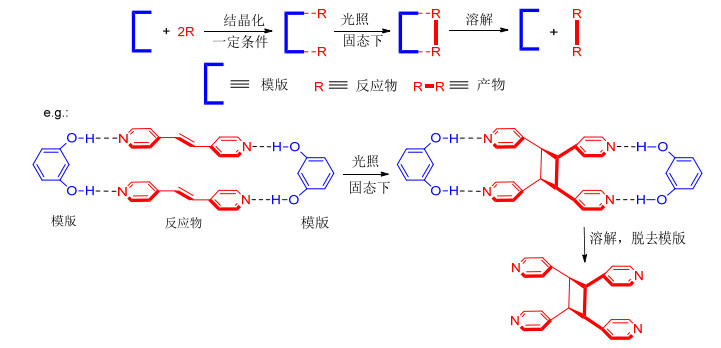
 图 1
模版分子与反应物分子自组装及[2+2]环加成反应过程
Figure 1.
Self-assembly of template and reactant molecular and process of [2+2] photodimerizations
图 1
模版分子与反应物分子自组装及[2+2]环加成反应过程
Figure 1.
Self-assembly of template and reactant molecular and process of [2+2] photodimerizations
引用本文:
欧亚平, 张静, 庾江喜, 朱小明. 1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物的合成、光谱性质及理论研究[J]. 有机化学,
2017, 37(2): 394-402.
doi:
10.6023/cjoc201606040

Citation: Ou Yaping, Zhang Jing, Yu Jiangxi, Zhu Xiaoming. Synthesis, Spectral Properties and Theoretical Studies of 1, 4-Bis[2-(4-pyridyl) ethenyl]benzene Derivatives[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 394-402. doi: 10.6023/cjoc201606040
Citation: Ou Yaping, Zhang Jing, Yu Jiangxi, Zhu Xiaoming. Synthesis, Spectral Properties and Theoretical Studies of 1, 4-Bis[2-(4-pyridyl) ethenyl]benzene Derivatives[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 394-402. doi: 10.6023/cjoc201606040
1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物的合成、光谱性质及理论研究
摘要:
通过经典的Witting-Horner反应及Sonogashira交叉偶联反应以较高的产率合成得到4个不对称型氢键受体分子1,4-双[2-(4-吡啶基)乙烯基]苯衍生物,并对其结构及光谱性质开展了研究.系列化合物均通过核磁、元素分析及质谱的表征,晶体结构表明由于苯环邻位芳环乙炔基的空间效应导致氢键组装的构型发生改变.紫外可见光谱结合含时密度泛函理论(TDDFT)计算表明目标化合物在紫外区域均呈现出强烈的π→π*及ICT跃迁吸收.荧光光谱表明随着共轭体系的逐渐增大也导致了它们的发射峰有一个红移的倾向,且有利于分子之间形成π-π堆积.以上结果将为后续的氢键自组装的区域选择性及光照环化实验研究提供了一定的合成基础及理论依据.
-
关键词:
- 不对称氢键受体分子
- / 1, 4-双[2-(4-吡啶基) 乙烯基]苯
- / 晶体结构
- / 光谱性质
English
Synthesis, Spectral Properties and Theoretical Studies of 1, 4-Bis[2-(4-pyridyl) ethenyl]benzene Derivatives
Abstract:
Four asymmetric hydrogen-bond acceptors of 1, 4-bis[2-(4-pyridyl) ethenyl]benzene derivatives with higher yields were synthesized through the classical Witting-Horner and Sonogashira cross-coupling reactions, and their structures and spectral properties were investigated. The series of compounds all have been characterized by NMR, MS and element analysis. Crystal structures indicated that the conformation of hydrogen-bond self-assemble may be change due to the hindrance effect from the arylacetyl group in o-position of benzene. UV-Vis spectra combined with time-dependent density functional theory (TDDFT) calculation results showed that target compounds all display a strong π→π* and ICT transition absorptions. Fluorescence spectrum indicated that gradual increasing of conjugate systems make their emission peak tend to a red shift, which is conducive to π-π accumulation. Above results will provide certain synthetic basis and theoretical foundation for subsequent studies about regioselectivity of hydrogen-bond self-assemble and cyclization reactions from light.
-
1987年, C. J. Pedersen (佩德森)、J. M. Lehn (莱恩)、D. J. Cram (克来姆) 三位化学家被授予诺贝尔化学奖以表彰他们在超分子化学领域的杰出贡献, 自此以后, 超分子化学作为一门新兴的科学快速蓬勃发展起来, 并以其强大的生命力和广阔的应用前景逐渐发展成为许多科学家们的主要研究领域和前沿课题.超分子化学主要研究的是弱相互作用 (即非共价作用), 如氢键作用、π-π作用、疏溶剂作用、偶极作用、静电作用即范德华力等.其内容更为丰富, 主要有分子识别、分子自组装、超分子光化学、超分子催化以及超分子与生物科学相结合等, 目前已经渗透到生物、环境、医学、材料、农业等众多学科领域[1~4].
分子自组装是超分子化学的一个重要研究内容.所谓分子自组装, 即分子与分子之间通过弱相互作用 (非共价作用) 自发形成的稳定的分子聚集体过程, 其本质就是分子识别作用, 在生命体系中普遍存在, 许多复杂且有生物学功能的超分子系统如蛋白质、核酸等都是通过分子自组装形成的[5].特别的是, 它也作为合成一些分子的重要手段, 可以合成常规合成手段无法得到的化合物[6~7]. MacGillivray等[8~12]已经在这方面做了大量的研究工作, 目的是利用这种弱相互作用 (非共价作用) 来指导共价键的形成, 尤其通过小的有机分子或者金属有机配合物作为模版分子与反应物 (一般为刚性的烯烃分子) 经过分子识别和自组装来诱导固态 (共晶) 条件下符合光化学活性的[2+2]环加成反应, 如图 1所示.该方法能够合成不同立体构型的四元环状化合物且具有较高的产率, 得到的环丁烷衍生物可以作为天然产物分子的一个重要构建单元以及其他反应的合成子.同时模版分子可以回收再利用, 这样为有机合成策略提供了一条新途径.
图 1
模版分子与反应物分子自组装及[2+2]环加成反应过程
Figure 1.
Self-assembly of template and reactant molecular and process of [2+2] photodimerizations
分子自组装后共晶, 在固态下发生[2+2]环加成反应必须要达到Schmidt光致环化条件.那么这依赖于模版分子的选择以及反应物本身的性质, 通常所用的模版分子有 (含金属有机模版) 几种[8], 如图 2所示.也正是由于模版分子的不同从而可以控制两分子中双键之间的距离.此外, 通过研究肉桂酸在固态下发生光致环化的条件发现, 烯烃发生[2+2]环加成两个双键处在平行的位置且它们之间的距离应小于4.2 ,如图 3所示.由此可见在固态条件下, 只要分子在堆积形态下的双键距离小于4.2 且处于平行的位置, 那么在光照条件下就可能发生环加成反应.
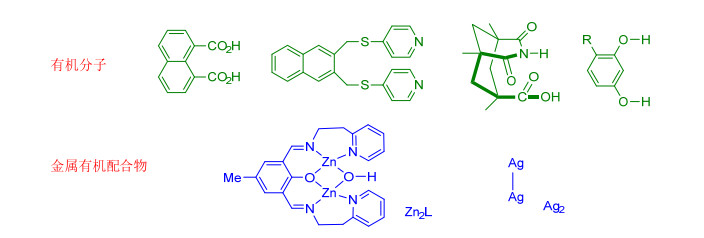 图 2
不同类型的模版分子 (有机小分子及金属模版)
Figure 2.
Different templates molecules (small organic molecular and metal-organic templates)
图 2
不同类型的模版分子 (有机小分子及金属模版)
Figure 2.
Different templates molecules (small organic molecular and metal-organic templates)
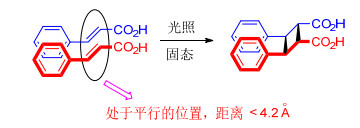 图 3
分子在固态条件下发生光致环化的条件
Figure 3.
Conditions of photodimerization to molecular in the solid state
图 3
分子在固态条件下发生光致环化的条件
Figure 3.
Conditions of photodimerization to molecular in the solid state
随着研究的不断深入, 科学家们不断研究影响分子自组装、共晶以及光致环化的各种因素, 从而有利于多元环状化合物的合成, 2003年, MacGillivray等[13]通过间苯二酚衍生物为模版与反应物1, 4-双[2-(4-吡啶基) 乙烯基]苯进行自组装, 然后共晶, 固态条件下再经过光照关环, 脱去模版分子得到了[2.2]对环芳衍生物, 如图 4所示. [2.2]对环芳结构单元本身就作为一个具有面面堆积作用的例子, 其衍生物已广泛应用于不对称合成、生物医药、纳米技术以及催化等[14~17], 因此该方法对[2.2]对环芳衍生物的合成提供了一条新的途径.
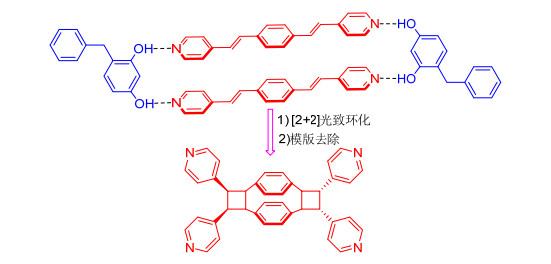 图 4
1, 4-双[2-(4-吡啶基) 乙烯基]苯与间苯二酚衍生物共晶光照下固态关环形成[2.2]对环芳衍生物
Figure 4.
Cocrystallization of 1, 4-bis[2-(4-pyridyl) ethenyl]benzene with resorcinol derivatives, and forming the [2.2]paracyclophane derivatives by UV-irradiation
图 4
1, 4-双[2-(4-吡啶基) 乙烯基]苯与间苯二酚衍生物共晶光照下固态关环形成[2.2]对环芳衍生物
Figure 4.
Cocrystallization of 1, 4-bis[2-(4-pyridyl) ethenyl]benzene with resorcinol derivatives, and forming the [2.2]paracyclophane derivatives by UV-irradiation
通过文献调研发现, 以上的分子自组装主要驱动力是氢键作用及π-π作用, 且氢键受体分子均是对称的乙烯基吡啶衍生物, 如1, 4-双[2-(4-吡啶基) 乙烯基]苯, 如果在其中间的苯环上引入具有共轭体系的取代基, 那么两分子氢键受体与两分子氢键给体在进行自组装时, 可能会出现两种情况 (交叉式和重叠式)(如图 5所示), 产生了区域选择性, 然后经过强紫外光照, 分子间平行双键之间发生关环, 从而合成具有区域选择性的[2.2]对环芳衍生物.究竟出现那种自组装形式, 依赖于取代基之间π-π堆积作用强弱、电子性质以及空间效应等因素.
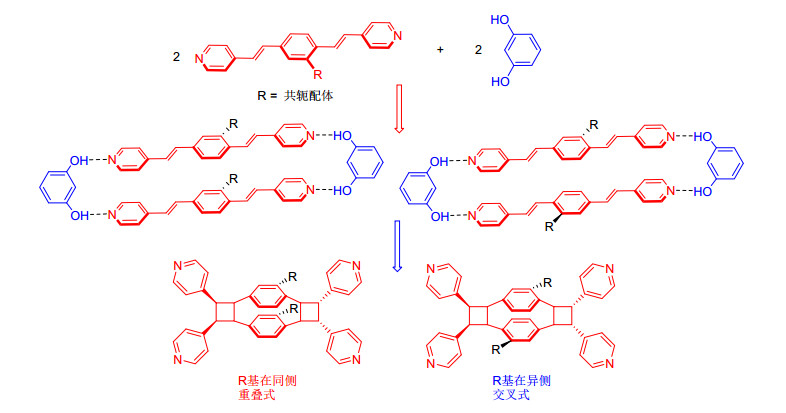 图 5
不对称氢键受体分子与间苯二酚两种氢键自组装情况及两种不同的光致环化产物
Figure 5.
Hydrogen-bond self-assemble situations of asymmetric hydrogen-bond acceptor with resorcinol and two different photodimerization products
图 5
不对称氢键受体分子与间苯二酚两种氢键自组装情况及两种不同的光致环化产物
Figure 5.
Hydrogen-bond self-assemble situations of asymmetric hydrogen-bond acceptor with resorcinol and two different photodimerization products
基与以上分析, 为了探索氢键组装下, π-π堆积作用、电子效应及空间效应所形成的区域选择性组装情况, 本论文特设计合成如图 6所示一系列不对称的氢键受体分子1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物, 并对其结构及性质进行表征.希望为后续探索区域选择性自组装情况提供合成基础和理论依据.
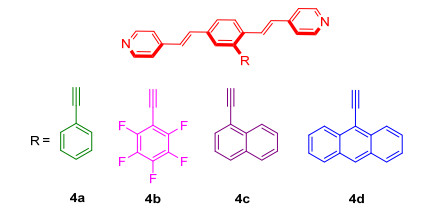 图 6
不对称的氢键受体分子1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物4a~4d
Figure 6.
Asymmetric hydrogen-bond acceptor 1, 4-bis[2-(4-pyridyl) ethenyl]benzene derivatives 4a~4d
图 6
不对称的氢键受体分子1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物4a~4d
Figure 6.
Asymmetric hydrogen-bond acceptor 1, 4-bis[2-(4-pyridyl) ethenyl]benzene derivatives 4a~4d
1 结果与讨论
1.1 目标化合物的合成
目标化合物的合成经历了两个重要的人名反应, 即Witting-Horner反应及Sonogashira交叉偶联反应, 其合成路线如图 7所示.
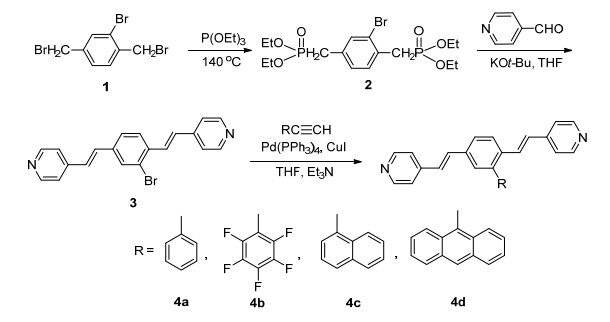 图 7
目标化合物4a~4d的合成路线示意图
Figure 7.
Synthetic route diagram of target compounds 4a~4d
图 7
目标化合物4a~4d的合成路线示意图
Figure 7.
Synthetic route diagram of target compounds 4a~4d
中间体2通过类似文献[20]的方法以70%的收率合成得到, 其在常温下为粘稠液体, 若将其置于冰箱冷冻, 则为白色固体.从中间体2至中间体3的经典的Witting-Horner反应, 关键是通过强碱来控制形成双键的顺反.在该反应中, 我们选择的强碱是KOBu-t, 生成的产物绝大多数为反式产物, 且顺式产物可以通过重结晶的方法除去, 从而以65%的收率得到中间体3.生成的溴代产物3再分别与末端炔 (苯乙炔、五氟苯乙炔、α-萘乙炔及9-蒽乙炔) 在Pd (PPh3)4及CuI的作用下发生Sonogashira交叉偶联反应, 分别以中等以上的收率得到4a~4d.
1.2 目标化合物4b的X射线晶体结构
目标化合物4b经过了X射线单晶衍射表征, 该晶体是通过扩散法 (氯仿/甲醇) 在室温下培养 (放置近30 d后) 得到.其为棕黄色块状晶体.化合物4b的分子结构图如图 8所示, 对应的部分键长、键角及二面角被列在表 1中.从键长数据中可以看出, C (14)-C (15) 及C (21)-C (22) 的键长各为1.292和1.191 ,分别对应于碳碳双键及叁键键长范围. C (6)-C (7)-C (8) 键角接近120°, 也可以很明显看出形成的为反式双键, C (10)-C (21)-C (22) 键角接近180°, 也进一步证实了叁键的存在.平面[C (9), C (10), C (21)]与平面[C (22), C (23), C (28)]之间的夹角接近于0°, 表明该分子是一个高度共平面的分子, 从而使得分子与分子之间可能存在一定的π-π堆积, 从图 8(b)可以看出这种堆积作用. CCDC号为1488285.
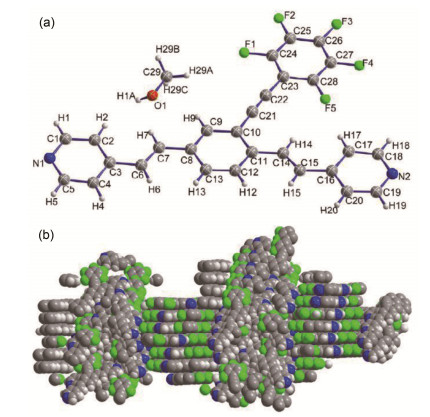 图 8
目标化合物4b的分子结构图
Figure 8.
Molecular structure of target compound 4b
图 8
目标化合物4b的分子结构图
Figure 8.
Molecular structure of target compound 4b
 表 1
目标化合物4b的晶体结构对应的部分键长 ( )、键角 (°) 和二面角 (°)
Table 1.
Selected bond lengths ( ), angles (°) and dihedral angle (°) from crystal structure of 4b
表 1
目标化合物4b的晶体结构对应的部分键长 ( )、键角 (°) 和二面角 (°)
Table 1.
Selected bond lengths ( ), angles (°) and dihedral angle (°) from crystal structure of 4b
键长/ N (2)-C (18) 1.311 C (11)-C (10) 1.394 C (18)-C (17) 1.370 C (10)-C (21) 1.442 C (17)-C (16) 1.384 C (21)-C (22) 1.191 C (16)-C (15) 1.489 C (22)-C (23) 1.426 C (15)-C (14) 1.292 C (23)-C (28) 1.392 C (14)-C (11) 1.456 C (28)-F (5) 1.335 键角/(°) C (6)-C (7)-C (8) 126.52 C (9)-C (10)-C (21) 119.54 C (10)-C (21)-C (22) 178.25 二面角/(°) [C (6), C (7), C (8)]-[C (13), C (8), C (9)] 4.82 [C (11), C (14), C (15)]-[C (10), C (11), C (12)] 2.10 [C (9), C (10), C (21)]-[C (22), C (23), C (28)] 0.38 表 1 目标化合物4b的晶体结构对应的部分键长 ( )、键角 (°) 和二面角 (°)
Table 1. Selected bond lengths ( ), angles (°) and dihedral angle (°) from crystal structure of 4b1.3 光谱及理论计算研究
1.3.2 荧光光谱分析
为了更好了解系列化合物的发射光谱特点, 我们对其进行了荧光光谱实验, 图 11为中间体3及目标化合物4a~4d在CH2Cl2溶液中的荧光光谱图.从图中可以看出, 中间体3在411 nm处呈现出最强的荧光发射, 随着中间体3苯环上的溴原子被苯乙炔基、五氟苯乙炔基、萘乙炔基蒽乙炔基取代分别得到4a~4d, 原有的发射峰分别成两个, 这可能由于共轭体系的增大, 使π-π作用增强, 促使其形成激基缔合物.这在另一方面说明它们的堆积作用增强, 从而使得荧光强度比中间体3要弱[22].此外, 随着共轭体系的逐渐增大导致了它们的发射峰有一个红移的倾向.从图中可以看出化合物4d在500 nm左右有一个较弱的发射峰, 因此该化合物在紫外灯下 (365 nm) 呈现黄光.从以上分析可以看出, 共轭体系的增大有利于分子之间形成π-π堆积, 从而有望实现区域性组装.
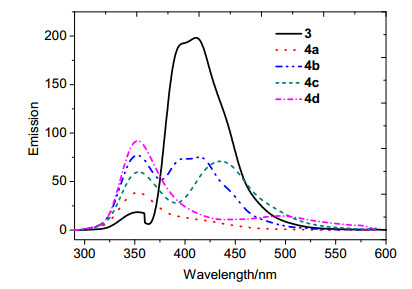 图 11
中间体3及目标化合物4a~4d在CH2Cl2溶液中的荧光光谱图
Figure 11.
Fluorescence emission spectrum of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution
图 11
中间体3及目标化合物4a~4d在CH2Cl2溶液中的荧光光谱图
Figure 11.
Fluorescence emission spectrum of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution
1.3.1 紫外可见吸收光谱研究及含时密度泛函理论计算
为了考察目标化合物的电子跃迁情况, 目标化合物4a~4d及中间体3均被进行了紫外可见光谱测试 (浓度为2.0×10-5 mol/L的CH2Cl2溶液), 对应的电子跃迁吸收光谱如图 9所示.
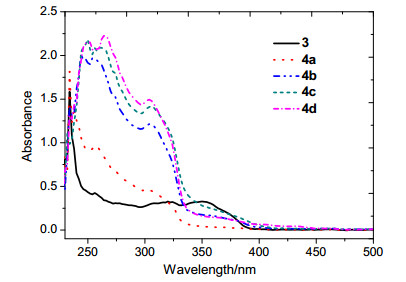 图 9
中间体3及目标化合物4a~4d在CH2Cl2溶液中的紫外光谱图
Figure 9.
UV-Vis absorption spectra of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution
图 9
中间体3及目标化合物4a~4d在CH2Cl2溶液中的紫外光谱图
Figure 9.
UV-Vis absorption spectra of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution
从图中可以看出, 中间体3在350 nm处呈现一个中等强度的吸收峰, 而在250~300 nm之间呈现很弱的吸收.随着中间体3上的溴原子被不同的共轭体所取代, 目标化合物4a~4d在250~325 nm之间的吸收逐渐增强, 这主要是由于分子的共轭体增大, 导致由π→π*跃迁的K带吸收更强, 甚至能够淹没350~400 nm之间的B带.当然这些跃迁吸收主要来自于π→π的跃迁及分子内电荷转移跃迁 (ICT) 吸收[21], 通过后面的含时密度泛函理论 (TDDFT) 得到证实.比较目标分子4a~4d, 其在250~330 nm之间的吸收呈现出相似的吸收峰型, 在350 nm以后的矮而宽的吸收呈现出微弱的红移, 这主要由于分子共轭体系的增大所致.
为了更好地理解系列目标化合物的电子跃迁性质, 我们对代表性目标化合物4b展开了密度泛函理论计算, 图 10为目标化合物4b主要跃迁设计的部分分子轨道图, 对应的主要跃迁及其归属见表4.从图 10可以看出, 该化合物在紫外区域的跃迁分别来自于HOMO→LUMO、HOMO-1→LUMO及HOMO-2→ LUMO+1, 主要归属于π→π*及分子内电荷转移 (ICT) 跃迁吸收.
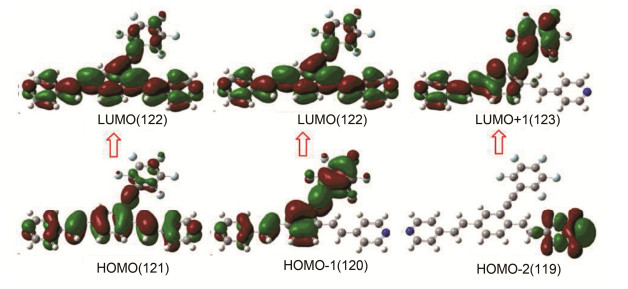 图 10
目标化合物4b主要跃迁涉及的部分分子轨道图
Figure 10.
Selected frontier orbitals involved in the transition for target compound 4b
图 10
目标化合物4b主要跃迁涉及的部分分子轨道图
Figure 10.
Selected frontier orbitals involved in the transition for target compound 4b
2 结论
通过经典的Witting-Horner反应及Sonogashira交叉偶联反应以较高的收率合成了4个不对称型的氢键受体分子1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物4a~4d, 并对其结构及性质进行了研究.该系列分子结构均通过了核磁、质谱及元素分析结构表征.此外化合物4b的结构通过X射线单晶衍射确认.从化合物4b的晶体结构可以看出4-吡啶乙烯基在苯的同侧, 这主要与邻位上的芳环乙炔基的空间效应有关.四个氢键受体分子通过间苯二酚组装, 然后光致环化, 去模版得到图 12的结果.
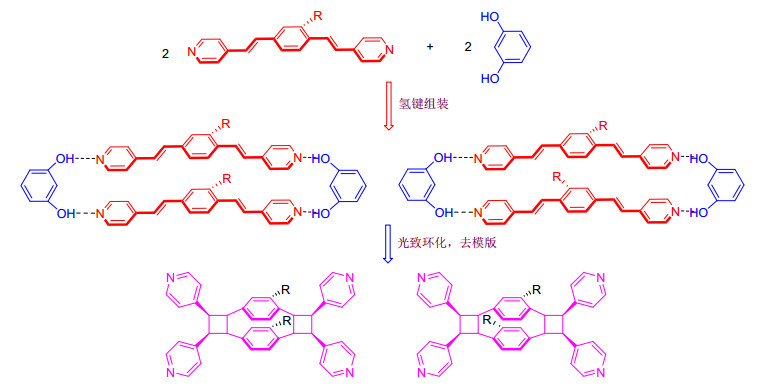 图 12
根据晶体结构分析实际过程中可能的氢键组装情况
Figure 12.
Possible process of hydrogen-bond self-assemble according to crystal structure analysis
表 2
TDDFT计算模型分子4b主要的电子跃迁及其归属
Table 2.
Major electronic excitations for 4b determined by TD-DFT methods (B3LYP/6-31G)
图 12
根据晶体结构分析实际过程中可能的氢键组装情况
Figure 12.
Possible process of hydrogen-bond self-assemble according to crystal structure analysis
表 2
TDDFT计算模型分子4b主要的电子跃迁及其归属
Table 2.
Major electronic excitations for 4b determined by TD-DFT methods (B3LYP/6-31G)
波长/nm 振子强度 (f) 跃迁 归属 457 0.9722 HOMO→LUMO (70%) π→π 365 1.3586 HOMO-1→LUMO (52%) ICT 283 0.2134 HOMO-2→LUMO+1 (37%) ICT 表 2 TDDFT计算模型分子4b主要的电子跃迁及其归属
Table 2. Major electronic excitations for 4b determined by TD-DFT methods (B3LYP/6-31G)紫外可见光谱结合TDDFT计算表明目标化合物4a~4d在紫外区域均呈现出强烈的π→π*及ICT跃迁吸收, 完全符合这类化合物的吸收特征.荧光光谱表明随着共轭体系的逐渐增大, 它们的发射峰有一个红移的倾向, 且有利于分子之间形成π-π堆积.以上结果将为后续的氢键自组装的区域选择性及光照环化实验研究提供一定的合成基础及理论依据.
3 实验部分
3.1 中间体2-溴-1, 4-二苯甲基膦酸二乙酯 (2) 的合成
氮气保护下, 在带有搅拌磁子的50 mL圆底烧瓶中依次加入化合物1 (1.6 g, 4.8 mmol)、亚磷酸三乙酯 (9 mL), 混合体系在140 ℃下加热回流4 h后, 冷却至室温, 加入30 mL正己烷, 析出固体 (在常温下易变成液体), 减压蒸馏除去过量的亚磷酸三乙酯及溴乙烷, 得到1.5 g米白色粘稠固体, 产率70%. m.p. 25~30 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.26 (s, 12H, CH3), 3.09 (d, J=20 Hz, 2H, Ar-CH2), 3.37 (d, J=20 Hz, 2H, Ar-CH2), 4.05 (s, 8H, CH3), 7.23 (s, 1H, Ar-H), 7.41 (s, 1H, Ar-H), 7.52 (s, 1H, Ar-H); 13C NMR (100 MHz, CDCl3) δ: 15.94, 15.99, 31.86 (d, J=17 Hz, O=PCH2), 33.25 (d, J=17 Hz, O=PCH2), 61.81 (OCH2), 61.87 (OCH2), 124.39, 128.56, 130.07, 131.16, 132.04, 132.09, 133.57; MS m/z: 456.01 (M+). Anal. calcd for C16H27BrO6P2: C 42.03, H 5.95; found C 42.10, H 5.88.
3.1 仪器与试剂
实验过程中大部分反应都是在氮气或者氩气保护下进行的, 采用的是标准的Schlenk双排管真空线技术.溶剂四氢呋喃经钠/二苯甲酮处理, 二氯甲烷和正己烷用氢化钙处理, 三乙胺用KOH干燥处理, 使用前将处理好的溶剂新蒸出并脱气.试剂2, 5-二甲基溴苯、4-吡啶甲醛、叔丁醇钾、碘化亚铜、亚磷酸三乙酯、三苯基膦、溴苯、五氟代碘苯、α-溴代萘、9-溴蒽等从中国医药集团上海试剂公司购买, 溶剂均为国产分析纯; 2, 5-二溴甲基溴苯[18]、苯乙炔[19]、五氟苯乙炔[19]、α-乙炔基萘[19]、9-乙炔基蒽[19]均按文献方法合成得到.
元素分析 (C、H、N) 经华中师大分析测试中心Vario ElIII CHNSO元素分析仪测得; 质谱 (MS) 使用Firmigan Trace质谱仪测定; 1H NMR、13C NMR和31P NMR经Varian MERCURY Plus 400 MHz或Varian MERCURY Plus 600 MHz核磁共振仪测得, 其中1H NMR、13C NMR谱化学位移以TMS作为内标, 31P NMR谱化学位移以85% H3PO4作为外标.
化合物的单晶在Bruker Apex CCD X射线衍射仪下收集, 其晶体衍射数据经过SAINT v 6.26还原, 然后经SADABS软件修正晶胞参数.晶体结构由直接法 (SHELXTL-97) 程序解出.以差值傅立叶合成法定出非氢原子坐标, 并以各项异性热参数用于全矩阵最小二乘法对其进行修正, 氢原子用理论加氢的方式产生.
紫外可见光谱使用日本岛津公司生产的UV-3600紫外-可见-近红外光谱仪测定; 荧光光谱在Fluoromax-P荧光光谱仪 (HORIBA JOBIN YVON INC) 测定.
DFT计算通过高斯09程序在B3LYP/6-31G*理论水平下进行; 几何优化在没有任何对称性限制下完成的, 频率计算基于几何优化结果; 电子跃迁来自于含时密度泛函理论计算 (TDDFT) 结果; 分子轨道贡献及可视化分别通过Multiwfn2.6.1_bin_Win软件包及GaussView 5.0程序完成.
3.2 中间体2-溴-1, 4-双[2-(4-吡啶基) 乙烯基]苯 (3) 的合成
氮气保护下, 向含有磁子的50 mL圆底烧瓶中加入叔丁醇钾 (381 mg, 3.40 mmol), 经恒压滴液漏斗再往烧瓶中逐渐滴加入含有中间体2 (457 mg, 1.00 mmol) 及4-吡啶甲醛 (252 mg, 2.35 mmol) 的四氢呋喃 (THF) 溶液, 混合体系室温反应1 h后, 向体系中滴加入HCl (2 mol/L, 20 mL), 蛋黄色沉淀析出, 然后加入饱和的NaHCO3溶液, 最后体系用CH2Cl2萃取, 有机相依次用饱和NaHCO3溶液, 饱和NaCl溶液洗涤, 无水Na2SO4干燥, 旋干, 粗产品经柱层析分离[硅胶200~300目, V(THF):V(石油醚)=1:2]得到0.20 g黄色固体, 产率55%. m.p. 192~196 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.01 (t, J=16 Hz, 2H, CH=), 7.20 (d, J=16 Hz, 1H, CH=), 7.38 (dd, J=8 Hz, 4H, Py-H), 7.49~7.52 (m, 1H), 7.68 (t, J=16 Hz, 2H, Py-CH=), 7.70 (s, 1H), 8.58~8.62 (m, 4H, Py-H); 13C NMR (100 MHz, CDCl3) δ: 120.79, 120.95, 124.93, 126.02, 126.95, 127.64, 128.82, 130.71, 130.99, 131.41, 135.72, 137.78, 143.74, 143.89, 150.21; MS m/z: 362.02 (M+). Anal. calcd for C20H15BrN2: C 66.13, H 4.16; found C 66.20, H 4.02.
3.3 目标化合物4a~4d的合成
3.3.3 2-(1-萘乙炔基)-1, 4-双[2-(4-吡啶基) 乙烯基]苯 (4c) 的合成
化合物4c的合成步骤与化合物4a相似, 投料量:中间体3 (100 mg, 0.275 mmol), CuI (6 mg, 0.24 mmol), Pd (PPh3)4 (0.28 g, 0.24 mmol), 三乙胺 (5 mL) 及四氢呋喃 (10 mL), 1-乙炔基萘 (83 mg, 0.55 mmol).粗产品经柱层析分离[硅胶200~300目, V(丙酮):V(石油醚)=1:1作为洗脱剂]得到83 mg亮黄色固体, 产率69%. m.p. 256~260 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.10 (d, J=20 Hz, 1H, CH=), 7.18 (d, J=16 Hz, 1H, CH=), 7.29 (d, J=4 Hz, 1H), 7.41 (dd, J=4 Hz, 4H, Py-H), 7.50~7.59 (m, 4H), 7.77 (d, J=12 Hz, 1H), 7.83 (d, J=8 Hz, 2H), 7.91 (d, J=8 Hz, 2H), 8.06 (d, J=16 Hz, 1H, CH=), 8.44 (d, J=8 Hz, 1H), 8.60 (s, 4H).由于在常用氘代试剂中溶解度较差, 没有收集到该化合物的碳谱. MS m/z: 434.20 (M+). Anal. calcd for C32H22N2: C 88.45, H 5.10; found C 88.39, H 5.31.
3.3.1 2-苯乙炔基-1, 4-双[2-(4-吡啶基) 乙烯基]苯 (4b) 的合成
氮气保护下, 向含有磁子的25 mL二颈瓶中依次加入中间体3(100 mg, 0.275 mmol), Pd (PPh3)4 (32 mg, 0.028 mmol) 及CuI (6 mg, 0.041 mmol), 然后注入已脱气的THF (10 mL) 及Et3N (5 mL), 室温搅拌30 min后, 加入苯乙炔 (140 mg, 1.38 mmol), 体系加热回流反应48 h冷却至室温.过滤 (经硅藻土) 得滤液, 旋干, 柱层析分离[硅胶200~300目, V(丙酮):V(石油醚)=1:1作为洗脱剂]得到68 mg黄色固体, 产率64%. m.p. 198~203 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.08 (d, J=16 Hz, 1H, CH=), 7.16 (d, J=16 Hz, 1H, CH=), 7.27 (d, J=16 Hz, 1H, CH=), 7.37~7.42 (m, 7H), 7.52 (d, J=8 Hz, 1H), 7.59~7.61 (m, 2H), 7.74 (d, J=12 Hz, 2H), 7.90 (d, J=16 Hz, 1H, CH=), 8.59~8.61 (m, 4H); 13C NMR (100 MHz, CDCl3) δ: 87.02 (Ph-C≡), 95.19 (Ar-C≡), 120.86, 122.73, 123.36, 125.56, 126.94, 127.06, 127.80, 128.54, 128.76, 130.42, 131.24, 131.46, 136.12, 137.35, 144.06, 144.37, 150.21; MS m/z: 384.11 (M+). Anal. calcd for C28H20N2: C 87.47, H 5.24; found C 87.23, H 5.36.
3.3.4 2-(9-蒽乙炔基)-1, 4-双[2-(4-吡啶基) 乙烯基]苯 (4d) 的合成
化合物4d的合成步骤与化合物4a相似, 投料量:中间体3 (200 mg, 0.45 mmol), CuI (12 mg, 0.48 mmol), Pd (PPh3)4 (0.56 g, 0.48 mmol), 三乙胺 (10 mL) 及四氢呋喃 (20 mL), 9-乙炔基蒽 (182 mg, 0.90 mmol).粗产品经柱层析分离[硅胶200~300目, V(丙酮):V(石油醚)=1:1作为洗脱剂]得到175 mg黄色固体, 产率: 66%. m.p. 300~303 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.14 (d, J=16 Hz, 1H, CH=), 7.21 (d, J=16 Hz, 1H, CH=), 7.34 (d, J=16 Hz, 1H, CH=), 7.43 (dd, J=4 Hz, 4H, Py-H), 7.54~7.61 (m, 5H), 7.83 (d, J=8 Hz, 1H), 7.95 (s, 1H), 8.06~8.07 (m, 2H), 8.20 (d, J=16 Hz, 1H, CH=), 8.50 (s, 1H), 8.59~8.67 (m, 6H).由于在常用氘代试剂中溶解度较差, 没有收集到该化合物的碳谱. MS m/z: 484.23 (M+). Anal. calcd for C36H24N2: C 89.23, H 4.99; found C 89.30, H 4.92.
辅助材料 (Supporting Information) 最终产物和部分中间体的1H NMR及部分13C NMR谱图, 化合物4b的晶体数据, 以及化合物4a和4c经DFT方法 (B3LYP/ 6-31G*) 优化下的构型.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.3.2 2-五氟苯乙炔基-1, 4-双[2-(4-吡啶基) 乙烯基]苯 (4b) 的合成
化合物4b的合成步骤与化合物4a相似, 投料量:中间体3 (100 mg, 0.275 mmol), CuI (6 mg, 0.24 mmol), Pd (PPh3)4 (0.28 g, 0.24 mmol), 三乙胺 (5 mL) 及四氢呋喃 (10 mL), 五氟苯乙炔 (106 mg, 0.55 mmol).粗产品经柱层析分离[硅胶200~300目, V(丙酮):V(石油醚)=1:1作为洗脱剂]得到101 mg紫黑色固体, 产率78%. m.p. 229~233 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.11 (d, J=16 Hz, 1H, CH=), 7.19 (d, J=16 Hz, 1H, CH=), 7.30 (d, J=4 Hz, 1H), 7.42 (dd, J=4 Hz, 4H, Py-H), 7.63 (d, J=8 Hz, 1H), 7.80 (t, J=8 Hz, 2H), 7.90 (d, J=16 Hz, 1H, CH=), 8.62 (t, J=4 Hz, 4H, Py-H); 13C NMR (100 MHz, CDCl3) δ: 102.06 (Ar-C≡), 115.89 (Ar-C≡), 115.96, 116.18, 131.42, 131.52, 136.15, 137.17, 143.23, 163.70, 166.21, 173.81; MS m/z: 474.14 (M+). Anal. calcd for C28H15F5N2: C 70.89, H 3.19; found C 70.70, H 3.23.
-
-
[1]
Tozawa, T.; Jones, J. T. A.; Swamy, S. I.; Jiang, S.; Adams, D. J.; Shakespeare, S.; Clowes, R.; Bradshaw, D.; Hasell, T.; Chong, S. Y.; Tang, C.; Thompson, S.; Parker, J.; Trewin, A.; Bacsa, J.; Slawin, A. M. Z.; Steiner, A.; Cooper, A. I. Nat. Mater. 2009, 8, 973. doi: 10.1038/nmat2545
-
[2]
Yan, X.; Xu, D.; Chi, X.; Chen, J.; Dong, S.; Ding, X.; Yu, Y.; Huang, F. Adv. Mater. 2012, 24, 362. doi: 10.1002/adma.201103220
-
[3]
Hu, Q. D.; Tang, G. P.; Chu, P. K. Acc. Chem. Res. 2014, 47, 2017. doi: 10.1021/ar500055s
-
[4]
Kuehl, C. J.; Huang, S. D.; Stang, P. J. J. Am. Chem. Soc. 2001, 123, 9634. doi: 10.1021/ja0114355
-
[5]
Ensslen, P and Wagenknecht, H. Acc. Chem. Res., 2015, 48, 2724. doi: 10.1021/acs.accounts.5b00314
-
[6]
Koshti, V. S.; Mote, N. R.; Gonnade, R. G. and Chikkali, S. H. Organometallics 2015, 34, 4802. doi: 10.1021/acs.organomet.5b00664
-
[7]
Elemans, J. A. A. W.; Slangen, R. R. J.; Rowan, A. E.; Nolte, R. J. M. J. Org. Chem., 2003, 68, 9040. doi: 10.1021/jo035130f
-
[8]
MacGillivray, L. R.; Papaefstathiou, G. S.; Friscic, T.; Hamilton, T. D.; Bucar, D-K.; Chu Q.; Varshney, D. B.; Georgiev, I. V. Acc. Chem. Res. 2008, 41, 280. doi: 10.1021/ar700145r
-
[9]
Sokolov, A. N.; Friscic, T.; MacGillivray, L. R. J. Am. Chem. Soc. 2006, 128, 2806. doi: 10.1021/ja057939a
-
[10]
Sokolov, A. N.; Bucar, D-K.; Baltrusaitis, J.; Gu, S. X.; MacGillivray, L. R. Angew. Chem. 2010, 122, 4369. doi: 10.1002/ange.v122:25
-
[11]
MacGillivray, L. R. J. Org. Chem. 2008, 73, 3311. doi: 10.1021/jo8001563
-
[12]
Friscic, T.; Drab, D. M.; MacGillivray, L. R. Org. Lett. 2004, 6, 4647. doi: 10.1021/ol0484052
-
[13]
Frišcic, T.; MacGillivray, L. R. Chem. Commun. 2003, 1306.
-
[14]
Jiang, B.; Lei, Y.; Zhao, X.-L. J. Org. Chem. 2008, 73, 7833. doi: 10.1021/jo801373r
-
[15]
Lahann, J.; Höcher, H.; Langer, R. Angew. Chem., Int. Ed. 2001, 40, 726. doi: 10.1002/1521-3773(20010216)40:4<>1.0.CO;2-X
-
[16]
Sillen, C.; Liu, N.; Ho, W.; Maddox, J. B.; Mukamel, S.; Liu, B.; Bazan, G. C. Nano Lett. 2008, 8, 208. doi: 10.1021/nl072493c
-
[17]
Bolm, C.; Whelligan, D. K. Adv. Synth. Catal. 2006, 348, 2093. doi: 10.1002/(ISSN)1615-4169
-
[18]
Kay, K.-Y.; Baek, Y. G. Chem. Ber. /Recl. 1997, 130, 581. doi: 10.1002/(ISSN)1099-0682
-
[19]
Wu, X. H.; Jin, S.; Liang, J. H.; Li, Z. Y.; Yu, G.-A.; Liu, S. H. Organometallics 2009, 28, 2450. doi: 10.1021/om900018y
-
[20]
Wang, L.; Tao, X-L.; Yang, J-X.; Yu, W-T.; Ren, Y.; Xin, Y.; Liu, Z.; Jiang, M-H. J Solid State Chem. 2004, 177, 4293. doi: 10.1016/j.jssc.2004.08.036
-
[21]
Kaminker, R.; Lahav, M.; Motiei, L.; Vartanian, M.; Popo-vitz-Biro, R.; Iron, M. A.; vander Boom, M. E. Angew. Chem., Int. Ed. 2010, 49, 1218. doi: 10.1002/anie.200906636
-
[22]
Hrobarikova, V.; Hrobarik, P.; Gajdos, P.; Fitilis, I.; Fakis, M.; Persephonis, P.; Zahradnık, P. J. Org. Chem. 2010, 75, 3053. doi: 10.1021/jo100359q
-
[1]
-

图 1 模版分子与反应物分子自组装及[2+2]环加成反应过程
Figure 1 Self-assembly of template and reactant molecular and process of [2+2] photodimerizations

图 2 不同类型的模版分子 (有机小分子及金属模版)
Figure 2 Different templates molecules (small organic molecular and metal-organic templates)

图 3 分子在固态条件下发生光致环化的条件
Figure 3 Conditions of photodimerization to molecular in the solid state

图 4 1, 4-双[2-(4-吡啶基) 乙烯基]苯与间苯二酚衍生物共晶光照下固态关环形成[2.2]对环芳衍生物
Figure 4 Cocrystallization of 1, 4-bis[2-(4-pyridyl) ethenyl]benzene with resorcinol derivatives, and forming the [2.2]paracyclophane derivatives by UV-irradiation

图 5 不对称氢键受体分子与间苯二酚两种氢键自组装情况及两种不同的光致环化产物
Figure 5 Hydrogen-bond self-assemble situations of asymmetric hydrogen-bond acceptor with resorcinol and two different photodimerization products

图 6 不对称的氢键受体分子1, 4-双[2-(4-吡啶基) 乙烯基]苯衍生物4a~4d
Figure 6 Asymmetric hydrogen-bond acceptor 1, 4-bis[2-(4-pyridyl) ethenyl]benzene derivatives 4a~4d

图 8 目标化合物4b的分子结构图
Figure 8 Molecular structure of target compound 4b
(a) Top view; (b) side view

图 9 中间体3及目标化合物4a~4d在CH2Cl2溶液中的紫外光谱图
Figure 9 UV-Vis absorption spectra of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution

图 10 目标化合物4b主要跃迁涉及的部分分子轨道图
Figure 10 Selected frontier orbitals involved in the transition for target compound 4b
Contour values (0.04 (e/b3)1/2

图 11 中间体3及目标化合物4a~4d在CH2Cl2溶液中的荧光光谱图
Figure 11 Fluorescence emission spectrum of intermediate 3 and target compounds 4a~4d in CH2Cl2 solution

图 12 根据晶体结构分析实际过程中可能的氢键组装情况
Figure 12 Possible process of hydrogen-bond self-assemble according to crystal structure analysis
表 1 目标化合物4b的晶体结构对应的部分键长 ( )、键角 (°) 和二面角 (°)
Table 1. Selected bond lengths ( ), angles (°) and dihedral angle (°) from crystal structure of 4b
键长/ N (2)-C (18) 1.311 C (11)-C (10) 1.394 C (18)-C (17) 1.370 C (10)-C (21) 1.442 C (17)-C (16) 1.384 C (21)-C (22) 1.191 C (16)-C (15) 1.489 C (22)-C (23) 1.426 C (15)-C (14) 1.292 C (23)-C (28) 1.392 C (14)-C (11) 1.456 C (28)-F (5) 1.335 键角/(°) C (6)-C (7)-C (8) 126.52 C (9)-C (10)-C (21) 119.54 C (10)-C (21)-C (22) 178.25 二面角/(°) [C (6), C (7), C (8)]-[C (13), C (8), C (9)] 4.82 [C (11), C (14), C (15)]-[C (10), C (11), C (12)] 2.10 [C (9), C (10), C (21)]-[C (22), C (23), C (28)] 0.38  下载: 导出CSV
下载: 导出CSV
表 2 TDDFT计算模型分子4b主要的电子跃迁及其归属
Table 2. Major electronic excitations for 4b determined by TD-DFT methods (B3LYP/6-31G)
波长/nm 振子强度 (f) 跃迁 归属 457 0.9722 HOMO→LUMO (70%) π→π 365 1.3586 HOMO-1→LUMO (52%) ICT 283 0.2134 HOMO-2→LUMO+1 (37%) ICT
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 18
- 文章访问数: 2588
- HTML全文浏览量: 542
 下载:
下载:
