C. M.; Keener, M.; Callou, F. Org. Lett. 2015, 17, 3968. doi: 10.1021/acs.orglett.5b01812
-
[26]
Thiemann, T.; Jasem, Y. A.; Soom, N. A. C. R. Chimie 2016, 1.
-
[27]
Kawahata, N. H.; Brookes, J.; Makara, G. M. Tetrahedron Lett. 2002, 43, 7221. doi: 10.1016/S0040-4039(02)01657-X
-
[28]
Hosmane, R. S.; Ujjinamatada, R. K. Tetrahedron Lett. 2005, 46, 6005. doi: 10.1016/j.tetlet.2005.07.024
-
[29]
An, D. K.; Jeon, A. R.; Kim, M. E.; Park, J. K.; Shin, W. K. Tetrahedron 2014, 70, 4420. doi: 10.1016/j.tet.2014.03.045
-
[30]
Mecinovic, J.; Lenstra, D. C.; Nguyen, D. T. Tetrahedron 2015, 71, 5547. doi: 10.1016/j.tet.2015.06.066
-
[31]
Wang, Q. W.; Yang, Y.; Fu, R. Z.; Ma, Y. S.; Yang, F.; Li, J. J.; Chai, W.; Yuan, R. X. Tetrahedron Lett. 2015, 56, 4527. doi: 10.1016/j.tetlet.2015.05.118
-
[32]
Du, G. F.; Guo, H.; Wang, Y.; Dai, B. U.; He, L. Tetrahedron 2015, 71, 3472. doi: 10.1016/j.tet.2015.03.071
-
[33]
Mizuno, N.; Yamaguchi, K.; Matsushita, M. Angew. Chem., Int. Ed. 2004, 43, 1576. doi: 10.1002/(ISSN)1521-3773
-
[34]
Varma, R. S.; Polshettimar, V. Chem. Eur. J. 2009, 15, 1582. doi: 10.1002/chem.v15:7
-
[35]
Kim, A. Y.; Bae, H. S.; Park, S. H; Park, S. K.; Park, R. H. Catal. Lett. 2011, 141, 685. doi: 10.1007/s10562-011-0561-y
-
[36]
Shimizu, K. Z.; Imaiida, N.; Sawabe, K.; Satsuma. A. Appl. Catal., A 2012, 421~422, 114. http://www.researchgate.net/publication/257373405_Hydration_of_nitriles_to_amides_in_water_by_SiO2-supported_Ag_catalysts_promoted_by_adsorbed_oxygen_atoms
-
[37]
Tyler, D. R.; Knapp, S. M. M.; Sherbow, T. J.; Yelle, R. B.; Juliette, J. Organometallics 2013, 32, 3744. doi: 10.1021/om400380j
-
[38]
Qu, J. P.; Tong, P.; Yang, D. W.; Li, Y.; Wang, B. W. Organometallics 2015, 34, 3571. doi: 10.1021/acs.organomet.5b00387
-
[39]
Jin, J. Y.; Etzion, R.; Santoso, B.; Yanaranop, P. Polymer 2016, 98, 244. doi: 10.1016/j.polymer.2016.06.041
-
[40]
Alper, H.; Larksarp, C. J. Org. Chem. 2000, 65, 2773. doi: 10.1021/jo991922r
-
[41]
Inoue, Y.; Tsukada, N.; Ohba, Y. J. Org. Chem. 2003, 687, 436. doi: 10.1016/j.jorganchem.2003.08.014
-
[42]
Li, Z.; Ding, R. B.; Lu, Z.; Xiao, S. X.; Ma, X. L. J. Mol. Catal. A: Chem. 2006, 250, 100. doi: 10.1016/j.molcata.2006.01.056
-
[43]
Crabtree, R. H.; Hull, J. F.; Hilton, S. T. Inorg. Chim. Acta 2010, 363, 1243. doi: 10.1016/j.ica.2009.08.022
-
[44]
(a) Giacomelli, G.; Luca, L. D.; Porcheddu, A. J. Org. Chem. 2002, 67, 6272;
(b) Li, Q.; Yan, L. Y.; Xia, D.; Shen, Y. C. Chin. J. Org. Chem. 2011, 31, 2034 (in Chinese).(李倩, 严罗一, 夏定, 申永存, 有机化学, 2011, 31, 2034.)
(c) Dong, D.; Yan, P.; Yu, M. J. Chem. Sci. 2016, 128, 951. -
[45]
Deng, Y. Q.; Yin, S. F.; Au, C. T. lnd. Eng. Chem. Res. 2012, 51, 9492. doi: 10.1021/ie3001277
-
[46]
Maronna, M. M.; Kruissink, E. C.; Tinge, J. T.; Agar, D. W.; Hoelderich, W. F. Ind. Eng. Chem. Res. 2016, 55, 1202. doi: 10.1021/acs.iecr.5b04240
-
[47]
Shin, Y. Y.; Jung, D. J.; Lee, S. G. Catal. 2013, 3, 525.
-
[48]
Mahajan, P. S.; Humne, V. T.; Tanpure, S. D.; Mhaske, S. B. Org. Lett. 2016, 18, 3450. doi: 10.1021/acs.orglett.6b01634
-
[49]
Zhang, F.-L.; Wang, Y.-F.; Lonca, G. H.; Zhu, X.; Chiba, S. An-gew. Chem., Int. Ed. 2014, 53, 4390. doi: 10.1002/anie.201400938
-
[50]
Zhu, X.; Wang, Y.-F.; Zhang, F.-L.; Chiba, S. Chem. Asian J. 2014, 9, 2458. doi: 10.1002/asia.201402421
-
[51]
Zhang, F. L.; Zhu, X.; Chiba, S. Org. Lett. 2015, 17, 3138. doi: 10.1021/acs.orglett.5b01458
-
[52]
Howell, A. R.; Chen, D. Tetrahedron Lett. 2015, 56, 3583. doi: 10.1016/j.tetlet.2015.02.133
-
[53]
Lüdtke, D. S.; Silva, L.; Affeldt, R. F. J. Org. Chem. 2016, 81, 5464. doi: 10.1021/acs.joc.6b00832
-
[54]
Babu, V. V. S.; Vasant, G-R.; Tantry, S. J. Tetrahedron Lett. 2005, 46, 4099. doi: 10.1016/j.tetlet.2005.04.007
-
[55]
Li, Z. F.; Lai, G. Q.; Chen, W. F.; Li, K. B.; Hu, Z. Q.; Wang, L. L. Organometallics 2011, 30, 2026. doi: 10.1021/om200080f
-
[56]
Liguori, A.; Siciliano, C.; Leggio, A.; Belsito, E. L.; Di, M. L.; Leotta, G. V.; Romio, E. Tetrahedron Lett. 2015, 56, 199. doi: 10.1016/j.tetlet.2014.11.067
-
[57]
Gooßen, L. J.; Rauhaus, J. E.; Deng, G. Angew, Chem, Int. Ed. 2005, 44, 4042. doi: 10.1002/(ISSN)1521-3773
-
[58]
Chang, S.; Kim, S. K.; Do, Y.; Lee, J.; Jung, D. Y.; Ahn, D.-S.; Lee, J. M. J. Am. Chem. Soc. 2006, 128, 12954. doi: 10.1021/ja0639315
-
[59]
Takai, K.; Kuninobu, Y.; Salprima, Y. S. Org. Lett. 2007, 9, 5609. doi: 10.1021/ol702564e
-
[60]
Beller, M.; Fang, X. J.; Jackstell, R. Angew, Chem., Int. Ed. 2013, 52, 14089. doi: 10.1002/anie.201308455
-
[61]
Stephenson, N. A.; Zhu, J.; Gellman, S. H.; Stahl, S. S. J. Am. Chem. Soc. 2009, 131, 10003. doi: 10.1021/ja8094262
-
[62]
Srinivas, M.; Hudwekar, A. D.; Venkateswalu, V.; Reddy, G. L. Tetrahedron Lett. 2015, 56, 4775. doi: 10.1016/j.tetlet.2015.06.052
-
[63]
Janousek, Z.; Collard, J.; Viehe, H. G. Angew. Chem., Int. Ed. Engl. 1972, 11, 917. doi: 10.1002/(ISSN)1521-3773
-
[64]
Jin, X. J.; Ka, Z. Y.; Mizuno, N. Chem. Lett. 2012, 41, 866. doi: 10.1246/cl.2012.866
-
[65]
Xu, S. X.; Liu, J. Q.; Hu, D. H.; Bi, X. H. Green Chem. 2015, 17, 184. doi: 10.1039/C4GC01328J
-
[66]
Huang, H.; Tang, L. N.; Xi, Y.; He, G. K.; Zhu, H. J. Tetrahedron Lett. 2016, 57, 1873. doi: 10.1016/j.tetlet.2016.03.052
-
[67]
Ohmura, R.; Takahata, M.; Togo, H. Tetrahedron Lett. 2010, 51, 4378. doi: 10.1016/j.tetlet.2010.06.051
-
[68]
Yamaguchi, K.; Kobayashi, H.; Oishi, T.; Mizuno, N. Angew. Chem. 2012, 124, 559. doi: 10.1002/ange.201107110
-
[69]
Wu, A.; Cao, L.; Ding, J.; Gao, M.; Wang, Z.; Li, J. Org. Lett. 2009, 11, 3810. doi: 10.1021/ol901250c
-
[70]
Peng, Y. Q.; Song, G. H. Org. Prep. Proced. Int. 2002, 34, 95. doi: 10.1080/00304940209355747
-
[71]
Ahmed, M.; Baburajan, P.; Sureshm, A. S. Tetrahedron Lett. 2015, 56, 4864. doi: 10.1016/j.tetlet.2015.06.054
-
[72]
Wang, Y.; Yamaguchi, K.; Mizuno, N. Angew. Chem., Int. Ed. 2012, 51, 7250. doi: 10.1002/anie.201203098
-
[73]
Wang, T.; Yuan, L.; Zhao, Z.; Shao, A.; Gao, M.; Huang, Y.; Xiong, F.; Zhang, H.; Zhao, J. Green Chem. 2015, 17, 2741. doi: 10.1039/C5GC00299K
-
[74]
Zhao, J.; Zhao, Z.; Wang, T.; Yuan, L.; Hu, X.; Xiong, F. Adv. Synth. Catal. 2015, 357, 2566. doi: 10.1002/adsc.201500310
-
[75]
Griffin, J. M.; Atherton, J. H.; Page, M. I.; Powles, N. T. J. Phys. Org. Chem. 2016, 29, 768. doi: 10.1002/poc.v29.12
-
[76]
Batra, S.; Dighe, S. U. Adv. Synth. Catal. 2016, 358, 500. doi: 10.1002/adsc.201500906
-
[1]
-

图 1 酰胺的化学结构和合成酰胺键的传统化学方法
Figure 1 Chemical structure of amides and the conventional chemical method for amide bond synthesis

图式3 (Fe-TEMPO) 催化氧化氨化机理
Scheme 3 A mechanism for (Fe-TEMPO)-catalysed tandem oxidative amidation

图式5 高温高压反应流程示意图
Scheme 5 Schematic flow setup for high-temperature and high-pressure reactions

图式16 硫桥双核铁复合物促进腈水解成酰胺
Scheme 16 Hydration of nitriles to amides by thiolate-bridged diiron complexes

图式18 钌催化肟重排反应机理
Scheme 18 Proposed mechanism for the Ru (Ⅱ) catalyzed rearrangement of oximes

图式26 酰胺和胺转换机理
Scheme 26 Mechanism for the formation of transamidated product from amide with aniline

图式29 甲基酮转变成一级酰胺的反应机理
Scheme 29 Mechanism for the transformation of methyl ketone to primary amide
-


 下载:
下载:






























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 822
- 文章访问数: 31088
- HTML全文浏览量: 9244
 下载:
下载:
