 图 图式 1
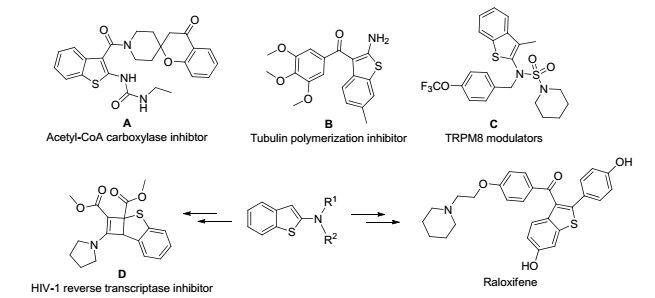
含有2-氨基苯并噻吩结构的生物活性化合物
Figure 图式 1.
Selected bioactive compounds containing 2-amino benzo[b]thiophene
图 图式 1
含有2-氨基苯并噻吩结构的生物活性化合物
Figure 图式 1.
Selected bioactive compounds containing 2-amino benzo[b]thiophene
引用本文:
李红亮, 王正林, 邓卫平. Cu(I)-催化邻卤硫代苯乙酰胺的分子内环化合成2-氨基苯并噻吩[J]. 有机化学,
2016, 36(10): 2419-2425.
doi:
10.6023/cjoc201605030

Citation: Li Hongliang, Wang Zhenglin, Deng Weiping. Cu(I)-Catalyzed Intramolecular Cyclization of Ortho-halogenated Phenylthioacetamides for the Synthesis of 2-Amino Benzo[b]thiophenes[J]. Chinese Journal of Organic Chemistry, 2016, 36(10): 2419-2425. doi: 10.6023/cjoc201605030
Citation: Li Hongliang, Wang Zhenglin, Deng Weiping. Cu(I)-Catalyzed Intramolecular Cyclization of Ortho-halogenated Phenylthioacetamides for the Synthesis of 2-Amino Benzo[b]thiophenes[J]. Chinese Journal of Organic Chemistry, 2016, 36(10): 2419-2425. doi: 10.6023/cjoc201605030
Cu(I)-催化邻卤硫代苯乙酰胺的分子内环化合成2-氨基苯并噻吩
English
Cu(I)-Catalyzed Intramolecular Cyclization of Ortho-halogenated Phenylthioacetamides for the Synthesis of 2-Amino Benzo[b]thiophenes
Abstract:
The Cu(I)-catalyzed intramolecular cyclization of ortho-halogenated phenylthioacetamides to form 2-amino ben-zo[b]thiophenes in water was studied. This method employed water instead of organic solvent, and the catalyst-loading can be as low as to 5 mol% and the yield of products is up to 98%, which overcomes the drawbacks such as harsh reaction conditions and low yields in the previous methods. In addition, the reaction can be scaled up to gram-scale and catalyst can be reused with a maintained yield of products. This reaction provided a simple and environmentally friendly protocol for the preparaion of 2-amino benzo[b]thiophene and their derivatives.
-
苯并噻吩类化合物是一类重要的含硫杂环化合物,在医药、农药以及材料化学等诸多领域具有广阔的应用前景,显示着巨大的开发价值,因而受到人们的广泛关注[1]. 其中,2-氨基苯并噻吩类化合物作为一种具有特殊结构苯并噻吩衍生物,也多见于众多药物和活性分子中,在苯并噻吩的大家族中扮演着重要的角色[2]. 如含2-氨基苯并噻吩结构的化合物A是一种乙酰辅酶A羧化酶抑制剂[2a],化合物B具有很显著的微管蛋白聚合抑制活性[2b]. 化合物C具有调节离子通道TRPM8的活性[2c]. 此外,2-氨基苯并噻吩骨架 也是合成HIV-1逆转录酶抑制剂化合物D[2d]以及选择性雌激素受体调节剂雷洛昔芬及其类似物[2e~2g]的关键中间体(Scheme 1). 因此,近年来,2-氨基苯并噻吩及其衍生物的合成受到了越来越多化学家的关注.
图 图式 1
含有2-氨基苯并噻吩结构的生物活性化合物
Figure 图式 1.
Selected bioactive compounds containing 2-amino benzo[b]thiophene
然而到目前为止,用于合成2-氨基苯并噻吩的方法还不是很多,最早的合成方法是通过对已有的苯并噻吩环的2-位进行氨基化修饰,但是该方法需在高温条件进行,并且只能用来制备简单的2-氨基苯并噻吩结构化合物[Scheme 2,(a)] [3]. 随后研究的重点主要集中在通过分子内环化C—S键的构建来实现2-氨基苯并噻吩的合成. 如1987年,Hopkinson等[4]将二芳基甲酮与N,N-二甲基硫代甲酰胺在强碱二异丙基氨基锂(LDA)的作用下,经过两步反应得到了2-氨基苯并噻吩类化合物,但该方法反应步骤繁琐并且需要低温条件[Scheme 2,(b)].2007年,Neckers课题组[5]将Willgerodt-Kindler反应应用到2-氨基苯并噻吩的合成,实现了三组分“一锅法”合成4-硝基-2氨基苯并噻吩. 然而该反应的产率普遍偏低,最高产率只有47%,并且底物的普适性很差,底物中氯原子的对位必须含有硝基[Scheme 2,(c)]. 最近,杨春皓课题组[6]报道了钯催化的2-氨基苯并噻吩类化合物的合成方法,为合成取代的2-氨基苯并噻吩提供了一条简便的路径,但贵金属催化剂钯以及大量醋酐的使用极大地限制了该方法的应用,此外,底物氨基氮上取代基仅局限于氢和甲基取代,并且产率也不高[Scheme 2,(d)]. 通过以上的文献调研,我们发现,通过廉价金属铜催化的分子内C—S键构建来实现2-氨基苯并噻吩的合成方法尚未见报道. 由此可见,开发一种简便、高效的合成方法来实现2-氨基苯并噻吩的合成具有突出的优势和潜在的价值.
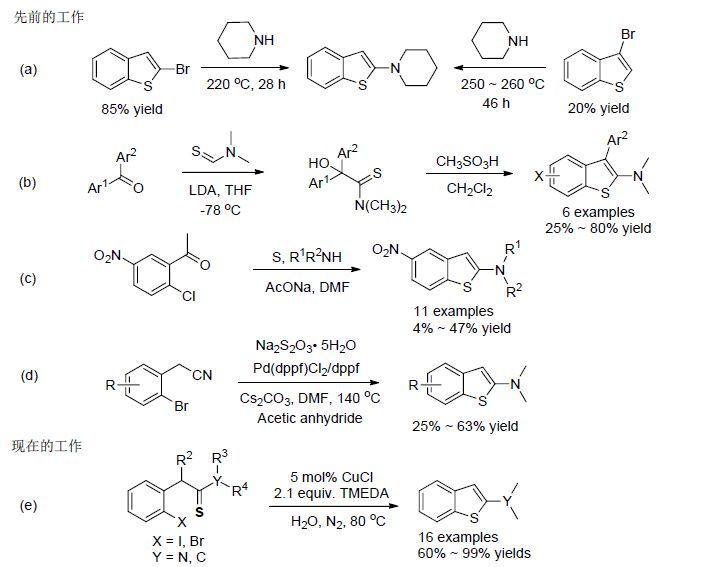 图 图式 2
2-氨基苯并噻吩的合成方法
Figure 图式 2.
Synthetic methods for 2-amino benzo[b]thiophenes
图 图式 2
2-氨基苯并噻吩的合成方法
Figure 图式 2.
Synthetic methods for 2-amino benzo[b]thiophenes
2006年,Willis课题组[7]开发了一条钯催化的α-(邻卤代芳基)硫代环酮类化合物发生分子内C—S键的偶联反应合成苯并噻吩的方法. 由此可见,过渡金属在催化该类型反应时并不存在硫原子对金属的毒化现象. 通过文献调研,我们发现以金属钯或铜等催化剂催化C—S键偶反应来构建硫醚及苯并噻唑类化合物的方法已有了大量的文献报道[8]. 另外,在我们课题组通过以含硫代酰胺结构底物合成一系列2-氨基噻吩环衍生物的研究工作中,发现此类硫代酰胺的稳定性很好,并且底物中硫原子的烯醇化能力较酮羰基更强,因而更容易发生C—S键的生成[9]. 另一方面,随着绿色化学的理念深入人心,近年来水相中Ullmann类型的C—S键偶联反应也得到了广泛的报道,水作为一种无毒无害的天然反应介质在此类反应过程中展现了强大的实用价值[10]. 基于此,本文报道采用廉价CuCl为催化剂,开发水相中邻卤硫代苯乙酰胺类化合物合成2-氨基苯并噻吩类化合物的新方法. 该反应具有易操作、产率高、绿色实用等优点[Scheme 2,(e)].
1 结果与讨论
1.1 底物扩展
在获得最佳反应条件后,对反应的底物进行了普适性考察,结果如Eq. 1所示. 首先对卤素原子进行考察,以产物2a所用底物为研究对象,当卤原子为溴时,2a的产率为95%; 当卤原子为碘时,反应几乎可以定量得到产物2a; 遗憾的是,氯原子取代的底物在此反应中并不适用,推测可能是由于C—Cl键的键能较强,铜催化时氧化加成步骤难以实现.
接下来选择以反应活性适中的溴代底物为考察对象,如Eq. 1所示,底物中硫代酰胺部分的氮原子上有各种取代基,如N,N-二甲基、N,N-二乙基、N,N-二异丙基、N-甲基-N-苯基、N-甲基-N-苄基、吡咯烷、哌啶以及吗啉取代时均能以良好到优异的产率获得相应目标产物(2b~2h),其中,以N,N-二异丙基氨基取代时产率明显较其他相似底物低,这可能是由于位阻的原因导致底物的反应活性有所降低. 另外,硫代酰胺的α-位有取代基时,无论是供电子甲基或吸电子取代基氰基取代均能以高产率得到相应的目标产物(2i,2j). 接着考察苯环上取代基对反应的影响,结果显示,苯环上电性对反应的影响较大,当以吸电子取代基取代时,如氯或氟取代时,产物可以保持在优秀的收率(2i,2j),然而当以供电子甲氧基取代时,产率仅为65% (2m). 最后考察了该催化体系对Willis课题组[7]在合成苯并噻吩时所用的底物2-(2-溴代苯基)-硫代环己酮和2-(2-溴代苯基)-硫代环庚酮是否也适应,令人高兴的是,这两个底物在此反应催化体系下也能得到良好的收率得到目标产物2n和2o.
最后,对反应的实用性进行了系统的研究,放大量实验结果显示,底物1a的用量放大到10 mol (2.67 g),产物2a的收率也能保持在87%. 以放大量的实验为基础,对催化体系的循环使用情况进行了研究. 该催化体系在第一次的催化反应完成后经过萃取,补加2.1 equiv.的TMEDA,随即进行第二次循环使用,结果显示第二次催化所得的产物2a产率仍可保持到83%. 通过放大实验和催化剂循环使用实验可以确定该反应具有潜在的工业应用价值(Eq. 2).
1.1 条件筛选
以化合物1a为模型底物,参照Willis课题组[7]合成苯并噻吩的方法,当以Pd2(dba)3为催化剂时,无任何反应发生(表 1,Entry 1). 以Pd(OAc)2为催化剂时,也仅得到8%产率的目标产物2a (Entry 2). 随后,转向尝试以金属铜为催化剂,参照Batey课题组合成苯并噻唑的方法[10],我们以5 mol%的CuI为催化剂,10 mol%的1,10-Phen为配体,2 equiv. Cs2CO3为碱,2 mL的1,4-dioxane为反应溶剂,氮气氛围中80 ℃下反应,令人惊喜的是,反应3 h后,原料1a全部消失,并以92%的收率得到了2-(N,N-二甲氨基)苯并噻吩(2a) (Entry 3). 接下来进一步地考察了不同催化剂、反应溶剂、碱、配体、反应温度、催化剂及配体用量对产物2a的影响,以期望能获得更高效、简洁的反应模式.
 表 1
反应条件的优化
Table 1.
Optimization of reaction conditions
表 1
反应条件的优化
Table 1.
Optimization of reaction conditions

Entry Cat. Ligand Base Solvent Temp./℃ Yieldb/% 1 Pd2(dba)3 DPEphos Cs2CO3 Toluene 80 0 2 Pd(OAc)2 DPEphos Cs2CO3 Toluene 80 8 3 CuI 1,10-phen Cs2CO3 Dioxane 80 92 4 CuBr 1,10-phen Cs2CO3 Dioxane 80 90 5 CuCl 1,10-phen Cs2CO3 Dioxane 80 93 6 Cu(OAc)2•H2O 1,10-phen Cs2CO3 Dioxane 80 72 7c CuCl2 1,10-phen Cs2CO3 Dioxane 80 85 8 CuCl 1,10-phen Cs2CO3 Dioxane 80 82 9 CuCl 1,10-phen Cs2CO3 DMSO 80 81 10 CuCl 1,10-phen Cs2CO3 THF 80 76 11 CuCl 1,10-phen Cs2CO3 Toluene 80 65 12 CuCl 1,10-phen Cs2CO3 CH3CN 80 68 13 CuCl 1,10-phen Cs2CO3 CH3CH2OH 80 15 14 CuCl 1,10-phen Cs2CO3 H2O 80 92 15 CuCl 1,10-phen Cs2CO3 H2O 60 73 16 CuCl 1,10-phen Cs2CO3 H2O 100 91 17 CuCl Glycol Cs2CO3 H2O 80 0 18 CuCl L-Proline Cs2CO3 H2O 80 35 19 CuCl Edamine Cs2CO3 H2O 80 79 20 CuCl TMEDA Cs2CO3 H2O 80 8 21 CuCl DMEDA Cs2CO3 H2O 80 92 22 CuCl DMEDA Et3N H2O 80 18 23 CuCl DMEDA K2CO3 H2O 80 49 24 CuCl DMEDA NaOtBu H2O 80 67 25d CuCl DMEDA DBU H2O 80 47 26d CuCl DMEDA — H2O 80 67 27 CuCl TMEDA — H2O 80 95 a Reaction conditions: 1a (0.2 mmol),catalyst (0.01 mmol),ligand (0.02 mmol),base (0.40 mmol),solvent (2.0 mL) under N2; b Yields of isolated products.; c Used 2 mol% CuCl as the catalyst;. d With 0.42 mmol ligands. 表 1 反应条件的优化
Table 1. Optimization of reaction conditions首先,对反应的催化剂进行进一步考察,其它的一价铜盐(如CuBr,CuCl)均表现出很高的催化活性(Entry 4),而二价铜盐,如Cu(OAc)2•H2O和CuCl2,催化效果稍有减低(Entries 6,7). 将CuCl的用量降低至2 mol%时,产物2a的产率相应降低至83% (Entry 8). 接着对反应的溶剂进行了考察,当以DMSO、THF、甲苯、乙腈为溶剂时,产物收率均有明显的减低(Entries 9~12); 当以乙醇为溶剂时,原料大量剩余,2a的产率只有15% (Entriy 13); 当我们尝试以H2O为溶剂时,令人高兴的是,产物2a的收率保持与使用1,4-dioxane相当(Entry 14). 于是我们选择了这一环境友好的溶剂进而对温度进行考察,降低温度至60 ℃时反应也可发生,但产率仅为73%,升高温度至100 ℃对收率并无较大影响(Entries 15,16). 配体筛选方面我们考察了几种常用于与金属铜配位的氮、氧类的双齿配体. 当使用O,O-二齿配体乙二醇时,反应不能发生(Entry 17); 当使用N,O-二齿配体L-脯氨酸,产率也仅为35% (Entry 18); 当使用N,N-二齿配体乙二胺时,产物2a的产率为79% (Entry 19),反应效果明显高于O,O-二齿配体及N,O-二齿配体. 由此推测该反应中N,N-二齿配体的使用有利于反应的进行. 于是又考察了四甲基乙二胺(TMEDA),但奇怪的是只能得到8%的产物收率(Entry 200); 而当以N,N'-二甲基乙二胺(DMEDA)为配体时,反应产率与使用1,10-phen产率相同(Entry 21),于是选择以廉价的DMEDA为反应的配体对碱进行考察. 当使用弱碱如Et3N,K2CO3时,只能以中等以下的收率得到目标产物(Entries 22,23),而当使用碱性稍强的NaOBu-t和DBU时,也只得到中等收率(Entries 24,25). 由于乙二胺类配体既能与铜配位又有碱性,因此,我们设想是否可以只加入一种物质就能起到配体和碱的双重作用,于是我们向体系中加入2.1 equiv.的DMEDA,遗憾的是只能得到67%收率的目标产物(Entry 26); 然而,当体系中加入2.1 equiv.的TMEDA时,反应非常顺利,3 h后即能以95%的收率得到目标产物2a (Entry 27).
通过以上反应条件的筛选,确定了最佳的反应条件为: 5 mol% CuI为催化剂,2.1 equiv. TMEDA为配体和碱,2 mL的H2O为溶剂,在80 ℃下氮气氛围中反应.
2 结论
发展了一价铜催化的2-氨基苯并噻吩的绿色合成方法. 以廉价的CuCl为催化剂,TMEDA为碱和配体,以H2O为反应溶剂,邻卤硫代苯乙酰胺经分子内 C—S键偶联环化反应,合成了一系列具有潜在应用价值的2-氨基苯并噻吩类化合物,产率最高可达98%,克服了以往合成方法中产率低、反应条件苛刻等缺点. 此外,该反应放大到克级后产率基本得到保持,催化体系也可实现循环使用. 总之,我们发展了一条廉价、高效、绿色、实用的2-氨基苯并噻吩类化合物的合成方法,为精细化工材料及药物中间体的合成提供了一种新的思路.
3 实验部分
3.1 仪器与试剂
反应底物均购自韶远、安耐吉、百灵威等公司,所有药品和试剂均为分析纯. 核磁共振氢谱(1H NMR)和碳谱(13C NMR)是通过Bruker公司的Bruker DPX-400核磁共振波谱仪测定,内标物为四甲基硅烷(TMS),溶剂为氘代氯仿(CDCl3); 高分辨质谱(EI)是通Finigann MAT8401质谱仪测定; 熔点是通过上海精密科学仪器有限公司所生产的GW X-4显微熔点仪进行测定,熔点仪未经过校正.
3.2 化合物2的制备方法
在氮气保护的条件下,将1.0 mg (0.01 mmol) CuCl,48.8 mg (0.42 mmol)四甲基乙二胺和化合物1 (0.2 mmol) 加入到10 mL的反应管中,并加入2 mL蒸馏水. 然后在80 ℃条件下反应,TLC监测反应进程. 待反应结束后,用二氯甲烷萃取三次,合并有机相,用饱和的食盐水洗涤后,再用无水硫酸钠干燥,减压浓缩得粗产物,经柱层析分离得纯苯并噻吩产物2.
N,N-二甲氨基苯并噻吩(2a): 黄色油状物. X=Br,33.3 mg,产率94%; X=I,34.8 mg,产率98%. Rf=0.50 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.59 (d,J=7.9 Hz,1H),7.44 (d,J=7.9 Hz,1H),7.24 (dd,J=7.7,1.1 Hz,1H),7.06 (dd,J=7.7,1.1 Hz,1H),5.99 (s,1H),3.02 (s,6H); 13C NMR (100 MHz,CDCl3) δ: 157.7,141.4,132.47,124.4,121.4,120.3,120.1,96.5,42.5. HRMS (EI-TOF) calcd for C10H11NS 177.0612,found 177.0613.
N,N-二乙氨基苯并噻吩(2b): 黄色油状物,36.1 mg,产率88%,Rf=0.40 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.55 (d,J=7.8 Hz,1H),7.38 (d,J=7.7 Hz,1H),7.19 (t,J=7.3 Hz,1H),6.99 (t,J=7.3 Hz,1H),5.94 (s,1H),3.37 (q,J=6.9 Hz,4H),1.24 (t,J=7.0 Hz,6H); 13C NMR (100 MHz,CDCl3) δ: 155.6,141.7,131.6,124.4,121.2,119.9,119.6,95.2,46.8,12.4. HRMS (EI-TOF) calcd for C12H15NS 205.0925,found 205.0926.
2-(N,N-二异丙基氨基)苯并噻吩(2c): 黄色油状物,30.3 mg,产率65%,Rf=0.45 [V(石油醚):V(乙酸乙 酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.57 (d,J=7.8 Hz,1H),7.43 (d,J=7.8 Hz,1H),7.20 (t,J=7.4 Hz,1H),7.04 (t,J=7.4 Hz,1H),6.21 (s,1H),3.81~3.74 (m,2H),1.30 (d,J=6.7 Hz,12H); 13C NMR (100 MHz,CDCl3) δ: 153.5,140.5,132.5,124.1,121.1,120.6,120.2,102.2,50.8,20.8. HRMS (EI-TOF) calcd for C14H19NS 233.1238,found 2 33.1240.
2-(N-甲基-N-苯基-2-氨基)苯并噻吩(2d): 黄色固体,34.9 mg,产率73%,Rf=0.35 [V(石油醚):V(乙酸乙酯)=10:1]. m.p. 75~77 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.58 (d,J=7.9 Hz,1H),7.49 (d,J=7.9 Hz,1H),7.32 (t,J=7.5 Hz,2H),7.20~7.24 (m,3H),7.13 (t,J=7.6 Hz,1H),7.05 (t,J=7.2 Hz,1H),6.52 (s,1H),3.41 (s,3H); 13C NMR (100 MHz,CDCl3) δ: 154.1,148.3,139.9,134.2,129.2,124.4,123.0,122.2,121.8,121.4,120.8,106.8,42.0. HRMS (EI-TOF) calcd for C15H13NS 239.0769,found 239.0770.
2-(N-甲基-N-苄基-2-氨基)苯并噻吩(2e): 黄色油状物,40.0 mg,产率79%,Rf=0.30 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.56 (d,J=7.8 Hz,1H),7.40 (d,J=7.7 Hz,1H),7.26~7.33 (m,5H),7.21 (t,J=7.5 Hz,1H),7.03 (t,J=7.3 Hz,1H),6.02 (s,1H),4.52 (s,2H),2.99 (s,3H); 13C NMR (100 MHz,CDCl3) δ: 157.1,1 41.5,137.3,132.0,128.6,127.4,127.4,124.5,121.4,120.4,120.0,96.4,59.2,39.5. HRMS (EI-TOF) calcd. for C16H15NS 253.0925,found 253.0924.
2-(1-吡咯烷基)苯并噻吩(2f): 黄色固体,32.9 mg,产率81%,Rf=0.50 [V(石油醚):V(乙酸乙酯)=10:1]. m.p. 70~72 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.57 (d,J=7.9 Hz,1H),7.40 (d,J=7.9 Hz,1H),7.20 (t,J=7.6 Hz,1H),6.99 (t,J=7.5 Hz,1H),5.84 (s,1H),3.38~3.35 (m,4H),2.07~2.04 (m,4H); 13C NMR (100 MHz,CDCl3) δ: 153.8,141.7,131.7,124.4,121.4,119.6,119.5,93.9,50.5,25.8. HRMS (EI-TOF) calcd for C12H13NS 203.0769,found 203.0770.
2-(1-哌啶基)苯并噻吩(2g): 白色固体,36.1 mg,产率83%,Rf=0.45 [V(石油醚):V(乙酸乙酯)=10:1]. m.p. 72~74 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.58 (d,J=7.9 Hz,1H),7.43 (d,J=7.8 Hz,1H),7.21 (t,J=7.6 Hz,1H),7.05 (t,J=7.6 Hz,1H),6.15 (s,1H),3.25 (t,J=4.0,4H),1.71~1.76 (m,4H),1 .63~1.59 (m,2H); 13C NMR (100 MHz,CDCl3) δ: 158.5,140.9,132.5,124.3,121.4,120.8,120.5,98.3,51.9,25.2,23.9. HRMS (EI-TOF) calcd. for C13H15NS 217.0925,found 217.0924.
2-(4-吗啉基)苯并噻吩(2h): 白色固体,39.9 mg,产率91%,Rf=0.40 [V(石油醚):V(乙酸乙酯)=10:1]. m.p. 78~90 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.61 (d,J=7.9 Hz,1H),7.48 (d,J=7.9 Hz,1H),7.29~7.22 (m,1H),7.11 (t,J=7.6 Hz,1H),6.23 (s,1H),3.88~3.86 (m,4H),3.26~3.23 (m,4H); 13C NMR (100 MHz,CDCl3) δ: 157.7,140.3,132.6,124.5,121.6,121.5,121.0,99.4,66.3,50.9. HRMS (EI-TOF) calcd for C12H15NOS 219.0718,found 219.0712.
2-(N,N-二甲氨基)-3-甲基苯并噻吩(2i): 黄色油状物,34.8 mg,产率91%,Rf=0.50 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.70 (d,J=7.9 Hz,1H),7.56 (d,J=7.9 Hz,1H),7.33 (t,J=7.5 Hz,1H),7.23 (t,J=7.6 Hz,1H),2.80 (s,6H),2.30 (s,3H); 13C NMR (100 MHz,CDCl3) δ: 153.9,140.1,134.7,123.9,123.0,122.4,120.7,118.51,46.1,11.2. HRMS (EI-TOF) calcd for C11H13NS 191.0769,found 191.0770.
2-(N,N-二甲氨基)-3-氰基苯并噻吩(2j): 黄色固体,37.2 mg,产率92%,Rf=0.40 [V(石油醚):V(乙酸乙 酯)=10:1]. m.p. 74~76 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.54 (d,J=8.0 Hz,1H),7.50 (d,J=7.9 Hz,1H),7.35 (t,J=8.0 Hz,1H),7.14 (t,J=8.0 Hz,1H),3.35 (s,6H); 13C NMR (100 MHz,CDCl3) δ: 164.9,139.7,128.3,125.8,122.4,121.1,119.3,117.6,43.3. HRMS (EI-TOF) calcd for C11H10N2S 202 .0565,found 202.0566.
2-(N,N-二甲氨基)-5-氯苯并噻吩(2k): 白色固体,33.9 mg,产率80%,Rf=0.35 [V(石油醚):V(乙酸乙 酯)=10:1]. m.p. 87~89 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.45(d,J=8.3 Hz,1H),7.36 (s,1H),6.97 (d,J=8.4 Hz,1H),5.87 (s,1H),3.01 (s,6H); 13C NMR (100 MHz,CDCl3) δ: 159.1,142.8,130.6,130.2,122.3,120.2,119.5,95.4,42.3. HRMS (EI-TOF) calcd for C10H10ClNS 211.0222,found 211.0221.
2-(N,N-二甲氨基)-6-氟苯并噻吩(2l): 灰白色固体,34.8 mg,产率89%,Rf=0.40 [V(石油醚):V(乙酸乙 酯)=10:1]. m.p. 82~84 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.34~7.27 (m,2H),7.00~6.92 (m,1H),5.93 (s,1H),2.98 (s,6H); 13C NMR (100 MHz,CDCl3) δ: 158.1 (d,J=277.3 Hz),157.3 (d,J=2.7 Hz),137.6 (d,J=1.7 Hz),133.0 (d,J=10.0 Hz),120.7 (d,J=8.4 Hz),112.6 (d,J=23.2 Hz),107.9 (d,J=25.7 Hz),96.0,42.5. HRMS (EI- TOF) calcd for C10H10FNS 195.0518,found 195.0519.
2-(N,N-二甲氨基)-5-甲氧基苯并噻吩(2m): 灰白色固体,26.9 mg,产率65%,m.p. 93~95 ℃,Rf=0.30 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.32 (d,J=8.6 Hz,1H),7.26 (s,1H),7.14 (s,1H),6.86 (d,J=8.6 Hz,1H),5.93 (s,1H),3.82 (s,3H),2.96 (s,6H); 13C NMR (100 MHz,CDCl3) δ: 156.0,154.8,135.0,133.7,120.9,113.2,105.6,96.8,55.7,42.7. HRMS (EI-TOF) calcd for C11H13NOS 207.0718,found 207.0719.
1,2,3,4-四氢二苯并[b,d]噻吩(2n): 无色油状物,22.6 mg,产率60%,Rf=0.60 [V(石油醚):V(乙酸乙酯)=10:1]. 1H NMR (400 MHz,CDCl3) δ: 7.76 (d,J=7.9 Hz,1H),7.57 (d,J=7.8 Hz,1H),7.35~7.31 (m,1H),7.28~7.24 (m,1H),2.86 (t,J=4.9 Hz,2H),2.80~2.72 (m,2H),2.03~1.85 (m,4H); 13C NMR (100 MHz,CDCl3) δ: 139.8,138.4,137.0,129.4,123.7,123.5,122.2,120.4,25.7,23.6,23.6,22.3. HRMS (EI-TOF) calcd for C12H12S 188.0660,found 188.0661.
7,8,9,10-四氢-6H-苯并[b]环庚基[d]噻吩(2o): 黄色固体,30.3 mg,产率75%,Rf=0.55 [V(石油醚):V(乙酸乙酯)=10:1]. m.p. 64~66 ℃; 1H NMR (400 MHz,CDCl3) δ: 7.75 (d,J=8.0 Hz,1H),7.62 (d,J=8.0 Hz,1H),7.34 (t,J=7.6 Hz,1H),7.26~7.23 (m,1H),3.01~2.83 (m,4H),2.00~1.89 (m,2H),1.83~1.69 (m,4H); 13C NMR (125 MHz,CDCl3) δ: 140.9,140.6,138.0,134.2,123.7,123.1,122.1,120. 8,32.3,30.3,27.8,27.2,27.0. HRMS (EI-TOF) calcd for C13H14S 202.0816,found 202.0817.
3.3 1a的克级放大实验和催化剂的循环使用
在氮气保护的条件下,将0.05 g (0.5 mmol) CuCl,2.44 g (21 mmol)四甲基乙二胺和2.58 g化合物1a (10 mmol)加入到200 mL的反应瓶中,并加入80 mL蒸馏水. 然后在80 ℃条件下反应,TLC监测反应进程. 3 h后,用二氯甲烷萃取三次,分离水相和有机相. 有机相用饱和的食盐水洗涤后,再用无水硫酸钠干燥,减压浓缩得粗产物,经柱层析分离得1.54 g苯并噻吩化合物2a,产率为87%. 另外,向水相中补加2.44 g四甲基乙二胺,进行再次催化反应,3 h后,分离得到二次催化反应的产物2a 1.47 g,产率为83%.
辅助材料(Supporting Information) 产物2a~2o的核磁共振谱图. 这些材料可以免费从本刊网站(http://sioc- journal.cn/)上下载.
-
-
[1]
Perepichka, I. F.; Perepichka, D. F. Handbook of Thio-phene-Based Materials, Wiley-VCH Verlag, New York, 2009. (b) Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893. (c) Zhang, T. Y.; O'Toole, J.; Proctor, C. S. Sulfur Reports 1999, 22, 1. (d) Guo, H. F.; Shao, H. Y.; Yang, Z. Y.; Xue, S. T.; Li, X.; Liu, Z. Y.; He, X. B.; Jiang, J. D.; Zhang, Y. Q.; Si, S. Y.; Li, Z. R. J. Med. Chem. 2010, 53, 1819. (c) Berrade, L.; Aisa, B.; Ramirez, M.; Galiano, S.; Guccione, S.; Moltzau, L. R.; Levy, F. O.; Nicoletti, F.; Battaglia, G.; Molinaro, G.; Aldana, I.; Monge, A.; Perez-Silandes, S. J. Med. Chem. 2011, 54, 3086.
-
[2]
Chonan, T.; Wakasugi, D.; Yamamoto, D.; Yashiro, M.; Oi, T.; Tanaka, H.; Ohoka-Sugita, A.; Io, F.; Koretsune, H.; Hiratate, A. Biorg. Med. Chem. 2011, 19, 1580. (b) Romagnoli, R.; Baraldi, P. G.; Carrion, M. D.; Cara, C. L.; Preti, D.; Fruttarolo, F.; Pavani, M. G.; Tabrizi, M. A.; Tolomeo, M.; Grimaudo, S. J. Med. Chem. 2007, 50, 2273. (c) Matthews, J. M. US 201001603372010[Chem. Abstr. 2010, 153, 116343]. (d) Krajewski, K.; Zhang, Y.; Parrish, D.; Deschamps, J.; Roller, P. P.; Pathak, V. K. Bioorg. Med. Chem. Lett. 2006, 16, 3034. (e) Duan, Z.; Ranjit, S.; Liu, X. Org. Lett. 2010, 12, 2430. (f) Chalmers, M. J.; Wang, Y.; Novick, S.; Sato, M.; Bryant, H. U.; Montrose-Rafizdeh, C.; Griffin, P. R.; Dodge, J. A. ACS Med. Chem. Lett. 2012, 3, 207. (g) Qin, Z.; Kastrati, I.; Chandrasena, R. E. P.; Liu, H.; Yao, P.; Petukhov, P. A.; Bolton, J. L.; Thatcher, G. R. J. Med. Chem. 2007, 50, 2682. doi: 10.1016/j.bmc.2011.01.041
-
[3]
Brower, K. R.; Amstutz, E. J. Org. Chem. 1954, 19, 411. doi: 10.1021/jo01368a019
-
[4]
Ablenas, F.; George, B.; Maleki, M.; Jain, R.; Hopkinson, A.; Lee-Ruff, E. Can. J. Chem. 1987, 65, 1800.
-
[5]
Solovyev, A. Y.; Androsov, D. A.; Neckers, D. C. J. Org. Chem. 2007, 72, 3122. doi: 10.1021/jo062141a
-
[6]
Hou, C.; He, Q.; Yang, C. Org. Lett. 2014, 16, 5040.
-
[7]
Willis, M. C.; Taylor, D.; Gillmore, A. T. Tetrahedron 2006, 62, 11513. doi: 10.1016/j.tet.2006.05.004
-
[8]
Kosugi, M.; Shimizu, T.; Migita, T. Chem. Lett. 1978, 7, 13. (b) Li, G. Angew Chem, Int Ed. 2001, 40, 1513. (c) Suzuki, H.; Abe, H.; Osuka, A. Chem. Lett. 1980, 9, 1363. (d) Joyce, L. L.; Evindar, G.; Batey, R. A. Chem. Commun. 2004, 40, 446. (e) Qin, Y.; Peng, Q. Chin. J. Org. Chem. 2011, 31, 1169(in Chinese). (秦元成, 彭强, 有机化学, 2011, 31, 1169.) (f) Zhu, N.; Zhang, Z.; Gao, M.; Han, L.; Suo, Q.; Hong, H. Chin. J. Org. Chem. 2013, 33, 1423(in Chinese). (竺宁, 薛士徐, 高敏, 韩利民, 索全伶, 洪海龙, 有机化学, 2013, 33, 1423.)
-
[9]
Wang, Z.; Li, H.; Ge, L.; An, X.; Zhang, Z.; Luo, X.; Fossey, J. S.; Deng, W. J. Org. Chem. 2014, 79, 1156. (b) Ge, L.; Wang, Z.; An, X.; Luo, X.; Deng, W. Org. Biomol. Chem. 2014, 42, 8473. (c) Luo, X.; Ge, L.; An, X.; Jin, J.; Wang, Y.; Sun, P.; Deng, W. J. Org. Chem. 2015, 80, 4611. doi: 10.1021/jo4026034
-
[10]
Carril, M.; San Martin, R.; Domínguez, E.; Tellitu, I. Chem. Eur. J. 2007, 13, 5100. (b) Rout, L.; Saha, P.; Jammi, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2008, 640. (c) Ke, F.; Qu, Y.; Jiang, Z.; Li, Z.; Zhou, X. Org. Lett. 2011, 13, 454. (d) Wang, F.; Chen, C.; Deng, G.; Xi, C. J. Org. Chem. 2012, 77, 4148. (e) Li, Z.; Wu, Z.; Deng, H.; Zhou, X. Chin. J. Org. Chem. 2013, 33, 760(in Chinese). (李正凯, 吴之清, 邓杭, 周向葛, 有机化学, 2013, 33, 760.) (f) Wen, L.-R.; Li, S.-L.; Zhang, J.; Li, M. Green Chem. 2015, 17, 1581. (g) Wen, L.-R.; Yuan, W.-K. Li, M. J. Org. Chem. 2015, 80, 4942. doi: 10.1002/(ISSN)1521-3765
-
[11]
Evindar, G.; Batey, R. A. J. Org. Chem. 2006, 71, 1802. doi: 10.1021/jo051927q
-
[1]
-

图式 1 含有2-氨基苯并噻吩结构的生物活性化合物
Scheme 1 Selected bioactive compounds containing 2-amino benzo[b]thiophene
表 1 反应条件的优化
Table 1. Optimization of reaction conditions
Entry Cat. Ligand Base Solvent Temp./℃ Yieldb/% 1 Pd2(dba)3 DPEphos Cs2CO3 Toluene 80 0 2 Pd(OAc)2 DPEphos Cs2CO3 Toluene 80 8 3 CuI 1,10-phen Cs2CO3 Dioxane 80 92 4 CuBr 1,10-phen Cs2CO3 Dioxane 80 90 5 CuCl 1,10-phen Cs2CO3 Dioxane 80 93 6 Cu(OAc)2•H2O 1,10-phen Cs2CO3 Dioxane 80 72 7c CuCl2 1,10-phen Cs2CO3 Dioxane 80 85 8 CuCl 1,10-phen Cs2CO3 Dioxane 80 82 9 CuCl 1,10-phen Cs2CO3 DMSO 80 81 10 CuCl 1,10-phen Cs2CO3 THF 80 76 11 CuCl 1,10-phen Cs2CO3 Toluene 80 65 12 CuCl 1,10-phen Cs2CO3 CH3CN 80 68 13 CuCl 1,10-phen Cs2CO3 CH3CH2OH 80 15 14 CuCl 1,10-phen Cs2CO3 H2O 80 92 15 CuCl 1,10-phen Cs2CO3 H2O 60 73 16 CuCl 1,10-phen Cs2CO3 H2O 100 91 17 CuCl Glycol Cs2CO3 H2O 80 0 18 CuCl L-Proline Cs2CO3 H2O 80 35 19 CuCl Edamine Cs2CO3 H2O 80 79 20 CuCl TMEDA Cs2CO3 H2O 80 8 21 CuCl DMEDA Cs2CO3 H2O 80 92 22 CuCl DMEDA Et3N H2O 80 18 23 CuCl DMEDA K2CO3 H2O 80 49 24 CuCl DMEDA NaOtBu H2O 80 67 25d CuCl DMEDA DBU H2O 80 47 26d CuCl DMEDA — H2O 80 67 27 CuCl TMEDA — H2O 80 95 a Reaction conditions: 1a (0.2 mmol),catalyst (0.01 mmol),ligand (0.02 mmol),base (0.40 mmol),solvent (2.0 mL) under N2; b Yields of isolated products.; c Used 2 mol% CuCl as the catalyst;. d With 0.42 mmol ligands.  下载: 导出CSV
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 748
- HTML全文浏览量: 105
 下载:
下载:
