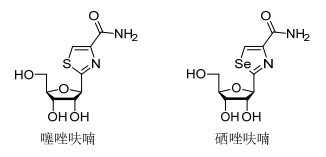
 图 1
含有噻唑呋喃和硒唑呋喃的分子
Figure 1.
Molecules containing thiazole furan and selenazole furan
图 1
含有噻唑呋喃和硒唑呋喃的分子
Figure 1.
Molecules containing thiazole furan and selenazole furan
引用本文:
张成路, 孙晓娜, 蒲雨昕, 李传银, 孙丽杰, 王静, 李益政. 1, 3-硒唑为模板多种杂环修饰的新型分子的设计、合成及活性研究[J]. 有机化学,
2017, 37(2): 440-454.
doi:
10.6023/cjoc201607024

Citation: Zhang Chenglu, Sun Xiaona, Pu Yuxin, Li Chuanyin, Sun Lijie, Wang Jing, Li Yizheng. Design, Synthesis and Activities of Multiheterocyclic Modified Novel Molecules Using 1, 3-Selenazole as Template[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 440-454. doi: 10.6023/cjoc201607024
Citation: Zhang Chenglu, Sun Xiaona, Pu Yuxin, Li Chuanyin, Sun Lijie, Wang Jing, Li Yizheng. Design, Synthesis and Activities of Multiheterocyclic Modified Novel Molecules Using 1, 3-Selenazole as Template[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 440-454. doi: 10.6023/cjoc201607024
1, 3-硒唑为模板多种杂环修饰的新型分子的设计、合成及活性研究
English
Design, Synthesis and Activities of Multiheterocyclic Modified Novel Molecules Using 1, 3-Selenazole as Template
Abstract:
Six kinds, twenty-two novel target molecules were first designed and synthesized by using substituted 1, 3-selenazole as a template, which were modified by 1, 2, 4-triazole, tetrazole, oxadiazole, pyrazole, 1, 2, 4-triazine and succinic imide respectively. Their structures were confirmed by IR, NMR and HRMS. The inhibitory activities of the target molecules against cell division cycle 25B phosphatase (Cdc25B) were evaluated. As a result, thirteen compounds exhibited good inhibitory activities. The IC50 values of five compounds were lower than the positive reference Na3VO4 and were expected to be anticancer drugs leading compounds. The analysis of structure-activity relationship found that the introduction of multinitrgon-heterocyclic triazole, tetrazole and triazine, thiadiazole and oxadiazole containing amino or mercapto group onto 1, 3-selenazole were expected to obtain novel excellent bioactivity organic selenium containing molecules.
-
Key words:
- 1, 3-selenazole
- / multi-heterocycle
- / Cdc25B inhibitor
-
噁唑和噻唑是众多天然产物和临床药物中重要的药效基团.含噁唑和噻唑的分子具有广泛的生物活性, 如抗菌[1~3]和抗肿瘤[4~7]等.该类分子在生化反应中易代谢, 且本质是非致癌的[8], 因此具有噁唑和噻唑分子的设计合成已成为药物筛选的重要途径.硒是人体必需微量元素, 是重要的食物源抗氧化剂, 缺硒会引起如癌症、心脏病、关节炎和免疫系统紊乱等疾病, 流行病学调查结果表明, 适量补硒有益于人体健康等[9, 10].尤其是近年来的研究结果表明, 有机硒化物具有优良的抗肿瘤细胞增殖活性, 是蛋白激酶活化剂、抗氧化剂, 具有抗炎和抗菌活性[11~13]等, 因此新型有机硒化物的研究引起了广泛的关注.由于硒、硫和氧元素为同族元素, 理化性质相似, 用硒元素替代噻唑和噁唑结构中的硫元素和氧元素, 一方面可为人体补充必须的微量元素硒, 另一方面根据生物电子等排原理, 可以保持分子的构型和电子云分布基本不变, 同时硒元素的引入可以影响有机分子的化学性质并提高其生物活性, 如噻唑呋喃类似物硒唑呋喃 (图 1), 在体外及体内的抗肿瘤活性筛选中均优于噻唑呋喃[14], 表明1, 3-硒唑具有更为优异的化学和生物学特性, 因此设计合成含有硒唑结构基元的新型分子, 探究不同结构硒唑衍生物的合成策略, 无疑具有重要的理论创新意义, 并为筛选活性优良的硒唑衍生物奠定基础.
图 1
含有噻唑呋喃和硒唑呋喃的分子
Figure 1.
Molecules containing thiazole furan and selenazole furan
在药用活性分子的研究中, 含不同杂环结构分子的设计合成已成为药物创制的重要方法, 其中吡唑、三唑、噁二唑、噻唑和四唑等活性结构的引入, 已成为药用分子设计的重中之重.吡唑衍生物因其优良的抗肿瘤活性[15~17]等, 显示了化学治疗的潜力; 三唑衍生物因具有杀虫、除草、抗菌、促进和调节农作物生长功效, 以及抗炎、抗病毒和抗肿瘤等生物活性, 已在农业和医药等领域得到了广泛的应用[18~23]; 1, 3, 4-噁二唑衍生物也表现出优异的抗肿瘤活性[24~27]; 在富有优良活性的多种天然产物和大量与医学和生物学特性相关的有意义的药物中, 噻唑是其核心结构, 该类分子能有效治疗高血压、精神分裂症、炎症、过敏和HIV感染细菌等症[28~34]; 含四唑杂环的分子在抗癌、抗血压、抗过敏和抗菌性等方面得到广泛的应用, 使四唑类药物合成筛选具有很大的发展潜能[35~37]; 近年来, 环状亚胺类化合物因其广泛生物活性[38~43], 结构的多样性和广谱的生物活性已被确定为最有前途的骨架, 成为学科领域的研究热点[44].
为了构筑以硒唑为核心骨架的含不同杂环的新型分子, 研究同一模板下不同杂环药效基团同存于一分子中的合成策略, 本文以2-对甲氧苯基-1, 3-硒唑为模板, 将吡唑、三唑、噁二唑、噻唑和四唑等优良药效杂环结构通过经典的方法共筑于不同分子中, 创建22个不同结构的新型杂环分子, 并通过其对细胞分裂周期蛋白 (cell division cycle 25, 缩写为Cdc25B) 双位点磷酸酯酶抑制作用, 对不同结构的分子进行抗癌活性评价.
1 结果与讨论
1.1 目标化合物的合成与表征
通过1H NMR、13C NMR、IR和HRMS等波谱技术对22种目标分子进行结构表征:在IR谱图中, 3100和1600~1450 cm-1的吸收峰表明目标分子的结构中存在芳香环.在1H NMR测试中, δ 7.00~8.00出现芳香环的质子信号, 在δ 3.8左右处出现对甲氧基的特征峰, 在δ 2.7左右处出现与硒唑环相连接的甲基的特征峰.在13C NMR谱图中, 在δ 110~185范围内出现的信号峰为芳香环和硒唑环的碳信号, 在δ 55左右为对甲氧基碳信号峰, 在δ 18.2左右为硒唑环上甲基的碳信号峰.在HRMS谱图中, 目标分子均出现了[M+1]+峰.目标分子的合成路线如Schemes 1~7所示.
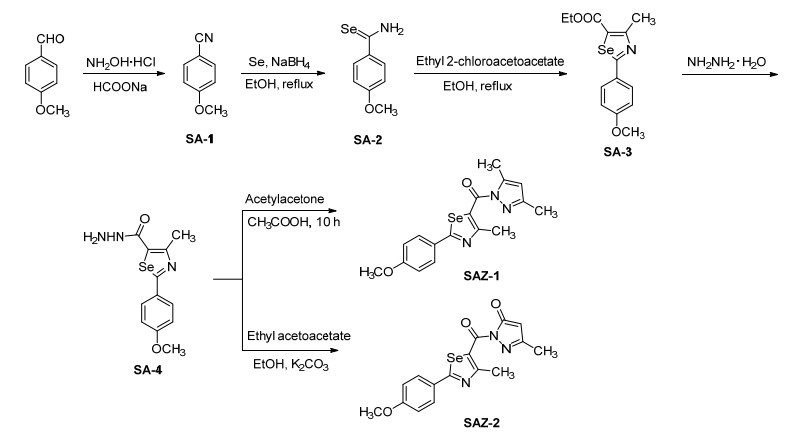 图 图式1
合成含吡唑的化合物SAZ-1~SAZ-2
Figure 图式1.
Synthesis of pyrazole containing compounds SAZ-1~SAZ-2
图 图式1
合成含吡唑的化合物SAZ-1~SAZ-2
Figure 图式1.
Synthesis of pyrazole containing compounds SAZ-1~SAZ-2
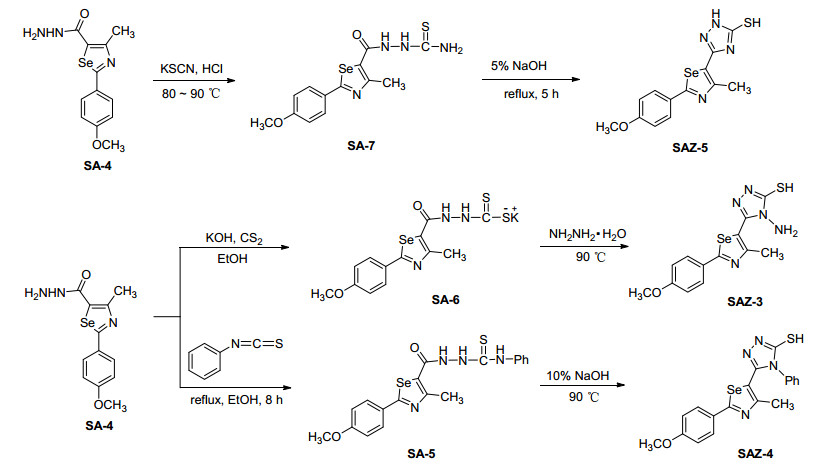 图 图式2
合成含1, 2, 4-三唑的化合物SAZ-3~SAZ-5
Figure 图式2.
Synthesis of 1, 2, 4-triazole containing compounds SAZ-3~SAZ-5
图 图式2
合成含1, 2, 4-三唑的化合物SAZ-3~SAZ-5
Figure 图式2.
Synthesis of 1, 2, 4-triazole containing compounds SAZ-3~SAZ-5
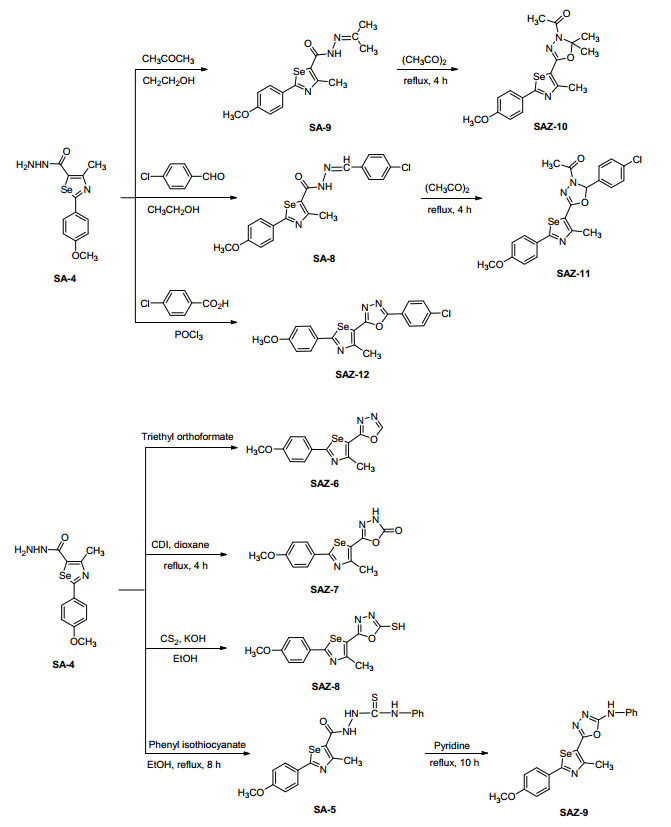 图 图式3
合成含噁二唑的化合物SAZ 6~SAZ-12
Figure 图式3.
Synthesis of oxadiazole containing compounds SAZ 6~SAZ-12
图 图式3
合成含噁二唑的化合物SAZ 6~SAZ-12
Figure 图式3.
Synthesis of oxadiazole containing compounds SAZ 6~SAZ-12
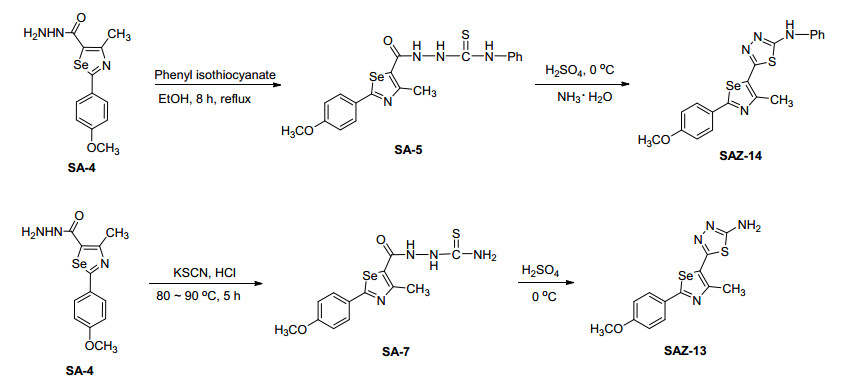 图 图式4
合成含噻二唑的化合物SAZ-13~SAZ-14
Figure 图式4.
Synthesis of thiadiazole containing compounds SAZ-13~SAZ-14
图 图式4
合成含噻二唑的化合物SAZ-13~SAZ-14
Figure 图式4.
Synthesis of thiadiazole containing compounds SAZ-13~SAZ-14
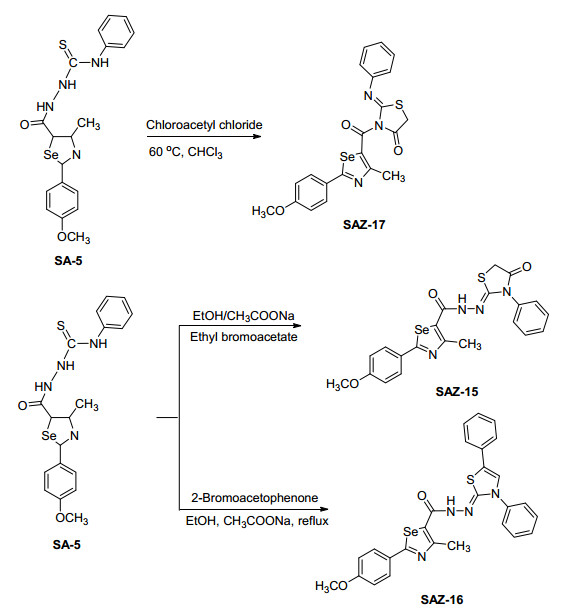 图 图式5
合成含噻唑环的化合物SAZ-15~SAZ-17
Figure 图式5.
Synthesis of the thiazole ring-containing compounds SAZ-15~SAZ-17
图 图式5
合成含噻唑环的化合物SAZ-15~SAZ-17
Figure 图式5.
Synthesis of the thiazole ring-containing compounds SAZ-15~SAZ-17
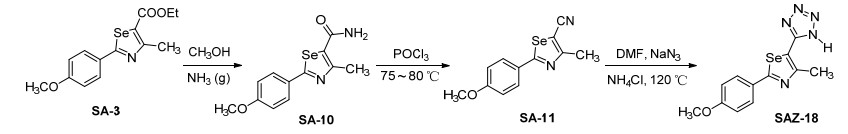 图 图式6
合成含四唑环的化合物SAZ-18
Figure 图式6.
Synthesis of the tetrazole ring-containing compounds SAZ-18
图 图式6
合成含四唑环的化合物SAZ-18
Figure 图式6.
Synthesis of the tetrazole ring-containing compounds SAZ-18
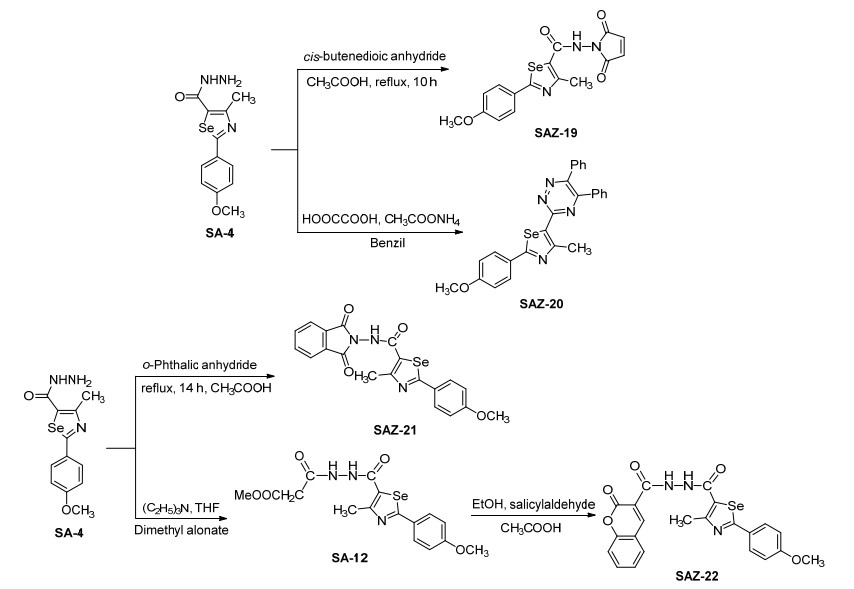 图 图式7
合成其他杂环化合物SAZ-19~SAZ-22
Figure 图式7.
Synthesis other heterocyclic compounds SAZ-19~SAZ-22
图 图式7
合成其他杂环化合物SAZ-19~SAZ-22
Figure 图式7.
Synthesis other heterocyclic compounds SAZ-19~SAZ-22
1.2 目标化合物的生物活性测试
应用大肠杆菌表达系统表达Cdc25B催化结构域, 以GST融合蛋白形式存在于清液中, 细胞裂解后经GSH-亲和柱纯化, 获得活性GST融合的Cdc25B蛋白.采用荧光底物OMFP考察不同化合物对重组酶的活性抑制情况. OMFP水解产物OMF被485 nm激发光激发后可发射出波长为530 nm的荧光检测信号, 从而考察酶的活性变化及化合物对其抑制情况.
目标分子对Cdc25B的抑制活性测试由国家新药筛选中心完成.在底物浓度为5 μg/mL时, 设置三个复孔, 对样品的活性进行初筛测试.选择抑制率大于50%的化合物进行复筛, 测试样品浓度为5 μg/mL时的抑制活性, 得出抑制活性剂量依赖关系, 即IC50值. IC50值通过样品活性对样品浓度进行非线性拟和得到, 筛选实验方法按照参考文献[45~47]的方法进行.测试结果如表 1所示.
 表 1
目标化合物对Cdc25B的抑制活性
Table 1.
Inhibition of target compounds against Cdc25B
表 1
目标化合物对Cdc25B的抑制活性
Table 1.
Inhibition of target compounds against Cdc25B
Compd. Cdc25B Inhibition ratea/% IC50b/(μg•mL-1) SAZ-1 84.30±4.52 1.82±0.65 SAZ-2 78.06±7.06 1.89±0.14 SAZ-3 96.07±6.88 1.95±0.34 SAZ-4 87.07±0.46 2.04±0.22 SAZ-5 73.30±5.05 4.85±0.29 SAZ-6 18.8±3.32 NA SAZ-7 25.6±3.08 NA SAZ-8 31.99±3.13 NA SAZ-9 87.23±7.64 1.46±0.45 SAZ-10 28.77±4.56 NA SAZ-11 20.76±1.12 NA SAZ-12 16.18±9.22 NA SAZ-13 75.15±4.16 1.75±0.39 SAZ-14 105.04±2.10 1.25±0.26 SAZ-15 77.62±7.62 3.25±0.45 SAZ-16 9.48±3.41 NA SAZ-17 15.86±3.89 NA SAZ-18 90.57±6.85 1.46±0.19 SAZ-19 83.23±1.03 3.93±0.16 SAZ-20 74.21±2.45 4.34±0.36 SAZ-21 81.27±3.94 2.66±0.14 SAZ-22 51.53±6.14 NA Na3VO4 — 1.86±0.24 a待测样品浓度为20 μg/mL时的抑制率; b IC50值通过待测样品在浓度为5 μg/mL时的抑制率非线性拟和计算得到 表 1 目标化合物对Cdc25B的抑制活性
Table 1. Inhibition of target compounds against Cdc25B对22种含硒唑的杂环化合物进行了抑制Cdc25B的活性的研究, 结果表明, 13种化合物对Cdc25B具有良好的抑制活性, 其中SAZ-14的抑制率最高, 达到105.04%.在进一步测试IC50值的结果中, SAZ-1、SAZ-9、SAZ-13、SAZ-14和SAZ-18共有5种目标分子的抑制活性高于阳性参照物Na3VO4 [(1.86±0.24) μg/mL], 其IC50值分别为1.82±0.65、1.46±0.45、1.75±0.39、1.25±0.26和1.46±0.19 μg/mL.分析化合物结构发现, 以对甲氧苯基取代的1, 3-硒唑为模板, 当在结构中分别引入吡唑、含氨基或巯基的1, 2, 4-三唑、含酰亚氨基的1, 3, 4-噻二唑、含亚氨基的1, 3, 4-噁二唑、1, 2, 4-三嗪和顺丁二烯酰亚胺等时, 目标分子均具有较高的抑制活性.
2 结论
由于Cdc25B的活性中心具有亲核基团, 易受到亲电子化合物的攻击.已报道的一类亲电子抑制剂[48], 其抑制作用产生的原因是通过它们的亲电子基团进攻活性中心的半胱氨酸残基.目标分子SAZ-19和SAZ-21中附加的环状酰亚胺基元是具有较好的亲电性, 因此表达出优良的抑制活性.在SAZ-16、SAZ-17和SAZ-22分子中, 虽然含有酰亚胺等亲电子基团, 但因其分子具有较大的空间位阻, 加大了亲电基团与活性基团的靶点的相互作用的难度, 所以表现出对Cdc25B的抑制活性较低.含多氮杂环1, 2, 4-三唑 (SAZ-3~SAZ-5)、四唑 (SAZ-18) 和1, 2, 4-三嗪 (SAZ-20), 以及含吡唑 (SAZ-18) 和噻二唑 (SAZ-13和SAZ-14) 药效基团的目标分子, 因与生物大分子中杂原子作用位点增多, 可以引起细胞的G2/M期阻滞, 从而抑制细胞增殖, 达到较好的抗癌活性效果.值得关注的是含有氨基、巯基和苯亚氨基的目标分子SAZ-3~SAZ-5、SAZ-9、SAZ-13和SAZ-5均表现出优良的活性, 其可能原因是目标分子与生物大分子之间产生了氢键的作用, 使活性得到了提高.
综上所述, 芳基取代的1, 3-硒唑分别与吡唑、含氨基或巯基的12, 4-三唑、含酰亚氨基的1, 3, 4-噻二唑、含苯亚氨基的1, 3, 4-噁二唑、1, 2, 4-三嗪、四唑和酰亚胺等拼合, 表现出较优良的抑制Cdc25B活性, 有望成为抗肿瘤药物的先导物, 研究结果为创建和筛选新型硒唑药物分子提供了重要的参考.
3 实验部分
3.1 仪器与试剂
对甲氧基苯甲醛、对氯苯甲醛、水杨醛、对氯苯甲酸、硒粉、硼氢化钠、2-氯乙酰乙酸乙酯、乙酰乙酸乙酯、原甲酸三乙酯、异硫氰酸苯酯、乙酰丙酮、氯乙酰氯、2-溴苯乙酮、三氯氧磷、邻苯二甲酸酐、顺丁烯二酸酐、水合肼、二硫化碳、N, N-羰基二咪唑 (CDI) 和叠氮化钠 (国药集团化学试剂有限公司), 所用试剂均为分析纯.
AVANCE 500 MHz NMR核磁共振波谱仪; Accu-rate-Mass TOF LC/MS型精确质量液相色谱飞行时间质谱联用仪; TENSOR 27傅立叶变换红外光谱仪; X-5型显微熔点测定仪, 温度未校正; WFH-203B型三用紫外分析仪.
3.2 化合物SA-1~SA-12的合成
中间体SA-1根据文献[49]的方法合成, 中间体SA-2根据文献[50]合成, 所得数值与文献值一致.
3.3 化合物SAZ-1~SAZ-22的合成
3.2.3 2-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羰基)-N-苯基硫代氨基脲 (SA-5) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入10 mL乙醇, 温度升至78 ℃, 搅拌至完全溶解, 加入0.2 mL (0.015 mol) 异硫氰酸苯酯, 搅拌, 体系为浅黄色澄清溶液.反应30 min后, 黄色固体逐渐析出, 继续回流反应, 用TLC监测反应, 反应时间约为8 h.反应完成后, 静置冷却至室温, 将固体抽滤, 得粗产品并用乙醇重结晶得浅黄色固体0.312 g, 收率70%. m.p. 211.5~212.3 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.03 (s, 1H, NH), 10.16 (s, 1H, NH), 9.81 (s, 1H, NH), 7.94 (d, J=8.75 Hz, 2H, PhH), 7.35 (t, J=7.75 Hz, 3H, PhH), 7.18 (d, J=7.70 Hz, 2H, PhH), 7.05 (t, J=8.70 Hz, 2H, PhH), 3.85 (s, 3H, OCH3), 2.65 (s, 3H, CH3); IR (KBr) ν: 3220, 3030, 2936, 1680, 1600, 1520, 1465, 1250, 845 cm-1. HRMS (positive-ESIMS) calcd for C19H18N4O2SSe (M+1)+446.0316, found 446.0322.
3.2.10 3-(2-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羰基) 肼)-3-甲酰乙酸甲酯 (SA-12) 的合成
取70 μL (0.0015 mol) 丙二酸二甲酯和8 mL THF于三颈瓶中, 充分搅拌, 温度升至66 ℃, 加入80 μL (0.0015 mol) 三乙胺, 搅拌30 min后, 加入0.31 g (0.001 mol) 中间体SA-4, 搅拌完全溶解, 体系为黄色溶液, 回流, 用TLC监测反应, 反应时间约为5 h.反应停止后, 静置冷却至室温, 将溶剂减压蒸去, 得橘黄色固体0.287 g, 收率: 70%. m.p. 276.8~277.9 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.12 (s, 1H, NH), 9.80 (s, 1H, NH), 7.72 (d, J=8.75 Hz, 2H, PhH), 7.05 (t, J=8.70 Hz, 2H, PhH), 3.85 (s, 3H, OCH3), 3.64 (s, 3H, CH3), 3.50 (s, 2H, CH2), 2.65 (s, 3H, CH3); IR (KBr) ν: 3267, 3030, 2940, 1680, 1600, 1500, 1455, 1390, 1250, 850, 730 cm-1. HRMS (positive-ESIMS) calcd for C16H17N3O5Se (M+1)+ 411.0333, found 411.0339.
3.2.5 2-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羰基) 硫代氨基脲 (SA-7) 的合成
取0.31 g (0.001 mol) 的中间体SA-4于三颈瓶中, 加入15 mL 7%的稀盐酸, 搅拌, 加入0.19 g (0.002 mol) KSCN, 温度升至90 ℃, 搅拌至完全溶解, 反应4 h后, 有固体析出, 用TLC监测反应.反应停止后, 体系静置冷却至室温, 抽滤, 得黄绿色固体, 用二氧六环重结晶得浅黄色棒状晶体0.221 g, 收率: 60%. m.p. 287.5~289.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.65 (s, 2H, NH2), 10.16 (s, 1H, NH), 9.81 (s, 1H, NH), 7.94 (d, J=8.75 Hz, 2H, PhH), 7.45 (d, J=8.70 Hz, 2H, PhH), 3.86 (s, 3H, OCH3), 2.67 (s, 3H, CH3); IR (KBr) ν: 3420, 3250, 3030, 2936, 1680, 1600, 1520, 1465, 1253, 840 cm-1. HRMS (positive-ESIMS) calcd for C13H14N4O2SSe (M+1)+ 370.0003, found 370.0010.
3.2.6 N'-(4-氯苯亚甲基)-2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-碳酰肼 (SA-8) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入10 mL乙醇搅拌至完全溶解, 温度升至78 ℃, 再加入0.17 g (0.011 mol) 对氯苯甲醛, 搅拌溶解, 反应4.5 h后, 加入3~4滴冰醋酸, 有黄绿色固体析出, 继续搅拌, 再反应1 h, 用TLC监测反应.反应停止后, 将混合物倒入40 mL冰水中, 搅拌, 有固体析出, 抽滤, 得黄色固体, 用二氧六环重结晶得黄色固体0.368 g, 收率: 85%. m.p. 245.7~247 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.65 (s, 1H, NH), 10.07 (s, 1H, CH), 7.86 (d, J=8.60 Hz, 4H, PhH), 7.04 (d, J=8.75 Hz, 4H, PhH), 3.84 (s, 3H, OCH3), 2.76 (s, 3H, CH3); IR (KBr) ν: 3260, 3030, 2936, 1673, 1603, 1500, 1465, 1253, 841, 720 cm-1. HRMS (positive-ESIMS) calcd for C19H16ClN3O2Se (M+1)+ 433.0096, found 433.0103.
3.2.4 2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-硫钾盐 (SA-6) 的合成
室温条件下, 取0.12 g (0.001 mol) KOH于三颈瓶中, 加入10 mL乙醇, 搅拌至完全溶解后, 加入0.31 g (0.001 mol) 中间体SA-4, 搅拌, 缓慢滴加用2 mL乙醇稀释的0.2 mL (0.0015 mol) CS2, 滴加完毕后, 体系为橘黄色的澄清溶液, 搅拌10 min后, 逐渐有黄色固体析出, 反应时间为10 h.将固体抽滤, 无水乙醇多次洗涤, 得橘黄色固体0.369 g, 收率: 87%. m.p.>300 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.95 (s, 1H, NH), 10.18 (s, 1H, NH), 11.07 (s, 1H, SH), 7.65 (d, J=8.60 Hz, 2H, PhH), 7.34 (d, J=8.75 Hz, 2H, PhH), 3.85 (s, 3H, OCH3), 2.66 (s, 3H, CH3); IR (KBr) ν: 3240, 3030, 2936, 2560, 1675, 1610, 1520, 1460, 1255, 840 cm-1. HRMS (positive-ESI-MS) calcd for C13H12KN3O2S2Se (M+1)+ 424.9173, found 424.9169.
3.2.2 2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-碳酰肼 (SA-4) 的合成
取0.972 g (0.003 mol) 的中间体SA-3于三颈瓶中, 加入10 mL乙醇搅拌溶解, 温度升至78 ℃, 缓慢滴加7 mL水合肼, 搅拌, 无色溶液逐渐变红, 回流, 溶液颜色最后为红色, 用TLC监测反应, 反应时间约为14 h.反应完成后趁热过滤, 滤液冷却至室温, 放入冰箱中冷冻一夜, 析出固体, 抽滤得灰色固体, 固体用20 mL乙醇重结晶得白色针状晶体0.558 g, 收率60%. m.p. 192.1~193.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.86 (d, J=8.80 Hz, 2H, PhH), 6.94 (d, J=8.80 Hz, 2H, PhH), 6.93 (s, 1H, NH), 4.82 (s, 2H, NH2), 3.86 (s, 3H, OCH3), 2.74 (s, 3H, CH3); IR (KBr) ν: 3440, 3288, 3010, 2924, 1673, 1599, 1500, 1465, 1240, 845 cm-1. HRMS (positive-ESI-MS) calcd for C12H13N3O2Se (M+1)+ 311.0173, found 311.0182.
3.2.1 2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羧酸乙酯 (SA-3) 的合成
在室温条件下, 取0.4 g (0.0019 mol) 的中间体SA-2于三颈瓶中, 加入15 mL绝对无水乙醇搅拌溶解后, 滴加0.265 mL (0.0019 mol) 2-氯乙酰乙酸乙酯, 滴加完毕后, 溶液为黄绿色, 搅拌15 min, 转入油浴中, 温度缓慢升至78 ℃, 回流, 用薄层色谱 (TLC) 监测反应, 反应时间约为2 h.反应停止后, 趁热过滤, 冷却至室温, 滤液为黄绿色, 倒入50 mL冰水混合物中, 搅拌, 有浅粉色固体析出, 抽滤, 固体用20 mL乙醇重结晶得浅粉色针状晶体0.505 g, 收率82%. m.p. 95.1~96.0 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.86 (d, J=8.80 Hz, 2H, PhH), 6.94 (d, J=8.80 Hz, 2H, PhH), 4.34 (q, J=7.10 Hz, 2H, CH2), 3.86 (s, 3H, OCH3), 2.74 (s, 3H, CH3), 1.38 (t, J=7.15 Hz, 3H, CH3); IR (KBr) ν: 3080, 2936, 1670, 1599, 1465, 1250, 841 cm-1. HRMS (positive-ESIMS) calcd for C14H15NO3Se (M+1)+ 325.0217, found 325.0226.
3.2.8 2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-甲酰胺 (SA-10) 的合成
在冰浴条件下, 向0.64 g (0.002 mol) 的中间体SA-3的甲醇浊液中通入氨气, 搅拌, 固体逐渐溶解, 体系为黄色澄清溶液, 继续通氨气, 时间约为6 h后, 转移至室温, 继续搅拌反应24 h.反应停止后, 将氨气减压抽出, 蒸去溶剂, 得粗产品并用3:1 (V石油醚:V乙酸乙酯) 的洗脱剂进行柱层析得白色固体0.295 g, 收率: 50%. m.p. 245.7~247.0 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.86 (d, J=8.80 Hz, 2H, Ph), 6.94 (d, J=8.80 Hz, 2H, Ph), 4.87 (s, 2H, NH2), 3.84 (s, 3H, OCH3), 2.72 (s, 3H, CH3); IR (KBr) ν: 3430, 3033, 2930, 1670, 1606, 1505, 1460, 1260, 845 cm-1. HRMS (positive-ESIMS) calcd for C12H12N2O2Se (M+1)+ 296.0064, found 296.0070.
3.2.7 2-(4-甲氧苯基)-4-甲基-N'-(丙烷-2-亚甲基)-1, 3-硒唑-5-碳酰肼 (SA-9) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入10 mL乙醇搅拌至完全溶解, 温度升至78 ℃, 加入0.07 mL (0.0015 mol) 丙酮, 搅拌溶解回流, 反应时间4 h后, 加入3~4滴冰醋酸, 继续搅拌, 再反应1 h.反应停止后, 将混合物倒入40 mL冰水中, 搅拌, 有黄色固体析出, 用二氧六环重结晶得黄色固体0.298 g, 收率: 85.2%. m.p. 223.2~224.8 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.65 (s, 1H, NH), 7.90~7.88 (m, 2H, PhH), 7.05~7.02 (m, 2H, PhH), 3.84 (s, 3H, OCH3), 2.72 (s, 3H, CH3), 1.79 (s, 6H, CH3); IR (KBr) ν: 3255, 3030, 2930, 1670, 1600, 1500, 1465, 1391, 1253, 845 cm-1. HRMS (positive-ESIMS) calcd for C15H17N3O2Se (M+1)+ 351.0486, found 351.0478.
3.2.9 2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-甲腈 (SA-11) 的合成
取0.296 g (0.001 mol) 中间体SA-10于三颈瓶中, 加入10 mL乙腈, 搅拌, 温度升至80 ℃, 加入3 mL干燥的三氯氧磷, 搅拌反应约为5 h.反应停止后, 混合物冷却至室温, 加入冰水, 搅拌, 固体析出, 抽滤, 干燥得白色固体0.175 g, 收率: 60%. m.p. 145.8~147.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.84 (d, J=8.80 Hz, 2H, PhH), 7.05 (d, J=8.80 Hz, 2H, PhH), 3.86 (s, 3H, OCH3), 2.74 (s, 3H, CH3); IR (KBr) ν: 3033, 2934, 2350, 1600, 1500, 1450, 1250, 845 cm-1. HRMS (positive-ESIMS) calcd for C12H10N2OSe (M+1)+ 277.9958, found 277.9965.
3.3.11 (2-(4-氯苯基)-5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噁二唑-3(2H)-基) 乙烯酮 (SAZ-11) 的合成
取0.43 g (0.001 mol) SA-8于三颈瓶中, 加入8 mL乙酸酐, 搅拌至完全溶解, 回流, 用TLC监测反应, 反应时间约为5 h.反应停止后, 冷却至室温, 将混合物倒入50 mL冰水混合物中, 搅拌, 有黄绿色固体析出, 抽滤, 固体粗产品用丙酮重结晶得黄绿色固体0.291 g, 收率为61.25%. m.p. 279.4~281.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 12.95 (s, 1H, CH), 7.90 (d, J=8.70 Hz, 2H, PhH), 7.52 (s, 2H, PhH), 7.19 (s, 2H, PhH), 7.05 (d, J=8.60 Hz, 2H, PhH), 3.83 (s, 3H, OCH3), 2.63 (s, 3H, CH3), 2.23 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.52 (硒唑环), 167.20 (C=O), 162.40 (硒唑环), 157.37 (硒唑环), 152.60 (噁二唑环), 135.80 (Ph), 135.07 (Ph), 129.40 (Ph), 129.25 (Ph), 129.04 (Ph), 128.19 (Ph), 117.95 (Ph), 115.25 (Ph), 91.64 (噁二唑环), 56.01 (OCH3), 21.62 (CH3), 18.20 (CH3); IR (KBr) ν: 3030, 2924, 1665, 1640, 1600, 1446, 1250, 835 cm-1; HRMS (positive-ESIMS) calcd for C21H18ClN3O3Se (M+1)+ 475.0202, found 475.0198.
3.3.21 N-(1, 3-二氧代异吲哚啉-2-基)-2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-甲酰胺 (SAZ-21) 的合成
取0.31 g (0.001mol) 中间体SA-4于三颈瓶中, 加入10 mL冰乙酸搅拌至完全溶解, 再加入0.2 g (0.001mol) 邻苯二甲酸酐搅拌完全溶解, 回流.用TLC监测反应, 反应时间约为10 h.反应停止后, 冷却至室温, 将混合物倒入冰水中, 搅拌有固体析出, 抽滤, 固体用乙醇重结晶, 得浅黄色棒状晶体0.322 g, 收率为73%. m.p. 227.8~229.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.05 (s, 1H, NH), 8.03~8.01 (m, 2H, PhH), 7.99~7.97 (m, 2H, PhH), 7.95 (d, J=8.70 Hz, 2H, PhH), 7.08 (d, J=8.75 Hz, 2H, PhH), 3.85 (s, 3H, OCH3), 2.65 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 175, 13 (硒唑环), 165.67 (硒唑环), 162.72 (硒唑环), 162.42 (C=O), 159.18 (异吲哚啉环), 135.95 (Ph), 129.88 (Ph), 129.31 (Ph), 128.26 (Ph), 125.57 (Ph), 124.40 (Ph), 115.26 (Ph), 56.01 (OCH3), 18.78 (CH3); IR (KBr) ν: 3207, 3034, 2975, 1738, 1643, 1600, 1520, 1432, 1250, 697 cm-1; HRMS (positive-ESIMS) calcd for C20H15N3O4Se (M+1)+ 441.0228, found 441.0230.
3.3.12 2-(4-氯苯基)-5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噁二唑 (SAZ-12) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入5 mL三氯氧磷, 温度升至90 ℃, 搅拌至完全溶解后, 加入0.12 g (0.0011 mol) 对氯苯甲酸, 搅拌至完全溶解, 30 min后, 逐渐有黄绿色固体析出.用TLC监测反应, 反应时间为7 h.反应停止后, 将混合物倒入60 mL冰水混合物中搅拌, 有黄绿色固体析出, 抽滤, 固体粗产品用乙醇重结晶得浅黄色固体0.301 g, 收率为70%[53]. m.p. 252.3~253.6 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.86 (d, J=8.60 Hz, 4H, PhH), 7.04 (d, J=8.75 Hz, 4H, PhH), 3.83 (s, 3H, OCH3), 2.56 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 172.94 (硒唑环), 163.31 (硒唑环), 161.96 (硒唑环), 155.03 (噁二唑环), 150.18 (噁二唑环), 128.87 (Ph), 128.63 (Ph), 115.13 (Ph), 55.93 (OCH3), 18.32 (CH3); IR (KBr) ν: 3092, 2924, 1686, 1592, 1483, 1250, 828, 762 cm-1; HRMS (positive-ESIMS) calcd for C19H14ClN3O2Se (M+1)+ 430.9940, found 430.9950.
3.3.17 3-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羰基)-2-(苯基亚氨基) 噻唑烷-4-酮 (SAZ-17) 的合成
在室温条件下, 取0.478 g (0.001 mol) 中间体SA-5于三颈瓶中, 加入3 mL (0.01 mol) 氯乙酰氯搅拌溶解, 体系为黄色溶液, 加入10 mL CHCl3并将体系转入油浴中, 在60 ℃条件下继续反应, 用TLC监测反应, 反应时间约为4 h.反应停止后, 将混合物倒入50 mL冰水混合物中, 搅拌, 分液, 有机相干燥, 并蒸去溶剂, 固体用15 mL无水乙醚洗涤3次, 得黄绿色固体0.230 g, 收率为50.2%. m.p. 235.3~237.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.94 (d, J=8.80 Hz, 2H, PhH), 7.56 (q, J=7.70 Hz, 5H, PhH), 7.06 (t, J=8.80 Hz, 2H, PhH), 4.76 (s, 2H, CH2), 3.83 (t, J=6.70 Hz 3H, OCH3), 2.57 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.97 (硒唑环), 166.48 (C=O), 162.50 (硒唑环), 160.03 (硒唑环), 158.99 (噻唑烷环), 156.76 (噻唑烷环), 137.56 (Ph), 130.28 (Ph), 129.89 (Ph), 129.38 (Ph), 128.05 (Ph), 116.71 (Ph), 115.26 (Ph), 115.18 (Ph), 56.04 (OCH3), 44.26 (噻唑烷环), 18.16 (CH3); IR (KBr) ν: 3034, 2931, 1723, 1640, 1600, 1548, 1446, 1250, 845 cm-1; HRMS (positive-ESI-MS) calcd for C21H18N2O3SSe (M+1)+ 458.0203, found 458.0200.
3.3.19 N'-(2, 5-二氧-2, 5-二氢-1H-吡咯-1-基)-2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-甲酰胺 (SAZ-19) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入冰乙酸搅拌至完全溶解, 再加0.15 g (0.001 mol) 顺丁烯二酸酐搅拌完全溶解, 回流.用TLC监测反应, 反应时间约为10 h.反应停止后, 冷却至室温, 将混合物倒入冰水中, 搅拌有固体析出, 抽滤, 固体粗产品用乙醇重结晶得浅黄色棒状晶体0.254 g, 收率为65%. m.p. 165.2~167.0 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.05 (s, 1H, NH), 8.03~8.01 (m, 1H, CH=CH), 7.99~7.97 (m, 1H, CH=CH), 7.95 (d, J=8.75 Hz, 2H, PhH), 7.08 (d, J=8.75 Hz, 2H, PhH), 3.85 (s, 3H, OCH3), 2.65 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 175.13 (硒唑环), 165.67 (硒唑环), 162.72 (硒唑环), 162.42 (C=O), 159.18 (C=O), 135.95 (吡咯环), 129.31 (吡咯环), 56.01 (OCH3), 18.75 (CH3); IR (KBr) ν: 3222, 3084, 2967, 2830, 1730, 1650, 1600, 1500, 1450, 1250, 820, 690 cm-1; HRMS (positive-ESIMS) calcd for C16H13N3O4Se (M+1)+ 391.0071, found 391.0069.
3.3.16 N'-(3, 5-二苯基噻唑-2(3H)-亚基)-2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-酰肼 (SAZ-16) 的合成
取0.478 g (0.001 mol) 中间体SA-5于三口瓶中, 加入12 mL乙醇, 温度升至78 ℃, 搅拌, 不完全溶解, 体系为浅绿色浊液, 加入0.148 g (0.0012 mol) 2-溴苯乙酮, 搅拌, 体系由浅绿色浊液变为红棕色浊液, 搅拌30 min后, 加入无水乙酸钠 (0.002 mol), 搅拌, 体系由红棕色浊液变为黄色澄清溶液.搅拌10 min后, 有黄色固体析出, 继续搅拌回流, 用TLC监测反应, 反应时间约为14 h.反应停止后, 静置冷却至室温, 将混合物倒入冰水中, 搅拌, 有黄绿色固体析出, 抽滤, 干燥, 将固体粗产品用丙酮重结晶得黄绿色固体0.274 g, 收率为50.2%[54]. m.p. 276.5~277.8 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.67 (s, 1H, NH), 7.93 (d, J=7.00 Hz, 2H, PhH), 7.61 (d, J=7.80 Hz, 2H, PhH), 7.39 (t, J=7.80 Hz, 3H, PhH), 7.26 (d, J=4.60 Hz, 3H, PhH), 7.07~7.01 (m, 4H, PhH), 6.60 (s, 1H, CH=C), 3.84 (d, J=6.70 Hz 3H, OCH3), 2.67 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.90 (硒唑环), 166.48 (C=O), 162.50 (硒唑环), 160.04 (硒唑环), 158.94 (噻唑烷环), 156.76 (噻唑烷环), 137.52 (Ph), 130.28 (Ph), 129.86 (Ph), 129.36 (Ph), 128.06 (Ph), 116.72 (Ph), 115.28 (Ph), 115.16 (Ph), 56.00 (OCH3), 18.18 (CH3); IR (KBr) ν: 3348, 3048.3, 2931, 1700, 1665, 1600, 1490, 1257, 755 cm-1; HRMS (positive-ESIMS) calcd for C27H22N4O2SSe (M+1)+ 546.0629, found 546.0635.
3.3.6 2-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噁二唑 (SAZ-6) 的合成
取0.31 g (0.001 mol) 中间体SA-4于三颈瓶中, 加入8 mL原甲酸三乙酯, 搅拌, 温度升至90 ℃, 固体全部溶解, 体系为无色澄清溶液, 用TLC监测反应, 反应时间为14 h.反应停止后, 静置冷却至室温, 放入冰箱中冷冻一夜, 有灰色固体析出, 抽滤, 固体用无水乙醇重结晶, 得灰白色针状晶体0.234 g, 收率为73%. m.p. 247.8~249.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.89 (s, 1H, CH), 8.07~7.89 (m, 2H, PhH), 7.08~6.99 (m, 2H, PhH), 3.84 (d, J=9.35 Hz, 3H, OCH3), 2.72 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 175.05 (硒唑环), 162.45 (硒唑环), 161.23 (硒唑环), 154.46 (噁二唑环), 150.18 (噁二唑环), 129.32 (Ph), 128.84 (Ph), 128.77 (Ph), 128.13 (Ph), 115.25 (Ph), 115.13 (Ph), 56.01 (OCH3), 18.37 (CH3); IR (KBr) ν: 3047, 1640, 1613, 1498, 1250, 842, 755 cm-1; HRMS (positive-ESIMS) calcd for C13H11-N3O2Se (M+1)+ 321.0016, found 321.0010.
3.3.18 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1H-四唑 (SAZ-18) 的合成
取0.28 g (0.001 mol) 中间体SA-11于三颈瓶中, 加入6 mL DMF搅拌至完全溶解, 加入0.107 g (0.002 mol) 氯化铵搅拌完全溶解, 温度缓慢升至120 ℃, 加入0.013 g (0.001 mol) 叠氮化钠, 搅拌, 用TLC监测反应, 反应时间约为3 h, 反应完成后, 冷却至室温, 倒入冰水混合物中, 搅拌, 有固体析出, 抽滤, 固体粗产品用无水乙醇重结晶得浅黄色棒状晶体为0.210 g, 收率为65%[55]. m.p. 245.7~247.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.95 (q, J=9.00 Hz, 2H, PhH), 7.07 (d, J=7.70 Hz, 2H, PhH), 3.84 (s, 3H, OCH3), 2.71 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.99 (硒唑环), 162.46 (硒唑环), 157.00 (四唑环), 129.28 (Ph), 127.97 (Ph), 116.34 (Ph), 115.17 (Ph), 55.99 (OCH3), 18.42 (CH3); IR (KBr) ν: 3326, 3035, 2924, 2203, 1600, 1520, 1461, 1265, 767 cm-1; HRMS (positive-ESIMS) calcd for C12H11N5OSe (M+1)+ 321.0129, found 321.0124.
3.3.1 (3, 5-二甲基-1H-吡唑-1-基)-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基) 甲酮 (SAZ-1) 的合成
向三颈瓶中加入0.31 g (0.001 mol) 的中间体SA-4和8 mL乙醇, 搅拌, 温度升至78 ℃, 完全溶解后, 加入57 mL (0.0012 mol) 乙酰丙酮, 搅拌回流, 用TLC监测反应, 反应时间约为10 h.反应停止后, 静置冷却至室温, 将混合物倒入冰水中, 搅拌, 固体析出, 抽滤, 固体粗产品用无水乙醇重结晶得黄色针状晶体0.319 g, 收率为85%. m.p. 166.8~168.0 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 8.00 (d, J=8.65 Hz, 2H, PhH), 6.96 (d, J=8.65 Hz, 2H, PhH), 6.04 (s, 1H, CH=C), 3.87 (s, 3H, OCH3), 2.91 (s, 3H, CH3), 2.65 (s, 3H, CH3), 2.30 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 182.95 (硒唑环), 166.07 (C=O), 162.53 (硒唑环), 162.30 (硒唑环), 151.35 (吡唑环), 144.80 (吡唑环), 129.10 (Ph), 128.71 (Ph), 115.22 (Ph), 112.39 (吡唑环), 55.99 (OCH3), 21.26 (CH3), 14.93 (CH3), 13.85 (CH3); IR (KBr) ν: 3030, 2960, 2840, 1665, 1606, 1520, 1483, 1250, 828 cm-1; HRMS (positive-ESIMS) calcd for C17H17N3O2Se (M+1)+ 375.0486, found 375.0496.
3.3.2 1-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-羰基)-3-甲基-1H-吡唑-5(4H)-酮 (SAZ-2) 的合成
向三颈瓶中加入0.31 g (0.001 mol) 中间体SA-4和8 mL乙醇, 搅拌, 温度升至78 ℃, 完全溶解后, 加入63 mL (0.0012 mol) 乙酰乙酸乙酯, 搅拌, 加入0.1 g (0.0012 mol) K2CO3回流, 用TLC监测反应, 反应时间约为14 h.反应停止后, 静置冷却至室温, 将混合物倒入冰水中, 搅拌, 固体析出, 抽滤, 用乙醇对固体进行重结晶得浅黄色针状晶体0.283 g, 收率为75%. m.p. 172.5~174.0 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.94 (d, J=8.70 Hz, 2H, PhH), 7.04 (q, J=8.70 Hz, 2H, PhH), 4.14 (d, J=7.10 Hz, 2H, CH2), 3.83 (s, 3H, OCH3), 2.73 (s, 3H, CH3), 2.07 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 182.90 (硒唑环), 166.07 (C=O), 162.54 (硒唑环), 162.31 (硒唑), 154.46 (吡唑环), 151.34 (吡唑环), 144.80 (吡唑环), 129.10 (Ph), 128.71 (Ph), 115.22 (Ph), 112.38 (Ph), 55.99 (OCH3), 21.24 (CH3), 14.90 (CH3); IR (KBr) ν: 3047, 1723, 1636, 1432, 1257, 830 cm-1; HRMS (positive-ESIMS) calcd for C16H15N3O3Se (M+1)+ 377.0279, found 377.0289.
3.3.4 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-4-苯基-4H-1, 2, 4-三唑-3-硫醇 (SAZ-4) 的合成
取0.478 g (0.001 mol) 的中间体SA-5于三颈瓶中, 加入15 mL的10%的NaOH水溶液, 升温至90 ℃, 搅拌, 固体逐渐溶解完全, 溶液为黄色.用TLC监测反应, 反应时间约为7 h.趁热过滤, 滤液冷却至室温, 倒入50 mL冰水混合物中, 冰完全融化后, 加入2 mol/L的稀盐酸调节PH值约为6, 有淡黄色固体析出, 抽滤, 用无水乙醇重结晶得淡黄色针状晶体0.299 g, 收率为70%. m.p. 206.8~208.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 14.22 (s, 1H, SH), 7.72 (q, J=6.85 Hz, 2H, PhH), 7.53 (t, J=6.85 Hz, 3H, PhH), 7.43~7.41 (m, 2H, PhH), 6.98 (q, J=6.85 Hz, 2H, PhH), 3.80 (s, 3H, OCH3), 2.41 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 175.23 (硒唑环), 168.55 (硒唑环), 162.07 (硒唑环), 156.42 (三唑环), 147.14 (三唑环), 134.18 (Ph), 130.45 (Ph), 129.91 (Ph), 129.59 (Ph), 128.81 (Ph), 128.13 (Ph), 118.50 (Ph), 115.16 (Ph), 55.93 (OCH3), 18.20 (CH3); IR (KBr) ν: 3069, 2931, 1606, 1512, 1453, 1250, 828 cm-1; HRMS (positive-ESI-MS) calcd for C19H16N4OSSe (M+1)+ 428.0210, found 428.0200.
3.3.5 3-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1H-1, 2, 4-三唑-5-硫醇 (SAZ-5) 的合成
取0.37 g (0.001 mol) 的中间体SA-7于三颈瓶中, 加入20 mL (20 equiv.) 5% NaOH水溶液, 搅拌, 温度升至90 ℃, 体系为澄清黄色溶液, 用TLC监测反应, 反应时间为6.5 h.反应停止后, 趁热过滤, 滤液冷却至室温, 用2 mol/L的稀盐酸溶液调节PH值为5, 搅拌, 有黄色固体析出, 抽滤并用乙醇重结晶得黄色固体0.217 g, 收率为61.7%. m.p. 252.6~254.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 13.76 (s, 1H, SH), 13.71 (s, 1H, NH), 7.90 (t, J=8.70 Hz, 2H, PhH), 7.05~7.02 (m, 2H, PhH), 3.84 (s, 3H, OCH3), 2.71 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.57 (硒唑环), 162.32 (硒唑环), 160.33 (硒唑环), 154.64 (三唑环), 146.63 (三唑环), 129.15 (Ph), 128.59 (Ph), 128.41 (Ph), 127.18 (Ph), 119.57 (Ph), 115.14 (Ph), 55.94 (OCH3), 18.33 (CH3); IR (KBr) ν: 3290, 3069, 2931, 1640, 1600, 1512, 1453, 1250, 828 cm-1; HRMS (positive-ESIMS) calcd for C13H12N4OSSe (M+1)+ 351.9897, found 351.9887.
3.3.14 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-N-苯基-1, 3, 4-噻二唑-2-胺 (SAZ-14) 的合成
在0 ℃下, 取0.478 g (0.001 mol) 中间体SA-5于三颈瓶中, 向体系中逐滴加入4 mL浓硫酸, 搅拌, 溶液为绿色粘稠状液体, 滴加完毕后, 继续搅拌30 min, 将反应体系转移至室温, 继续搅拌2.5 h.反应停止后, 将混合物倒入50 mL冰水中, 搅拌, 并用浓氨水调节PH值约为7, 继续搅拌, 有黄绿色固体析出.将固体抽滤, 固体粗产品用乙醇重结晶得黄绿色固体0.306 g, 收率为71.4%. m.p. 282.5~284.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 0.68 (s, 1H, NH), 7.90 (d, J=8.45 Hz, 2H, PhH), 7.65 (d, J=7.80 Hz, 2H, PhH), 7.39 (t, J=7.80 Hz, 2H, PhH), 7.06 (q, J=8.45 Hz, 3H, PhH), 3.84 (s, 3H, OCH3), 2.62 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.26 (硒唑环), 169.95 (硒唑环), 162.23 (硒唑环), 155.21 (噻二唑环), 154.18 (噻二唑环), 138.95 (Ph), 129.57 (Ph), 129.16 (Ph), 128.28 (Ph), 122.44 (Ph), 117.81 (Ph), 117.58 (Ph), 115.20 (Ph), 55.98 (OCH3), 18.26 (CH3); IR (KBr) ν: 3194, 3047, 2917, 1640, 1606, 1500, 1450, 1270, 747, 682 cm-1; HRMS (positive-ESI-MS) calcd for C19H16N4OSSe (M+1)+428.0210, found 428.0220.
3.3.20 3-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-5, 6-二苯基-1, 2, 4-三嗪 (SAZ-20) 的合成
向三颈瓶中加入0.0983 g (4.68×10-4mol) 联苯甲酰和10 mL乙醇, 搅拌, 温度升至78 ℃, 完全溶解后加入0.145 g (4.68×10-4mol) 的中间体SA-4, 再次完全溶解后加入醋酸铵0.3605 g (4.68×10-3mol), 完全溶解后加入草酸0.0118 g (4.68×10-3mol), 溶液由澄清变浑浊, 回流, 用TLC监测反应, 反应时间约为7 h.反应停止后, 静置冷却至室温, 过滤, 得黄绿色的滤液, 蒸去溶剂, 得固体粗产品, 并用3:1 (V石油醚:V乙酸乙酯) 的洗脱剂进行柱层析得黄绿色固体0.136 g, 收率为60%. m.p. 284.2~285.6 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.97 (d, J=8.00 Hz, 2H, PhH), 7.89 (q, J=8.00 Hz, 2H, PhH), 7.67~7.63 (m, 3H, PhH), 7.60~7.54 (m, 1H, PhH), 7.49 (d, J=7.70 Hz, 2H, PhH), 7.47~7.42 (m, 2H, PhH), 6.98 (d, J=7.70 Hz, 2H, PhH), 3.88 (d, J=4.30 Hz, 3H, OCH3), 2.83 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.90 (硒唑环), 162.50 (硒唑环), 160.04 (硒唑环), 158.98 (三嗪环), 156.78 (三嗪环), 137.52 (Ph), 130.28 (Ph), 129.86 (Ph), 129.36 (Ph), 128.06 (Ph), 116.72 (Ph), 115.28 (Ph), 115.16 (Ph), 56.00 (OCH3), 18.18 (CH3); IR (KBr) ν: 3157, 3047, 2924, 1621, 1600, 1510, 1450, 1250, 845, 690 cm-1; HRMS (positive-ESIMS) calcd for C26H20N4OSe (M+1)+ 484.0802, found 484.0808.
3.3.7 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噁二唑-2(3H)-酮 (SAZ-7) 的合成
向三颈瓶中加入0.31 g (0.001 mol) 的中间体SA-4和8 mL二氧六环, 搅拌至完全溶解, 体系为黄色澄清溶液, 加入白色粉末状的N, N-羰基二咪唑 (CDI), 搅拌完全溶解, 回流, TLC监测反应, 反应时间约为4 h.反应停止后, 静置冷却至室温, 减压蒸去溶剂, 得黄色固体.向固体中加入30 mL水, 充分搅拌, 抽滤, 固体干燥得黄色固体0.286 g, 收率为85%. m.p. 218.5~219.2 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 12.69 (s, 1H, NH), 7.96 (d, J=8.75 Hz, 2H, PhH), 7.11 (d, J=8.75 Hz, 2H, PhH), 3.89 (s, 3H, OCH3), 2.66 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.64 (硒唑环), 162.29 (硒唑环), 162.19 (硒唑环), 155.12 (噁二唑环), 151.67 (噁二唑环), 129.17 (Ph), 129.10 (Ph), 128.18 (Ph), 128.14 (Ph), 118.43 (Ph), 115.21 (Ph), 55.99 (OCH3), 18.24 (CH3); IR (KBr) ν: 3244, 3011, 1796, 1600, 1447, 1257, 820 cm-1; HRMS (positive-ESIMS) calcd for C13H11N3O3-Se (M+1)+ 336.9966, found 336.9950.
3.3.3 4-氨基-5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-4H-1, 2, 4-三唑-3-硫醇 (SAZ-3) 的合成
取0.5 g (0.0015 mol) 中间体SA-6完全溶解于8 mL水合肼中, 升温至90 ℃, 搅拌, 用TLC监测反应, 反应时间为6 h.将混合物倒入50 mL冰水混合物中搅拌, 冰完全溶解后, 加入稀盐酸调节PH值约为3~4, 有橘红色固体析出, 抽滤, 固体粗产品用乙醇重结晶得橘红色棒状晶体0.347 g, 收率为63%[51]. m.p. 256.8~257.9 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 13.98 (s, 1H, SH), 7.94 (d, J=8.70 Hz, 2H, PhH), 7.05 (d, J=8.70 Hz, 2H, PhH), 5.90 (s, 2H, NH2), 3.83 (d, J=3.45 Hz, 3H, OCH3), 2.69 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.50 (硒唑环), 162.33 (硒唑环), 160.33 (硒唑环), 154.60 (三唑环), 146.63 (三唑环), 129.16 (Ph), 128.94 (Ph), 128.40 (Ph), 127.18 (Ph), 119.57 (Ph), 115.14 (Ph), 55.92 (OCH3), 18.20 (CH3); IR (KBr) ν: 3424, 3106, 2924, 1650, 1600, 1250, 845 cm-1; HRMS (positive-ESIMS) calcd for C13H13N5OSSe (M+1)+ 367.0006, found 367.0015.
3.3.22 N-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-酰肼-香豆素甲酮 (SAZ-22) 的合成
取0.41 g (0.001mol) 中间体SA-12于三颈瓶中, 加入10 mL乙醇搅拌并完全溶解, 温度升至78 ℃, 加入水杨醛95 mL (0.0015mol), 搅拌, 加入2滴冰乙酸, 搅拌约20 min后, 有黄色固体析出, 继续回流, 用TLC监测反应, 反应时间约8 h.反应停止后, 静置冷却至室温, 将混合物倒入冰水中, 搅拌, 有黄色固体析出, 抽滤, 固体粗产品用乙醇重结晶得黄色针状晶体0.382 g, 收率为79%. m.p. 226.1~227.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 11.88 (s, 1H, NH), 10.07 (s, 1H, NH), 8.58 (s, 1H, CH=C), 7.96 (q, J=7.80 Hz, 3H, PhH), 7.27 (s, 1H, PhH), 7.07 (q, J=8.80 Hz, 2H, PhH), 6.95 (t, J=7.80 Hz, 2H, PhH), 3.84 (s, 3H, OCH3), 2.76 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 178.75 (硒唑环), 163.94 (硒唑环), 162.13 (硒唑环), 157.12 (C=O), 140.91 (吡喃环), 131.85 (Ph), 128.96 (Ph), 126.83 (Ph), 120.82 (Ph), 120.20 (Ph), 116.71 (Ph), 115.18 (Ph), 55.93 (OCH3), 20.48 (CH3); IR (KBr) ν: 3150, 3011, 2924, 2822, 1636, 1600, 1556, 1461, 1279, 820, 755 cm-1; HRMS (positive-ESIMS) calcd for C22H17N3O5Se (M+1)+483.0333, found 483.0330.
辅助材料 (Supporting Information) 所有目标化合物的核磁共振氢谱和碳谱.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.3.13 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噻二唑-2-胺 (SAZ-13) 的合成
在0 ℃下, 取0.37g (0.001 mol) 中间体SA-7于三颈瓶中, 向体系中缓慢滴加5 mL浓硫酸, 搅拌, 溶液为绿色粘稠状液体, 滴加完毕, 继续反应30 min, 将反应体系转移至室温, 继续反应2.5 h.反应停止后, 将混合物倒入40 mL冰水中, 搅拌, 并用浓氨水调节PH值约为7, 有黄绿色固体析出, 抽滤, 固体粗产品用乙醇重结晶得黄绿色固体0.263 g, 收率为75%. m.p. 277.6~278.8 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.90~7.86 (m, 2H, PhH), 7.46 (s, 2H, NH2), 7.05 (t, J=8.80 Hz, 2H, PhH), 3.83 (s, 3H, OCH3), 2.65 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.96 (硒唑环), 162.48 (硒唑环), 158.27 (噻二唑环), 157.08 (噻二唑环), 129.28 (Ph), 127.97 (Ph), 116.34 (Ph), 115.17 (Ph), 55.99 (OCH3), 18.42 (CH3); IR (KBr) ν: 3426, 3030, 2924, 1665, 1600, 1520, 1432, 1286, 842, 667 cm-1; HRMS (positive-ESI-MS) calcd for C13H12N4OSSe (M+1)+ 351.9897, found 351.9910.
3.3.8 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-1, 3, 4-噁二唑-2-硫醇 (SAZ-8) 的合成
在室温条件下, 取0.028 g (0.0015 mol) KOH于三口瓶中, 加入10 mL乙醇, 搅拌至完全溶解, 加入中间体0.31 g (0.001 mol) SA-4, 搅拌至完全溶解后, 再缓慢滴加0.2 mL (0.0015 mol) CS2, 体系由黄色变为橘黄色溶液, 滴加完毕后, 继续搅拌3 min, 逐渐有黄色固体析出, 继续搅拌4 h.将体系转移至油浴中, 加热回流约5 h.反应停止后, 将混合物倒入冰水中, 搅拌, 为黄色澄清溶液, 加稀盐酸调节PH约为4~5, 有黄色固体析出, 抽滤, 用乙醇对固体进行重结晶得黄色棒状晶体0.268 g, 收率为76.1%[52]. m.p. 268.3~269.6 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 14.80 (s, 1H, SH), 7.94 (d, J=8.70 Hz, 2H, PhH), 7.06 (d, J=8.70 Hz, 2H, PhH), 3.84 (s, 3H, OCH3), 2.66 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 177.21 (硒唑环), 174.99 (硒唑环), 162.46 (硒唑环), 158.21 (噁二唑环), 157.00 (噁二唑环), 129.28 (Ph), 127.978 (Ph), 116.34 (Ph), 115.17 (Ph), 55.99 (OCH3), 18.42 (CH3); IR (KBr) ν: 3034, 2917, 1640, 1610, 1500, 1450, 1373, 1250, 845 cm-1; HRMS (positive-ESIMS) calcd for C13H11N3O2SSe (M+1)+ 352.9737, found 352.9730.
3.3.10 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-2, 2-二甲基-1, 3, 4-噁二唑-3(2H)-基)-乙烯酮 (SAZ-10) 的合成
取0.35 g (0.001 mol) 中间体SA-9于三颈瓶中, 加入8 mL乙酸酐, 搅拌至完全溶解, 回流, 用TLC监测反应, 反应时间约为4 h.反应停止后, 冷却至室温, 将混合物倒入冰水混合物中, 搅拌, 有黄色固体析出, 抽滤, 干燥, 得黄色固体0.239 g, 收率为63%. m.p. 251.2~252.9 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.90~7.88 (m, 2H, PhH), 7.06~7.03 (m, 2H, PhH), 3.84 (d, J=6.60 Hz, 3H, OCH3), 2.71 (s, 3H, CH3), 2.18 (s, 3H, CH3), 1.79 (s, 6H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.97 (硒唑环), 166.18 (C=O), 162.30 (硒唑环), 156.77 (硒唑环), 150.76 (噁二唑环), 129.15 (Ph), 128.61 (Ph), 128.26 (Ph), 118.64 (Ph), 115.22 (Ph), 115.15 (Ph), 100.74 (噁二唑环), 55.97 (OCH3), 24.80 (CH3), 22.47 (CH3), 18.51 (CH3), 18.32 (CH3); IR (KBr) ν: 3030, 2931, 2850, 1701, 1600, 1526, 1453, 1257, 820 cm-1; HRMS (positive-ESI-MS) calcd for C16H17N3O3Se (M+1)+ 379.0435, found 379.0430.
3.3.15 2-(4-甲氧苯基)-4-甲基-N'-(4-羰基-3-苯基噻唑烷-2-亚基)-1, 3-硒唑-5-酰肼 (SAZ-15) 的合成
取0.478 g (0.001 mol) 中间体SA-5于三颈瓶中, 加入10 mL的绝对无水乙醇搅拌溶解, 温度升至78 ℃, 再加入0.167 mL (0.001 mol) 溴乙酸乙酯, 搅拌充分后, 加入4.5 g (0.01 mol) 无水乙酸钠, 搅拌, 5 min后, 溶液变浑浊, 有黄色固体析出, 继搅拌回流, 用TLC监测反应, 反应时间约为6 h.反应停止后, 将混合物冷却至室温, 倒入50 mL冰水混合物中, 搅拌, 有固体析出, 将固体抽滤, 得黄绿色固体0.389 g, 收率为80%. m.p. 235.3~236.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.69 (s, 1H, NH), 7.93 (d, J=8.70 Hz, 2H, PhH), 7.61 (d, J=7.75 Hz, 2H, PhH), 7.39 (t, J=7.90 Hz, 2H, PhH), 7.06 (q, J=7.90 Hz, 3H, PhH), 4.27 (s, 2H, CH2), 3.83 (s, 3H, OCH3), 2.68 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 174.94 (硒唑环), 166.48 (C=O), 162.50 (硒唑环), 160.04 (硒唑环), 158.94 (噻唑烷环), 156.70 (噻唑烷环), 137.58 (Ph), 130.28 (Ph), 129.89 (Ph), 129.38 (Ph), 128.06 (Ph), 116.72 (Ph), 115.26 (Ph), 115.18 (Ph), 56.02 (OCH3), 44.20 (噻唑烷环), 18.18 (CH3); IR (KBr) ν: 3233, 3055, 2975, 2844, 1738, 1665, 1600, 1475, 1250, 835, 690 cm-1; HRMS (positive-ESIMS) calcd for C21H18N4O3SSe (M+1)+ 486.0265, found 486.0259.
3.3.9 5-(2-(4-甲氧苯基)-4-甲基-1, 3-硒唑-5-基)-N-苯基-1, 3, 4-噁二唑-2-胺 (SAZ-9) 的合成
向三颈瓶中加入0.478 g (0.001 mol) 中间体SA-5, 加入8 mL吡啶, 搅拌, 体系为黄色溶液, 回流, 用TLC监测反应, 反应时间约为8 h.反应停止, 静置冷却至室温, 将混合物倒入冰水中, 搅拌, 有黄色固体析出, 抽滤, 固体粗产品用无水乙醇重结晶的黄色棒状晶体0.314 g, 收率为76.1%. m.p. 265.9~267.1 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 10.68 (s, 1H, NH), 7.93 (d, J=8.45 Hz, 2H, PhH), 7.61 (d, J=7.80 Hz, 2H, PhH), 7.38 (t, J=7.80 Hz, 2H, PhH), 7.07 (q, J=8.45 Hz, 3H, PhH), 3.84 (s, 3H, OCH3), 2.71 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 173.28 (硒唑环), 162.23 (硒唑环), 159.95 (硒唑环), 155.21 (噁二唑环), 154.78 (噁二唑环), 138.95 (Ph), 129.57 (Ph), 129.15 (Ph), 128.28 (Ph), 122.46 (Ph), 117.81 (Ph), 117.59 (Ph), 115.20 (Ph), 55.99 (OCH3), 18.26 (CH3); IR (KBr) ν: 3270, 3040, 1641, 1616, 1498, 1440, 1250, 747, 682 cm-1; HRMS (positive-ESIMS) calcd for C19H16N4O2Se (M+1)+ 412.0438, found 412.0430.
-
-
[1]
Karegoudar, P.; Karthikeyan, M. S.; Prasad, D. J.; Mahalinga, M.; Holla, B. S.; Kumari, N. S. Eur. J. Med. Chem. 2008, 43(2), 261. doi: 10.1016/j.ejmech.2007.03.014
-
[2]
Bharti, S. K.; Nath, G.; Tilak, R.; Singh, S. K. Eur. J. Med. Chem. 2010, 45(2), 651. doi: 10.1016/j.ejmech.2009.11.008
-
[3]
Mohammad, H.; Mayhoub, A. S.; Cushman, M.; Seleem, M. N. J. Antibiot. 2015, 68, 259. doi: 10.1038/ja.2014.142
-
[4]
de Sa, N. P.; Lino, C. I.; Fonseca, N. C.; Borelli, B. M.; Ramos, J. P.; Souza-Fagundes, E. M.; Rosa, C. A.; Santos, D. A.; Oliveira, R. B.; Johann, S. Eur. J. Med. Chem. 2015, 102, 233. doi: 10.1016/j.ejmech.2015.07.032
-
[5]
Chimenti, F.; Bizzarri, B.; Maccioni, E.; Secci, D.; Bolasco, A.; Fioravanti, R.; Chimenti, P.; Granese, A.; Carradori, S.; Rivanera, D.; Lilli, D.; Zicari, A.; Distinto, S. Bioorg. Med. Chem. Lett. 2007, 17(16), 4635. doi: 10.1016/j.bmcl.2007.05.078
-
[6]
Youssef, A. M.; Malki, A.; Badr, M. H. Med. Chem. 2012, 8, 151. doi: 10.2174/157340612800493719
-
[7]
Łaczkowski, K. Z.; Misiura, K.; Switalska, M. Anti-Cancer Agents Med. Chem. 2014, 14, 1271.
-
[8]
Taori, K.; Paul, V. J.; Luesch, H. J. Am. Chem. Soc. 2008, 130(6), 1806. doi: 10.1021/ja7110064
-
[9]
Stadtman, T. C. Ann. N. Y. Acad. Sci. 2000, 899, 399.
-
[10]
Metanis, N.; Keinan, E.; Dawson, P. E. J. Am. Chem. Soc. 2006, 128, 16684. doi: 10.1021/ja0661414
-
[11]
Nishina, A.; Kimura, H.; Kozawa, K.; Sommen, G.; Nakamura, T.; Heimgartner, H.; Koketsu, M.; Furukawa, S. Toxicol. Appl. Pharmacol. 2011, 257(3), 388. doi: 10.1016/j.taap.2011.09.022
-
[12]
Venardos, K. M.; Perkins, A.; Headrick, J. Curr. Med. Chem. 2007, 14(14), 1539. doi: 10.2174/092986707780831078
-
[13]
Sanmartin, C.; Plano, D.; Font, M. Curr. Med. Chem. 2011, 18(30), 4635. doi: 10.2174/092986711797379249
-
[14]
Ninomiya, M.; Garud, D. R.; Koketsu, M. Coord. Chem. Rev. 2011, 255(23), 2968.
-
[15]
Farag, A. M.; Mayhoub, A. S.; Barakat, S. E.; Bayomi, A. H. Bioorg. Med. Chem. 2008, 16(2), 881. doi: 10.1016/j.bmc.2007.10.015
-
[16]
Xia, Y.; Fan, C. D.; Zhao, B. X.; Zhao, J.; Shin, D. S.; Miao, J. Y. Eur. J. Med. Chem. 2008, 43(11), 2347. doi: 10.1016/j.ejmech.2008.01.021
-
[17]
Xia, Y., Dong, Z. W.; Zhao, B. X.; Ge, X.; Meng, N.; Shin, D. S.; Miao, J. Y. Bioorg. Med. Chem. 2007, 15(22), 6893. doi: 10.1016/j.bmc.2007.08.021
-
[18]
Simlot, R.; Izydore, R. A.; Wong, O. T. J. Pharm. Sci. 1994, 83, 367. doi: 10.1002/jps.2600830320
-
[19]
Kanagarajan, V.; Thanusu, J.; Ezhilarasi, M. R. Chem. Hetero-cycl. Compd. 2011, 47, 60. doi: 10.1007/s10593-011-0720-5
-
[20]
Mallakpour, S.; Rafiee, Z. Polym. Adv. Technol. 2008, 19(8), 1015. doi: 10.1002/pat.v19:8
-
[21]
Mallakpour, S.; Rafiee, Z. Polym. Bull. 2008, 60(4), 507. doi: 10.1007/s00289-008-0888-8
-
[22]
Mallakpour, S.; Rafiee, Z. Polymer 2007, 48(19), 5530. doi: 10.1016/j.polymer.2007.07.036
-
[23]
Mallakpour, S.; Rafiee, Z. Synlett 2007 (8), 1255.
-
[24]
Abu-Zaied, M. A. Z.; Nawwar, G. A. M.; Swellem, R. H.; El-Sayed, S. H. Pharmacol. Pharm. 2012, 3, 254. doi: 10.4236/pp.2012.32034
-
[25]
Sahu, V. K. R.; Singh, A. K.; Yadav, D. Int. J. ChemTech. Res. 2011, 3, 1362.
-
[26]
Jain, S. K.; Yadav, A. K.; Nayak, P. Int. J. Pharm. Sci. Drug Res. 2011, 3, 230.
-
[27]
Ouyang, X.; Piatnitski, E. L.; Pattaropong, V.; Chen, X. L.; He, H. Y.; Kiselyov, A. S.; Velankar, A.; Kawakami, J.; Labelle, M.; Smith, L.; Lohman, J.; Lee, S. P.; Malikzay, A.; Fleming, J.; Gerlak, J.; Wang, Y.; Rosler, R. L.; Zhou, K.; Mitelman, S.; Camara, M.; Surguladze, D.; Doody, J. F.; Tuma, M. C. Bioorg. Med. Chem. Lett. 2006, 16(22), 1191.
-
[28]
Lewis, J. R. Nat. Prod. Rep. 1999, 16, 389. doi: 10.1039/a802500b
-
[29]
Dorville, A.; McCort-Tranchepain, I.; Vichard, D.; Sather, W.; Maroun, R.; Ascher, P.; Roques, B. P. J. Med. Chem. 1992, 35(14), 2551. doi: 10.1021/jm00092a005
-
[30]
Jaen, J. C.; Wise, L. D.; Caprathe, B. W.; Tecle, H.; Bergmeier, S.; Humblet, C. C.; Heffner, T. G.; Meltzer, L. T.; Pugsley, T. A. J. Med. Chem. 1990, 33(1), 311. doi: 10.1021/jm00163a051
-
[31]
Clemence, F.; Martet, O. L.; Delevallee, F.; Benzoni, J.; Jonanen, A.; Jouquey, S.; Mouren, M.; Deraedt, R. J. Med. Chem. 1988, 31(7), 1453. doi: 10.1021/jm00402a034
-
[32]
Hargrave, K. D.; Hess, F. K.; Oliver, J. T. J. Med. Chem. 1983, 26(8), 1158. doi: 10.1021/jm00362a014
-
[33]
Tsuji, K.; Ishikawa, H. Bioorg. Med. Chem. Lett. 1994, 4(13), 1601. doi: 10.1016/S0960-894X(01)80574-6
-
[34]
Bell, F. W.; Cantrell, A. S.; Hoberg, M.; Jaskunas, S. R.; Johansson, N. J.; Jordan, C. L.; Kinnick, M. D.; Lind, P.; MorinJr, J. M. J. Med. Chem. 1995, 38(25), 4929. doi: 10.1021/jm00025a010
-
[35]
My Hang, V. H.; Michael, D. C.; Thomas, J. M.; Modi, W. Proc. Natl. Acad. Sci. 2006, 103(27), 10322. doi: 10.1073/pnas.0604241103
-
[36]
My Hang, V. H. WO 2008054538, 2008 [Chem. Abstr. 2008, 148, 474992].
-
[37]
Gao, H. X.; Shreeve, J. M. Chem. Rev. 201l, 111(11), 7377. doi: 10.1021/cr200039c
-
[38]
Huang, M. Z.; Luo, F. X.; Mo, H. B.; Ren, Y. G.; Wang, X. G.; Ou, X. M.; Lei, M. X.; Liu, A. P.; Huang, L.; Xu, M. C. J. Agric. Food Chem. 2009, 57(20), 9585. doi: 10.1021/jf901897f
-
[39]
Lawson, E. C.; Luci, D. K.; Ghosh, S.; Kinney, W. A.; Reynolds, C. H.; Qi, J.; Smith, C. E.; Wang, T. P.; Minor, L. K.; Haertlein, B. J.; Parry, T. J.; Damiano, B. P.; Maryanoff, B. E. J. Med. Chem. 2009, 52(23), 7432. doi: 10.1021/jm900683d
-
[40]
Iwai, Y.; Takahashi, H.; Hatakeyama, D.; Motoshima, K.; Ishikawa, M.; Sugita, K.; Hashimoto, Y.; Harada, Y.; Itamura, S.; Odagiri, T.; Tashiro, M.; Sei, Y.; Yamaguchi, K.; Kuzuhara, T. Bioorg. Med. Chem. 2010, 18(14), 5379. doi: 10.1016/j.bmc.2010.05.035
-
[41]
Kishida, K.; Aoyama, A.; Hashimoto, Y. Chem. Pharm. Bull. 2010, 58(11), 1525. doi: 10.1248/cpb.58.1525
-
[42]
Kolukisaoglu, Ü.; Wendler, C.; Goerdes, D. J. Enzyme Inhib. Med. Chem. 2010, 25(6), 876. doi: 10.3109/14756360903508448
-
[43]
Kaminski, K.; Obniska, J.; Wiklik, B.; Atamanyuk, D. Eur. J. Med. Chem. 2011, 46(8), 4634.
-
[44]
Bansode, T. N.; Shelke, J. V.; Dongre, V. G. Eur. J. Med. Chem. 2009, 44(12), 5094. doi: 10.1016/j.ejmech.2009.07.006
-
[45]
Huang, W. G.; Jiang, Y. Y.; Li, Q.; Li, J.; Li, J. Y.; Lu, W.; Cai, J. C. Tetrahedron 2005, 61(7), 1863. doi: 10.1016/j.tet.2004.12.033
-
[46]
Sun, L. P.; Shen, Q.; Piao, H. H.; Ma, W. P.; Gao, L. X.; Zhang, W.; Nan, F. J.; Li, J.; Piao, H. R. Eur. J. Med. Chem. 2011, 46(9), 3630. doi: 10.1016/j.ejmech.2011.05.027
-
[47]
Shi, L.; Yu, H. P.; Zhou, Y. Y.; Du, J. Q.; Shen, Q.; Li, J. Y.; Li, J. Acta Pharmacol. Sin. 2008, 29, 278. doi: 10.1111/aphs.2008.29.issue-2
-
[48]
Cao, S.; Foster, C.; Lazo, J. S.; Kingston, D. G. I. Bioorg. Med. Chem. 2005, 13(17), 5094. doi: 10.1016/j.bmc.2005.04.070
-
[49]
Suzanne, F.; Stephen, J. E.; Anthony, F. H. J. Org. Chem. 2004, 69(14), 4663. doi: 10.1021/jo049748g
-
[50]
Hua, G.; Li, Y.; Slawin, A. M. Z.; Woollines, J. D. Org. Lett. 2006, 8(23), 5251. doi: 10.1021/ol062053c
-
[51]
Li, C.; Liu, J. C.; Li, Y. R.; Gou, C.; Zhang, M. L.; Liu, H. Y.; Li, X. Z.; Zheng, C. J.; Piao, H. R. Bioorg. Med. Chem. Lett. 2015, 25, 3052. doi: 10.1016/j.bmcl.2015.04.081
-
[52]
Puthiyapurayil, P.; Poojarya, B.; Chikkanna, C.; Buridipad, S. K. Eur. J. Med. Chem. 2012, 53, 203. doi: 10.1016/j.ejmech.2012.03.056
-
[53]
Banerjee, A. G.; Das, N.; Shengule, S. A.; Srivastava, R. S.; Shrivastava, S. K. Eur. J. Med. Chem. 2015, 101, 81. doi: 10.1016/j.ejmech.2015.06.020
-
[54]
Bondock, S.; Fadaly, W.; Metwally, M. A. Eur. J. Med. Chem. 2010, 45, 3692. doi: 10.1016/j.ejmech.2010.05.018
-
[55]
Faria, J. V.; dos Santos, M. S.; Bernardino, A. M. R.; Becker, K. M.; Machado, G. M. C.; Rodrigues, R. F.; Canto-Cavalheiro, M. M.; Leon, L. L. Bioorg. Med. Chem. Lett. 2013, 23, 6310. doi: 10.1016/j.bmcl.2013.09.062
-
[1]
-

图式1 合成含吡唑的化合物SAZ-1~SAZ-2
Scheme 1 Synthesis of pyrazole containing compounds SAZ-1~SAZ-2

图式2 合成含1, 2, 4-三唑的化合物SAZ-3~SAZ-5
Scheme 2 Synthesis of 1, 2, 4-triazole containing compounds SAZ-3~SAZ-5

图式3 合成含噁二唑的化合物SAZ 6~SAZ-12
Scheme 3 Synthesis of oxadiazole containing compounds SAZ 6~SAZ-12

图式4 合成含噻二唑的化合物SAZ-13~SAZ-14
Scheme 4 Synthesis of thiadiazole containing compounds SAZ-13~SAZ-14

图式5 合成含噻唑环的化合物SAZ-15~SAZ-17
Scheme 5 Synthesis of the thiazole ring-containing compounds SAZ-15~SAZ-17

图式6 合成含四唑环的化合物SAZ-18
Scheme 6 Synthesis of the tetrazole ring-containing compounds SAZ-18

图式7 合成其他杂环化合物SAZ-19~SAZ-22
Scheme 7 Synthesis other heterocyclic compounds SAZ-19~SAZ-22
表 1 目标化合物对Cdc25B的抑制活性
Table 1. Inhibition of target compounds against Cdc25B
Compd. Cdc25B Inhibition ratea/% IC50b/(μg•mL-1) SAZ-1 84.30±4.52 1.82±0.65 SAZ-2 78.06±7.06 1.89±0.14 SAZ-3 96.07±6.88 1.95±0.34 SAZ-4 87.07±0.46 2.04±0.22 SAZ-5 73.30±5.05 4.85±0.29 SAZ-6 18.8±3.32 NA SAZ-7 25.6±3.08 NA SAZ-8 31.99±3.13 NA SAZ-9 87.23±7.64 1.46±0.45 SAZ-10 28.77±4.56 NA SAZ-11 20.76±1.12 NA SAZ-12 16.18±9.22 NA SAZ-13 75.15±4.16 1.75±0.39 SAZ-14 105.04±2.10 1.25±0.26 SAZ-15 77.62±7.62 3.25±0.45 SAZ-16 9.48±3.41 NA SAZ-17 15.86±3.89 NA SAZ-18 90.57±6.85 1.46±0.19 SAZ-19 83.23±1.03 3.93±0.16 SAZ-20 74.21±2.45 4.34±0.36 SAZ-21 81.27±3.94 2.66±0.14 SAZ-22 51.53±6.14 NA Na3VO4 — 1.86±0.24 a待测样品浓度为20 μg/mL时的抑制率; b IC50值通过待测样品在浓度为5 μg/mL时的抑制率非线性拟和计算得到  下载: 导出CSV
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 3271
- HTML全文浏览量: 333
 下载:
下载:
