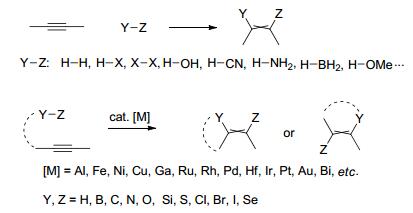
 图 图式1
传统的简单小分子对炔键的加成反应以及当代金属催化的碳-杂键对炔键的加成反应
Figure 图式1.
Traditional
addition of simple molecules to alkynes and modern metal-catalyzed
addition of carbon-hetero bonds to alkynes
图 图式1
传统的简单小分子对炔键的加成反应以及当代金属催化的碳-杂键对炔键的加成反应
Figure 图式1.
Traditional
addition of simple molecules to alkynes and modern metal-catalyzed
addition of carbon-hetero bonds to alkynes
引用本文:
赵飞, 贾秀稳, 王东萍, 费朝丽, 吴成林, 王江, 柳红. 金属催化的碳-杂键对炔键的加成反应研究进展[J]. 有机化学,
2017, 37(2): 284-300.
doi:
10.6023/cjoc201607035

Citation: Zhao Fei, Jia Xiuwen, Wang Dongping, Fei Chaoli, Wu Chenglin, Wang Jiang, Liu Hong. Research Progress in Metal-Catalyzed Addition of Carbon-Hetero Bonds to Alkynes[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 284-300. doi: 10.6023/cjoc201607035
Citation: Zhao Fei, Jia Xiuwen, Wang Dongping, Fei Chaoli, Wu Chenglin, Wang Jiang, Liu Hong. Research Progress in Metal-Catalyzed Addition of Carbon-Hetero Bonds to Alkynes[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 284-300. doi: 10.6023/cjoc201607035
金属催化的碳-杂键对炔键的加成反应研究进展
摘要:
碳-杂键对炔键的加成反应已经成为对碳碳叁键进行官能团化的一种重要手段,此类反应可以一步快速构建两个化学键即一个碳-碳键和一个碳-杂键,因而具有反应效率高、原子经济性高的特点.近年来,Al,Fe,Ni,Cu,Ga,Ru,Rh,Pd,Hf,Ir,Pt,Au,Bi等催化的诸多类型的碳-杂键对炔键的加成反应取得了重要进展.根据对炔键进行加成的碳-杂键的类型分为C-H,C-B,C-N,C-O,C-Si,C-S,C-X(X=Cl,Br,I),C-Se键这8类逐一进行介绍,并对各类加成反应的反应条件、反应选择性(区域选择性和立体化学选择性)以及反应机理进行了讨论和总结.
English
Research Progress in Metal-Catalyzed Addition of Carbon-Hetero Bonds to Alkynes
Abstract:
The addition of carbon-hetero bonds to alkynes has become an important apporach for the functionalization of carbon-carbon triple bonds. It can construct two bonds, namely one carbon-carbon bond and one carbon-hetero bond, in a single one step. These addition reactions feature high efficiency and high atom-economy. In recent years, metal-catalyzed (Al, Fe, Ni, Cu, Ga, Ru, Rh, Pd, Hf, Ir, Pt, Au, Bi, etc.) addition of many various kinds of carbon-hetero bonds to alkynes has achieved lots of important developments. According to the types of carbon-hetero bonds, the addition of 8 different kinds of carbon-hetero bonds [C-H, C-B, C-N, C-O, C-Si, C-S, C-X (X=Cl, Br, I), C-Se] to alkynes is reviewed in this paper, and the reaction conditions, reaction selectivities (regioselectivities and stereoselectivities) and reaction mechanisms are also discussed and summarized.
-
Key words:
- metal-catalyzed
- / carbon-hetero bonds
- / alkynes
- / addition reactions
-
炔键的加成反应是快速而高效地实现对炔键官能团化的一种重要有机合成策略[1], 然而传统的加成反应仅局限于简单的小分子, 如H2, HX (X=F, Cl, Br), X2 (X=F, Cl, Br), H2O, HCN, NH3, BH3, MeOH等 (Scheme 1), 而且加成反应的形式仅局限于分子间加成, 这从一定程度上限制了加成反应的应用范围.近年来, 随着合成方法学的不断发展, 有机化学家们发展了金属催化的更加复杂的小分子对炔键的加成反应 (Scheme 1), 反应既可以发生在分子间, 也可以发生在分子内, 这极大程度地丰富了加成反应的内涵并拓展了加成反应的应用范围.从断键和成键的角度看, 金属催化的碳-杂键对炔键的加成反应可以一步快速构建两个化学键, 即一个碳-碳键和一个碳-杂键, 高效地实现对炔键的官能团化, 而且此类反应具有操作简单、催化效率高、原子经济性高、底物适用范围广等特点.近年来, 有机化学家们发展了多种金属如Al, Fe, Ni, Cu, Ga, Ru, Rh, Pd, Hf, Ir, Pt, Au, Bi等催化的诸多类型的碳-杂键对炔键的加成反应.本文根据对炔键进行加成的碳-杂键的类型分为C—H, C—B, C—N, C—O, C—Si, C—S, C—X (X=Cl, Br, I), C—Se键这8类逐一进行介绍.
图 图式1
传统的简单小分子对炔键的加成反应以及当代金属催化的碳-杂键对炔键的加成反应
Figure 图式1.
Traditional
addition of simple molecules to alkynes and modern metal-catalyzed
addition of carbon-hetero bonds to alkynes
1 碳-杂键对炔键的加成
1.1 C—H键对炔键的加成
C—H键的键能较大, 要实现其对炔键的加成具有一定的挑战性, 往往需要金属催化剂对C—H键进行活化后方能实现对炔键的加成. 2005年, Takemoto课题组[2]首次实现了铑催化的C—H键 (醛氢键) 对炔键的分子内加成 (Scheme 2).当以含有炔键的脂肪链状甲酰胺类化合物1为底物, 以Rh4(CO)12为催化剂时, 于二甲苯中在130 ℃反应10 h即可以较好的收率只得到E式目标产物2(Scheme 2).该反应同样适用于邻炔基苯胺类底物3, 这类底物的反应活性更高, 以Rh4(CO)12为催化剂, 只需要在甲苯中于100 ℃反应6 h即可以较高的收率得到目标产物4, 但此类底物参与反应时立体选择性较底物1稍差, E式产物和Z式产物的比例在2.4:1到12.5:1之间 (Scheme 2).此反应可能的反应机理如Scheme 3所示, 铑催化剂首先与底物中的炔键配合生成中间体A1, 然后铑对C—H键进行氧化插入生成中间体B1, 随后Rh—H键对炔键进行加成 (顺式加成为主) 得到中间体C1, C1经过还原消除即可得到产物并释放出催化剂, 进行下一轮的催化循环. Takemoto报道的合成方法具有操作简单、立体选择性高等特点, 为合成吡咯烷酮和氧化吲哚类化合物提供了新思路.
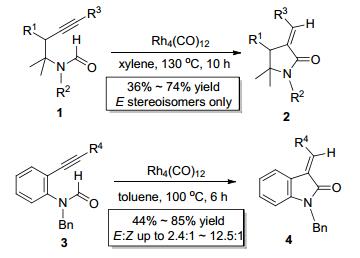 图 图式2
铑催化C—H键对炔键的分子内加成
Figure 图式2.
Rh-catalyzed
intramolecular addition of C—H bond to alkynes
图 图式2
铑催化C—H键对炔键的分子内加成
Figure 图式2.
Rh-catalyzed
intramolecular addition of C—H bond to alkynes
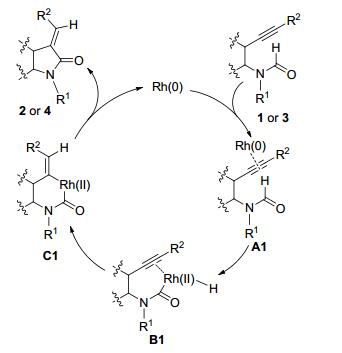 图 图式3
铑催化C—H键对炔键的分子内加成反应机理
Figure 图式3.
Proposed
reaction mechanism of Rh-catalyzed intramolecular addition of C—H bond to alkynes
图 图式3
铑催化C—H键对炔键的分子内加成反应机理
Figure 图式3.
Proposed
reaction mechanism of Rh-catalyzed intramolecular addition of C—H bond to alkynes
2009年, Hiyama小组[3]报道了Ni (COD)2和Lewis酸 (AlMe3或BPh3) 共催化的甲酰胺类化合物5中的醛氢键对炔6的分子间加成 (Eq. 1), 该反应区域选择性极好, 当使用非对称炔烃时, 酰胺基团只加成到位阻较小的炔碳上; 该反应的立体化学选择性也非常好, 主要以顺式加成的形式得到E式α, β-不饱和酰胺类化合物7.
继Hiyama小组的工作之后, Tsuji小组[4]几乎同时独立报道了甲酰胺类化合物8中的醛氢键对炔9的分子间加成 (Scheme 4).该反应以PdCl2(PhCN)2为催化剂, Xantphos为配体, PhCOCl为添加剂, 在三甲苯中于140 ℃反应20 h即可以高收率得到目标产物10.值得一提的是, 添加剂PhCOCl在反应中扮演着至关重要的角色, 不可或缺, 这可能是因为PhCOCl与甲酰胺底物8发生反应进而产生HCl, HCl进一步与钯催化剂反应产生“Pd-H”物种, 该“Pd-H”物种能推动整个催化循环[4, 5].此反应最大特点是区域选择性好、立体选择性高 (E: Z=94:6~100:0).当使用非末端对称炔烃时, 反应几乎只得到E式产物; 当使用非末端不对称炔烃时, 反应仅得到单一区域选择性产物, 酰胺基团只加成到位阻较小的炔碳上, 且几乎只得到E式产物 (Scheme 4); 当使用末端炔烃12时, 反应主要得到端烯加成产物13(Scheme 4).此反应的加成产物是α, β-不饱和酰胺类化合物, 是一类重要的Michael受体, 也是一类重要的有机中间体.
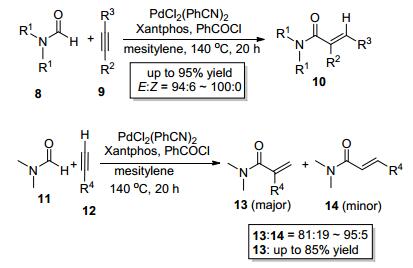 图 图式4
钯催化C—H键对炔键的分子间加成
Figure 图式4.
Pd-catalyzed
intermolecular addition of C—H bond to alkynes
图 图式4
钯催化C—H键对炔键的分子间加成
Figure 图式4.
Pd-catalyzed
intermolecular addition of C—H bond to alkynes
2011年, Trost小组[6, 7]以Pd (OAc)2为催化剂实现了炔15中的C—H键对炔丙酸酯16的分子间加成 (Scheme 5), 该反应具有专一的区域选择性和立体化学选择性, 以顺式加成的形式得到了高度官能团化的烯炔类产物17, 产物17可以进一步关环得到多取代的吡咯类化合物.随后, Xu小组[8]也报道了类似的工作, 他们以[Rh (COD) Cl]2为催化剂实现了炔18中的C—H键对炔丙醇 (醚)19或炔丙胺21的分子间加成 (Scheme 5), 以专一的区域选择性和立体化学选择性分别得到了烯炔类产物20或22.
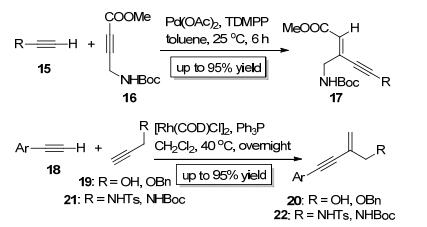 图 图式5
钯或铑催化C—H键对炔键的分子间加成
Figure 图式5.
Pd
or Rh-catalyzed intermolecular addition of C—H bond to alkynes
图 图式5
钯或铑催化C—H键对炔键的分子间加成
Figure 图式5.
Pd
or Rh-catalyzed intermolecular addition of C—H bond to alkynes
2014年, Zhu等[9]报道了Pd2(dba)3催化的炔23中的C—H键对炔胺类底物24的分子间加成 (Scheme 6), 该反应具有专一的区域选择性, 炔基只加成到与氮原子相连的炔碳上; 该反应的立体化学选择性也非常好, 几乎只得到反式加成产物25. 2015年, Reddy小组[10]也报道了类似的反应, 他们以Pd (PPh3)2Cl2为催化剂实现了炔26中的C—H键对炔基苯基醚类底物27的分子间加成 (Scheme 6), 并以专一的区域选择性和立体化学选择性得到了顺式加成产物28. Zhu小组和Reddy小组[11]发展的方法所合成的产物都是高度官能团化的烯炔类化合物, 很容易发生多种后续的化学转化, 是十分有用的有机合成中间体.
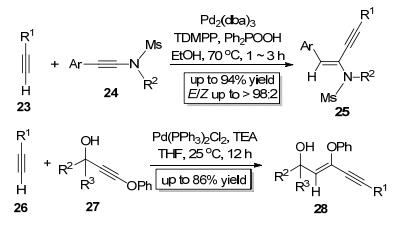 图 图式6
钯催化C—H键对炔键的分子间加成
Figure 图式6.
Pd-catalyzed
intermolecular addition of C—H bond to alkynes
图 图式6
钯催化C—H键对炔键的分子间加成
Figure 图式6.
Pd-catalyzed
intermolecular addition of C—H bond to alkynes
1.2 C—B键对炔键的加成
2003年, Suginome小组[12]首次报道了钯或镍催化的C—B键对炔键的分子内加成 (Scheme 7), 该反应根据反应底物29的不同选取Pd2(dba)3, Pd (PPh3)4或Ni (COD)2为催化剂, 在甲苯中于80~110 ℃反应2~43 h即可得到目标产物30.此反应以极好的区域选择性和立体化学选择性 (E:Z=100:0) 以顺式加成的形式得到E式五元环关环产物, 该产物可以在不同条件下进一步转化为多种重要的有机中间体.紧接着他们[13]又实现了钯催化的C—B键对炔键的分子间加成 (Scheme 7), 该反应区域选择性较好, 硼原子主要加成到位阻较小的炔碳上, 该反应还具有专一的立体化学选择性, 只得到顺式加成产物.他们还将此反应运用到了角鲨烯合成酶抑制剂P-3622[14]的合成中.随后, 他们[15]于2006年以Ni (COD)2为催化剂实现了炔基硼酸酯类底物35中的C—B键对炔键的分子间加成 (Scheme 7), 该反应能以高收率和区域选择性主要得到顺式加成的烯基硼酸酯类产物37. Suginome小组发展的方法为合成官能团化的烯基硼酸酯类化合物提供了有效的路径.
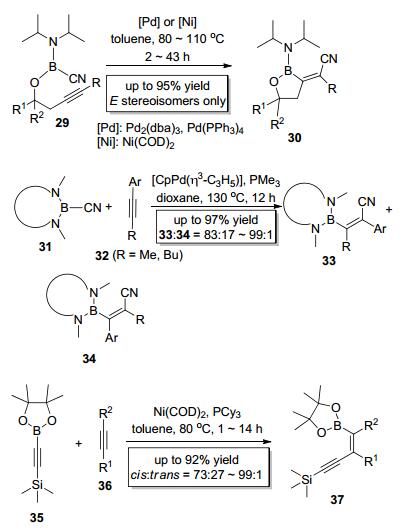 图 图式7
钯或镍催化C—B键对炔键的加成
Figure 图式7.
Pd or Ni-catalyzed addition of C—B bond to alkynes
图 图式7
钯或镍催化C—B键对炔键的加成
Figure 图式7.
Pd or Ni-catalyzed addition of C—B bond to alkynes
1.3 C—N键对炔键的加成
C—N键对炔键的加成反应一直是有机化学家们的研究热点, 因为此类反应可以高效地构建吲哚骨架.早期开发的催化体系具有一些缺点, 如催化剂昂贵、反应收率低等, 然而随着新型催化体系的不断涌现, 这些缺点都被逐一克服, 这使C—N键对炔键的加成反应成为构建高度官能团化的吲哚类化合物的一种重要策略.
2004年, Yamamoto课题组[16]首次报道了铂催化的C—N键对炔键的分子内加成 (Scheme 8), 他们采用38为底物, 以PtCl2为催化剂在苯甲醚中加热至80 ℃即可得到C—N键对炔键的分子内加成产物39, 该反应在C—N键对炔键进行加成的同时伴随着酰基的分子内1, 3-迁移.但该方法存在两个不足之处:一是催化剂PtCl2十分昂贵; 二是副产物较多.大部分底物38参与反应时除了得到目标产物39外, 还会得到脱酰基副产物40, 而且部分实例中40的含量高达30%, 这两个缺点从一定程度上降低了此反应的实用价值. Nakamura课题组[17]于2009年进一步研究了Yamamoto发展的催化体系并拓展了该反应的适用范围 (Scheme 8), 他们以PtI4为催化剂实现了底物41中的酰胺基的分子内1, 3-迁移, 合成了吲哚-3-酰胺类化合物42, 该反应仍然伴随着一定比例的脱酰胺基副产物43; 当使用PtCl2或PtCl4为催化剂时, 可以高效地实现底物44结构中的酯基的分子内1, 3-迁移, 得到吲哚-3-羧酸酯类化合物45, 有趣的是, 该反应中并未检测到脱酯基副产物46(Scheme 8).
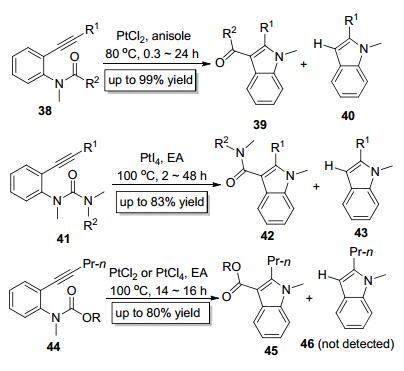 图 图式8
铂催化C—N键对炔键的分子内加成
Figure 图式8.
Pt-catalyzed
intramolecular addition of C—N bond to alkynes
图 图式8
铂催化C—N键对炔键的分子内加成
Figure 图式8.
Pt-catalyzed
intramolecular addition of C—N bond to alkynes
Li课题组[18]于2012年报道了钌催化的底物47中的C—N键对炔键的分子内加成 (Scheme 9), 该反应能高效地构建吲哚-3-甲醛类化合物48.但该反应时间较长, 且底物适用范围较窄, 迁移基团仅限于位阻较小的甲酰基, 位阻稍大的迁移基团如乙酰基等参与反应时收率低, 这可能是由于钌催化剂的催化活性较低而引起的.我们课题组[19]于2014年报道了钯催化的C—N键对炔键的加成, 采用49为底物, 以PdCl2(CH3CN)2为催化剂即可快速而高效地构建高度官能团化的吲哚类化合物50 (Scheme 9).该反应的迁移基团不但适用于位阻较小的甲酰基, 也适用于位阻较大的乙酰基、丙酰基、异丁酰基、特戊酰基、苯甲酰基、苯乙酰基等.此外, 酰胺基、酮酰基均可以作为迁移基团参与此反应, 值得一提的是, 酮酰基是被首次证明可以作为迁移基团参与此类反应.紧接着, 我们[20]进一步设计了酮酰胺类底物51, 并研究了酮酰胺中的C—N键对炔键的加成, 此反应能以高收率得到吲哚-3-酮酰基类化合物52(Scheme 9), 为吲哚-3-酮酰基类化合物的制备提供了新方法.
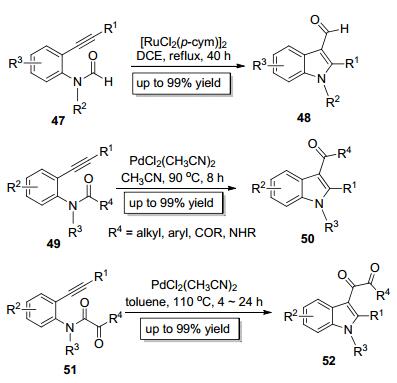 图 图式9
钌或钯催化C—N键对炔键的分子内加成
Figure 图式9.
Ru
or Pd-catalyzed intramolecular addition of C—N bond to alkynes
图 图式9
钌或钯催化C—N键对炔键的分子内加成
Figure 图式9.
Ru
or Pd-catalyzed intramolecular addition of C—N bond to alkynes
Scheme 10为以上铂、钌、钯催化的C—N键对炔键的加成反应可能的反应机理, 金属催化剂[M]首先与底物53结构中的炔键配合并活化炔键从而生成中间体A2, 紧接着氮原子对炔键进行亲核进攻生成中间体B2, 然后迁移基团 (MG) 发生分子内1, 3-迁移形成中间体C2, 最后再消除催化剂得到产物54, 催化剂再进入下一轮催化循环.
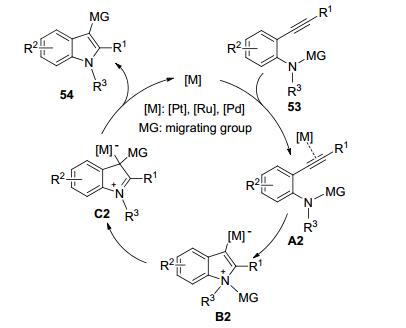 图 图式10
铂、钌、钯催化C—N键对炔键的分子内加成反应机理
Figure 图式10.
Proposed reaction
mechanism of Pt, Ru, Pd-catalyzed intramolecular addition of C—N bond to alkynes
图 图式10
铂、钌、钯催化C—N键对炔键的分子内加成反应机理
Figure 图式10.
Proposed reaction
mechanism of Pt, Ru, Pd-catalyzed intramolecular addition of C—N bond to alkynes
基于之前的工作基础, 我们课题组[21]进一步设计了N-烯丙基取代的磺酰胺类底物55, 并使用不同价态的金属钯实现了C—N键和S—N键对炔键的选择性加成 (Scheme 11).当以Pd (PPh3)4为催化剂时, 高选择性地实现C—N键对炔键的加成, 此时烯丙基作为迁移基团发生分子内1, 3-迁移; 当以PdCl2(CH3CN)2为催化剂时, 高选择性地实现S—N键对炔键的加成, 此时磺酰基作为迁移基团发生分子内1, 3-迁移.因此, 我们通过同一种底物55成功构建了1-磺酰基-3-烯丙基吲哚 (56) 和1-烯丙基-3-磺酰基吲哚 (57) 这两种吲哚骨架的化合物.值得注意的是, 实现C—N键和S—N键对炔键的选择性加成的两种催化体系除了钯催化剂不同之外, 其他反应条件如溶剂、温度等均相同.之所以仅通过改变钯催化剂就能实现C—N键和S—N键对炔键的选择性加成, 这可能是由Pd (PPh3)4和PdCl2(CH3CN)2这两种不同价态的催化剂因不同的催化机理而导致的截然不同的反应路径所决定的 (Scheme 12).零价钯Pd (PPh3)4首先对C—N键进行氧化加成生成中间体A3, 紧接着在钯的作用下生成离子对中间体B3, 然后氮原子对钯活化的炔键进行亲核加成生成中间体C3, 最后还原消除催化剂而得到产物56, 释放出的钯催化剂再进入下一轮催化循环.二价钯PdCl2(CH3CN)2首先与底物55结构中的炔键配合并活化炔键从而生成中间体A4, 紧接着氮原子对炔键进行亲核进攻生成中间体B4, 然后磺酰基发生分子内1, 3-迁移形成中间体C4, 最后再消除催化剂得到产物57, 催化剂再进入下一轮催化循环.此方法既符合当代多样性合成的要求, 也为构建高度官能团化的吲哚类化合物提供了新路径.
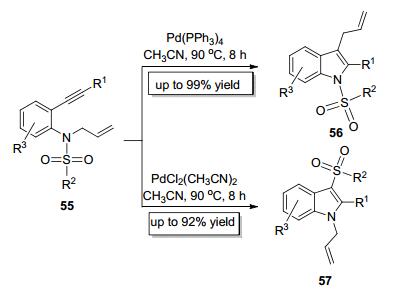 图 图式11
钯催化C—N键和S—N键对炔键的选择性分子内加成
Figure 图式11.
Pd-catalyzed
selective intramolecular addition of C—N and S—N bonds to alkynes
图 图式11
钯催化C—N键和S—N键对炔键的选择性分子内加成
Figure 图式11.
Pd-catalyzed
selective intramolecular addition of C—N and S—N bonds to alkynes
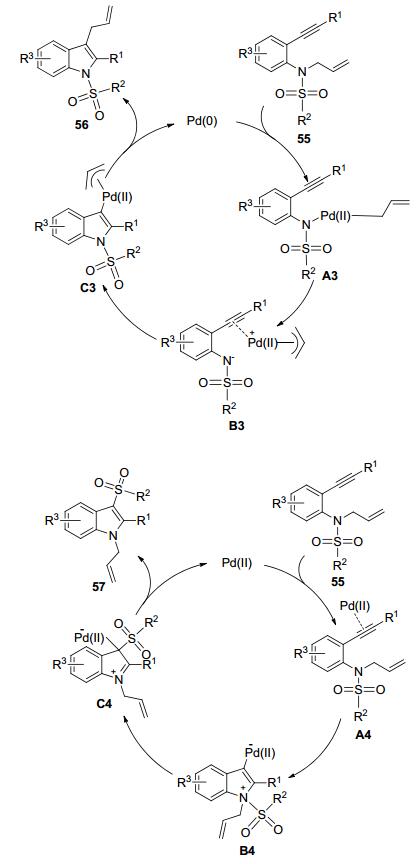 图 图式12
零价钯和二价钯分别催化的C—N键和S—N键对炔键的加成反应机理
Figure 图式12.
Proposed
reaction mechanisms of Pd (0)-catalyzed intramolecular addition of C—N bond to alkynes and Pd (Ⅱ)-catalyzed
intramolecular addition of S—N bond to alkynes
图 图式12
零价钯和二价钯分别催化的C—N键和S—N键对炔键的加成反应机理
Figure 图式12.
Proposed
reaction mechanisms of Pd (0)-catalyzed intramolecular addition of C—N bond to alkynes and Pd (Ⅱ)-catalyzed
intramolecular addition of S—N bond to alkynes
2002年, Yamamoto等[22]以Pd (PPh3)4为催化剂实现了烯丙基苯胺类底物58中的C—N键对炔键的分子内加成 (Scheme 13), 在该反应中, 烯丙基作为迁移基团发生分子内1, 3-迁移, 从而构建了3位烯丙基取代的吲哚类化合物59. Roy小组[23]于2011年也报道了类似的反应, 他们以60为底物, AuCl3为催化剂合成了结构更加复杂的吲哚类衍生物61(Scheme 13).值得一提的是, Stevens等[24]也以AuCl3为催化剂实现了底物62中的C—N键对炔键的分子内加成 (Scheme 13), 该反应伴随着烯丙基的重排和芳构化反应, 构建了1-氰基取代的异吲哚类化合物63.
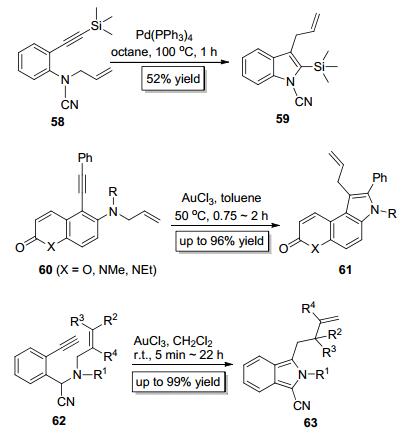 图 图式13
钯或金催化C—N键对炔键的分子内加成
Figure 图式13.
Pd
or Au-catalyzed intramolecular addition of C—N bond to alkynes
图 图式13
钯或金催化C—N键对炔键的分子内加成
Figure 图式13.
Pd
or Au-catalyzed intramolecular addition of C—N bond to alkynes
2007年, Nakamura小组[25]报道了PdBr2催化的C—N键对炔键的分子内加成 (Scheme 14), 尽管反应产率较低, 但是烷氧烷基在该反应中作为迁移基团成功地实现了分子内1.3-迁移, 这进一步增加了迁移基团的种类, 也扩大了C—N键对炔键的加成反应的适用范围. 2010年, Bertrand小组[26]以AuCl (CAAC) 为催化剂实现了苯胺类底物66中的C—N键对炔键的加成, 成功地实现了甲基的分子内1, 3-迁移, 以较高的产率构建了1, 2, 3-三烃基取代的吲哚类化合物67(Scheme 14). Pozharskii小组[27]于2015年也报道了类似的反应, 以Pd2dba3为催化剂实现了底物68中的C—N键对炔键的分子内加成, 从而构建了苯并[g]吲哚类化合物69(Scheme 14). Bertrand和Pozharskii等报道的反应的最大特点是实现了甲基的分子内1, 3-迁移, 而以往同类型反应的迁移基团都是酰基、酯基、酰胺基、酮酰基等官能团, 这也是为数不多的能实现甲基迁移的加成反应[28].
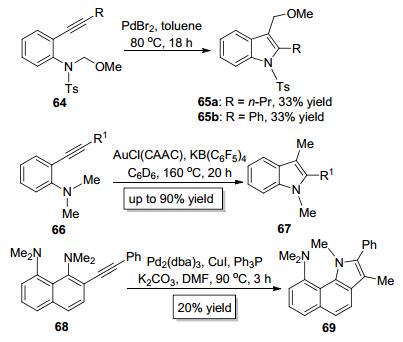 图 图式14
钯或金催化C—N键对炔键的分子内加成
Figure 图式14.
Pd
or Au-catalyzed intramolecular addition of C—N bond to alkynes
图 图式14
钯或金催化C—N键对炔键的分子内加成
Figure 图式14.
Pd
or Au-catalyzed intramolecular addition of C—N bond to alkynes
1.4 C—O键对炔键的加成
1998年, Cacchi等[29]以70为底物, Pd (PPh3)4为催化剂实现了C—O键对炔键的分子内加成, 成功合成了苯并呋喃类化合物71和72(Scheme 15), 但该反应缺乏选择性, 大多数情况下得到71和72的混合物, 而且产率较低, 这些缺点都限制了此反应的实际应用. 2005年, Yamamoto小组[30]设计了缩醛类底物73, 并以PtCl2为催化剂实现了其结构中的C—O键对炔键加成, 以高收率构建了3-烷氧烷基取代的苯并呋喃类化合物74(Scheme 15). Fürstner小组[31]同时独立报道了与Yamamoto小组类似的工作, 他们也以PtCl2为催化剂实现了底物75中的C—O键对炔键加成, 但Fürstner等发展的催化体系底物适用范围更广, 底物75不仅可以是缩醛, 还可以是苯基烷基醚、苯基烯丙基醚和苯基苄基醚, 这些底物都可以高效地参与反应而得到相应的苯并呋喃类产物76 (Scheme 15).值得一提的是, Fürstner等还运用其开发的催化体系以77为底物成功构建了含有异香豆素骨架的产物78(Scheme 15).有趣的是, Yamamoto小组和Fürstner小组在《美国化学学会会刊》上以“背靠背”的形式相继发表了他们的工作.此外, 麻生明课题组[32]于2007年报道了钯催化的C—O键对炔键的分子内加成 (Scheme 16), 他们以79和82为底物分别合成了苯并呋喃衍生物80和83, 尽管此反应伴随着脱烯丙基副产物 (81或84), 但此方法为构建复杂的苯并呋喃类衍生物提供了新的研究思路.
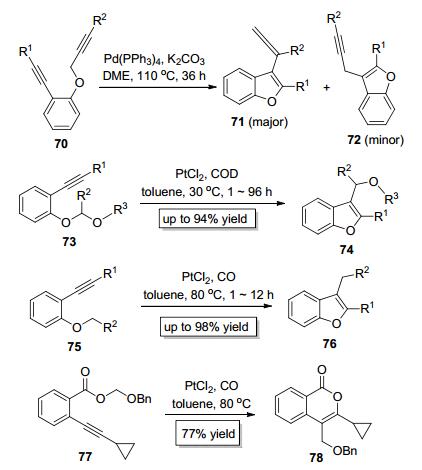 图 图式15
钯或铂催化C—O键对炔键的分子内加成
Figure 图式15.
Pd
or Pt-catalyzed intramolecular addition of C—O bond to alkynes
图 图式15
钯或铂催化C—O键对炔键的分子内加成
Figure 图式15.
Pd
or Pt-catalyzed intramolecular addition of C—O bond to alkynes
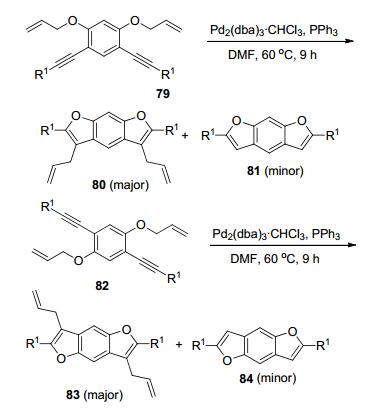 图 图式16
钯催化C—O键对炔键的分子内加成
Figure 图式16.
Pd-catalyzed
intramolecular addition of C—O bond to alkynes
图 图式16
钯催化C—O键对炔键的分子内加成
Figure 图式16.
Pd-catalyzed
intramolecular addition of C—O bond to alkynes
2006年, Toste小组[33]报道了金催化的苄醚类底物85中的C—O键对炔键的分子内加成 (Scheme 17), 在该反应中, 甲氧基作为迁移基团发生了分子内1, 3-迁移, 以高收率获得了茚类化合物86.有趣的是, 当使用缩酮类底物87时, 该反应得到的是甲基迁移而非甲氧基迁移的多取代茚类产物88(Scheme 17), 这可能是因为该反应经历了碳正离子中间体的缘故.此外, 当使用链状化合物89为底物时, 反应仍可顺利发生, 从而构建多取代的环己烯类产物90(Scheme 17).值得一提的是, 手性底物91 (99% ee) 参与反应后, 可以92%的产率得到手性产物92(95% ee), 该反应的手性可以得到保持 (Scheme 17).
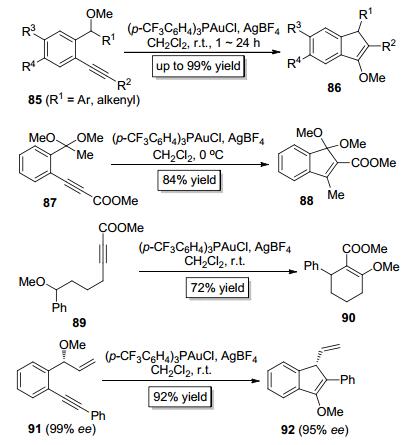 图 图式17
金催化C—O键对炔键的分子内加成
Figure 图式17.
Au-catalyzed
intramolecular addition of C—O bond to alkynes
图 图式17
金催化C—O键对炔键的分子内加成
Figure 图式17.
Au-catalyzed
intramolecular addition of C—O bond to alkynes
2008年, Nakamura小组[34]以PtCl2为催化剂实现了缩醛类底物93中的C—O键对炔键的分子内加成 (Scheme 18), 该反应具有专一的区域选择性, 只以5-exo关环的方式得到五元环产物.有趣的是, 该反应的立体化学选择性受酯基上的R1取代基的影响较大, 当R1为2, 2, 2-三氯乙基时, 反应主要以反式加成的方式得到Z式产物; 当R1为苯基时, 反应主要以顺式加成的方式得到E式产物[35].此外, 他们在后续的研究中还进一步发现该反应使用的配体对立体化学选择性也存在明显的影响[36], 其发展的方法为构建多取代的四氢呋喃类衍生物提供了有力工具.
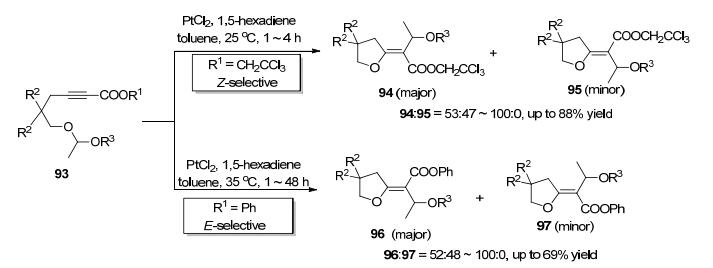 图 图式18
铂催化C—O键对炔键的分子内加成
Figure 图式18.
Pt-catalyzed intramolecular addition of C—O bond to alkynes
图 图式18
铂催化C—O键对炔键的分子内加成
Figure 图式18.
Pt-catalyzed intramolecular addition of C—O bond to alkynes
Takaki等[37]于2008年以Bi (OTf)3为催化剂实现了炔酸酯类底物98中的C—O键对炔键的分子内加成 (Scheme 19), 该反应主要以6-endo关环的方式得到反式加成的六元内酯类产物99. Blum小组[38]也报道了邻炔基苯甲酸烯丙基酯类底物100中的C—O键对炔键的分子内加成反应, 该反应在PPh3AuCl和Pd2(dba)3的共同催化下, 能以高收率获得异香豆素骨架的产物101(Scheme 19).此外, Miyata课题组[39]于2010年报道了肟醚类底物102中的C—O键对炔键的分子内加成反应, 该反应以AuCl3为催化剂, 以1, 2-二氯乙烷为溶剂, 回流2 h即可高收率地得到多取代的异噁唑类化合物103(Scheme 19). 2016年, Mino小组[40]报道了苯基烯丙基醚类底物104结构中的C—O键对炔键的分子内加成 (Scheme 19), 该反应以Pd2(dba)3为催化剂在含水溶剂体系中即可构建多取代的苯并呋喃类化合物105.由此可见, 金属催化的C—O键对炔键的加成反应可以高效而快速地构建含氧的结构丰富多样的有机杂环类化合物[41], 具有重要的应用价值.
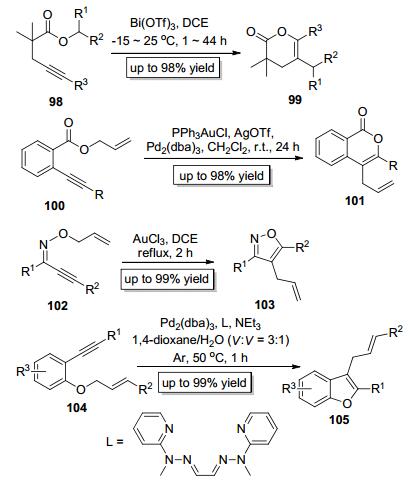 图 图式19
铋、金、钯催化C—O键对炔键的分子内加成
Figure 图式19.
Bi, Au, Pd-catalyzed intramolecular addition of C—O bond to alkynes
图 图式19
铋、金、钯催化C—O键对炔键的分子内加成
Figure 图式19.
Bi, Au, Pd-catalyzed intramolecular addition of C—O bond to alkynes
1.5 C—Si键对炔键的加成
Jung小组[42]于1995年以AlCl3为催化剂实现了烯丙基三甲基硅烷106中的C—Si键对苯炔107的分子间加成, 该反应具有很好的立体化学选择性和区域选择性.当使用的苯炔107中的R取代基为位阻较小的基团如氢、甲基时, 三甲基硅基主要加成到位阻较小的炔碳上, 烯丙基主要加成到位阻较大的炔碳上, 而且以顺式加成的方式得到E式产物 (Scheme 20); 当使用的苯炔为位阻较大的对称的二苯基乙炔110时, 反应主要以反式加成的方式得到Z式产物111 (Scheme 20).尽管该反应产率中等, 但属首次实现烯丙基和硅基对炔键的加成.紧接着, Yamamoto小组[43, 44]以EtAlCl2为催化剂实现了类似的反应 (Scheme 20), 该反应与Jung小组报道的反应具有相同的区域选择性, 但该反应只得到反式加成产物, 而且反应产率大幅度提高, 这使得该类反应更加实用和高效, Yamamoto等[45]还将此反应用于有机硅聚合物的合成中.
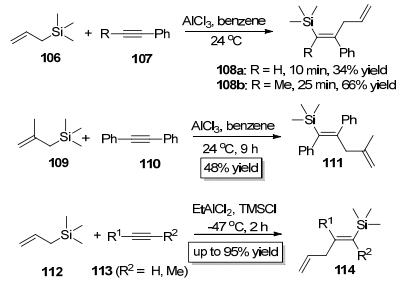 图 图式20
铝催化C—Si键对炔键的分子间加成
Figure 图式20.
Al-catalyzed intermolecular
addition of C—Si
bond to alkynes
图 图式20
铝催化C—Si键对炔键的分子间加成
Figure 图式20.
Al-catalyzed intermolecular
addition of C—Si
bond to alkynes
继实现烯丙基和硅基对炔键的加成之后, Yamamoto小组[46, 47]紧接着实现了烯基和硅基对炔键的加成.当使用炔键和烯键由碳原子连接的底物115时, 以EtAlCl2或AlCl3为催化剂, 反应只以exo关环的方式只得到反式加成产物116; 当使用炔键和烯键由碳原子连接的底物117时, 以EtAlCl2为催化剂, 反应只以edo关环的方式只得到反式加成产物118; 当使用炔键和烯键由硅原子连接的底物119时, 以EtAlCl2为催化剂, 反应只以6-edo关环的方式只得到反式加成产物120(Scheme 21).随后, 他们[47]又实现了苯基和硅基对炔键的加成, 当使用苯基和炔基由碳原子相连的底物121或123时, 分别以HfCl4或EtAlCl2为催化剂, 反应只以exo关环的方式只分别得到反式加成产物122或124; 当使用苯基和炔基由硅原子相连的底物125时, 反应只以edo关环的方式只得到反式加成产物126(Scheme 21). Yamamoto小组发展的方法可以便捷地合成常规方法难以得到的环己烯、环庚烯、苯并环己烷、苯并环庚烷骨架的有机硅类化合物、硅杂环戊烯和硅杂环己烯类化合物, 具有重要的研究价值.
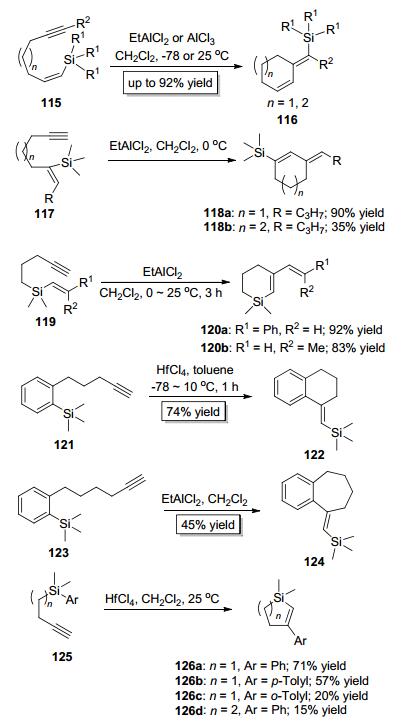 图 图式21
铝或铪催化C—Si键对炔键的分子内加成
Figure 图式21.
Al or Hf-catalyzed intramolecular
addition of C—Si
bond to alkynes
图 图式21
铝或铪催化C—Si键对炔键的分子内加成
Figure 图式21.
Al or Hf-catalyzed intramolecular
addition of C—Si
bond to alkynes
2008年, Murakami小组[48]报道了烯丙基硅类化合物127中的C—Si键对炔键的分子内加成 (Scheme 22), 该反应以AuNTf2为催化剂, 在二氯甲烷中于室温反应2~9 h即可以较高的产率得到3位烯丙基取代的苯并噻咯 (硅茚) 类产物128, 该反应的产率受金催化剂的配体影响较大, 配体以2-(二叔丁基膦) 联苯为最佳.此外, 当 (R=t-Bu) 时, 反应不发生.有趣的是, 他们还以129和底物127是末端炔烃 (R=H) 或位阻较大的非末端炔烃131为底物分别构建了复杂的二聚苯并噻咯类化合物130和132(Scheme 22).紧接着, 他们[49, 50]又设计了烯基硅类底物133, 并实现了其结构中的C—Si键对炔键的分子内加成 (Scheme 22), 成功构建了3位烯基取代的苯并噻咯类化合物134, Murakami等开发的方法为合成苯并噻咯类化合物提供了新路径[51].
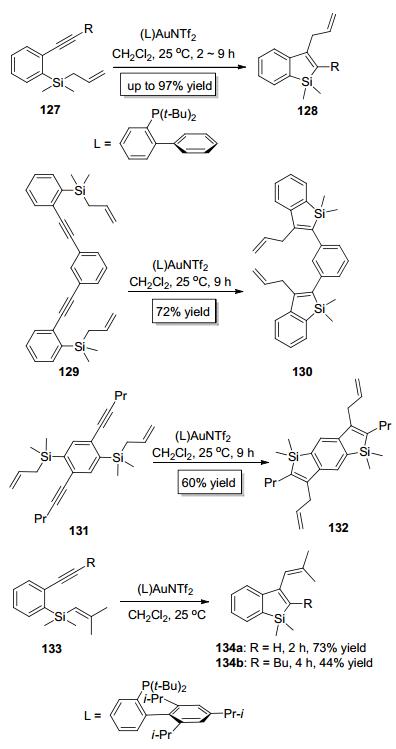 图 图式22
金催化C—Si键对炔键的分子内加成
Figure 图式22.
Au-catalyzed
intramolecular addition of C—Si bond to alkynes
图 图式22
金催化C—Si键对炔键的分子内加成
Figure 图式22.
Au-catalyzed
intramolecular addition of C—Si bond to alkynes
1.6 C—S键对炔键的加成
Yamamoto小组[52]于2004年以PdI2为催化剂实现了硫缩醛类底物135中的C—S键对炔键的分子内加成 (Scheme 23), 以高产率得到了1, 3-双硫代的茚类产物136.紧接着他们[53]采用AuCl为催化剂实现了底物137中的C—S键对炔键的分子内加成 (Scheme 23), 此反应仅需使用2%的AuCl为催化剂, 以二氯甲烷为溶剂在室温即可发生.虽然硫原子上的烷基取代基发生了分子内1, 3-迁移, 但绝大部分手性底物参与反应后, 反应的手性基本可以得到保持.
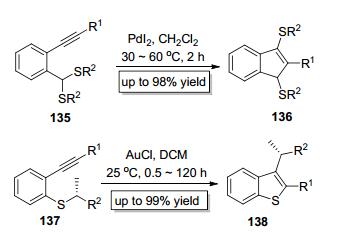 图 图式23
钯或金催化C—S键对炔键的分子内加成
Figure 图式23.
Pd
or Au-catalyzed intramolecular addition of C—S bond to alkynes
图 图式23
钯或金催化C—S键对炔键的分子内加成
Figure 图式23.
Pd
or Au-catalyzed intramolecular addition of C—S bond to alkynes
2005年, Kambe小组[54]报道了Pd (PPh3)4催化的C—S键对炔键的分子内加成 (Scheme 24), 该反应以139为底物, 只需要5%的Pd (PPh3)4作为催化剂, 在甲苯中回流1 h即可以72%的产率得到加成产物, 该反应具有专一的区域选择性和立体化学选择性, 只得到Z式四元内酰胺目标产物140. 2006年, Ogawa小组[55]也以Pd (PPh3)4为催化剂实现了硫氰酸酯141中的C—S键对端炔142的分子间加成 (Scheme 24), 该反应具有专一的区域选择性, 氰基只加成到末端炔碳上, 该反应还具有较高的立体化学选择性, 主要得到顺式加成产物143.该反应首次成功地将硫原子和氰基分别引入到炔基碳的两端, 从而构建了高度官能团化的β位硫取代的α, β-不饱和腈类化合物.
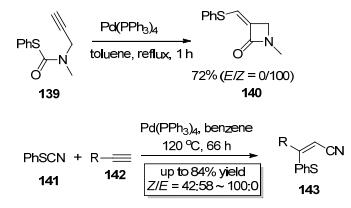 图 图式24
钯催化C—S键对炔键的加成
Figure 图式24.
Pd-catalyzed addition of C—S bond to alkynes
图 图式24
钯催化C—S键对炔键的加成
Figure 图式24.
Pd-catalyzed addition of C—S bond to alkynes
2006年, Nakamura课题组[56]实现了烷氧烷基苯基硫醚类底物144中的C—S键对炔键的分子内加成 (Scheme 25), 该反应以AuCl为催化剂, 以甲苯为溶剂在室温反应2 h即可高产率得到苯并噻吩产物145, 在该反应中, 烷氧烷基作为迁移基团发生了分子内1, 3-迁移.值得一提的是, 该反应同样适用于苄基苯基硫醚类底物146和烯丙基苯基硫醚类底物148(Scheme 25), Nakamura小组发展的方法产率高、底物适用范围广, 为合成官能团化的2, 3-双取代的苯并噻吩类化合物提供了便捷的方法.
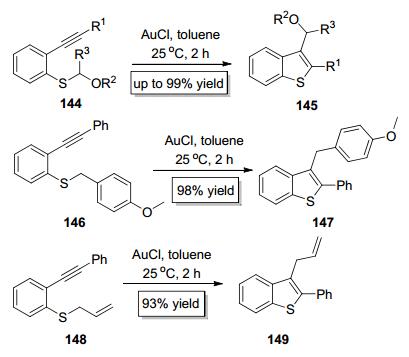 图 图式25
金催化C—S键对炔键的分子内加成
Figure 图式25.
Au-catalyzed
intramolecular addition of C—S bond to alkynes
图 图式25
金催化C—S键对炔键的分子内加成
Figure 图式25.
Au-catalyzed
intramolecular addition of C—S bond to alkynes
2012年, Willis小组[57]实现了铑催化的苯基甲基硫醚150中的C—S键对端炔151的分子间加成 (Eq. 2), 该反应具有专一的区域选择性, 硫原子只加成到非末端炔碳上, 该反应还具有专一的立体化学选择性, 只得到顺式加成产物152.该方法具有选择性好、产率高、底物适用范围广等特点, 能高效地构建官能团化的烯基硫醚类化合物[58].
1.7 C—X (X=Cl, Br, I) 键对炔键的加成
1.8 C—Se键对炔键的加成
2005年, Kambe课题组[54]报道了Pd (PPh3)4催化的硒代酯202中C—Se键对1-辛炔的分子间加成, 此反应具有区域选择性和立体选择性高 (顺式加成) 的特点, 几乎只获得单一构型的目标产物204, 该反应可以实现对炔碳的酰胺化从而构建α, β-不饱和酰胺类化合物 (Scheme 33).他们进一步设计了底物205来考察C—Se键对炔键的分子内加成的可能性, 实验证明C—Se键对炔键的分子内加成同样可行, 该反应不但区域选择性高, 而且立体选择性好 (E:Z=11:89~0:100), 主要生成Z式构型四元内酰胺产物206(Scheme 33).当增加炔基和氮原子之间的碳原子个数至3或4时, 反应仍然可以顺利进行, 只生成Z式构型的六元或七元内酰胺产物 (Scheme 33):有趣的是, 当使用氮原子上连接两个不同碳链长度的炔基取代基的化合物209为底物时, 反应优先生成四元内酰胺产物210而非六元内酰胺产物211 (Scheme 33). Kambe课题组还推测了此反应可能的反应机理 (Scheme 34), 与一价、二价、四价金属催化的类似反应不同, 此类反应可能涉及Pd对C—Se键的氧化加成以及Pd与炔基π电子的配位, Pd或先与C—Se键进行氧化加成生成中间体A5再与炔基π电子配位生成中间体B5, 或Pd先与炔基π电子配位生成中间体A5'再与C—Se键进行氧化加成生成中间体B5, 然后Pd-Se键再对炔键进行加成生成中间体C5, 最后经过还原消除得到产物206并释放出催化剂.
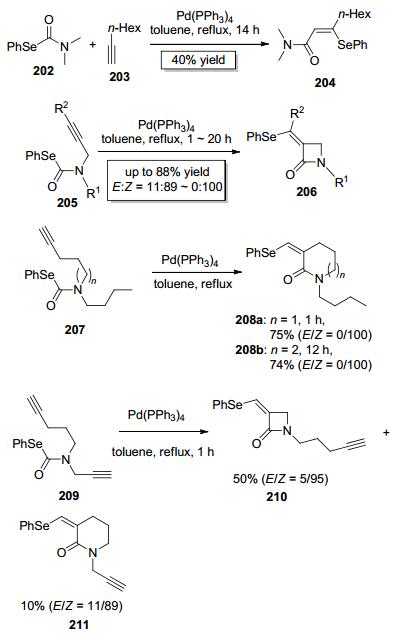 图 图式33
钯催化C—Se键对炔键的加成
Figure 图式33.
Pd-catalyzed addition of C—Se bond to alkynes
图 图式33
钯催化C—Se键对炔键的加成
Figure 图式33.
Pd-catalyzed addition of C—Se bond to alkynes
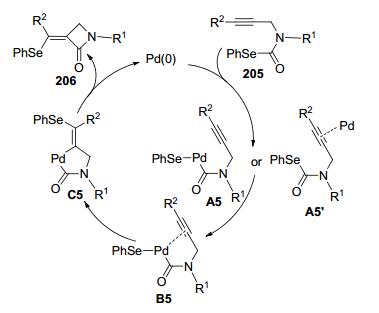 图 图式34
钯催化C—Se键对炔键的加成反应机理
Figure 图式34.
Proposed
reaction mechanism of Pd-catalyzed addition of C—Se bond to alkynes
图 图式34
钯催化C—Se键对炔键的加成反应机理
Figure 图式34.
Proposed
reaction mechanism of Pd-catalyzed addition of C—Se bond to alkynes
1.7.1 C (sp)—Br键对炔键的加成
2010年, Feng小组[59]报道了炔基溴类底物153中的C (sp)—Br键对炔154的分子间加成 (Scheme 26), 该反应以Pd (OAc)2为催化剂, 在乙腈中于30 ℃反应8 h即可以较高收率得到顺式加成产物155, 当使用的是非对称炔时, 反应的区域选择性受炔底物上的取代基影响较大. 2011年, Ohe小组[60]报道了GaCl3催化的溴化氰156中的C (sp)—Br键对炔键的分子间加成 (Scheme 26), 该反应区域选择性极好, 氰基只加成到位阻较小的炔碳上, 该反应立体化学选择性也非常好, 主要以顺式加成的形式得到Z式产物158(Z:E>90:10).该反应成功地在炔基碳的两端分别引入了溴和氰基, 从而构建了高度官能团化的β-溴-α, β-不饱和腈类化合物.
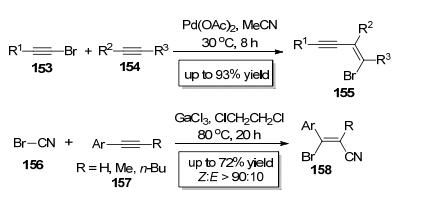 图 图式26
钯或镓催化C (sp)—Br键对炔键的分子间加成
Figure 图式26.
Pd
or Ga-catalyzed intermolecular addition of C (sp)—Br bond to alkynes
图 图式26
钯或镓催化C (sp)—Br键对炔键的分子间加成
Figure 图式26.
Pd
or Ga-catalyzed intermolecular addition of C (sp)—Br bond to alkynes
1.7.2 C (sp2)—X (X=Cl, Br, I) 键对炔键的加成
1998年, Tanaka小组[61]首次实现了氯甲酸甲酯159中的C (sp2)—Cl键对端炔160的分子间加成 (Scheme 27), 该反应以RhCl (COD)(PPh3) 为催化剂, 在甲苯中于110 ℃即可顺利进行, 该反应区域选择性极好, 氯原子只加成到非末端炔碳上; 该反应立体化学选择性也非常好, 主要以顺式加成的形式得到Z式产物161(Z:E=94:6~100:0).随后他们[62]又报道了Rh (acac)(CO)-(AsPh3) 催化的氯乙酰氯162中的C (sp2)—Cl键对端炔163的分子间加成 (Scheme 27), 以相同的区域选择性主要得到了顺式加成产物164(Z:E=53:47~100:0). Tanaka小组发展的方法可以快速而高效地构建高度官能团化的β-氯-α, β-不饱和酯和β-氯-α, β-不饱和酮类化合物, 具有重要的实用价值.
 图 图式27
铑催化C (sp2)—Cl键对炔键的分子间加成
Figure 图式27.
Rh-catalyzed
intermolecular addition of C (sp2)—Cl bond to alkynes
图 图式27
铑催化C (sp2)—Cl键对炔键的分子间加成
Figure 图式27.
Rh-catalyzed
intermolecular addition of C (sp2)—Cl bond to alkynes
2000年, Grigg小组[63]以Pd (OAc)2为催化剂实现了底物165中的C (sp2)—Cl键对炔键的加成, 顺式加成产物不经分离直接与有机锡试剂166发生Stille偶联, 即可一锅快速构建吡咯烷酮或氧化吲哚类化合物167(Scheme 28).当该反应以168为底物时, 可以一锅快速构建七元内酰胺化合物170(Scheme 28).
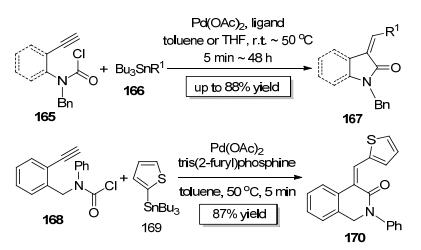 图 图式28
钯催化C (sp2)—Cl键对炔键的分子内加成
Figure 图式28.
Pd-catalyzed
intramolecular addition of C (sp2)—Cl bond to alkynes
图 图式28
钯催化C (sp2)—Cl键对炔键的分子内加成
Figure 图式28.
Pd-catalyzed
intramolecular addition of C (sp2)—Cl bond to alkynes
2009年, Tsuji课题组[64]以IrCl (COD)(IPr) 为催化剂实现了酰氯底物171中C (sp2)—Cl键对端炔172的分子间加成 (Scheme 29).该反应区域选择性极好, 只选择性地生成一种区域加成产物, 此反应的立体选择性也非常好, 几乎只生成顺式加成产物173.有趣的是, 当铱催化剂的配体变成RuPhos后, 该反应会释放出一分子一氧化碳而生成产物174.此外, 他们还采用175和176为反应底物成功构建了2, 5-双取代的呋喃类化合物177(Scheme 29). Tsuji小组开发的方法实现了相同底物在不同催化剂作用下生成不同结构骨架的两种产物, 是值得借鉴的反应条件控制的多样性合成策略范例.
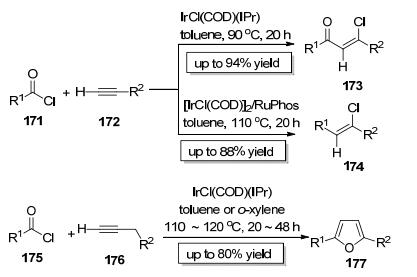 图 图式29
铱催化C (sp2)—Cl键对炔键的分子间加成
Figure 图式29.
Ir-catalyzed
intermolecular addition of C (sp2)—Cl bond to alkynes
图 图式29
铱催化C (sp2)—Cl键对炔键的分子间加成
Figure 图式29.
Ir-catalyzed
intermolecular addition of C (sp2)—Cl bond to alkynes
2015年, Lautens课题组[65]报道了钯催化芳卤类化合物178中的C (sp2)—X (X=Cl, Br, I) 键对非末端炔键的分子内加成 (Scheme 30).当反应温度为50 ℃时, 反应主要选择性得到顺式加成的五元环产物cis-179(Scheme 30), 有趣的是, 当反应温度升高至100 ℃时, 由于顺式加成产物向反式加成产物发生异构, 反应主要选择性得到反式加成的五元环产物trans-180(Scheme 30), 如此, 通过控制反应温度即可选择性得到两个立体异构体.此反应的反应历程涉及钯催化剂对C—X键的氧化加成、C—Pd键对炔键的分子内加成和钯催化剂的还原消除三步, 然而钯催化剂对C—X键的氧化加成一般是受加热促进而且是不可逆的, 也就是说要实现钯催化剂从ArPdⅡX复合物的还原消除是非常具有挑战性的[66]. Lautens等正是在这种挑战下, 利用大位阻的催化剂和大位阻的底物实现了钯催化剂从ArPdⅡX复合物的还原消除, 从而实现芳卤类化合物中的C—X键对炔键的分子内加成, 合成了交叉偶联反应常用的烯基卤类合成砌块.
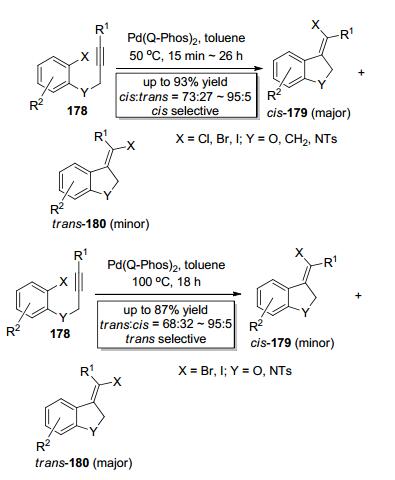 图 图式30
钯催化C (sp2)—X (X=Cl, Br, I) 键对炔键的分子内加成
Figure 图式30.
Pd-catalyzed
intramolecular addition of C (sp2)—X (X=Cl, Br, I) bond to alkynes
图 图式30
钯催化C (sp2)—X (X=Cl, Br, I) 键对炔键的分子内加成
Figure 图式30.
Pd-catalyzed
intramolecular addition of C (sp2)—X (X=Cl, Br, I) bond to alkynes
1.7.3 C (sp3)—X (X=Cl, Br, I) 键对炔键的加成
2009年, Liu课题组[67]报道了苄卤类化合物181中的C (sp3)—X (X=Cl, Br) 键对炔182的分子间加成 (Scheme 31), 该反应以价廉易得的FeCl3•6H2O为催化剂, 在二氯甲烷中于50 ℃反应12 h即可以较高产率主要获得反式加成产物183, 该反应系首次实现铁催化C (sp3)—X (X=Cl, Br) 键对炔键的分子间加成.随后, Hu小组[68]于2015年报道了Cu (OTf)2催化的C—I键对炔键的分子间加成 (Scheme 31), 羰基α位溴代的含有一个叔碳中心的酰胺或酮类底物184先与KI发生卤素置换反应生成羰基α位碘代的中间体, 该中间体结构中的C—I键进一步在Cu (OTf)2催化下与端炔185发生加成反应.该反应区域选择性高, 碘原子只加成到非末端炔碳上, 但该反应立体化学选择性整体上欠佳. Hu小组开发的方法为合成β, γ-不饱和酮和酰胺这两类化合物提供了便捷的路径, 他们还将开发的方法用于组蛋白去乙酰化酶抑制剂 (±)-trichostatin A的合成中.继Hu小组的工作之后, Zhu课题组[69]紧接着报道了CuBr催化的含有一个季碳中心的底物187中的C (sp3)—X (X=Cl, Br) 键对端炔188的分子间加成 (Scheme 31), 值得注意的是, 他们在底物中引入了1~2个强吸电子基团用于活化C (sp3)—X键.该反应的区域选择性极好, 卤原子只加成到非末端炔碳上; 该反应的立体化学选择性非常好, 而且与Hu小组所报道方法的立体化学选择性非相反, 几乎只得到反式加成产物189(trans:cis≥94:6).
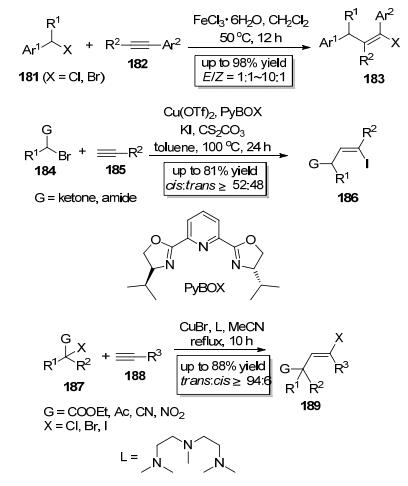 图 图式31
铁或铜催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Figure 图式31.
Fe or Cu-catalyzed intermolecular
addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes
图 图式31
铁或铜催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Figure 图式31.
Fe or Cu-catalyzed intermolecular
addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes
2011年, Zhu课题组[70]以Pd (OAc)2为催化剂实现了烯丙基卤190中的C (sp3)—X (X=Cl, Br) 键对炔基卤191的分子间加成 (Scheme 32), 该反应不但操作简单, 且底物适用范围广、产率高, 而且区域选择性和立体化学选择性极好, 几乎只以顺式加成的形式得到E式产物192.同年他们又报道了PdCl2催化的烯丙基氯193中的C (sp3)—Cl键对炔基乙基醚194的分子间加成 (Scheme 32)[71], 以较高的收率主要得到了顺式加成产物195. 2014年, Poisson小组[72]报道了Cu (OTf)2催化的多氟代底物二氟溴乙酸乙酯196中的C (sp3)—Br键对炔197的分子间加成 (Scheme 32), 尽管该反应立体化学选择性欠佳, 但是成功地将二氟亚甲基引入到了炔基碳上.值得一提的是, Hu小组同时独立地报道了FeBr2催化的多氟取代的碘代烷烃199中的C—I键对炔键的分子间加成 (Scheme 32)[73], 与Poisson小组报道的方法相比, 该方法立体化学选择性高, 主要得到多氟代的反式加成产物201.以上方法都可以快速构建可用于交叉偶联反应的烯基卤这一重要的合成砌块, 尤其是Hu小组发展的方法为合成多氟代的烯基碘提供了实用而高效的方法[74].
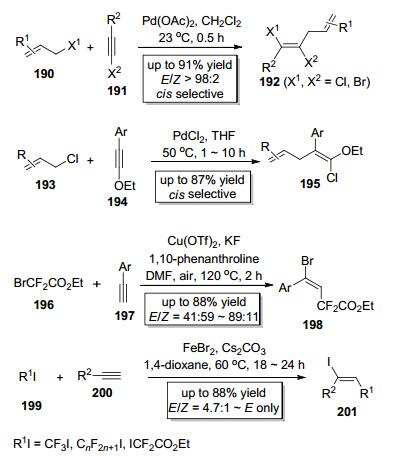 图 图式32
钯、铜、铁催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Figure 图式32.
Pd, Cu, Fe-catalyzed intermolecular addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes
图 图式32
钯、铜、铁催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Figure 图式32.
Pd, Cu, Fe-catalyzed intermolecular addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes
2 碳-杂键对炔键的加成反应在合成天然产物中的应用
Vibsanol (213) 是从植物Viburnum awabuki中分离得到的具有脂质过氧化作用抑制活性的天然产物[75~77], 其母核结构为2, 3, 5, 7位四取代的苯并呋喃, 而且其结构中存在4个裸露的羟基, 羟基被保护基团保护的化合物212是合成Vibsanol的关键前体 (Eq. 3). 2005年, Yamamoto小组[30]构建了底物216并采用铂催化的C—O键对炔键的分子内加成反应一步高效地构建了合成Vibsanol所必需的关键前体217.与之前文献报道的合成Vibsanol的关键前体的方法相比, 该方法避免了羟基的反复保护与脱保护, 缩短了反应步骤, 提高了总收率 (Scheme 35).
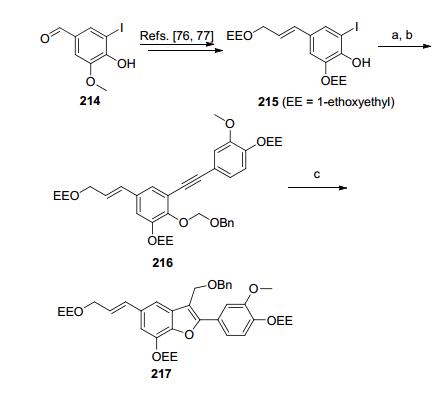 图 图式35
Vibsanol关键前体的合成
Figure 图式35.
Synthesis of the key precursor of Vibsanol
图 图式35
Vibsanol关键前体的合成
Figure 图式35.
Synthesis of the key precursor of Vibsanol
3 结论与展望
随着合成方法学的不断发展, 有机化学家们发展了多种金属, 如Al, Fe, Ni, Cu, Ga, Ru, Rh, Pd, Hf, Ir, Pt, Au, Bi等催化的诸多类型的碳-杂键对炔键的加成反应, 这些加成反应已经超出了传统的简单小分子对炔键的分子间加成反应的范畴, 极大地丰富了加成反应的内涵并拓展了加成反应的应用范围.根据对炔键进行加成的碳-杂键的类型分为C—H, C—B, C—N, C—O, C—Si, C—S, C—X (X=Cl, Br, I), C—Se键这8类并对各类加成反应的反应条件、反应选择性 (区域选择性和立体化学选择性) 和反应机理进行了讨论和总结, 阐述了该类合成方法在天然产物合成中的应用.
尽管金属催化的碳-杂键对炔键的加成反应在近年来取得了重要进展, 然而在此研究领域仍然存在许多挑战.如目前使用的大部分金属催化剂仍是价格较昂贵的Ru, Rh, Pd, Ni, Ir, Pt, Au等催化剂, 而且目前报道的许多类型的碳-杂键对炔键的加成反应仅局限于分子内而不能适用于分子间, 此外还有许多类型的碳-杂键对炔键的加成反应尚未实现.我们相信在不久的将来在此领域会有更多突破性的进展, 使得金属催化的碳-杂键对炔键的加成反应成为对炔键进行官能团化的一种重要策略, 并在有机合成中发挥更加重要的作用.
-
-
[1]
(a) Chinchilla, R.; Nájera, C. Chem. Rev. 2014, 114, 1783.
(b) Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L. J. Chem. Rev. 2015, 115, 2596.
(c) Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873.
(d) Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894.
(e) Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev. 2013, 113, 4905.
(f) Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev. 2016, 116, 3086.
(g) Daniel, F.; Harvey, D. M. S. Chem. Rev. 1996, 96, 271.
(h) Krause, N.; Winter, C. Chem. Soc. Rev. 2011, 40, 1994.
(i) Hirner, J. J.; Shi, Y.; Blum, S. A. Acc. Chem. Res. 2011, 44, 603.
(j) Pina, C. D.; Falletta, E.; Rossi, M. Chem. Soc. Rev. 2012, 41, 350.
(k) Stratakis, M.; Garcia, H. Chem. Rev. 2012, 112, 4469.
(l) Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448.
(m) Liu, L.-P.; Hammond, G. B. Chem. Soc. Rev. 2012, 41, 3129.
(n) Platon, M.; Amardeil, R.; Djakovitch, L.; Hierso, J. C. Chem. Soc. Rev. 2012, 41, 3929.
(o) Shiri, M. Chem. Rev. 2012, 112, 3508.
(p) Abbiati, G.; Marinelli, F.; Rossi, E.; Arcadi, A. Isr. J. Chem. 2013, 53, 856.
(q) Cariou, K.; Ronan, B.; Mignani, S.; Fensterbank, L.; Malacria, M. Angew. Chem., Int. Ed. 2007, 46, 1881.
(r) Hashmi, A. S. K.; Graf, K.; Ackermann, M.; Rominger, F. ChemCatChem 2013, 5, 1200.
(s) Kolundžić, F.; Murali, A.; Pérez-Galán, P.; Bauer, J. O.; Strohmann, C.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2014, 53, 8122.
(t) Tokimizu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Angew. Chem., Int. Ed. 2015, 54, 7862.
(u) Zhu, H.; Yan, B.-Y.; Cao, Y.; Chen, Z.-Y. Chin. J. Org. Chem. 2015, 35, 509 (in Chinese).(朱辉, 严冰玉, 曹杨, 陈知远, 有机化学, 2015, 35, 509.)
(v) Wang, M.-M.; Wang, Z.-X.; Shang, M.; Dai, H.-X. Chin. J. Org. Chem. 2015, 35, 570 (in Chinese).(王明明, 王子潇, 商明, 戴辉雄, 有机化学, 2015, 35, 570.)
(w) Wu, C.-R.; Yang, Y.-P.; Shi, F. Chin. J. Org. Chem. 2015, 35, 770 (in Chinese).(吴春蕊, 杨玉坡, 史峰, 有机化学, 2015, 35, 770.)
(x) Zhou, P.; Qiu, H.-H.; Shi, J.-C. Chin. J. Org. Chem. 2016, 36, 425 (in Chinese).(周鹏, 邱会华, 施继成, 有机化学, 2016, 36, 425.)
(y) Wen, Z.-G.; Tian, C.; Borzov, M. V.; Nie, W.-L Acta Chim. Sinica 2016, 74, 498 (in Chinese).(温志国, 田冲, Borzov, M. V., 聂万丽, 化学学报, 2016, 74, 498.)
(z) Wang, J.; Cui, D.-M. Chin. J. Org. Chem. 2016, 36, 1163 (in Chinese).(王剑, 崔冬梅, 有机化学, 2016, 36, 1163.) -
[2]
Kobayashi, Y.; Kamisaki, H.; Yanada, K.; Yanada, R.; Takemoto, Y. Tetrahedron Lett. 2005, 46, 7549. doi: 10.1016/j.tetlet.2005.08.133
-
[3]
Nakao, Y.; Idei, H.; Kanyiva, K. S.; Hiyama, T. J. Am. Chem. Soc. 2009, 131, 5070. doi: 10.1021/ja901153s
-
[4]
Fujihara, T.; Katafuchi, Y.; Iwai, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2010, 132, 2094. doi: 10.1021/ja910038p
-
[5]
(a) Lindhardt, A. T.; Mantel, M. L. H.; Skrydstrup, T. Angew. Chem., Int. Ed. 2008, 47, 2668.
(b) Hills, I. D.; Fu, G. C. J. Am. Chem. Soc. 2004, 126, 13178.
(c) Grushin, V. V. Chem. Rev. 1996, 96, 2011. -
[6]
Trost, B. M.; Lumb, J. P.; Azzarelli, J. M. J. Am. Chem. Soc. 2011, 133, 740. doi: 10.1021/ja110117g
-
[7]
Trost, B. M.; Taft, B. R.; Masters, J. T.; Lumb, J. P. J. Am. Chem. Soc. 2011, 133, 8502. doi: 10.1021/ja203171x
-
[8]
Xu, H.-D.; Zhang, R.-W.; Li, X.; Huang, S.; Tang, W.; Hu, W.-H. Org. Lett. 2013, 15, 840. doi: 10.1021/ol303531m
-
[9]
Liu, G.; Kong, W.; Che, J.-W.; Zhu, G.-G. Adv. Synth. Catal. 2014, 356, 3314. doi: 10.1002/adsc.201400572
-
[10]
Babu, M. H.; Dwivedi, V.; Kant, R.; Reddy, M. S. Angew. Chem., Int. Ed. 2015, 54, 3783. doi: 10.1002/anie.201411261
-
[11]
For more examples of hydroalkynylation of alkynes, see:
(a) Katayama, H.; Yari, H.; Tanaka, M.; Ozawa, F. Chem. Commun. 2005, 4336.
(b) Bassetti, M.; Pasquini, C.; Raneri, A.; Rosato, D. J. Org. Chem. 2007, 72, 4558.
(c) Jahier, C.; Zatolochnaya, O. V.; N. Zvyagintsev, V. V.; Ananikov, P.; Gevorgyan, V. Org. Lett. 2012, 14, 2846.
(d) Chen, T.; Guo, C.; Goto, M.; Han, L.-B. Chem. Commun. 2013, 49, 7498.
(e) Xu, C.; Du, W.; Zeng, Y.; Dai, B.; Guo, H. Org. Lett. 2014, 16, 948.
(f) Platel, R. H.; Schafer, L. L. Chem. Commun. 2012, 48, 10609.
(g) Midya, G. C.; Parasar, B.; Dhara, K.; Dash, J. Org. Biomol. Chem. 2014, 12, 1812.
(h) Ventre, S.; Derat, E.; Amatore, M.; Aubert, C.; Petit, M. Adv. Synth. Catal. 2013, 355, 2584.
(i) Shen, R.-W.; Chen, K.; Deng, Q.-L.; Yang, J.-J.; Zhang, L.-X. Org. Lett. 2014, 16, 1208. -
[12]
Suginome, M.; Yamamoto, A.; Murakami, M. J. Am. Chem. Soc. 2003, 125, 6358. doi: 10.1021/ja0349195
-
[13]
Suginome, M.; Yamamoto, A.; Murakami, M. Angew. Chem., Int. Ed. 2005, 44, 2380. doi: 10.1002/(ISSN)1521-3773
-
[14]
(a) Harwood, H. J. Jr.; Barbacci-Tobin, E. G.; Petras, S. F.; Lindsey, S.; Pellarin, L. D. Biochem. Pharmacol. 1997, 53, 839.
(b) Morand, P.; Bagli, J. F.; Kraml, M.; Dubuc, J. J. Med. Chem. 1964, 7, 504. -
[15]
Suginome, M.; Shirakura, M.; Yamamoto, A. J. Am. Chem. Soc. 2006, 128, 14438. doi: 10.1021/ja064970j
-
[16]
Shimada, T.; Nakamura, I.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 10546. doi: 10.1021/ja047542r
-
[17]
Nakamura, I.; Sato, Y.; Konta, S.; Terada, M. Tetrahedron Lett. 2009, 50, 2075. doi: 10.1016/j.tetlet.2009.02.108
-
[18]
Wu, C.-Y.; Hu, M.; Liu, Y.; Song, R.-J.; Lei, Y.; Tang, B.-X.; Li, R.-J.; Li, J.-H. Chem. Commun. 2012, 48, 3197. doi: 10.1039/c2cc17887g
-
[19]
Zhao, F.; Zhang, D.-Y.; Nian, Y.; Zhang, L.; Yang, W.; Liu, H. Org. Lett. 2014, 16, 5124. doi: 10.1021/ol5024745
-
[20]
Wu, C.-L.; Zhao, F.; Shu, S.-J.; Wang, J.; Liu, H. RSC Adv. 2015, 5, 90396. doi: 10.1039/C5RA18813J
-
[21]
Wu, C.-L.; Zhao, F.; Du, Y.-L.; Zhao, L.; Chen, L.; Wang, J.; Liu, H. RSC Adv. 2016, 6, 70682. doi: 10.1039/C6RA15945A
-
[22]
Kamijo, S.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 1940. https://www.ncbi.nlm.nih.gov/pubmed/12358538
-
[23]
Majumdar, K. C.; Hazra, S.; Roy, B. Tetrahedron Lett. 2011, 52, 6697. doi: 10.1016/j.tetlet.2011.09.108
-
[24]
Heugebaert, T. S.; Stevens, C. V. Org. Lett. 2009, 11, 5018. doi: 10.1021/ol902031f
-
[25]
Nakamura, I.; Mizushima, Y.; Yamagishi, U.; Yamamoto, Y. Tetrahedron 2007, 63, 8670. doi: 10.1016/j.tet.2007.03.039
-
[26]
Zeng, X.-M.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 942. doi: 10.1002/anie.200905341
-
[27]
Filatova, E. A.; Pozharskii, A. F.; Gulevskaya, A. V.; Ozeryanskii, V. A. J. Org. Chem. 2015, 80, 872. doi: 10.1021/jo502363t
-
[28]
For more examples of addition of C-N bond to alkynes, see: (a) Nakamura, I.; Yamagishi, U.; Song, D.; Konta, S.; Yamamoto, Y. Chem. Asian J. 2008, 3, 285.
(b) Tobisu, M.; Ano, Y.; Chatani, N. Org. Lett. 2009, 11, 3250.
(c) Cacchi, S.; Fabrizi, G.; Pace, Paola. J. Org. Chem. 1998, 63, 1001.
(d) Li, G.-T.; Huang, X.-G.; Zhang, L.-M. Angew. Chem., Int. Ed. 2008, 47, 346. -
[29]
Cacchi, S.; Fabrizi, G.; Moro, L. Tetrahedron Lett. 1998, 39, 5101. doi: 10.1016/S0040-4039(98)00936-8
-
[30]
Nakamura, I.; Mizushima, Y.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 15022. doi: 10.1021/ja055202f
-
[31]
Fürstner, A.; Davies, P. W. J. Am. Chem. Soc. 2005, 127, 15024. doi: 10.1021/ja055659p
-
[32]
Liang, Z.-Q.; Ma, S.-M.; Yu, J.-H.; Xu, R.-R. J. Org. Chem. 2007, 72, 9219. doi: 10.1021/jo7016263
-
[33]
Dubé, P.; Toste, F. D. J. Am. Chem. Soc. 2006, 128, 12062. doi: 10.1021/ja064209+
-
[34]
Nakamura, I.; Chan, C. S.; Araki, T.; Terada, M.; Yamamoto, Y. Org. Lett. 2008, 10, 309. doi: 10.1021/ol702795u
-
[35]
(a) Miki, K.; Nishino, F.; Ohe, K.; Uemura, S. J. Am. Chem. Soc. 2002, 124, 5260.
(b) Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. Chem. Eur. J. 2003, 9, 2627.
(c) Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mourie`s, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656.
(d) Kusama, H.; Miyashita, Y.; Takaya, J.; Iwasawa, N. Org. Lett. 2006, 8, 289. -
[36]
Nakamura, I.; Chan, C. S.; Araki, T.; Terada, M. Yamamoto, Y. Adv. Synth. Catal. 2009, 351, 1089. doi: 10.1002/adsc.v351:7/8
-
[37]
Komeyama, K.; Takahashi, K.; Takaki, K. Org. Lett. 2008, 10, 5119. doi: 10.1021/ol8019567
-
[38]
Shi, Y.; Roth, K. E.; Ramgren, S. D.; Blum, S. A. J. Am. Chem. Soc. 2009, 131, 18022. doi: 10.1021/ja9068497
-
[39]
Ueda, M.; Sato, A.; Ikeda, Y.; Miyoshi, T.; Naito, T.; Miyata, O. Org. Lett. 2010, 12, 2594. doi: 10.1021/ol100803e
-
[40]
Watanabe, K.; Mino, T.; Ikematsu, T.; Hatta, C.; Yoshida, Y.; Sakamoto, M. Org. Chem. Front. 2016, 3, 979. doi: 10.1039/C6QO00112B
-
[41]
For more examples of addition of C-O bond to alkynes, see: (a) Ackermann, M.; Bucher, J.; Rappold, M.; Graf, K.; Rominger, F.; Hashmi, A. S. Chem. Asian J. 2013, 8, 1786.
(b) Hashmi, A. S. K.; Lothschütz, C.; Döpp, R.; Ackermann, M.; Becker, J. D. B.; Rudolph, M.; Scholz, C.; Rominger, F. Adv. Synth. Catal. 2012, 354, 133.
(c) Monteiro, N.; Balme, G. Synlett 1998, 746.
(d) Fürstner, A.; Heilmann, E. K.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 4760.
(e) Tsuda, T.; Ohashi, Y.; Nagahama, N.; Sumiya, R.; Saegusa, T. J. Org. Chem. 1988, 53, 2650.
(f) Obata, T.; Suzuki, S.; Nakagawa, A.; Kajihara, R.; Noguchi, K.; Saito, A. Org. Lett. 2016, 18, 4136. -
[42]
Yeon, S. Ho.; Han, J. S.; Hong, E.; Do, Y.; Jung, I. N. J. Organomet. Chem. 1995, 499, 159. doi: 10.1016/0022-328X(95)00323-I
-
[43]
Asao, N.; Yoshikawa, E.; Yamamoto, Y. J. Org. Chem. 1996, 61, 4874. doi: 10.1021/jo960779o
-
[44]
Yoshikawa, E.; Gevorgyan, V.; Asao, N.; Yamamoto, Y. J. Am. Chem. Soc. 1997, 119, 6781. doi: 10.1021/ja970443b
-
[45]
Asao, N.; Tomeba, H.; Yamamoto, Y. Tetrahedron Lett. 2005, 46, 27. doi: 10.1016/j.tetlet.2004.11.048
-
[46]
Asao, N.; Shimada, T.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3797. doi: 10.1021/ja990385p
-
[47]
Asao, N.; Shimada, T.; Shimada, T.; Yamamoto, Y. J. Am. Chem. Soc. 2001, 123, 10899. doi: 10.1021/ja011316p
-
[48]
Matsuda, T.; Kadowaki, S.; Yamaguchi, Y.; Murakami, M. Chem. Commun. 2008, 2744. http://www.ncbi.nlm.nih.gov/pubmed/18688296
-
[49]
Matsuda, T.; Yamaguchi, Y.; Shigeno, M.; Sato, S.; Murakami, M. Chem. Commun. 2011, 47, 8697. doi: 10.1039/c1cc12457a
-
[50]
Zhou, T.; Xia, Y.-Z. Organometallics 2014, 33, 4230. doi: 10.1021/om500499s
-
[51]
Imamura, K.; Yoshikawa, E.; Gevorgyan, V.; Yamamoto, Y. J. Am. Chem. Soc. 1998, 120, 5339. doi: 10.1021/ja9803409
-
[52]
Nakamura, I.; Bajracharya, G. B.; Wu, H.; Oishi, K.; Mizushima, Y.; Gridnev, I. D.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 15423. doi: 10.1021/ja044603c
-
[53]
Nakamura, I.; Sato, T.; Terada, M.; Yamamoto, Y. Org. Lett. 2008, 10, 2649. doi: 10.1021/ol8007556
-
[54]
Toyofuku, M.; Fujiwara, S.; Shinike, T.; Kuniyasu, H.; Kambe, N. J. Am. Chem. Soc. 2005, 127, 9706. doi: 10.1021/ja052175k
-
[55]
Kamiya, I.; Kawakami, J.; Yano, S.; Nomoto, A.; Ogawa, A. Organometallics. 2006, 25, 3562. doi: 10.1021/om0600442
-
[56]
Nakamura, I.; Sato, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2006, 45, 4473. doi: 10.1002/(ISSN)1521-3773
-
[57]
Hooper, J. F.; Chaplin, A. B.; González-Rodríguez, C.; Thompson, A. L.; Weller, A. S.; Willis, M. C. J. Am. Chem. Soc. 2012, 134, 2906. doi: 10.1021/ja2108992
-
[58]
For more examples of addition of C-S bond to alkynes, see:
(a) Lee, Y. T.; Choi, S. Y.; Chung, Y. K. Tetrahedron Lett. 2007, 48, 5673.
(b) Davies, P. W.; Albrecht, S. J. C. Chem. Commun. 2008, 238. -
[59]
Li, Y.-B.; Liu, X.-H.; Jiang, H.-F.; Feng, Z.-N. Angew. Chem., Int. Ed. 2010, 49, 3338. doi: 10.1002/anie.201000003
-
[60]
Murai, M.; Hatano, Ryo.; Kitabata, S.; Ohe, K. Chem. Commun. 2011, 47, 2375. doi: 10.1039/C0CC04385K
-
[61]
Hua, R.; Shimada, S.; Tanaka, M. J. Am. Chem. Soc. 1998, 120, 12365. doi: 10.1021/ja9822299
-
[62]
Kashiwabara, T.; Fuse, K.; Hua, R.; Tanaka, M. Org. Lett. 2008, 10, 5469. doi: 10.1021/ol802260w
-
[63]
Fielding, M. R.; Grigg, R.; Urchb, C. J. Chem. Commun. 2000, 2239.
-
[64]
Iwai, T.; Fujihara, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2009, 131, 6668. doi: 10.1021/ja901778y
-
[65]
Le, C. M.; Menzies, P. J. C.; Petrone, D. A.; Lautens, M. Angew. Chem., Int. Ed. 2015, 54, 254. doi: 10.1002/anie.201409248
-
[66]
(a) Legault, C. Y.; Garcia, Y.; Merlic, C. A.; Houk, K. N. J. Am. Chem. Soc. 2007, 129, 12664.
(b) Xue, L.; Lin, Z. Chem. Soc. Rev. 2010, 39, 1692.
(c) García-Melchor, M.; Braga, A. A. C.; Lledós, A.; Ujaque, G.; Maseras, F. Acc. Chem. Res. 2013, 46, 2626. -
[67]
Liu, Z.-Q.; Wang, J.-G.; Zhao, Y.-K.; Zhou, B. Adv. Synth. Catal. 2009, 351, 371. doi: 10.1002/adsc.200800708
-
[68]
Xu, T.; Hu, X. Angew. Chem., Int. Ed. 2015, 54, 1307. doi: 10.1002/anie.201410279
-
[69]
Che, C.; Zheng, H.-L; Zhu, G.-G. Org. Lett. 2015, 17, 1617. doi: 10.1021/acs.orglett.5b00546
-
[70]
Chen, X.-Y.; Kong, W.; Cai, H.-T.; Kong, L.-C.; Zhu, G.-G. Chem. Commun. 2011, 47, 2164. doi: 10.1039/c0cc04879h
-
[71]
Cai, H.-T.; Yuan, Z.-L.; Zhu, W.-D.; Zhu, G.-G. Chem. Commun. 2011, 47, 8682. doi: 10.1039/c1cc13424h
-
[72]
Belhomme, M.; Dru, D.; Xiong, H.; Cahard, D.; Besset, T.; Poisson, T.; Pannecoucke, X. Synthesis 2014, 46, 1859. doi: 10.1055/s-00000084
-
[73]
Xu, T.; Cheung, C. W.; Hu, X. Angew. Chem., Int. Ed. 2014, 53, 4910. doi: 10.1002/anie.201402511
-
[74]
For more examples of addition of C-X (X=Cl, Br, I) bonds to alkynes, see:
(a) Ichinose, Y.; Matsunaga, S.-I.; Fugami, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1989, 30, 3155.
(b) Mawson, S. D.; Weavers, R. T. Tetrahedron 1995, 51, 11257.
(c) Tang, Y.; Li, C.-Z. Org. Lett. 2004, 6, 3229.
(d) Kippo, T.; Fukuyama, T.; Ryu, I. Org. Lett. 2010, 12, 4006.
(e) Li, J.-X.; Yang, S.-R.; Wu, W.-Q.; Qi, C.-R.; Deng, Z.-X.; Jiang, H.-F. Tetrahedron 2014, 70, 1516.
(f). Yeh, M.-C. P.; Lin, H.-H.; Kuo, S.-F.; Chen, P.-J.; Hong, J.-W. Adv. Synth. Catal. 2014, 356, 3816.
(g) Wallentin, C.-J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. J. Am. Chem. Soc. 2012, 134, 8875.
(h) Arceo, E.; Montroni, E.; Melchiorre, P. Angew. Chem., Int. Ed. 2014, 53, 120648.
(i) Iwai, T.; Fujihara, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2012, 134, 1268.
(j) Goossen, L. J.; Rodríguez. N.; Goossen, K. Angew. Chem., Int. Ed. 2009, 48, 9592. -
[75]
Fukuyama, Y.; Nakahara, M.; Minami, H.; Kodama, M. Chem. Pharm. Bull. 1996, 44, 1418. doi: 10.1248/cpb.44.1418
-
[76]
Sakai, A.; Aoyama, T.; Shioiri, T. Tetrahedron Lett. 1999, 40, 4211. doi: 10.1016/S0040-4039(99)00714-5
-
[77]
Sasai, A.; Aoyama, T.; Shioiri, T. Heterocycles 2000, 52, 643. doi: 10.3987/COM-99-S63
-
[1]
-

图式1 传统的简单小分子对炔键的加成反应以及当代金属催化的碳-杂键对炔键的加成反应
Scheme 1 Traditional addition of simple molecules to alkynes and modern metal-catalyzed addition of carbon-hetero bonds to alkynes

图式2 铑催化C—H键对炔键的分子内加成
Scheme 2 Rh-catalyzed intramolecular addition of C—H bond to alkynes

图式3 铑催化C—H键对炔键的分子内加成反应机理
Scheme 3 Proposed reaction mechanism of Rh-catalyzed intramolecular addition of C—H bond to alkynes

图式4 钯催化C—H键对炔键的分子间加成
Scheme 4 Pd-catalyzed intermolecular addition of C—H bond to alkynes

图式5 钯或铑催化C—H键对炔键的分子间加成
Scheme 5 Pd or Rh-catalyzed intermolecular addition of C—H bond to alkynes

图式6 钯催化C—H键对炔键的分子间加成
Scheme 6 Pd-catalyzed intermolecular addition of C—H bond to alkynes

图式8 铂催化C—N键对炔键的分子内加成
Scheme 8 Pt-catalyzed intramolecular addition of C—N bond to alkynes

图式9 钌或钯催化C—N键对炔键的分子内加成
Scheme 9 Ru or Pd-catalyzed intramolecular addition of C—N bond to alkynes

图式10 铂、钌、钯催化C—N键对炔键的分子内加成反应机理
Scheme 10 Proposed reaction mechanism of Pt, Ru, Pd-catalyzed intramolecular addition of C—N bond to alkynes

图式11 钯催化C—N键和S—N键对炔键的选择性分子内加成
Scheme 11 Pd-catalyzed selective intramolecular addition of C—N and S—N bonds to alkynes

图式12 零价钯和二价钯分别催化的C—N键和S—N键对炔键的加成反应机理
Scheme 12 Proposed reaction mechanisms of Pd (0)-catalyzed intramolecular addition of C—N bond to alkynes and Pd (Ⅱ)-catalyzed intramolecular addition of S—N bond to alkynes

图式13 钯或金催化C—N键对炔键的分子内加成
Scheme 13 Pd or Au-catalyzed intramolecular addition of C—N bond to alkynes

图式14 钯或金催化C—N键对炔键的分子内加成
Scheme 14 Pd or Au-catalyzed intramolecular addition of C—N bond to alkynes

图式15 钯或铂催化C—O键对炔键的分子内加成
Scheme 15 Pd or Pt-catalyzed intramolecular addition of C—O bond to alkynes

图式16 钯催化C—O键对炔键的分子内加成
Scheme 16 Pd-catalyzed intramolecular addition of C—O bond to alkynes

图式17 金催化C—O键对炔键的分子内加成
Scheme 17 Au-catalyzed intramolecular addition of C—O bond to alkynes

图式18 铂催化C—O键对炔键的分子内加成
Scheme 18 Pt-catalyzed intramolecular addition of C—O bond to alkynes

图式19 铋、金、钯催化C—O键对炔键的分子内加成
Scheme 19 Bi, Au, Pd-catalyzed intramolecular addition of C—O bond to alkynes

图式20 铝催化C—Si键对炔键的分子间加成
Scheme 20 Al-catalyzed intermolecular addition of C—Si bond to alkynes

图式21 铝或铪催化C—Si键对炔键的分子内加成
Scheme 21 Al or Hf-catalyzed intramolecular addition of C—Si bond to alkynes

图式22 金催化C—Si键对炔键的分子内加成
Scheme 22 Au-catalyzed intramolecular addition of C—Si bond to alkynes

图式23 钯或金催化C—S键对炔键的分子内加成
Scheme 23 Pd or Au-catalyzed intramolecular addition of C—S bond to alkynes

图式25 金催化C—S键对炔键的分子内加成
Scheme 25 Au-catalyzed intramolecular addition of C—S bond to alkynes

图式26 钯或镓催化C (sp)—Br键对炔键的分子间加成
Scheme 26 Pd or Ga-catalyzed intermolecular addition of C (sp)—Br bond to alkynes

图式27 铑催化C (sp2)—Cl键对炔键的分子间加成
Scheme 27 Rh-catalyzed intermolecular addition of C (sp2)—Cl bond to alkynes

图式28 钯催化C (sp2)—Cl键对炔键的分子内加成
Scheme 28 Pd-catalyzed intramolecular addition of C (sp2)—Cl bond to alkynes

图式29 铱催化C (sp2)—Cl键对炔键的分子间加成
Scheme 29 Ir-catalyzed intermolecular addition of C (sp2)—Cl bond to alkynes

图式30 钯催化C (sp2)—X (X=Cl, Br, I) 键对炔键的分子内加成
Scheme 30 Pd-catalyzed intramolecular addition of C (sp2)—X (X=Cl, Br, I) bond to alkynes

图式31 铁或铜催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Scheme 31 Fe or Cu-catalyzed intermolecular addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes

图式32 钯、铜、铁催化C (sp3)—X (X=Cl, Br, I) 键对炔键的分子间加成
Scheme 32 Pd, Cu, Fe-catalyzed intermolecular addition of C (sp3)—X (X=Cl, Br, I) bond to alkynes

图式34 钯催化C—Se键对炔键的加成反应机理
Scheme 34 Proposed reaction mechanism of Pd-catalyzed addition of C—Se bond to alkynes
-


 下载:
下载:


































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 19
- 文章访问数: 1927
- HTML全文浏览量: 364
 下载:
下载:
