引用本文:
刘倩倩, 洪玉标, 郑煜, 薛明强. 二(β-二亚胺基)二价稀土配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应[J]. 有机化学,
2016, 36(9): 2168-2174.
doi:
10.6023/cjoc201603004

Citation: Liu Qianqian, Hong Yubiao, Zheng Yu, Xue Mingqiang. Bis(β-diketiminato) Lanthanide(Ⅱ) Complexes-Catalyzed Hydro-phosphonylation of Aldehydes/Ketones and Diethyl Phosphite[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2168-2174. doi: 10.6023/cjoc201603004
Citation: Liu Qianqian, Hong Yubiao, Zheng Yu, Xue Mingqiang. Bis(β-diketiminato) Lanthanide(Ⅱ) Complexes-Catalyzed Hydro-phosphonylation of Aldehydes/Ketones and Diethyl Phosphite[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2168-2174. doi: 10.6023/cjoc201603004
二(β-二亚胺基)二价稀土配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应
摘要:
研究了一系列二(β-二亚胺基)二价稀土配合物[Eu(L2, 6-ipr2)2·CH3C6H5, L2, 6-ipr2=[N(2, 6-iPr2C6H3)C(Me)]2CH-(1); Eu(L2, 6-Me2)2(THF), L2, 6-Me2=[N(2, 6-Me2C6H3)C(Me)]2CH-(2); Eu(L2, 4, 6-Me3)2(THF), L2, 4, 6-Me3=[N(2, 4, 6-Me3C6H2)-C(Me)]2CH-(3); Eu(LPh2, 6-ipr2)2, LPh2, 6-ipr2=[(2, 6-iPr2C6H3)NC(Me)CHC(Me)N(C6H5)]-(4); Sm(L2, 6-ipr2)2·CH3C6H5(5); Yb(LPh2, 6-ipr2)2(6); Yb(L2-Me)2(THF), L2-Me=[N(2-MeC6H4)C(Me)]2CH-(7)]对醛/酮与亚磷酸二乙酯的氢磷化反应的催化行为.发现所有配合物都可以在温和条件下,高效地催化芳醛以及杂环芳醛与亚磷酸二乙酯的氢磷化反应,在催化剂用量为0.08 mol%,25℃,无溶剂条件下反应5 min,α-羟基膦酸酯的收率可以达到90%~99%.催化活性有赖于β-二亚胺基的结构,其活性顺序为L2,6-Me2 < L2,4,6-Me3 < L2,6-ipr2≈LPh2,6-ipr2.该催化体系具有优秀的醛底物适应能力.这类二价稀土配合物也可以有效地催化未活化的酮与亚磷酸二乙酯的反应,并显示良好的底物适应能力.
English
Bis(β-diketiminato) Lanthanide(Ⅱ) Complexes-Catalyzed Hydro-phosphonylation of Aldehydes/Ketones and Diethyl Phosphite
Abstract:
The hydrophosphonylation of aldehydes/ketones was explored by use of seven bis(β-diketiminato) lanthanide(Ⅱ) complexes[Eu(L2, 6-ipr2)2·CH3C6H5, L2, 6-ipr2=[N(2, 6-iPr2C6H3)C(Me)]2CH- (1); Eu(L2, 6-Me2)2(THF), L2, 6-Me2=[N(2, 6-Me2C6H3)C(Me)]2CH- (2); Eu(L2, 4, 6-Me3)2(THF), L2, 4, 6-Me3=[N(2, 4, 6-Me3C6H2)C(Me)]2CH- (3); Eu(LPh2, 6-ipr2)2, LPh2, 6-ipr2=[(2, 6-iPr2C6H3)NC(Me)CHC(Me)N(C6H5)]-(4); Sm(L2, 6-ipr2)2·CH3C6H5 (5); Yb(LPh2, 6-ipr2)2 (6); Yb(L2-Me)2(THF), L2-Me=[N(2-MeC6H4)C(Me)]2CH- (7)] as the catalysts. All complexes were found to be able to catalyze the hydrophosphonylation between aromatic or heterecycle aldehyde and diethyl phosphite with high activity under wild conditions. All the reactions gave the products in 90%~99% yields using 0.08 mol% of complex 7 at 25℃ under solvent-free. The catalyst activity was found to depend on the β-diketiminato ligands with the sequence of L2, 6-Me2 < L2, 4, 6-Me3 < L2, 6-ipr2≈LPh2, 6-ipr2. This catalyst system showed a wide scope of aldehydes. Besides, bis(β-diketiminato) lanthanide(Ⅱ) complexes could also efficiently catalyze the hy-drophosphonylation of unactive ketones with diethyl phosphite and showed a good substrate scope.
-
Key words:
- β-diketiminato
- / lanthanide(Ⅱ) complexes
- / hydrophosphonylation
- / aldehydes/ketones
-
有机膦化合物在金属有机化学、配位化学及生物化学中都具有重要的应用价值.因此, C—P键的形成也就成了有机合成化学中的一个重要组成部分.α-羟基膦酸酯作为一类重要的有机膦化合物, 是一些抗病毒制剂、抗癌药物、及多种酶的结构单元[1], 具有多种生物功能, 在生物和药物化学工业等领域具有广泛的应用价值[2].
二烷基亚膦酸酯与醛、酮的加成反应(Pudovik反应)是合成α-羟基膦酸酯的直接的原子经济性的方法[3].因此, 开发有效的催化剂催化这一反应一直是有机合成化学中的研究热点之一.到目前为止, 已经开发了包括无机化合物[4]、有机小分子[5]、酸[6]、碱[7]、主族[8]和过渡金属[9]化合物等催化体系, 他们都可以有效地催化这一反应.最近报道的三价稀土金属有机配合物也是这类反应的有效催化剂[10].
二价稀土金属有机配合物是一类单电子转移试剂, 具有很强的还原能力, 它们在很多有机反应中都显示出独特的催化行为.然而, 有关它们催化醛和酮的氢磷化反应的报道却很少[11].最近我们在研究二(β-二亚胺基)二价稀土配合物的催化性能时发现, 它们不仅是催化胺与碳化二亚胺加成反应的高效催化剂[12], 而且也是一类催化醛和酮与亚磷酸酯的氢磷化反应的有效催化剂.本文将详细报道二(β-二亚胺基)二价稀土配合物催化醛和未活化的酮与亚磷酸二乙酯的氢磷化反应的研究结果.
1 结果与讨论
1.1 二(β-二亚胺基)二价稀土配合物催化苯甲醛与亚磷酸二乙酯氢磷化反应的活性
我们用以下7个二(β-二亚胺基)二价稀土金属配合物为催化剂, 考察了它们催化苯甲醛与亚磷酸二乙酯反应的活性.结果见表 1.
 表 1
二(β-二亚胺基)二价稀土配合物催化苯甲醛与亚磷酸二乙酯的氢磷化反应条件优化a
Table 1.
Hydrophosphonylation of benzaldehyde and diethyl phosphite catalyzed by complexes 1~7
表 1
二(β-二亚胺基)二价稀土配合物催化苯甲醛与亚磷酸二乙酯的氢磷化反应条件优化a
Table 1.
Hydrophosphonylation of benzaldehyde and diethyl phosphite catalyzed by complexes 1~7

Entry Cat. Loading/mol% Yieldb/% 1 1 0.08 97 2c 1 0.05 74 3 2 0.08 89 4 3 0.08 92 5 4 0.08 99 6 5 0.08 93 7 6 0.08 97 8 7 0.08 97 aReaction conditions: benzaldehyde (10.0 mmol), diethyl phosphite (12.0 mmol), neat, 5 min.bIsolated yields.c35 min. 表 1 二(β-二亚胺基)二价稀土配合物催化苯甲醛与亚磷酸二乙酯的氢磷化反应条件优化a
Table 1. Hydrophosphonylation of benzaldehyde and diethyl phosphite catalyzed by complexes 1~7从表 1结果可以看到, 所有这些二价稀土金属配合物都可以在温和的反应条件下, 有效地催化这一反应(Table 1, Entries 1, 3~8).如当催化剂用量为0.08 mol%时, 25 ℃, 无溶剂条件下苯甲醛和亚磷酸二乙酯的反应都能够很顺利地进行, 反应5 min后, 可以以很高的收率得到α-羟基膦酸酯.即使当催化剂用量降为0.05 mol%时, 反应35 min, 产物收率仍可高达74% (Table 1, Entry 2).
配合物的催化活性与β-二亚胺基配体的结构有关.当配体为对称结构的配合物时, 配体体积大的活性更高, 其活性顺序为L2, 6-Me2<L2, 4, 6-Me3<L2, 6-ipr2 (Table 1, Entries 3, 4, 1).相比于配体为对称结构的配合物时, 不对称结构的配合物的活性更高, 这与这些配合物在催化胍化反应的活性顺序基本一致[12].从实验结果可以看到, 中心金属对催化活性的影响不是很明显, 无论中心金属是Sm, Yb还是Eu, 都显示很高的催化活性.如在相同反应条件下, 在配体为L2, 6-ipr2时, 无论中心金属是Eu还是Sm, 产物收率分别达到93%和97% (Table 1, Entries 1, 6).当配体都为LPh2, 6-ipr2时, 中心金属为Eu和Yb时, 反应产物的产率可分别达到99%和97% (Table 1, Entries 5, 7).
1.2 醛氢磷化反应的底物拓展
为了进一步了解本催化体系的底物适应能力, 我们以配合物7为催化剂, 以催化剂用量为0.08 mol%, 25 ℃, 无溶剂作为反应条件, 考察了这类配合物对不同醛与亚磷酸二乙酯氢磷化反应的催化活性.反应结果见表 2.
表 2
醛氢磷化反应的底物拓展a
Table 2.
Extension of the substrate scope of the hydrophosphonylation of aldehydes
 表 2 醛氢磷化反应的底物拓展a
表 2 醛氢磷化反应的底物拓展a
Table 2. Extension of the substrate scope of the hydrophosphonylation of aldehydes从表 2的结果可以看到, 配合物7显示有很好的底物适应能力.苯环上取代基的电子效应对该反应没有明显的影响.无论苯环上含氟、氯、硝基等吸电子基团还是含甲氧基、甲基等给电子基团的芳醛, 反应都能很快进行并以90%以上的收率给出相应的α-羟基膦酸酯(表 2, 10i, 10e, 10h, 10c, 10d).苯环上取代基的位阻效应对该反应也没有明显的影响.如以邻甲基苯甲醛为底物的反应的产物收率与对甲基苯甲醛的反应收率相当(表 2, 10g, 10b).对于芳杂环的3-吡啶甲醛, 反应也能顺利进行并以高产率得到目标产物(表 2, 10j).
1.3 酮的氢磷化反应条件优化
由于未活化的酮的反应活性低, 因此对这类氢磷化反应的研究更引起了化学工作者的研究兴趣.鉴于二(β-二亚胺基)二价稀土配合物在催化醛的氢磷化反应中显示很高的催化活性, 我们进一步研究了它们对酮的氢磷化反应的催化行为.首先以苯乙酮11a和亚磷酸二乙酯的反应作为模板反应, 以配合物7为催化剂, 对苯乙酮氢磷化反应的条件进行了优化, 结果见表 3.可以看出, 虽然苯乙酮比苯甲醛的反应活性低, 但配合物7仍可以顺利地催化这一反应.在催化剂用量为0.5 mol%, 25 ℃反应20 min, 产物收率可以达到86%(表 3, Entry 2).但当反应时间缩短为5 min, 产物收率降为64%(表 3, Entry 3).将催化剂用量降低到0.2 mol%时, 在相同条件下反应, 产物的收率有所降低(表 3, Entry 1).从表 3还可以看到, 反应温度对产物的收率有较大影响.反应温度低于25 ℃, 产物的收率将明显降低(表 3, Entries 5, 6), 温度升高到40 ℃时, 产物的收率略有下降(表 3, Entry 7).这可能是因为高温有利于逆反应的进行, 即α-羟基磷酸酯分解成亚磷酸酯和苯乙酮.这与文献报道的结果相类似[11].溶剂对反应有较大影响.在非极性溶剂甲苯和正己烷中进行反应, 其活性明显高于在极性溶剂THF中的反应, 这可能由于THF对稀土金属的配位所致(表 3, Entries 8~10).无溶剂条件下反应的产率与在正己烷中反应所得的产率相当(表 3, Entry 9).于是, 我们选择催化剂用量为0.5 mol%, 25 ℃, 无溶剂作为反应条件, 进一步考察了配合物2、3、6的催化活性(表 3, Entries 11~13).可以看出, 这些配合物都能有效地催化这一反应, 只是配合物6的活性稍低.
表 3
二(β-二亚胺基)二价稀土配合物催化苯乙酮与亚磷酸二乙酯的氢磷化反应条件优化a
Table 3.
Optimization of reaction conditions for the hydrophosphonylation of acetophnone and diethyl phosphite

Entry Cat.(mol%) Temp./℃ Time/min Solvent Yieldb/% 1 7 (0.2) 25 20 Neat 64 2 7 (0.5) 25 20 Neat 86 3 7 (0.5) 25 5 Neat 64 4 7 (0.5) 25 10 Neat 81 5 7 (0.5) 0 20 Neat 57 6 7 (0.5) 10 20 Neat 73 7 7 (0.5) 40 20 Neat 81 8 7 (0.5) 25 20 Toluene 77 9 7 (0.5) 25 20 n-Hexane 84 10 7 (0.5) 25 20 THF 60 11 2 (0.5) 25 20 Neat 86 12 3 (0.5) 25 20 Neat 83 13 6 (0.5) 25 20 Neat 78 aReaction conditions: acetophenone (1.00 mmol), diethyl phosphite (1.20 mmol), solvent (0.2 mL), bNMR yields. 表 3 二(β-二亚胺基)二价稀土配合物催化苯乙酮与亚磷酸二乙酯的氢磷化反应条件优化a
Table 3. Optimization of reaction conditions for the hydrophosphonylation of acetophnone and diethyl phosphite1.4 酮的氢磷化反应的底物拓展
根据上面的结果, 我们以配合物7为催化剂, 催化剂用量为0.5 mol%, 25 ℃, 反应20 min作为反应条件, 对酮底物进行了拓展, 结果见表 4.表 4结果表明, 配合物7对酮底物也有良好的适应能力.苯环的邻位和对位含有吸电子取代基的芳酮都可以顺利地进行反应, 并以90%~99%的收率得到相应产物(表 4, 12b, 12c, 12f, 12j, 12k).含有共轭结构的底物, 如二苯甲酮, 2-萘乙酮都可以很快进行反应, 产物的收率可分别高达93%和95%(表 4, 12d, 12e).脂肪酮也可顺利地与亚磷酸二乙酯反应, 并显示很高的活性.如丙酮在相同反应条件下可以以98%的收率得到相应的α-羟基磷酸酯(表 4, 12i).然而, 当芳酮的苯环上带有供电子取代基时, 反应活性有所下降.如对甲基苯乙酮反应时, 在相同条件下, 产物的收率只有77%(表 4, 12g), 含杂环结构的酮活性也有所降低(表 4, 12h).
表 4
酮氢磷化反应的底物拓展a
Table 4.
Extension of the substrate scope of the hydrophosphonylation of ketones
 表 4 酮氢磷化反应的底物拓展a
表 4 酮氢磷化反应的底物拓展a
Table 4. Extension of the substrate scope of the hydrophosphonylation of ketones2 结论
本文研究了7个二(β-二亚胺基)二价稀土金属配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应.结果表明所有的配合物都可以在温和条件下高活性地催化芳醛以及杂环芳醛与亚磷酸二乙酯的反应, 在低催化剂用量下(0.08 mol%), 25 ℃反应5 min, 产物的产率可达到90%以上.反应具有优秀的底物适应能力.这类二价稀土配合物对未活化的酮与亚磷酸二乙酯的反应也有良好的催化活性及良好的底物适应能力.
为了进一步考察该氢磷化反应的机理, 同时基于配合物7具有抗磁性, 选择配合物7进行核磁管跟踪反应.首先, 将配合物7 (31.8 mg, 0.0365 mmol) (图 1, b)与亚磷酸二乙酯(0.0365 mmol, 4.70 μL) (图 1, a)按1:1的物质的量比在氘苯(0.6 mL)中进行反应.反应1.5 h后, 从1H NMR可看出与亚磷酸二乙酯的磷原子相连的质子在δ 7.45和5.75处的两组峰消失, 同时可以看到化学位移在δ 12.85处有新的信号峰产生, 这是中性β-二亚胺上NH的信号峰, 而亚磷酸二乙酯质子峰的消失以及这个中性配体的生成表明配合物7首先与亚磷酸二乙酯经过一个质子化作用的过程, 形成了相应的中间物种A (图 1, c).
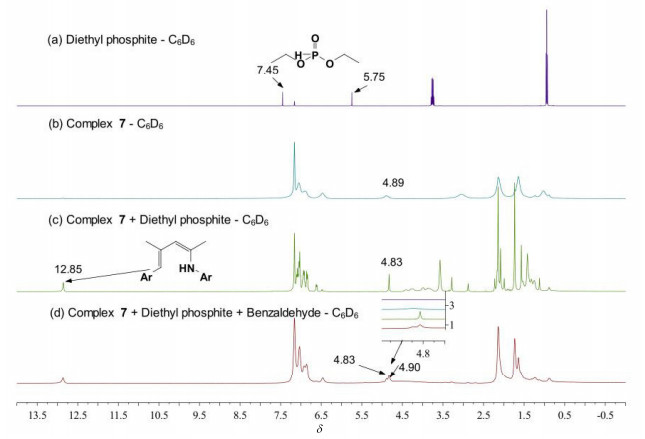 图 1
(a)亚磷酸二乙酯、(b)配合物7、(c)配合物7+亚磷酸二乙酯和(d)配合物7+亚磷酸二乙酯+苯甲醛的1H NMR谱图(氘苯)
Figure 1.
1H NMR spectra of (a) diethyl phosphite, (b) complex 7, (c) complex 7+diethylphosphite, and (d) complex 7+diethylphosphite+benzaldehyde
图 1
(a)亚磷酸二乙酯、(b)配合物7、(c)配合物7+亚磷酸二乙酯和(d)配合物7+亚磷酸二乙酯+苯甲醛的1H NMR谱图(氘苯)
Figure 1.
1H NMR spectra of (a) diethyl phosphite, (b) complex 7, (c) complex 7+diethylphosphite, and (d) complex 7+diethylphosphite+benzaldehyde
随后, 我们在此反应液中再加入1 equiv.的苯甲醛(0.0365 mmol, 3.70 μL).反应1.5 h后, 从1H NMR中可看出苯甲醛的质子在低场的信号峰消失了, 同时, 在δ 4.90处有新的信号峰生成, 这可能是α-碳原子上质子特征峰, 表明苯甲醛进一步与中间物种A发生了加成反应, 最终形成了该反应体系的活性中间体C, 这与文献[10i]报道的机理相类似(图 1, d).
基于以上实验结果, 并结合之前稀土催化方面的文献报道[10i, 11], 我们提出了该反应的一个可能机理(Scheme 1).首先, 亚磷酸二乙酯与二(β-二亚胺基)二价稀土金属配合物上的β-二亚胺基发生质子交换, 形成了中间物种A, 醛或酮与中间物种A可能经过渡态B形成活性物种C.最后, 活性物种C被另一分子亚磷酸二乙酯质解, 得到一分子α-羟基膦酸酯D并形成中间物种A, 从而完成整个催化循环.
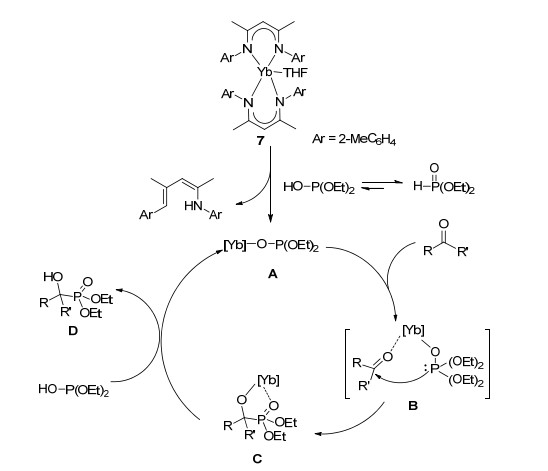 图 图式1
二(β-二亚胺基)镱(Ⅱ)配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应可能的机理
Figure 图式1.
Possible mechanism for the hydrophosphonylation of aldehydes/ketones and diethyl phosphite catalyzed by YbⅡ complex
图 图式1
二(β-二亚胺基)镱(Ⅱ)配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应可能的机理
Figure 图式1.
Possible mechanism for the hydrophosphonylation of aldehydes/ketones and diethyl phosphite catalyzed by YbⅡ complex
3 实验部分
3.1 仪器与试剂
1H NMR (400 MHz)检测采用Bruker Avance DRX-400核磁共振仪, 以TMS为内标, 氘代氯仿(CDCl3)为溶剂.所有操作除特别说明外, 均在无水无氧的氩气保护下或手套箱中进行.配合物1~7按文献[12]方法制备.四氢呋喃、甲苯和正己烷均在金属钠丝存在下干燥回流, 直至加入二苯甲酮呈紫黑色, 在氩气保护下蒸出封瓶备用; 亚磷酸二乙酯经氢化钙干燥数天后减压蒸馏封瓶备用; 液态醛、酮经氢化钙干燥2 d后蒸出备用; 固态醛、酮在结晶瓶中真空脱水脱氧8 h以上, 通入氩气封瓶备用.
3.2 实验方法
在手套箱中, 25 ℃下, 在20 mL Schlenk反应瓶中称取催化剂7 (0.008 mmol, 0.08 mol%), 然后往反应瓶中加入亚磷酸二乙酯(1.55 mL, 12 mmol), 搅拌5 min, 然后用注射器加入醛(10 mmol), 在指定温度下搅拌反应5 min后, 迅速转出手套箱, 暴露于空气中并往反应体系中加入适量去离子水终止反应, 再加入适量的乙酸乙酯溶解产物, 旋蒸除去溶剂, 残余固体经正己烷洗涤, 得到白色固体, 减压抽干后得到产物10, 称重, 计算产率.
在手套箱中, 在20 mL Schlenk反应瓶中称取催化剂7 (0.009 mmol, 0.5 mol%), 然后往反应瓶中加入亚磷酸二乙酯(0.28 mL, 1.2 mmol), 在25 ℃下搅拌5 min, 然后用注射器加入酮(1 mmol), 置于预先设定的温度条件下反应.待反应至一定时间后, 快速转出手套箱, 暴露于空气中并向反应体系中加入适量去离子水终止反应, 再加入适量的乙酸乙酯溶解固体, 旋干, 经硅胶柱层析[洗脱剂: V(乙酸乙酯):V(石油醚)=1:10]得到白色固体或无色油状产物12, 称重, 计算产率.对于苯乙酮的核磁收率:反应至设定时间后, 迅速移出手套箱, 取1~2滴反应液于核磁管中再加入氘代氯仿, 测定核磁(1H NMR), 计算产率.
10a~10j及12a~12k的核磁数据如下:
α-羟基-苄基膦酸二乙酯(10a)[7d]: 1H NMR (CDCl3, 400 MHz) δ: 1.21 (t, J=7.1 Hz, 3H), 1.26 (t, J=7.1 Hz, 3H), 3.78 (dd, J=9.6, 5.2 Hz 1H), 3.94~4.08 (m, 4H), 5.01 (dd, J=10.8, 5.2 Hz, 1H), 7.28~7.38 (m, 3H), 7.48~7.49 (m, 2H).
α-羟基-(4-甲基)-苄基膦酸二乙酯(10b)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.22 (t, J=7.1 Hz, 3H), 1.27 (t, J=7.1 Hz, 3H), 2.34 (d, J=1.7 Hz, 3H), 3.93~4.09 (m, 4H), 4.97 (d, J=10.4 Hz, 1H), 7.17 (d, J=8.0 Hz, 2H), 7.36 (dd, J=8.1, 2.0 Hz, 2H).
α-羟基-(4-硝基)-苄基膦酸二乙酯(10c)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.27 (m, 6H), 4.04~4.16 (m, 4H), 4.83 (s, 1H), 5.16 (d, J=12.2 Hz, 1H), 7.66 (dd, J=8.7, 2.2 Hz, 2H), 8.22 (d, J=8.4 Hz, 2H).
α-羟基-(3-硝基)-苄基膦酸二乙酯(10d)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.24~1.31 (m, 6H), 4.04~4.20 (m, 4H), 5.15 (dd, J=11.4, 5.3 Hz, 1H), 5.40 (s, 1H), 7.52 (t, J=8.0 Hz, 1H), 7.81 (d, J=7.6 Hz, 1H), 8.15 (d, J=7.8 Hz, 1H), 8.40 (m, 1H).
α-羟基-(3-氯)-苄基膦酸二乙酯(10e)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.23~1.30 (m, 6H), 3.82 (dd, J=8.9, 5.1 Hz, 1H), 4.08~4.14 (m, 4H), 4.99 (dd, J=11.1, 5.0 Hz, 1H), 7.28~7.29 (m, 2H), 7.36~7.37 (m, 1H), 7.50 (s, 1H).
α-羟基-(2-甲氧基)-苄基膦酸二乙酯(10f)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.16 (t, J=7.1 Hz, 3H), 1.29 (t, J=7.1 Hz, 3H), 3.84 (s, 3H), 3.87~4.15 (m, 4H), 5.40 (d, J=11.7 Hz, 1H), 6.88 (d, J=8.3 Hz, 1H), 6.98 (t, J=7.5 Hz, 1H), 7.25~7.30 (m, 1H), 7.52 (m, 1H).
α-羟基-(2-甲基)-苄基膦酸二乙酯(10g)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.20 (t, J=7.1 Hz, 3H), 1.28 (t, J=7.1 Hz, 3H), 2.38 (s, 3H), 3.89~4.10 (m, 4H), 5.25 (d, J=10.9 Hz, 1H), 7.14~7.25 (m, 3H), 7.62~7.64 (m, 1H).
α-羟基-(2-氯)-苄基膦酸二乙酯(10h)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.20 (t, J=7.0 Hz, 3H), 1.31 (t, J=7.0 Hz, 3H), 3.94~4.18 (m, 4H), 5.56 (d, J=11.6 Hz, 1H), 7.22~7.24 (m, 1H), 7.30~7.36 (m, 2H), 7.75 (d, J=7.5 Hz, 1H).
α-羟基-(4-氟)-苄基膦酸二乙酯(10i)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.23~1.27 (m, 6H), 3.95~4.09 (m, 4H), 4.99 (d, J=10.3 Hz, 1H), 7.05 (t, J=8.5 Hz, 2H), 7.44~7.48 (m, 2H).
α-羟基-(3-吡啶基)-苄基膦酸二乙酯(10j)[11]:1H NMR (CDCl3, 400 MHz) δ: 1.22~1.29 (m, 6H), 4.04~4.14 (m, 4H), 5.06 (d, J=11.3 Hz, 1H), 7.27~7.31 (m, 1H), 7.86~7.88 (m, 1H), 8.51~8.53 (m, 1H), 8.64 (s, 1H).
α-羟基-甲基-苄基膦酸二乙酯(12a)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.18 (t, J=7.1 Hz, 3H), 1.26 (t, J=7.1 Hz, 3H), 1.82 (d, J=15.3 Hz, 3H), 2.94~2.96 (m, 1H), 3.81~4.12 (m, 4H), 7.28~7.30 (m, 1H), 7.34~7.36 (m, 2H), 7.59~7.62 (m, 2H).
α-羟基-甲基-(2-氯)-苄基膦酸二乙酯(12b)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.21 (t, J=7.1 Hz, 3H), 1.28 (t, J=7.1 Hz, 3H), 1.98 (d, J=15.2 Hz, 3H), 3.96~4.15 (m, 4H), 4.17 (d, J=6.2 Hz, 1H), 7.18~7.28 (m, 2H), 7.34~7.36 (m, 1H), 7.75 (m, 1H).
α-羟基-甲基-(4-氟)-苄基膦酸二乙酯(12c)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.19~1.26 (m, 6H), 1.79 (d, J=15.4 Hz, 3H), 4.06~4.10 (m, 4H), 7.02 (t, J=8.6 Hz, 2H), 7.55~7.59 (m, 2H).
α-羟基-苯基-苄基膦酸二乙酯(12d)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.17 (t, J=7.1 Hz, 6H), 3.48~3.51 (m, 1H), 3.85~4.04 (m, 4H), 7.24~7.28 (m, 6H), 7.31~7.34 (m, 4H), 7.67~7.69 (m, 4H).
α-羟基-甲基-(2-萘甲基)-膦酸二乙酯(12e)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.19 (t, J=7.1 Hz, 3H), 1.26 (t, J=7.1 Hz, 3H), 1.92 (d, J=15.4 Hz, 3H), 3.61 (d, J=6.0 Hz, 1H), 3.87~4.15 (m, 4H), 7.45~7.49 (m, 2H), 7.72~7.75 (m, 1H), 7.82~7.87 (m, 3H), 8.09 (s, 1H).
α-羟基-甲基-(4-硝基)-苄基膦酸二乙酯(12f)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.26 (t, J=6.7 Hz, 3H), 1.85 (d, J=15.4 Hz, 3H), 4.03~4.16 (m, 4H), 4.31 (d, J=2.5 Hz, 1H), 7.79 (d, J=7.5 Hz, 2H), 8.20 (d, J=8.2 Hz, 2H).
α-羟基-甲基-(4-甲基)-苄基膦酸二乙酯(12g)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.20~1.26 (m, 6H), 1.80 (d, J=15.1 Hz, 3H), 2.34 (s, 3H), 3.25 (d, J=4.0 Hz, 1H), 3.91~4.08 (m, 4H), 7.16 (d, J=5.2 Hz, 2H), 7.47 (d, J=4.5 Hz, 2H).
α-羟基-甲基-(2-噻吩基)膦酸二乙酯(12h)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.31 (m, 6H), 1.90 (d, J=14.7 Hz, 3H), 4.10~4.17 (m, 4H), 4.27 (d, J=4.0 Hz, 1H), 7.03 (s, 1H), 7.18 (s, 1H), 7.30 (s, 1H).
α-羟基-甲基-乙基膦酸二乙酯(12i)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.27 (t, J=7.1 Hz, 6H), 1.37 (d, J=15.3 Hz, 6H), 4.08~4.15 (m, 4H).
α-羟基-甲基-(4-溴)-苄基膦酸二乙酯(12j)[7d]:1H NMR (CDCl3, 400 MHz) δ: 1.20~1.27 (m, 6H), 1.78 (d, J=15.4 Hz, 3H), 3.73 (d, J=5.3 Hz 1H), 3.92~4.13 (m, 4H), 7.47 (s, 4H).
α-羟基-甲基-(4-氯)-苄基膦酸二乙酯(12k)[11]:1H NMR (CDCl3, 400 MHz) δ: 1.22 (t, J=7.1 Hz, 3H), 1.26 (t, J=7.1 Hz, 3H), 1.79 (d, J=15.3 Hz, 3H), 3.41 (d, J=5.7 Hz, 1H), 3.90~4.14 (m, 4H), 7.32 (d, J=8.3 Hz, 2H), 7.52~7.55 (m, 2H).
辅助材料(Supporting Information) 所合成的化合物的1H NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
Karasik, A. A.; Sinyashin, O. G. Phosphorus Compounds:Ad-vanced Tools in Catalysis and Material Science, Vol. 37, Eds.:Peruzzini, M.; Gonsalvi, L, Kazan, 2011, pp. 375~444.
-
[2]
Costisella, B.; Keitel, I.; Ozegowski, S. Sulfur Silicon 1993, 84, 115. doi: 10.1080/10426509308034321
-
[3]
Moore, M. L.; Dreyer, G. B. Perspect. Drug Discovery Des. 1993, 1, 85.
(b) Stowassser, B.; Budt, K. H.; Li, J. Q.; Peyman, A.; Ruppert, D. Tetrahedron Lett. 1992, 33, 6625. -
[4]
Texier-Boullet, F.; Foucaud, A. Synthesis 1982, 165.
(b) Texier-Boullet, F.; Foucaud, A. Synthesis 1982, 916.
(c) de Noronha, R. G.; Costa, P. J.; Romão, C. C.; Calhorda, M. J.; Fernandes, A. C. Organometallics 2009, 28, 6206.
(d) Martínez-Castro, E.; López, Ó.; Maya, I.; Farnández-Bolaños, J. G.; Petrini, M. Green Chem. 2010, 12, 1171. -
[5]
Wang, F.; Liu, X. H.; Cui, X.; Xiong, Y.; Zhou, X.; Feng, X. M. Chem. Eur. J. 2009, 15, 589. doi: 10.1002/chem.v15:3
-
[6]
Yokomatsu, T.; Yamagishi, T.; Shibuya, S. Tetrahedron:Asymmetry 1993, 4, 1401.
(b) Groaning, M. D.; Rowe, B. J.; Spilling, C. D. Tetrahedron Lett. 1998, 39, 5485.
(c) Zhou, X.; Liu, Y. L.; Chang, L.; Zhao, J. N.; Shang, D. J.; Liu, X. H.; Lin, L. L.; Feng, X. M. Adv. Synth. Catal. 2009, 351, 2567. -
[7]
Simoni, D.; Invidiata, F. P.; Manferdini, M.; Lampronti, I.; Rondanin, R.; Roberti, M.; Pollini, G. P. Tetrahedron Lett. 1998, 39, 7615.
(b) Simoni, D.; Rondanin, R.; Morini, M.; Baruchello, R.; In-vidiata, F. P. Tetrahedron Lett. 2000, 41, 1607.
(c) Rulev, A. Y. Heteroat. Chem. 2013, 24, 187.
(d) Liu, C. W.; Zhang, Y.; Qian, Q. Q.; Yuan, D.; Yao, Y. M. Org. Lett. 2014, 16, 6172. -
[8]
Arai, T.; Bougauchi, M.; Sasai, H.; Shibasaki, M. J. Org. Chem. 1996, 61, 2926.
(b) Saito, B.; Katsuki, T. Angew. Chem., Int. Ed. 2005, 44, 4600.
(c) Ito, K.; Tsutsumi, H.; Setoyama, M.; Saito, B.; Katsuki, T. Synlett 2007, 1960.
(d) Zhou, X.; Liu, X. H.; Yang, X.; Shang, D. J.; Xin, J. G.; Feng, X. M. Angew. Chem., Int. Ed. 2008, 47, 392.
(e) Abell, J. P.; Yamamoto, H. J. Am. Chem. Soc. 2008, 130, 10521.
(f) Suyama, K.; Sakai, Y.; Matsumoto, K.; Saito, B.; Katsuki, T. Angew. Chem., Int. Ed. 2010, 49, 797.
(g) Liu, B.; Carpentier, J.; Sarazin, Y. Chem. Eur. J. 2012, 18, 13259. -
[9]
Yokomatsu, T.; Yamgishi, T.; Shibuya, S. Tetrahedron:Asymmetry 1993, 4, 1779.
(b) Yang, F.; Zhao, D. B.; Lan, J. B.; Xi, P. H.; Yang, L.; Xiang, S. H.; You, J. S. Angew. Chem., Int. Ed. 2008, 47, 5646.
(c) Xu, F.; Liu, Y. P.; Tu, J. X.; Lei, C.; Li, G. Q. Tetrahedron:Asymmetry 2015, 26, 891. -
[10]
Sasai, H.; Bougauchi, M.; Arai, T.; Shibasaki, M. Tetrahedron Lett. 1997, 38, 2717.
(b) Chen, W. L.; Hui, Y. H.; Zhou, X.; Jiang, J.; Cai, Y. F.; Liu, X. H.; Lin, L. L.; Feng, X. M. Tetrahedron Lett. 2010, 51, 4175.
(c) Wu, Q. M.; Zhou, J.; Yao, Z. G.; Xu, F.; Shen, Q. J. Org. Chem. 2010, 75, 7498.
(d) Zhu, X. C.; Wang, S. W.; Zhou, S. L.; Wei, Y.; Zhang, L. J.; Wang, F. H.; Feng, Z. J.; Guo, L. P.; Mu, X. L. Inorg. Chem. 2012, 51, 7134.
(e) Zhou, S. L.; Wang, H. Y.; Ping, J.; Wang, S. W.; Zhang, L. J.; Zhu, X. C.; Wei, Y.; Wang, F. H.; Feng, Z. J.; Gu, X. X.; Yang, S.; Miao, H. Organometallics 2012, 31, 1696.
(f) Miao, H.; Zhou, S. L.; Wang, S. W.; Zhang, L. J.; Wei, Y.; Yang, S.; Wang, F. H.; Chen, Z.; Chen, Y.; Yuan, Q. B. Sci. China Chem. 2013, 56, 329.
(g) Zhou, S. L.; Wu, Z. S.; Rong, J. W.; Wang, S. W.; Yang, G. S.; Zhu, X. C.; Zhang, L. J. Chem. Eur. J. 2012, 18, 2653.
(h) Yang, S.; Zhu, X. C.; Zhou, S. L.; Wang, S. W.; Feng, Z. J.; Wei, Y.; Miao, H.; Guo, L. P.; Wang, F. H.; Zhang, G. C.; Gu, X. X.; Mu, X. L. Dalton Trans. 2014, 43, 2521.
(i) Liu, C. W.; Qian, Q. Q.; Nie, K.; Wang, Y. R.; Shen, Q.; Yuan, D.; Yao, Y. M. Dalton Trans. 2014, 43, 8355. -
[11]
Zhao, L.; Ding, H.; Zhao, B.; Lu, C. R.; Yao, Y. M. Polyhedron 2014, 83, 50. doi: 10.1016/j.poly.2014.04.018
-
[12]
Xue, M. Q.; Zheng, Y.; Hong, Y. B.; Yao, Y. M.; Xu, F.; Zhang, Y.; Shen, Q. Dalton Trans. 2015, 44, 20075. doi: 10.1039/C5DT03674G
-
[1]
-

图 1 (a)亚磷酸二乙酯、(b)配合物7、(c)配合物7+亚磷酸二乙酯和(d)配合物7+亚磷酸二乙酯+苯甲醛的1H NMR谱图(氘苯)
Figure 1 1H NMR spectra of (a) diethyl phosphite, (b) complex 7, (c) complex 7+diethylphosphite, and (d) complex 7+diethylphosphite+benzaldehyde

图式1 二(β-二亚胺基)镱(Ⅱ)配合物催化醛/酮与亚磷酸二乙酯的氢磷化反应可能的机理
Scheme 1 Possible mechanism for the hydrophosphonylation of aldehydes/ketones and diethyl phosphite catalyzed by YbⅡ complex
表 1 二(β-二亚胺基)二价稀土配合物催化苯甲醛与亚磷酸二乙酯的氢磷化反应条件优化a
Table 1. Hydrophosphonylation of benzaldehyde and diethyl phosphite catalyzed by complexes 1~7
Entry Cat. Loading/mol% Yieldb/% 1 1 0.08 97 2c 1 0.05 74 3 2 0.08 89 4 3 0.08 92 5 4 0.08 99 6 5 0.08 93 7 6 0.08 97 8 7 0.08 97 aReaction conditions: benzaldehyde (10.0 mmol), diethyl phosphite (12.0 mmol), neat, 5 min.bIsolated yields.c35 min.  下载: 导出CSV
下载: 导出CSV
表 2 醛氢磷化反应的底物拓展a
Table 2. Extension of the substrate scope of the hydrophosphonylation of aldehydes
下载: 导出CSV
表 3 二(β-二亚胺基)二价稀土配合物催化苯乙酮与亚磷酸二乙酯的氢磷化反应条件优化a
Table 3. Optimization of reaction conditions for the hydrophosphonylation of acetophnone and diethyl phosphite
Entry Cat.(mol%) Temp./℃ Time/min Solvent Yieldb/% 1 7 (0.2) 25 20 Neat 64 2 7 (0.5) 25 20 Neat 86 3 7 (0.5) 25 5 Neat 64 4 7 (0.5) 25 10 Neat 81 5 7 (0.5) 0 20 Neat 57 6 7 (0.5) 10 20 Neat 73 7 7 (0.5) 40 20 Neat 81 8 7 (0.5) 25 20 Toluene 77 9 7 (0.5) 25 20 n-Hexane 84 10 7 (0.5) 25 20 THF 60 11 2 (0.5) 25 20 Neat 86 12 3 (0.5) 25 20 Neat 83 13 6 (0.5) 25 20 Neat 78 aReaction conditions: acetophenone (1.00 mmol), diethyl phosphite (1.20 mmol), solvent (0.2 mL), bNMR yields.
下载: 导出CSV
表 4 酮氢磷化反应的底物拓展a
Table 4. Extension of the substrate scope of the hydrophosphonylation of ketones
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1160
- HTML全文浏览量: 229
 下载:
下载:
