 图 图式1
“受阻”路易斯酸碱对可逆活化氢气的发现
Figure 图式1.
Discovery of reversible H2
activation by FLPs
图 图式1
“受阻”路易斯酸碱对可逆活化氢气的发现
Figure 图式1.
Discovery of reversible H2
activation by FLPs
引用本文:
王辉, 郑亿, 潘振涛, 傅鸿樑, 凌飞, 钟为慧. “受阻”路易斯酸碱对催化氢化反应的研究进展[J]. 有机化学,
2017, 37(2): 301-313.
doi:
10.6023/cjoc201607046

Citation: Wang Hui, Zheng Yi, Pan Zhentao, Fu Hongliang, Ling Fei, Zhong Weihui. Progress of Frustrated Lewis Pairs in Catalytic Hydrogenation[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 301-313. doi: 10.6023/cjoc201607046
Citation: Wang Hui, Zheng Yi, Pan Zhentao, Fu Hongliang, Ling Fei, Zhong Weihui. Progress of Frustrated Lewis Pairs in Catalytic Hydrogenation[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 301-313. doi: 10.6023/cjoc201607046
“受阻”路易斯酸碱对催化氢化反应的研究进展
摘要:
“受阻”路易斯酸碱对(FLPs)催化氢化反应是当前催化加氢领域的研究热点之一.该类反应具有环境友好、无重金属残留等特点,具有潜在的工业化应用前景.根据底物类别,对近几年”受阻”路易斯酸碱对在催化氢化领域的研究进展进行简要评述,并对FLPs不对称催化氢化进行简单介绍.
-
关键词:
- “受阻”路易斯酸碱对
- / 催化氢化
- / 不对称催化
- / 进展
English
Progress of Frustrated Lewis Pairs in Catalytic Hydrogenation
Abstract:
Frustrated Lewis pairs (FLPs) catalyzed hydrogenation reaction is one of the hotspots in the current hydrogenation field. This kind of reaction has the advantages of environment friendly, no metal residue, etc., and has a potential prospect for industrial application. According to the category of the substrate, a brief review of the recent progress in the field of the FLPs-catalyzed hydrogenation as well as the asymmetric hydrogenation is depicted.
-
Key words:
- frustrated Lewis pairs
- / catalytic hydrogenation
- / asymmetric catalysis
- / progress
-
催化氢化反应是指在催化剂作用下氢加成到有机化合物的不饱和基团上的反应.在过去的几十年中, 其催化剂往往采用以钌 (Ru)、铑 (Rh)、钯 (Pd)、铂 (Pt)、铱 (Ir)、锇 (Os) 等为核心的过渡金属.尽管过渡金属催化氢化反应硕果累累, 但逐渐暴露出价格昂贵, 重金属残留等问题.尤其是医药行业对食品药品中重金属标准的提高, 在一定程度上限制了贵金属催化剂在原料药及中间体合成中的应用.
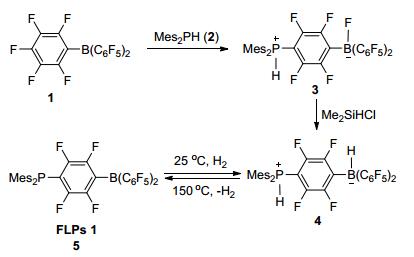
作为金属催化氢化的补充, 非金属参与的催化氢化反应由于缺少合适的催化剂而报道相对较少.直到2006年, Stephen等[1]在研究路易斯酸B (C6F5)3 (1) 与路易斯碱2反应时发现, 它们没有形成稳定的路易斯酸碱加合物, 而是发生亲核取代反应生成两性离子化合物3, 将3与二甲基氯硅烷反应得到空气以及湿度稳定的两性离子化合物4.化合物4加热到150 ℃时可以释放出H2生成化合物5, 而在25 ℃化合物5可以吸收H2重新生成4(Scheme 1), 过程伴随着明显的颜色变化 (图 1).随后, Stephan等提出“受阻”路易斯酸碱对 (Frustrated Lewis Pairs, FLPs) 的概念, 即当大位阻的路易斯酸与路易斯碱混合, 由于空间位阻大而不能形成经典加合物, 从而表现出一些特殊的反应活性.
图 图式1
“受阻”路易斯酸碱对可逆活化氢气的发现
Figure 图式1.
Discovery of reversible H2
activation by FLPs
 图 1
FLPs吸收及释放H2的颜色变化
Figure 1.
Color changed by FLPs absorb and release H2
图 1
FLPs吸收及释放H2的颜色变化
Figure 1.
Color changed by FLPs absorb and release H2
这个简单的概念一经提出, 迅速成为当下研究的热点, 受到越来越多的关注与重视, 也为无机化学领域开辟了新的研究方向.这种可逆活化H2方法的发现, 使FLP在催化氢化领域得到了前所未有的应用, 打开了FLPs催化剂在“非金属”催化氢化领域的大门.
随后, FLPs活化H2的研究层出不穷. Erker等[2]报道了乙基桥连的P/B体系分子内FLPs 2, 室温下在戊烷溶剂中与H2结合形成稳定的两性离子, 收率为60% (Eq. 1). Repo等[3]报道了N/B体系分子间FLPs 3, 与H2反应得到产物收率为95% (Eq. 2). Tamm等[4]报道了C/B体系分子间FLPs 4, 通过对H2异裂得到两性离子产物, 收率为82% (Eq. 3).除此之外, 通过改变Lewis碱性中心N/P/C上取代基以及硼试剂的酸碱性, 得到很多分子内或者分子间FLPs体系, 实现H2的活化[5~11].
经过10年的发展, FLPs在H2活化领域已经取得了许多突破性进展[12, 13].本文从底物官能团类型出发, 主要综述了近几年FLPs催化还原亚胺、烯烃、炔烃、羰基化合物、芳香族化合物等底物的研究与进展, 并对FLPs在不对称催化氢化中的应用进行了介绍, 以及对该理论未来的发展进行展望.
1 FLPs催化还原亚胺
亚胺的还原是制备饱和胺的最佳方法之一, 并已在制药和精细化工产业发挥巨大作用.由于过渡金属催化容易导致金属残留问题, 以FLP为代表的非金属催化还原亚胺的方法逐渐受到重视.
2007年, Stephen等[14]报道在甲苯溶剂中, 一定温度 (80~140 ℃) 及H2压力下 (101~505 kPa) 利用催化剂FLPs 1实现亚胺的还原, 得到57%~99%的收率 (Eq. 4).若底物为缺电子亚胺, 反应需要更高的压力及温度, 反应时间也更久.该反应为FLPs催化还原亚胺的首次报道, 拓展了FLPs的研究领域, 也预示着由NaBH4或LiAlH4等催化的还原反应中催化剂的用量可能由化学计量转变成催化量, 从而大大减少三废的排放.
经过研究发现, 亚胺的还原反应是由亚胺的质子化引发的, 可能机理:首先FLPs 1吸收H2生成两性离子化合物14, 接着14上质子转移到底物亚胺上形成中间体13, 最后与氢负离子再转移到底物上得到还原产物12, 并释放出催化剂 (Scheme 2).
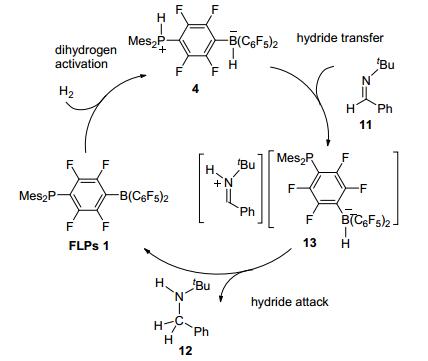 图 图式2
FLPs 1催化还原亚胺的可能机理
Figure 图式2.
Possible mechanism of imine catalytic reduction by FLPs 1
图 图式2
FLPs 1催化还原亚胺的可能机理
Figure 图式2.
Possible mechanism of imine catalytic reduction by FLPs 1
2008年, Stephen等[15]研究发现商品化的Lewis酸B (C6F5)3是一种高效的催化剂, 由于五氟苯强吸电子效应, 使得B (C6F5)3具有一个很强的酸性中心, 可以与亚胺原位生成FLP, 在氢气条件下实现亚胺的还原.他们以甲苯作溶剂, 利用1在120 ℃及H2(505 kPa) 氛围中反应41 h实现亚胺14的还原, 产物收率为94% (Eq. 5).如果在相同反应条件下, 加入额外5 mol%的P (C6H2-Me3)3, 反应时间缩短至8 h, 收率稍有提高 (Eq. 6).可能原因是P/B体系FLPs相对于N/B体系还原亚胺的活性更高, 因此加入P (C6H2Me3)3表现出促进反应的作用.
选择性还原肟亚胺的碳氮双键一直比较困难, 而采用均相的过渡金属催化肟亚胺类衍生物往往直接得到伯胺. 2014年, Oestreich等[16]报道了利用B (C6F5)3选择性还原肟亚胺而不断裂N—O键.以甲苯作溶剂, 10.1 MPa H2压力下, 5 mol%的1, 实现肟亚胺的还原, 收率为66%~99% (Eq. 7).
2016年, 杜海峰等[17]报道同样利用B (C6F5)3在H2(202 kPa) 下直接还原3位取代的1, 4-苯并恶嗪18, 收率为93%~99% (Eq. 8).该反应的突出优点是催化剂用量相对较少.
亚胺的还原往往采用H2作为氢源, 存在一定的危险性. 2011年, Stephan等[18]报道以二异丙胺为氢源, 1为催化剂, 实现亚胺的催化氢化, 收率为37%~98% (Eq. 9).随后, Oestreich等[19]以1, 5-二甲基-1, 4-环己二烯为氢源, 125 ℃甲苯作溶剂, 同样在B (C6F5)3的催化下还原亚胺, 收率最高可达99% (Eq. 10).该类反应无需高压, 避免了H2使用存在的高压危险, 为寻找开发新型氢源提供宝贵的参考.
通过对B (C6F5)3结构进行适当改造, 可以得到各种新型Lewis酸. 2009年, Berke等[20]报道利用1, 8-二五氟苯基硼基萘在d-甲苯溶剂中120 ℃下, H2(1.52 MPa) 氛围中实现亚胺的催化还原, 产物收率最高可达99% (Eq. 11).需要指出的是, 该反应的限速步在H2的活化, 尽管该Lewis酸具有两个酸性B中心, 但两个B中心共同活化H2的能垒要比单一B中心的高.因此, 该Lewis酸可能由一个B中心来活化H2, 而另一个B中心则表现出一定的吸电子效应.
2014年, Ashley等[21]报道利用1在甲苯中80 ℃还原亚胺, 当H2压力从1.0 MPa提高到3.0 MPa时, 产物收率从7%提高到了99%, 表明压力对反应有很大影响.随后他们设计合成了B (C6Cl5)(C6F5)2 (24), B (C6Cl5)2-(C6F5) (25)(图 2), 与1分别在[D8]THF中还原亚胺, 其中24的催化收率最高为99%.通过研究发现, 产生不同催化效果的最主要原因在于三种Lewis酸的酸性大小不同. Lewis酸24的B中心酸性介于1和25之间, 可能正好由于这种适中程度的酸性使得24表现出较好的催化效果.
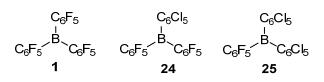 图 2
Lewis酸1、24和25
Figure 2.
Lewis acid 1, 24 and 25
图 2
Lewis酸1、24和25
Figure 2.
Lewis acid 1, 24 and 25
除了B (C6F5)3直接还原亚胺之外, 还有很多FLPs催化的报道. 2008年, Erker等[22]报道FLPs 2对亚胺进行还原, 在室温及253 kPa下得到70%的收率 (Eq. 12).该反应之所以能在温和的条件下进行, 得益于FLPs 2空间位阻和电子因素之间微妙的平衡关系. 2014年我们研究小组[23]以环蕃为骨架设计的FLPs 5在温和条件下还原亚胺得到93%的收率 (Eq. 13);而且考察了催化剂的回收套用问题, 成功实现三次回收套用且不影响反应收率.从结构可以看出, FLPs 5与FLPs 2的酸碱性中心B/P是通过两个碳原子相连, 都具有一个适宜空间位阻的“活性口袋”, 从而表现出较高的催化活性.
2011年, 为了考察空间位阻以及路易斯碱性对FLPs催化剂活性的影响, Repo等[24]设计了FLPs 6~8(图 3), 用于亚胺的还原, 其中FLPs 8表现出较佳的催化活性.可能是由于FLPs 8的胺部分具有适宜的空间位阻和碱性, 使活性中心N/B的受阻程度适中, 从而表现出较大的活性.
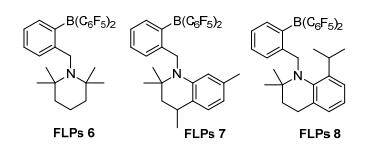 图 3
N/B催化剂FLPs 6~FLPs 8
Figure 3.
N/B catalysts FLPs 6~FLPs 8
图 3
N/B催化剂FLPs 6~FLPs 8
Figure 3.
N/B catalysts FLPs 6~FLPs 8
2015年, Stephan等[25]报道了C/B体系催化剂FLPs 9催化亚胺、烯胺的反应, 所得产物具有较高的收率 (Eq. 14).该反应催化剂对官能团具有一定耐受性, 底物适用范围相对较广.
在FLPs催化剂的组成中, 路易斯酸由于强吸电子与大位阻的要求, 其种类受到一定限制, 主要以多卤代芳基硼为主.最近, Krempner等[26]基于拓展路易斯酸种类的考虑, 发现提高路易斯碱的碱性, 可以适当降低路易斯酸的酸性.他们利用商业化的超级有机碱28, 与酸性较弱的路易斯酸29结合形成FLPs 10, 将它与亚胺共同暴露在H2氛围中, 发现亚胺被还原成饱和胺, 收率高达99% (Eq. 15).该反应拓展了路易斯酸碱的种类, 在温和条件下实现亚胺的高效还原.
通过上述报道可以发现, FLPs催化剂主要有P/B体系、N/B体系、C/B体系等.一般来说催化剂活性P/B>N/B>C/B.通过调控催化剂活性中心的空间位阻和酸碱性, 可以增大FLPs催化剂的活性.
2 FLPs催化还原烯烃及炔烃
2008年, Erker等[22]报道在室温及H2 (253 kPa) 条件下, 利用FLPs 2实现32的还原, 产物收率为70%~99% (Eq. 16). 2011年, 在FLPs 2催化剂基础之上, Erker等[27]将烯胺32与Piers硼[28]34结合形成分子内N/B FLPs 11, 在温和条件下异裂H2实现自身底物的还原 (Scheme 5).与FLPs 2相比, FLPs 11的催化效果相对较差, 可能是由于FLPs 11胺部位的碱性较弱和空间位阻较小所致.
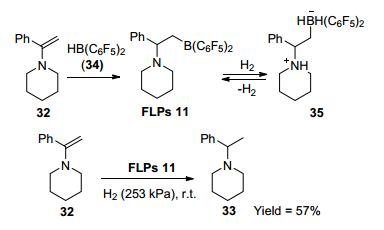 图 图式3
FLPs 11的合成及催化还原烯胺
Figure 图式3.
Synthesis of FLPs 11 and
catalytic reduction of enamine
图 图式3
FLPs 11的合成及催化还原烯胺
Figure 图式3.
Synthesis of FLPs 11 and
catalytic reduction of enamine
2008年, Erker等[29]利用1, 8-二二苯基膦萘 (36) 与1混合, 催化还原烯醇硅醚, 收率为85%~93% (Eq. 17).该反应较早地报道了FLPs对双键的催化还原, 为FLPs还原双键的研究奠定了基础. 2012年, Paradies等[30]报道了碱性稍弱的 (C6F5) Ph2P (37) 与 (C6F5) Ph2P (38), 与1混合用于还原烯烃.不同的是, 37/1的催化收率可达99%而38/1不能催化反应.从Lewis碱结构分析, 可能是38的取代基吸电子能力太强, 使38的碱性降低至不能实现催化还原的范围 (图 4).
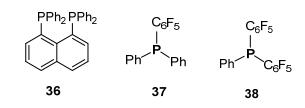 图 4
Lewis碱42~44
Figure 4.
Lewis
bases 42~44
图 4
Lewis碱42~44
Figure 4.
Lewis
bases 42~44
2010年, Soós等[31]提出了“排阻设计”(Size exclusion) 的概念, 他们认为增加对路易斯酸性中心的屏蔽作用, 可以改变构成的FLPs对H2的活化能力, 可能会提高FLPs的化学选择性.因此他们设计合成了新型Lewis酸B (C6F5)2(Mes), 与DABCO混合催化还原烯烃, 产物收率为92% (Eq. 18).
2012年, Alcarazo等[32]将1与DABCO混合, 80 ℃甲苯中选择性还原缺电子烯烃化合物, 产物收率为64%~94% (Eq. 19).他们报道了系列缺电子烯烃的还原, 但未见硝基烯烃的还原. 2013年, Paradies等[33]根据1设计合成了酸性稍弱的B (C6H3F2)3, 与7组成FLPs 12, 在室温下迅速还原硝基烯烃45, 产物收率可达99% (Eq. 20).该反应通过调节Lewis酸的酸性, 实现对硝基的耐受性, 预示着在官能团耐受性研究中可以考虑采用调节Lewis酸性的方法.
2013年, Stephan等[34]报道了1在乙醚溶剂中实现烯烃的氢化, 收率高达99% (Scheme 4).该反应利用溶剂作为路易斯碱, 参与FLPs催化反应, 大大提高了路易斯酸的反应活性.
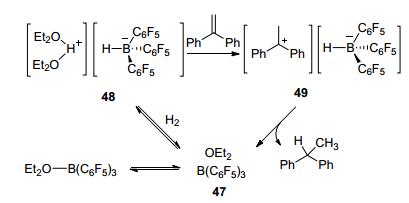 图 图式4
B (C6F5)3与Et2O组合催化还原烯烃
Figure 图式4.
Catalytic
reduction of olefins by B (C6F5)3 and Et2O
图 图式4
B (C6F5)3与Et2O组合催化还原烯烃
Figure 图式4.
Catalytic
reduction of olefins by B (C6F5)3 and Et2O
2014年, Erker等[35]基于具有较高催化活性的FLPs 2, 设计合成了FLPs 13和FLPs 14(图 5).其中FLPs 14活性较高, 可以催化还原烯烃等底物; 而FLPs 13的表现却比较惨淡.对比发现, 可能是FLPs 2和FLPs 14的“活性口袋”大小适中, 从而表现出较高的活性.
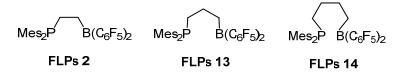 图 5
FLPs 2/13/14的结构
Figure 5.
Structures
of FLPs 2/13/14
图 5
FLPs 2/13/14的结构
Figure 5.
Structures
of FLPs 2/13/14
2013年, Repo等[36]报道了催化剂FLPs 15, 实现炔烃的顺式加氢.随后他们又拓展了20多个炔烃底物, 都得到了类似的结果, 表明FLPs催化剂在部分炔烃的催化还原中可作为Lindlar试剂的替代物.可能反应机理首先是50异裂H2得到51, 51受热后脱去C6F5H结构形成FLPs 15, 接着与底物炔烃加成得到52, 再吸收H2形成两性离子53, 随后发生H转移生成顺式加成产物并游离出催化剂 (Scheme 5).
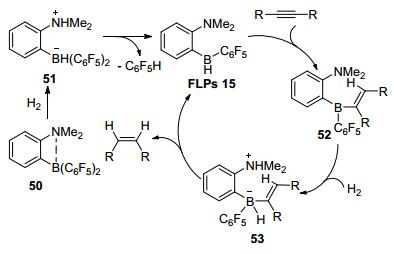 图 图式5
FLPs 15催化炔烃顺式加成可能机理
Figure 图式5.
Proposed
mechanism of the Z-selective reduction of alkynes by FLPs 15
图 图式5
FLPs 15催化炔烃顺式加成可能机理
Figure 图式5.
Proposed
mechanism of the Z-selective reduction of alkynes by FLPs 15
2016年, Taoufik等[37]报道了有机硅负载的催化剂FLPs 16和FLPs 17(图 6), 将它们暴露在H2氛围中可以很好地异裂H2.与FLPs 16不同的是, FLPs 17可实现炔烃的顺式加成, 得到较高的收率.可能反应机理是:有机硅负载的FLPs 17先与底物加成, 并且硼上的氢转移到不饱和碳上形成54, 接着将H2异裂形成两性离子55, 由于催化剂较大的空间位阻, 底物上的取代基被挤到了同侧, 随后路易斯碱上的氢转移到底物上得到顺式加成产物并且释放出FLPs 17(Scheme 6).
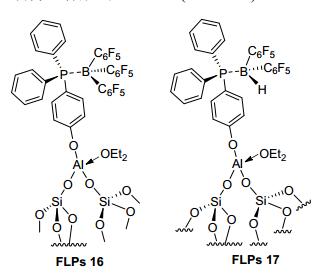 图 6
有机硅负载的催化剂FLPs 16/17
Figure 6.
Organic silicon supported
catalyst FLPs 16/17
图 6
有机硅负载的催化剂FLPs 16/17
Figure 6.
Organic silicon supported
catalyst FLPs 16/17
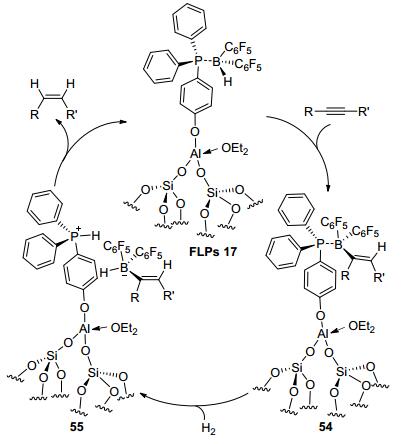 图 图式6
FLPs 17催化炔烃顺式加成可能机理
Figure 图式6.
Proposed
mechanism of the Z-selective reduction of
alkynes by FLPs 17
图 图式6
FLPs 17催化炔烃顺式加成可能机理
Figure 图式6.
Proposed
mechanism of the Z-selective reduction of
alkynes by FLPs 17
FLPs对烯烃及炔烃的还原效果直接与催化剂的结构相关.适当的Lewis酸碱性和适中的”活性口袋”有利于提高FLPs催化剂的活性; 通过调节多卤代硼的酸性强弱, 有可能提高催化剂的官能团耐受性.
3 FLPs催化还原羰基化合物
由于羰基易与硼试剂结合, 在一定程度上限制了FLPs催化羰基化合物的研究. 2012年, Erker等[38]报道了催化剂FLPs 18 (Scheme 7), 与α, β-不饱和羰基化合物共同暴露在H2氛围中, 发现只有双键被还原, 收率为74%~91%.同年, Paradies等[39]报道具有良好刚性和较大空间位阻的FLPs 19与FLPs 20(Scheme 7), 在HSiPh2Me与H2的环境中, 将α, β-不饱和羰基化合物的羰基和双键还原, 产物收率为58%~90%.其机理是首先HSiPh2Me与1催化56进行1, 4加成得到58, 接着FLPs 19/20在H2氛围中还原58得到产物59.
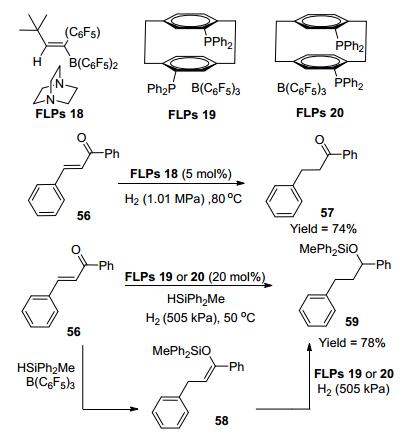 图 图式7
新型催化剂FLPs 18~20
Figure 图式7.
New catalysts FLPs 18~20
图 图式7
新型催化剂FLPs 18~20
Figure 图式7.
New catalysts FLPs 18~20
2014年, Stephan等[40]报道利用B (C6F5)3在甲苯中110 ℃和H2(404 kPa) 条件下, 将酮还原得到硼酯61, 收率为85%~99% (Eq. 21).随后, 他们[41]将溶剂替换成乙醚, 实现脂肪酮与芳香酮的还原, 产物收率为53%~99% (Eq. 22).将甲苯替换成醚类溶剂, 只需催化量的1就可将酮直接还原成醇.从反应机理可以发现, 醚类溶剂可以作为弱的Lewis碱参与催化反应.同样的结论再一次被Ashley等证明, 他们[42]以1, 4-二氧六烷作溶剂, 在较温和的条件下成功地将醛酮化合物还原, 产物收率为60%~99% (Eq. 23).然而在2015年, Stephan等[43]报道以甲苯作溶剂, 加入环糊精 (α-CD) 或者分子筛 (4 MS), 在60 ℃及H2(6.06 MPa) 下, 将羰基化合物还原成醇, 且得到较高的收率 (Eq. 24).该反应中B (C6F5)3与α-CD或4 MS可以组成FLPs催化剂, 从而实现催化反应.
2015年, Soós等[44]根据“排阻设计”(Size exclusion) 的概念, 设计合成了新型路易斯酸63(图 7), 以工业级THF作溶剂, 实现醛及酮的催化氢化, 催化剂表现出很好的耐湿性.该反应首次报道了FLPs在工业级溶剂中进行催化反应, 预示着适当减弱Lewis酸性可能会提高FLPs催化剂的耐湿性.
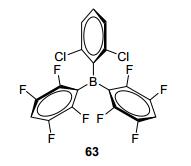 图 7
新型路易斯酸 (C6HF4)2B (C6H3Cl2)
Figure 7.
New Lewis acid (C6HF4)2B (C6H3Cl2)
图 7
新型路易斯酸 (C6HF4)2B (C6H3Cl2)
Figure 7.
New Lewis acid (C6HF4)2B (C6H3Cl2)
总的来说, FLPs对羰基化合物还原的报道相对较少.在醚类溶剂中利用B (C6F5)3可以实现羰基化合物的催化还原; 而减弱多卤代硼试剂的酸性有可能增强其对湿度的稳定性.
4 FLPs催化还原芳香族化合物
以往报道的FLPs催化剂主要应用于烯烃、炔烃、亚胺等的还原, 鲜有报道对芳香族化合物的还原. 2010年, Stephan等[45]发现在氢气氛围中, 将B (C6F5)3和N杂芳烃化合物接触, 可以选择性地将吡啶部分还原生成四氢喹啉, 收率为74%~88% (Eq. 25).该反应的缺点是必须在喹啉的2号或8号位引入取代基, 增大底物空间位阻, 而空间位阻较小的喹啉不适用.该反应首次报道了B (C6F5)3直接应用于N杂芳烃化合物的催化加氢反应, 拓展了底物范围, 为FLPs的研究开辟了新的方向.
可能的反应机理:首先发生H2的活化, 底物64和1与H2作用生成两性离子66, 接着发生H2的1, 4加成生成67或者1, 2加成生成68并游离出1, 接下来67或68异构化生成69, 然后又与1共同活化H2得到两性离子70, 最后再次发生H2加成得到还原产物65并释放出1(Scheme 8).
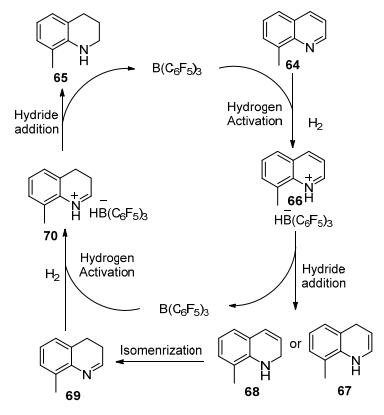 图 图式8
B (C6F5)3催化还原N杂芳烃化合物可能机理
Figure 图式8.
Proposed
mechanism of catalytic reduction of N heterocyclic aromatic compounds by B (C6F5)3
图 图式8
B (C6F5)3催化还原N杂芳烃化合物可能机理
Figure 图式8.
Proposed
mechanism of catalytic reduction of N heterocyclic aromatic compounds by B (C6F5)3
2012年, Soós等[46]根据“排阻设计”的概念报道了新型Lewis酸MesB (C6F4H)2(71), 在105 ℃下实现喹啉及衍生物的还原, 得到79%~99%的收率 (Eq. 26).相对于1而言, 71具有更大的空间位阻和更弱的Lewis酸性, 可能正是这些性质使71可以很好的还原空间位阻较小的喹啉及衍生物.
随后, 他们[47]将71应用到四氢喹啉生物碱枯杷仞合成中, 将中间体76还原得到77, 收率为91% (Scheme 9).这是FLPs催化剂首次用于生物碱的合成, 为FLPs催化剂的工业化应用奠定了基础.
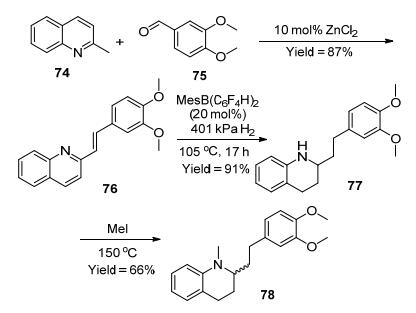 图 图式9
MesB (C6F4H)2应用于枯杷仞合成
Figure 图式9.
Application
of MesB (C6F4H)2in the synthesis of cuspareine
图 图式9
MesB (C6F4H)2应用于枯杷仞合成
Figure 图式9.
Application
of MesB (C6F4H)2in the synthesis of cuspareine
2012年, Stephan等[48]利用催化剂FLPs 21, 首次实现了蒽类及其衍生物的还原, 收率最高可以达到97%, 进一步拓展了FLPs催化的底物范围 (Eq. 27).值得注意的是, 该体系只有含蒽结构的多环芳烃可以被还原, 而萘、芘、菲、醌不能被还原, 可能是由于形成的碳正离子不够稳定, 从而不适用于该催化体系.
FLPs催化多环芳烃反应可能的机理与催化亚胺反应机理类似, 首先是FLPs 21吸收H2, 形成两性离子化合物81, 接着与多环芳烃底物反应, 质子转移到底物生成碳正离子中间体83, 然后83接收氢负离子得到还原产物, 并释放出FLPs催化剂 (Scheme 10).
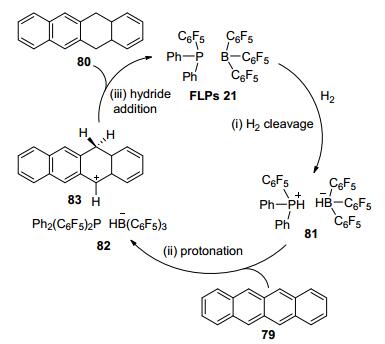 图 图式10
FLPs 21催化还原多环芳烃可能机理
Figure 图式10.
Proposed mechanism of catalytic
reduction poly-cyclic aromatic hydrocarbons by FLPs 21
图 图式10
FLPs 21催化还原多环芳烃可能机理
Figure 图式10.
Proposed mechanism of catalytic
reduction poly-cyclic aromatic hydrocarbons by FLPs 21
2012年, Stephan等[49]报道将1与芳胺84共同暴露在H2氛围中, 室温下发生H2的异裂得到两性离子化合物85.然而, 当温度升至110 ℃时, 发现可将苯环还原成环己烷, 收率良好为61%~93% (Scheme 11). 2015年, Stephan等[50]再次证实了这一现象.不同的是, 在还原对甲氧基苯胺时发现 (Eq. 28), 对位甲氧基可以被脱去, 而邻位甲氧基则不易被脱去.从反应机理 (Scheme 12) 可以发现, 对位甲氧基苯胺还原可以得到中间体92, 而邻位取代苯胺则生成中间体94, 可能是由于92的结构相对更加稳定, 从而表现出对位与邻位的差异.
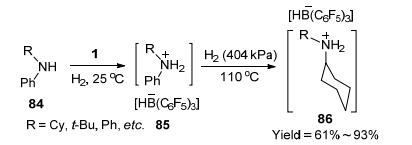 图 图式11
B (C6F5)3催化还原苯环
Figure 图式11.
B (C6F5)3 catalytic
reduction of benzene
图 图式11
B (C6F5)3催化还原苯环
Figure 图式11.
B (C6F5)3 catalytic
reduction of benzene
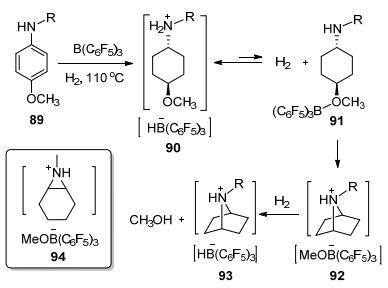 图 图式12
B (C6F5)3催化还原苯环可能机理
Figure 图式12.
Proposed
mechanism of catalytic reduction benzene by B (C6F5)3
图 图式12
B (C6F5)3催化还原苯环可能机理
Figure 图式12.
Proposed
mechanism of catalytic reduction benzene by B (C6F5)3
2013年, Stephan等[51]利用1在甲苯溶剂中, 115 ℃下与H2反应, 将吡啶衍生物还原得到两性离子化合物, 收率为54%~92% (Eq. 29).随后, 杜海峰等[52]将烯烃与Peris硼原位生成新型Lewis酸, 成功实现吡啶类衍生物的还原, 收率为80%~99% (Eq. 30).与Stephan报道使用化学计量的B (C6F5)3不同的是, 杜海峰报道只需催化量的1便可得到较高的收率.
在FLPs还原芳香族化合物中, 使用Lewis酸直接进行催化反应的报道较多.使用化学计量的B (C6F5)3还原N杂芳烃容易得到两性离子化合物, 而通过调节B (C6F5)3的酸性和空间位阻得到的新型Lewis酸可以在催化量的条件下直接还原N杂芳烃得到中性还原产物.
5 FLPs应用于不对称催化加氢反应
除了上述加氢反应, FLPs在不对称催化氢化领域也表现出独特的活性[53]. 2008年, Klankermayer等[54]报道了手性硼试剂98, 在甲苯中65 ℃下不对称还原亚胺, 转化率高达99%.尽管产物ee值仅13% (Eq. 31), 但为FLPs在不对称催化氢化领域的研究开辟了道路.
随后, 他们[55]以樟脑为骨架制备手性Lewis酸103a与103b, 由于它们不易分离, 通过直接加入路易斯碱与H2分离得到了104a和104b, 在温和的条件下实现亚胺的不对称催化还原反应, 产物收率为37%~99%, ee值为20%~83%(Scheme 13).由此表明, 适当骨架的手性FLPs催化剂有助于提高催化氢化反应的对映选择性.
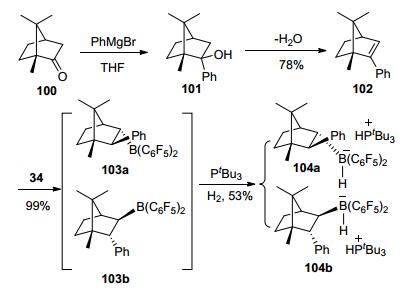 图 图式13
103a/103b的合成及H2活化
Figure 图式13.
Synthesis
of 103a/103b and activation of H2
图 图式13
103a/103b的合成及H2活化
Figure 图式13.
Synthesis
of 103a/103b and activation of H2
除了分子间P/B手性FLPs, Klankermayer等[56]又报道相同骨架的分子内手性P/B催化剂FLPs 22, 将其应用于亚胺的不对称催化加氢 (Eq. 32), 产物收率为21%~95%, ee值为70%~76%.该催化剂的突出优点是对空气稳定, 用量少且可回收使用四次. 2012年, 他们[57]报道了新型手性催化剂FLPs 23(图 8), 以Me2PhSiH为氢源实现亚胺的不对称还原, 得到90%的收率和84%的ee值 (Eq. 33).
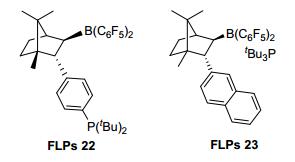 图 8
手性催化剂FLPs 22/23
Figure 8.
Chiral catalyst FLPs 22/23
图 8
手性催化剂FLPs 22/23
Figure 8.
Chiral catalyst FLPs 22/23
2011年, Repo等[18a]合成了分子内手性N/B催化剂FLPs 24, 首次实现了N杂环化合物109的不对称氢化, 拓宽了手性FLPs催化剂的底物范围, 反应转化率高达100% (Eq. 34), 但ee值仅37%, 且反应时间相对较长.反应ee值较低, 可能是由于催化剂手性中心距离较远或者空间位阻较小导致.
2015年, Repo等[58]报道合成手性连萘N/P体系催化剂FLPs 25, 在室温下甲基叔丁基醚中与H2(202 kPa) 作用实现亚胺的不对称还原, 产物收率为34%~92%, ee值为32%~83% (Eq. 35).随后他们又将该催化剂应用于烯胺的不对称还原, 产物的收率和ee值最高可达99%.该反应首次报道了在FLPs催化下, 实现烯胺化合物的不对称还原, 且反应条件温和, 催化剂合成步骤简单.
2013年, Oestreich等[59]报道了手性环硼路易斯酸113(图 9), 以Me2PhSiH为氢源实现亚胺的不对称还原, 得到54%的收率和62%的ee值 (Eq. 36). 2016年, 他们[60]根据113设计合成了新型Lewis酸114(图 9), 以PhSiH3为氢源, 在室温下实现羰基的手性还原, 得到最高87%的收率和99%的ee值 (Eq. 37).该反应无需额外加入Lewis碱便可实现还原, 为研究开发新型FLPs催化剂提供参考.
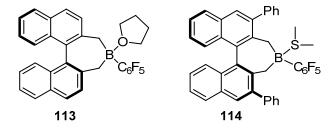 图 9
手性催化剂FLPs 113/114
Figure 9.
Chiral catalyst FLPs 113/114
图 9
手性催化剂FLPs 113/114
Figure 9.
Chiral catalyst FLPs 113/114
2013年, 杜海峰等[61]合成了手性联萘骨架二烯化合物, 与34加成得到118, 在H2环境中实现亚胺的不对称还原, 产物收率为63%~99%, ee值为74%~89% (Eq. 38).该反应催化剂原位生成, 无需分离提纯, 操作简便, 可直接投入反应, 快速而高效地筛选手性烯烃“配体”, 大大提高反应活性, 为FLPs催化剂的合成及应用于不对称催化氢化提供新的思路.
2014年, 杜海峰等[62]报道合成了FLPs 26, 将其应用于烯醇硅醚的还原, 得到89%~99%的收率和42%~98%的ee值 (Eq. 39). 2015年, 他们[63]又设计了手性催化剂FLPs 27, 在H2氛围实现不对称催化还原, 产物收率为82%~98%, ee值为87%~99% (Eq. 40).该催化剂适宜的空间结构和适中的路易斯酸性使其在不对称还原双键反应中表现出超高的活性 (图 10).
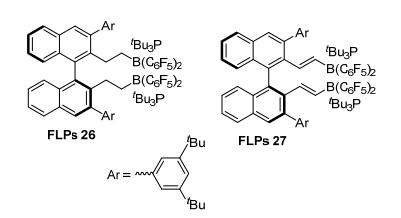 图 10
手性催化剂FLPs 26/27
Figure 10.
Chiral catalyst FLPs 26/27
图 10
手性催化剂FLPs 26/27
Figure 10.
Chiral catalyst FLPs 26/27
2016年, 杜海峰等[64]报道了催化剂FLPs 28, 以PhMe2SiH为氢源, 对1, 2-二羰基化合物实现不对称还原, 产物收率为52%~98%, ee值为86%~99% (Eq. 41).值得一提的是, 增加氢源的量, 并没有得到二醇还原副产物, 表现出较好的化学选择性.
由于FLPs催化剂对空间位阻要求较高, 在不对称还原反应中, 催化剂骨架的选择尤为重要.目前研究较多的主要有联萘、樟脑等骨架, 设计具有较大刚性的骨架对手性FLPs催化剂的合成具有重要意义.基于上述文献, 我们可以预测:二茂铁骨架具有平面及中心手性、足够的结构刚性、合适的立体位阻等诸多优点, 如果将其作为核心骨架引入FLPs催化剂中, 有可能提高反应的活性和对映选择性, 从而丰富手性FLPs催化剂的类型.
6 其它FLPs催化氢化反应
除了上述FLPs催化剂之外, 还有其它FLPs催化剂的报道, 拓展了FLPs的应用范围.
2009年, Ashley和O'Hare等[65]将FLPs 3与CO2/H2混合, 在160 ℃下反应6 d得到甲醇, 收率为17%~25% (Scheme 14).该反应首次报道了CO2在FLPs作用下转化成甲醇, 尽管收率不高, 但拓展了FLPs领域的新应用, 为FLPs在CO2还原反应的研究奠定了基础.
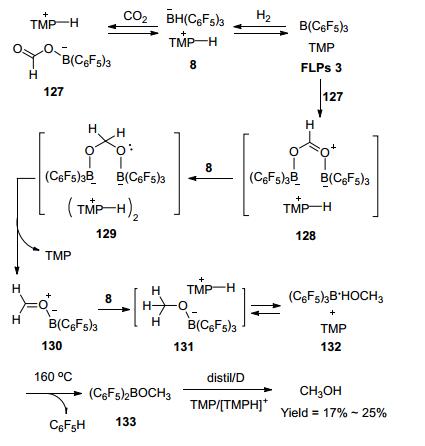 图 图式14
FLPs 3还原CO2成甲醇
Figure 图式14.
Reduction of CO2 to methanol by FLPs 3
图 图式14
FLPs 3还原CO2成甲醇
Figure 图式14.
Reduction of CO2 to methanol by FLPs 3
典型的FLPs路易斯酸, 例如多卤代硼, 由于其自身的缺电子性而具有特殊的反应活性. 2013年Stephan等[66, 67]报道了电子饱和的P[V]中心的新型路易斯酸.他们以134为原料和XeF2反应得到P[V] 135, 再与[Et3Si+]反应脱去一分子氟形成P[V]离子, 最后与[B (C6F5)4-]组成电子饱和的新型FLPs 29(Scheme 15).随后, 他们以Et3SiH为氢源, 利用该催化剂实现氟代化合物脱氟加氢反应 (Eq. 42).该反应打破常规思维, 创造性地设计了新型P[V]路易斯酸, 丰富了路易斯酸的种类, 为开发新型FLPs路易斯酸提供有益的启示.
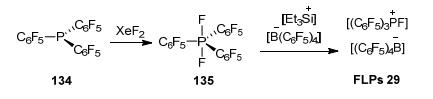 图 图式15
FLPs 29的合成
Figure 图式15.
Synthesis
of FLPs 29
图 图式15
FLPs 29的合成
Figure 图式15.
Synthesis
of FLPs 29
可能反应机理:首先FLPs 29与氟代底物反应, 夺取一分子氟生成135, 底物失去一分子氟后与硼负离子结合生成两性离子138, 接着与Et3SiH反应生成还原产物137和两性离子139, 最后139夺取物质135中的一分子氟形成Et3SiF和FLPs 29(Scheme 16).
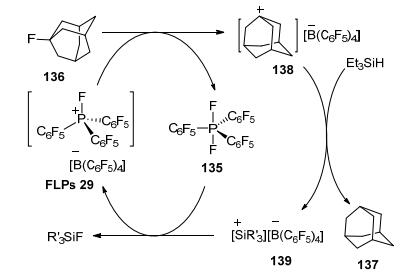 图 图式16
FLPs 29催化脱氟加氢反应的可能机理
Figure 图式16.
Proposed
mechanism of defluorination reaction catalyzed by FLPs 29
图 图式16
FLPs 29催化脱氟加氢反应的可能机理
Figure 图式16.
Proposed
mechanism of defluorination reaction catalyzed by FLPs 29
2016年, Oestreich等[68]报道在无需溶剂100 ℃条件下, 利用1和Et3SiH实现硝基化合物的还原, 得到43%~92%的收率 (Eq. 43).该反应突破常规, 实现硝基的还原, 拓展了FLPs底物范围, 不足之处是硅氢用量较大, 且官能团耐受性一般.
7 总结与展望
本文主要根据底物类别, 综述了近年来FLPs在催化氢化领域的研究进展, 并对不对称催化氢化领域进行了介绍.总的来说, FLPs催化剂的活性与自身空间位阻和酸碱性有很大的关系.适当的空间位阻和酸碱性可以使催化剂表现出较高活性; 而P/B体系FLPs相对于N/B体系活性更高; 通过调节Lewis酸的强弱, 有可能提高催化剂的官能团耐受性、耐湿性和底物适用性.在不对称还原中, 选择具有合适刚性的骨架有可能提高催化剂活性.
FLPs催化反应的主要特点是环境友好, 无重金属残留, 符合绿色化学的要求, 具有潜在的工业化应用前景.相比于过渡金属催化, FLP化学正处于起步阶段, 寻求制备简便、稳定性好、化学选择性高的手性FLPs催化剂可能是未来研究的热点.相信在不久的将来, FLP会在材料科学、有机催化、不对称合成等诸多领域发挥独特作用.因此, 在这个年轻领域的研究过程中, 需要不断突破常规思维, 积极探索, 提出和发展FLP领域的一些新方法和新概念, 为FLP领域的科学研究走向工业应用提供理论基础.
-
-
[1]
Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124. doi: 10.1126/science.1134230
-
[2]
Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Fröhlich, R.; Grimme, S.; Stephan, D.W. Chem. Commun. 2007, 5072. https://www.ncbi.nlm.nih.gov/pubmed/18049757
-
[3]
Sumerin, V.; Schulz, F.; Nieger, M.; Leskelä, M.; Repo, T.; Rieger, B. Angew. Chem. Int. Ed. 2008, 47, 6001. doi: 10.1002/anie.200800935
-
[4]
Holschumacher, D.; Bannenberg, T.; Hrib, C. G.; Jones, P. G.; Tamm, M. Angew. Chem. Int. Ed. 2008, 47, 7428. doi: 10.1002/anie.v47:39
-
[5]
Lu, Z.-P.; Cheng, Z.-H.; Chen, Z.-X.; Weng, L.-H.; Li, Z.-H.; Wang, H.-D. Angew. Chem., Int. Ed. 2011, 50, 12227. doi: 10.1002/anie.v50.51
-
[6]
Zaher, H.; Ashley, A. E.; Irwin, M.; Thompson, A. L.; Gutmann, M. J.; Krämer, T.; O'Hare, D. Chem. Commun., 2013, 49, 9755. doi: 10.1039/c3cc45889j
-
[7]
Caputo, C. B.; Zhu, K.-L.; Vukotic, V. N.; Loeb, S. J.; Stephan, D. W. Angew. Chem. Int. Ed. 2013, 52, 960. doi: 10.1002/anie.201207783
-
[8]
Chernichenko, K.; Kótai, B.; Pápai, I.; Zhivonitko, V.; Nieger, M.; Leskelä, M.; Repo; T. Angew. Chem., Int. Ed. 2015, 54, 1749. doi: 10.1002/anie.201410141
-
[9]
Samigullin, K.; Georg, I.; Bolte, M.; Lerner, H. W.; Wagner, M. Chem. Eur. J. 2016, 22, 3478. doi: 10.1002/chem.v22.10
-
[10]
Zheng, J.-H.; Lin, Y.-J.; Wang, H.-D. Dalton Trans. 2016, 45, 6088. doi: 10.1039/C5DT03815D
-
[11]
Mo, Z.-B.; Rit, A.; Campos, J.; Kolychev, E. L.; Aldridge, S. J. Am. Chem. Soc. 2016, 138, 3306. doi: 10.1021/jacs.6b01170
-
[12]
王平安, 孙晓莉, 高鹏, 有机化学, 2011, 31, 1369. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340408.shtmlWang, P.-A.; Sun, X.-L.; Gao, P. Chin. J. Org. Chem. 2011, 31, 1369 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340408.shtml
-
[13]
徐莹莹, 李钊, Maxim B., 聂万丽, 化学进展, 2012, 24, 1526. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201208012.htmXu, Y.-Y.; Li, Z.; Maxim B.; Nie, W.-L. Prog. Chem. 2012, 24, 1526 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201208012.htm
-
[14]
Chase, P. A.; Welch, G. C.; Jurca, T.; Stephan, D. W. Angew. Chem., Int. Ed. 2007, 46, 8050. doi: 10.1002/(ISSN)1521-3773
-
[15]
Chase, P. A.; Jurca, T.; Stephan, D. W. Chem. Commun. 2008, 1701. http://europepmc.org/abstract/MED/18368170
-
[16]
Mohr, J.; Oestreich, M. Angew. Chem., Int. Ed. 2014, 53, 13278. doi: 10.1002/anie.201407324
-
[17]
Wei, S.-M.; Feng, X.-Q.; Du, H.-F. Org. Biomol. Chem. 2016, 14, 8026. doi: 10.1039/C6OB01556E
-
[18]
Farrell, J. M.; Heiden, Z. M.; Stephan, D. W. Organometallics 2011, 30, 4497. doi: 10.1021/om2005832
-
[19]
Chatterjee, I.; Oestreich, M. Angew. Chem., Int. Ed. 2015, 54, 1965. doi: 10.1002/anie.201409246
-
[20]
Jiang, C.-F.; Blacque, O.; Berke, H. Chem. Commun. 2009, 5518. http://europepmc.org/abstract/med/19753342
-
[21]
Scott, D. J.; Fuchter, M. J.; Ashley, A. E. Angew. Chem., Int. Ed. 2014, 53, 10218. doi: 10.1002/anie.201405531
-
[22]
Spies, P.; Schwendemann, S.; Lange, S.; Kehr, G.; Fröhlich, R.; Erker, G. Angew. Chem., Int. Ed. 2008, 47, 7543. doi: 10.1002/anie.v47:39
-
[23]
Wang, G.; Chen, C.; Du, T.-Y.; Zhong, W.-H. Adv. Synth. Catal. 2014, 356, 1747. doi: 10.1002/adsc.201301007
-
[24]
(a) Sumerin, V.; Chernichenko, K.; Nieger, M.; Leskelä, M.; Rieger, B.; Repo, T. Adv. Synth. Catal. 2011, 353, 2093.
(b) Chernichenko, K.; Nieger, M.; Leskelä, M.; Repo, T. Dalton Trans. 2012, 41, 9029.
(c) Sumerin, V.; Schulz, F.; Atsumi, M.; Wang, C.; Nieger, M.; Leskelä, M.; Repo, T.; Pyykkö, P.; Rieger, B. J. Am. Chem. Soc. 2008, 130, 14117. -
[25]
Farrell, J. M.; Posaratnanathan, R. T.; Stephan, D. W. Chem. Sci. 2015, 6, 2010. doi: 10.1039/C4SC03675A
-
[26]
Mummadi, S.; Unruh, D. K.; Zhao, J.-Y.; Li, S.-H.; Krempner, C. J. Am. Chem. Soc. 2016, 138, 3286. doi: 10.1021/jacs.5b13545
-
[27]
Schwendemann, S.; Frölich, R.; Kehr, G.; Erker, G. Chem. Sci. 2011, 2, 1842. doi: 10.1039/c1sc00124h
-
[28]
(a) Parks, D. J.; Spence, R. E. v. H.; Piers, W. E. Angew. Chem., Int. Ed. Engl. 1995, 34, 809.
(b) Parks, D. J.; Piers, W. E.; Yap, G. P. A. Organometallics 1998, 17, 5492. -
[29]
Wang, H.-D.; Fröhlich, R.; Kehr, G.; Erker, G. Chem. Commun. 2008, 5966. http://pubs.rsc.org/en/Content/ArticleLanding/CC/2008/B813286K
-
[30]
Greb, L.; Oña-Burgos, P.; Schirmer, B.; Grimme, S.; Stephan, D. W.; Paradies, J. Angew. Chem., Int. Ed. 2012, 51, 10164. doi: 10.1002/anie.201204007
-
[31]
Erõs, G.; Mehdi, H.; Pápai, I.; Rokob, T. A.; Király, P.; Tárkányi, G.; Soós, T. Angew. Chem., Int. Ed. 2010, 49, 6559. doi: 10.1002/anie.201001518
-
[32]
Inés, B.; Palomas, D.; Holle, S.; Steinberg, S.; Nicasio, J. A.; Alcarazo, M. Angew. Chem., Int. Ed. 2012, 51, 12367. doi: 10.1002/anie.v51.49
-
[33]
(a) Greb, L.; Daniliuc, C. G.; Bergander, K.; Paradies, J. Angew. Chem., Int. Ed. 2013, 52, 5876.
(b) Paradies, J. Angew. Chem., Int. Ed. 2014, 53, 3552. -
[34]
Hounjet, L. J.; Bannwarth, C.; Garon, C. N.; Caputo, C. B.; Grimme, S.; Stephan, D. W. Angew. Chem., Int. Ed. 2013, 52, 7492. doi: 10.1002/anie.201303166
-
[35]
Wang, X.-W.; Kehr, G.; Daniliuc, C. G.; Erker, G. J. Am. Chem. Soc. 2014, 136, 3293. doi: 10.1021/ja413060u
-
[36]
Chernichenko, K.; Madarász, Á.; Pápai, I.; Nieger, M.; Leskelä, M.; Repo, T. Nat. Chem. 2013, 5, 718. doi: 10.1038/nchem.1693
-
[37]
Szeto, K. C.; Sahyoun, W.; Merle, N.; Castelbou, J. L.; Popoff, N.; Lefebvre, F.; Raynaud, J.; Godard, C.; Claver, C.; Delevoye, L.; Gauvinc, R. M.; Taoufik, M. Catal. Sci. Technol. 2016, 6, 882. doi: 10.1039/C5CY01372K
-
[38]
Reddy, J. S.; Xu, B.-H.; Mahdi, T.; Fröhlich, R.; Kehr, G.; Stephan, D. W.; Erker, G. Organometallics 2012, 31, 5638. doi: 10.1021/om3006068
-
[39]
Greb, L.; Oña-Burgos, P.; Kubas, A.; Falk, F. C.; Breher, F.; Finkc, K.; Paradies, J. Dalton Trans. 2012, 41, 9056. doi: 10.1039/c2dt30374d
-
[40]
Longobardi, L. E.; Tang, C.; Stephan, D. W. Dalton Trans. 2014, 43, 15723. doi: 10.1039/C4DT02648A
-
[41]
Mahdi, T.; Stephan, D. W. J. Am. Chem. Soc. 2014, 136, 15809. doi: 10.1021/ja508829x
-
[42]
Scott, D. J.; Fuchter, M. J.; Ashley, A. E. J. Am. Chem. Soc. 2014, 136, 15813. doi: 10.1021/ja5088979
-
[43]
Mahdi, T.; Stephan, D. W. Angew. Chem., Int. Ed. 2015, 54, 8511. doi: 10.1002/anie.201503087
-
[44]
Gyömöre, Á.; Bakos, M.; Földes, T.; Pápai, I.; Domján, A.; Soós, T. ACS Catal. 2015, 5, 5366. doi: 10.1021/acscatal.5b01299
-
[45]
Geier, S. J.; Chase, P. A.; Stephan, D. W. Chem. Commun. 2010, 46, 4884. doi: 10.1039/c0cc00719f
-
[46]
Erös, G.; Nagy, K.; Mehdi, H.; Pápai, I.; Nagy, P.; Király, P.; Tárkányi, G.; Soós, T. Chem. Eur. J. 2012, 18, 574. doi: 10.1002/chem.v18.2
-
[47]
Chen, B.-L.; Wang, B.; Lin, G.-Q. J. Org. Chem. 2010, 75, 941. doi: 10.1021/jo902424m
-
[48]
Segawa, Y.; Stephan, D. W. Chem. Commun. 2012, 48, 11963. doi: 10.1039/c2cc37190a
-
[49]
Mahdi, T.; Heiden, Z. M.; Grimme, S.; Stephan, D. W. J. Am. Chem. Soc. 2012, 134, 4088. doi: 10.1021/ja300228a
-
[50]
Longobardi, L. E.; Mahdi, T.; Stephan, D. W. Dalton Trans. 2015, 44, 7114. doi: 10.1039/C5DT00921A
-
[51]
Mahdi, T.; Castillo, J. N. D.; Stephan, D. W. Organometallics 2013, 32, 1971. doi: 10.1021/om4000727
-
[52]
Liu, Y.-B.; Du, H.-F. J. Am. Chem. Soc. 2013, 135, 12968. doi: 10.1021/ja406761j
-
[53]
刘勇兵, 杜海峰, 化学学报, 2014, 72, 771. doi: 10.6023/A14040344Liu Y. -B., Du H. -F. Acta Chim. Sinica 2014, 72, 771 (in Chinese) doi: 10.6023/A14040344
-
[54]
Chen, D.-J.; Klankermayer, J. Chem. Commun. 2008, 2130. http://www.ncbi.nlm.nih.gov/pubmed/18438491
-
[55]
Chen, D.-J.; Wang, Y.-T.; Klankermayer, J. Angew. Chem., Int. Ed. 2010, 49, 9475. doi: 10.1002/anie.201004525
-
[56]
Ghattas, G.; Chen, D.-J.; Pan, F.-F.; Klankermayer, J. Dalton Trans. 2012, 41, 9026. doi: 10.1039/c2dt30536d
-
[57]
Chen, D.-J.; Leich, V.; Pan, F.-F.; Klankermayer, J. Chem. Eur. J. 2012, 18, 5184. doi: 10.1002/chem.v18.17
-
[58]
Lindqvist, M.; Borre, K.; Axenov, K.; Kótai, B.; Nieger, M.; Leskelä, M.; Pápai, I.; Repo, T. J. Am. Chem. Soc. 2015, 137, 4038. doi: 10.1021/ja512658m
-
[59]
(a) Mewald, M.; Oestreich, M. Chem. Eur. J. 2012, 18, 14079.
(b) Hermeke, J.; Mewald, M.; Oestreich, M. J. Am. Chem. Soc. 2013, 135, 17537. -
[60]
Süsse, L.; Hermeke, J.; Oestreich, M. J. Am. Chem. Soc. 2016, 138, 6940. doi: 10.1021/jacs.6b03443
-
[61]
Liu, Y.-B.; Du, H.-F. J. Am. Chem. Soc. 2013, 135, 6810. doi: 10.1021/ja4025808
-
[62]
Wei, S.-M.; Du, H.-F. J. Am. Chem. Soc. 2014, 136, 12261. doi: 10.1021/ja507536n
-
[63]
Ren, X.-Y.; Li, G.; Wei, S.-M.; Du, H.-F. Org. Lett. 2015, 17, 990. doi: 10.1021/acs.orglett.5b00085
-
[64]
Ren, X.-Y.; Du, H.-F. J. Am. Chem. Soc. 2016, 138, 810. doi: 10.1021/jacs.5b13104
-
[65]
Ashley, A. E.; Thompson, A. L.; O'Hare, D. Angew. Chem., Int. Ed. 2009, 48, 9839. doi: 10.1002/anie.v48:52
-
[66]
Caputo, C. B.; Hounjet, L. J.; Dobrovetsky, R.; Stephan, D. W. Science 2013, 341, 1374. doi: 10.1126/science.1241764
-
[67]
Hounjet, L. J.; Caputo, C. B.; Stephan, D. W. Dalton Trans. 2013, 42, 2629. doi: 10.1039/C2DT32711B
-
[68]
Porwal, D.; Oestreich, M. Eur. J. Org. Chem. 2016, 3307.
-
[1]
-

图式2 FLPs 1催化还原亚胺的可能机理
Scheme 2 Possible mechanism of imine catalytic reduction by FLPs 1

图式3 FLPs 11的合成及催化还原烯胺
Scheme 3 Synthesis of FLPs 11 and catalytic reduction of enamine

图式4 B (C6F5)3与Et2O组合催化还原烯烃
Scheme 4 Catalytic reduction of olefins by B (C6F5)3 and Et2O

图式5 FLPs 15催化炔烃顺式加成可能机理
Scheme 5 Proposed mechanism of the Z-selective reduction of alkynes by FLPs 15

图式6 FLPs 17催化炔烃顺式加成可能机理
Scheme 6 Proposed mechanism of the Z-selective reduction of alkynes by FLPs 17

图式8 B (C6F5)3催化还原N杂芳烃化合物可能机理
Scheme 8 Proposed mechanism of catalytic reduction of N heterocyclic aromatic compounds by B (C6F5)3

图式9 MesB (C6F4H)2应用于枯杷仞合成
Scheme 9 Application of MesB (C6F4H)2in the synthesis of cuspareine

图式10 FLPs 21催化还原多环芳烃可能机理
Scheme 10 Proposed mechanism of catalytic reduction poly-cyclic aromatic hydrocarbons by FLPs 21

图式12 B (C6F5)3催化还原苯环可能机理
Scheme 12 Proposed mechanism of catalytic reduction benzene by B (C6F5)3
-


 下载:
下载:

























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 43
- 文章访问数: 3173
- HTML全文浏览量: 918
 下载:
下载:
