 图 1
含氟抗肿瘤药
Figure 1.
Antitumor drugs containing fluorine
图 1
含氟抗肿瘤药
Figure 1.
Antitumor drugs containing fluorine
引用本文:
李恭铭, 孙德群. 分子内直接引入含氟基团以及18F-标记方法研究进展[J]. 有机化学,
2016, 36(8): 1715-1740.
doi:
10.6023/cjoc201601009

Citation: Li Gongming, Sun Dequn. Recent Advances of Direct Incorporation of Fluorine-Containing Groups and 18F-Labeling Methods[J]. Chinese Journal of Organic Chemistry, 2016, 36(8): 1715-1740. doi: 10.6023/cjoc201601009
Citation: Li Gongming, Sun Dequn. Recent Advances of Direct Incorporation of Fluorine-Containing Groups and 18F-Labeling Methods[J]. Chinese Journal of Organic Chemistry, 2016, 36(8): 1715-1740. doi: 10.6023/cjoc201601009
分子内直接引入含氟基团以及18F-标记方法研究进展
摘要:
含氟基团拥有强吸电子性和高亲脂性,有机分子适当地引入含氟基团可以改善其原有的功能. 因此,向分子内引入含氟基团的方法成为了当前的研究热点. 由于三氟甲硫基拥有突出的性能,其引入方法成为主要研究热点之一,三氟甲硒基、三氟甲氧基以及三氟乙氧基的引入方法也受到了科研人员的关注. 18F拥有几乎理想的原子核性能,是临床成像使用最广泛的正电子发射断层显像(PET)放射性核素. 因此,新型[18F]放射性示踪剂的研发成为很多科研人员的研究方向. 研发新型[18F]放射性示踪剂的关键在于用18F对生物分子进行标记. 介绍了最近报道的向分子内直接引入CF3X(X=S,Se,O,CH2O)基团的新方法以及一些18F-标记方法的研究成果. 此外,总结了一些该领域值得关注的研究方向.
English
Recent Advances of Direct Incorporation of Fluorine-Containing Groups and 18F-Labeling Methods
Abstract:
Fluorine-containing groups have strong electron-withdrawing effect and high lipophilicity, organic molecules incorporated fluorine-containing groups can improve its function. Therefore, the incorporation of fluorine-containing groups into molecules has become the current research hotspot. The method for incorporation of CF3S group becomes one of the hot research field due to its excellent nature. The strategy for incorporation of CF3Se, CF3O and CF3CH2O groups has also been the attention of researchers. 18F has nearly ideal nuclear properties and is the most widely used positron emission tomography (PET) radioisotope for clinical imaging. Therefore, the development of new [18F]radiotracers becomes the research focus of many researchers. The method for 18F-labeling of biomolecules is the key to the development of new [18F]radiotracers. This paper mainly introduce the recently reported methodology of direct incorporating CF3X (X=S, Se, O, CH2O) groups into molecules and some research results of 18F-labeling methods. Furthermore, some noteworthy research directions in this field are summarized.
-
氟是地球上最为丰富的卤素[1],然而自然界中存在的含氟有机化合物只有小部分被鉴定出来,大多数有机氟化合物是由有机化学工作者合成的[2].
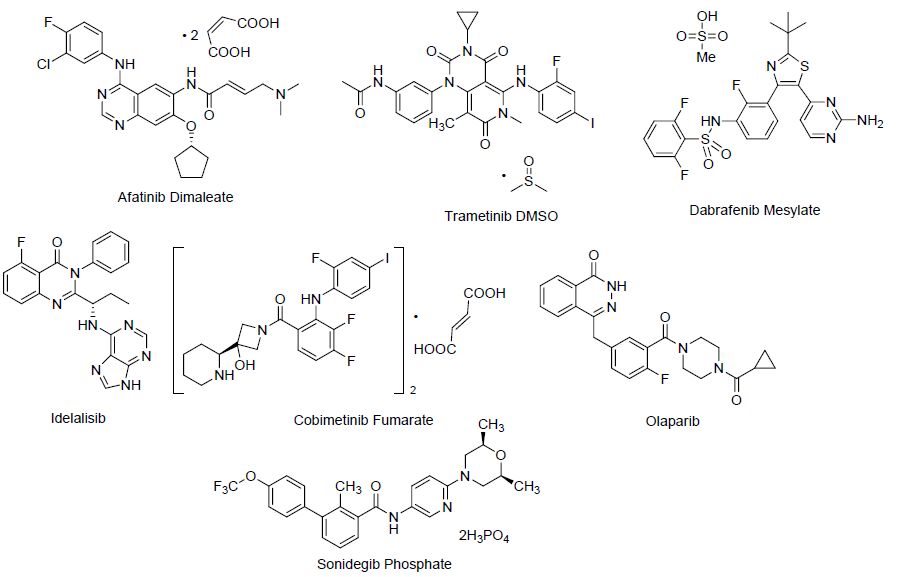
氟原子是具有最强电负性的元素,同时拥有接近氢原子的原子半径. 在分子中适当地引入氟原子或含氟基团后,可以明显地影响分子的构象、降低pKa、提高分子本身固有的能效和膜通透性,同时也影响了分子的代谢途径和药代动力学特征[3]. 因此,含氟药物的研发形成了一股热潮. 从2013年到截稿日期间,获得美国食品药品监督管理局(FDA)批准上市的化学分子单体共有72个,而含氟药物占21个. 其中具有抗癌功效的药物有7个[2b~2d],分别为激酶抑制剂类药物马来酸阿法替尼(Afatinib Dimaleate)[4]、二甲基亚砜曲美替尼(Trametinib Dimethyl Sulfoxide)[5]、甲磺酸达拉非尼(Dabrafenib Mesylate)[6]、艾代拉里斯(Idelalisib)[7]、Cobimetinib Fumarate[8],聚腺苷二磷酸-核糖聚合酶抑制剂类药物奥拉帕尼(Olaparib)[9],以及Hedgehog信号通路抑制剂类药物磷酸索尼吉步(Sonidegib Phosphate)[10] (图 1).
图 1
含氟抗肿瘤药
Figure 1.
Antitumor drugs containing fluorine
向分子内引入氟原子的研究热点主要为氟化反应、氟烷基化反应和三氟甲硫基化反应. 三氟甲硒基化反应和三氟烷氧化反应也引起了科研人员的关注. 作为研究热点,最近有多篇综述针对氟化反应[11]和氟烷基化反应[11c~11e,12]作了总结.
在此,本文主要介绍了直接三氟甲硫基化反应、三氟甲硒基化反应、三氟甲氧基化反应以及三氟乙氧基化反应的研究进展. 同时,由于18F同位素的半衰期为109.08 min,与11C (20.4 min)和124I (4.2 d)相比更加优越,氟原子逐渐成为了在正离子发射断层显像(PET)应用中最卓越的原子之一[3a]. 因此,本文介绍了最近的一些用18F对分子进行放射性标记的研究成果.
1 直接三氟甲硫基化反应
在各种含氟基团中,三氟甲硫基团(CF3S)是亲脂性最强的基团之一,Hansch参数πx=1.44[13]. 高的亲脂性有利于提高分子的膜渗透性从而增强吸收率,因此含三氟甲硫基的化合物在医药、农药研发领域有着广阔的前景. 三氟甲硫基化反应成功地引起了众多科研工作者的兴趣,多篇已经发表的综述[11e,12d,14]涉及到三氟甲硫基化反应,如卿凤翎课题组[14c]曾在本刊就直接三氟甲硫基化反应进行了详细报道. 为了避免报道的重复性,本文将主要介绍直接三氟甲硫基化反应最近的研究成果.
1.1 CF3S自由基参与的三氟甲硫基化反应
三氟甲硫银(AgSCF3)稳定无毒,是一个运用广泛的三氟甲硫基化试剂. AgSCF3在K2S2O8的存在下,可以被氧化得到CF3S自由基. 2014年,卿凤翎课题组[15]报道了制备烯丙基三氟甲基硫化物的新方法. 随后,该课题组[16]对其工作进行延伸,报道了醌类的三氟甲硫基化反应. 醌类结构在许多生物活性化合物中存在,并且许多天然的或合成的醌类化合物表现出广泛的药理特 性[16]. 该反应可能是由K2S2O8氧化AgSCF3得到CF3S自由基,随后CF3S自由基加成到醌类底物,得到的中间体经过进一步氧化和去质子化,最终得到含三氟甲硫基的目标产物. 该反应是直接在醌类化合物上引入三氟甲硫基的第一例报道(Scheme 1).
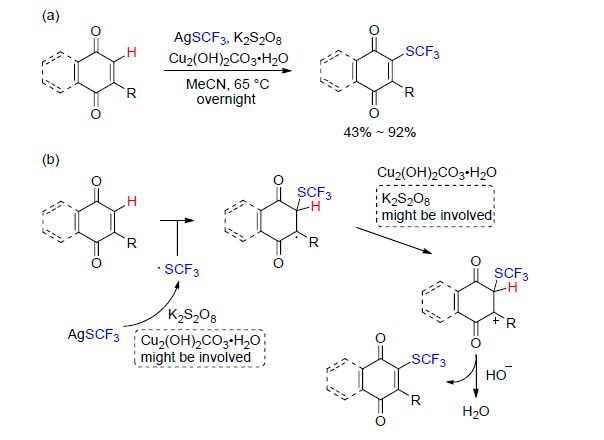 图 图式 1
醌的三氟甲硫基化反应以及其可能的机理
Figure 图式 1.
Trifluoromethylthiolation of quinines and its proposed mechanism
图 图式 1
醌的三氟甲硫基化反应以及其可能的机理
Figure 图式 1.
Trifluoromethylthiolation of quinines and its proposed mechanism
陈庆云和刘超等[17]报道了未活化的C(sp3)—H键的直接三氟甲硫基化反应. 反应过程中,K2S2O8发挥了关键作用,既活化C(sp3)—H键又氧化AgSCF3得到CF3S自由基(Scheme 2). 该反应表现出较好的区域选择性,叔碳C—H键比仲碳C—H键优先被转化为C—SCF3. 需要注意的是,环戊酮和环己酮不适于该反应,仅得到痕量目标产物. 然而环庚酮和环辛酮顺利得到了相应产物,收率分别为21%和48%.
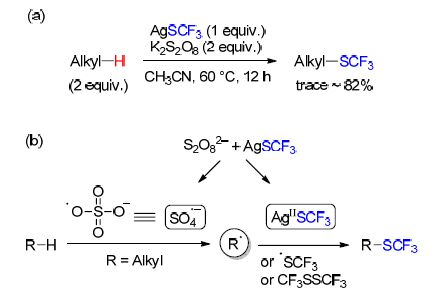 图 图式 2
未活化的C(sp3)—H键的直接三氟甲硫基化反应以及其可能的机理
Figure 图式 2.
Direct trifluoromethylthiolation of unactivated C(sp3)—H bond and its proposed mechanism
图 图式 2
未活化的C(sp3)—H键的直接三氟甲硫基化反应以及其可能的机理
Figure 图式 2.
Direct trifluoromethylthiolation of unactivated C(sp3)—H bond and its proposed mechanism
同时,汤平平课题组[18]也报道了银参与的,未活化的脂肪族C—H键的三氟甲硫基化反应(Eq. 1). 三氟甲硫基化被观察到选择性地发生在远离吸电子基的次甲基或亚甲基上. 该反应条件温和、操作简单,可以扩展至克级,在药物化学和农业化学领域有很大的应用前景.
多环吲哚类化合物多数具有好的生理或药理活性,拥有广泛的研究价值[19]. Nevado等[20]报道了由N-芳基- N-(2-乙炔基芳基磺酰基)丙烯酰胺合成吲哚[2,1-a]异喹啉类化合物的反应. 反应起初可能是由CF3S自由基选择性地与底物丙烯酸部分结合,形成新的C(sp3)—SCF3键以及一个α-羰基自由基中间体. 该中间体经过分子内的环化反应以及重新芳构化反应,脱去SO2得到酰胺基自由基中间体. 随后经过一系列的分子内环化反应以及重新芳构化反应,酰胺基自由基中间体转化成为目标化合物(Scheme 3).
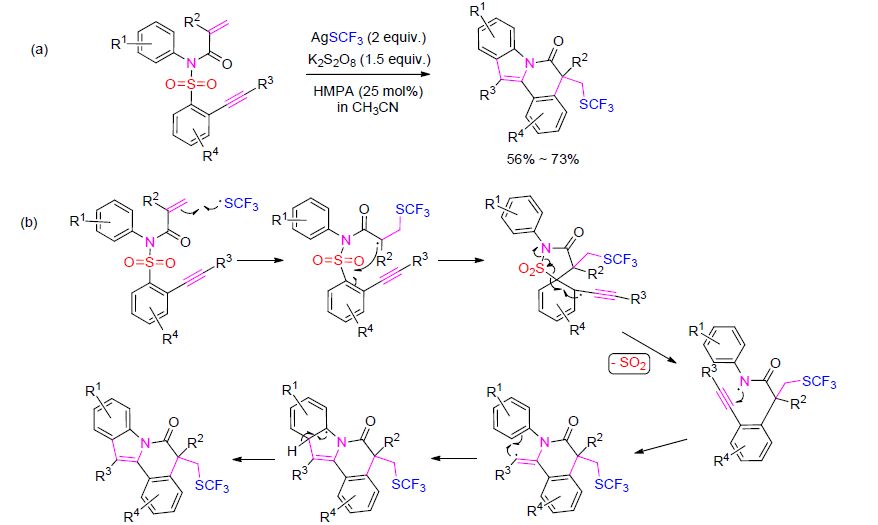 图 图式 3
吲哚[2,1-a]异喹啉类化合物的合成以及其可能的机理
Figure 图式 3.
Synthesis of indolo[2,1-a]isoquinolines and its proposed mechanism
图 图式 3
吲哚[2,1-a]异喹啉类化合物的合成以及其可能的机理
Figure 图式 3.
Synthesis of indolo[2,1-a]isoquinolines and its proposed mechanism
芴类化合物在生物医药、光电材料、太阳能电池等领域有广阔的前景[21]. 梁永民课题组[22]报道了三氟甲硫基化1,6-烯炔类化合物制备芴类化合物的反应. CF3S自由基进攻C≡C触发三氟甲硫基化-环化反应,一步形成一个C—SCF3键以及两个C—C键,构建出三氟甲硫取代的多环芴体系(Scheme 4).
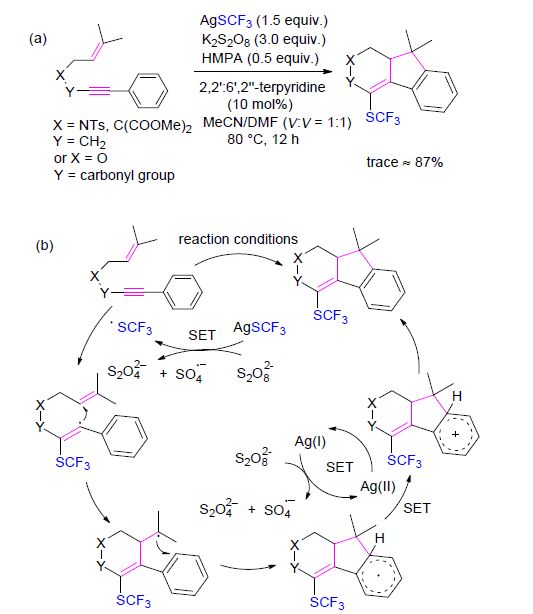 图 图式 4
1,6-烯炔的三氟甲硫基化反应以及其可能的机理
Figure 图式 4.
Trifluoromethylthiolation of 1,6-enynes and its proposed mechanism
图 图式 4
1,6-烯炔的三氟甲硫基化反应以及其可能的机理
Figure 图式 4.
Trifluoromethylthiolation of 1,6-enynes and its proposed mechanism
由于香豆素类化合物具有广谱的生物活性,如抗HIV[23]、抗菌[24]、抗炎症[25]、抗肿瘤[26]以及抗疟疾[27]活性,香豆素类化合物在药物化学研究中占有特殊地位. 2015年,王洪根等[28]报道了炔基甲酸芳酯通过氧化的自由基环合作用,合成3-三氟甲硫基香豆素类化合物的反应(Eq. 2). 同时,他们通过一系列的对照实验推论出了可信的反应机理: 首先AgSCF3被K2S2O8氧化产生CF3S自由基,后者与底物发生区域选择性加成反应得到乙烯基自由 基中间体. 随后乙烯基自由基中间体经过分子内的环化反应以及氧化作用得到Wheland中间体,再经过进一步去质子化/重新芳构化反应得到目标产物. 该反应条件温和、操作简单、起始底物易得且拥有极好的官能团普适性. 值得注意的是,底物芳环上酯基的邻位有甲基存在时,没有检测到目标产物的生成.
在大量催化剂 CuSCN存在下,芳环或芳杂环重氮盐与Me4NSCF3反应可以有效地转化成相应的三氟甲基硫醚类化合物[29] (Eq. 3). 该反应属于桑德迈尔反应类型,反应条件温和,所需时间短(少于1 h),并且兼容于多种官能团,适用于最后一步在类药分子中引入三氟甲硫基.
由于羰基易于进行衍生化反应,α-三氟甲硫基酮的合成在三氟甲基硫醚类化合物的研究中拥有重要意义. 2014年,翁志强课题组[30]使用易获得的单质硫以及三氟甲基三甲基硅烷(CF3SiMe3),在铜催化下对α-溴代酮进行三氟甲硫基化反应,得到了相应的α-三氟甲硫基酮类化合物(Eq. 4). 值得注意的是,反应过程中可能涉及到CF3自由基中间体. 该反应适用底物范围广,并且可以扩展至克级. α-溴代酮的芳环上,溴的间位或对位上带有硝基或氰基时,反应收率将减少,同时伴随有副产物三氟甲基醇的生成.
1.2 CF3S阳离子参与的三氟甲硫基化反应
PhNHSCF3是一个操作简单的,用于分子内直接引入CF3S基团的试剂. 2014年,吴劼课题组[31]报道了铋Ⅲ催化的PhNHSCF3与三甲基炔基硅烷之间发生的三氟甲硫基化反应(Eq. 5). 反应中,三氯化铋(BiCl3)的存在可以活化PhNHSCF3以提供CF3S阳离子,后者与三甲基炔基硅烷反应得到三氟甲硫基炔.
陈知远和李光明等[32]使用该试剂在BiCl3或TsOH的参与下,以1,2-二炔基苯为底物,选择性合成了三氟甲硫基苯并富烯类化合物(Scheme 5). 典型的苯并富烯衍生物Sulindac是一种抗炎药剂,拥有退热止痛功 效[33]. CF3S阳离子的存在将会促进亲电子的闭环作用,同时有利于氯化物或TsOH的亲核进攻.
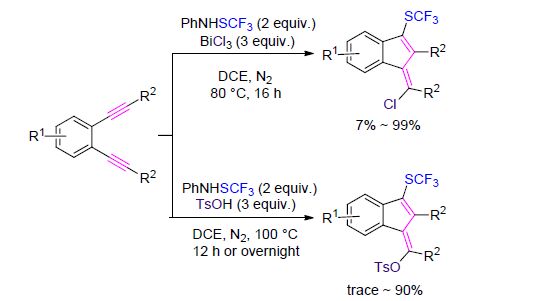 图 图式 5
1,2-二炔基苯与PhNHSCF3以及BiCl3或TsOH之间的反应
Figure 图式 5.
Reaction of 1,2-bis(alkynyl)benzene and PhNHSCF3 with BiCl3 or TsOH
图 图式 5
1,2-二炔基苯与PhNHSCF3以及BiCl3或TsOH之间的反应
Figure 图式 5.
Reaction of 1,2-bis(alkynyl)benzene and PhNHSCF3 with BiCl3 or TsOH
最近,Jereb课题组[34]报道了用PhNHSCF3在酸性条件下对酚类化合物进行亲电的三氟甲硫基化反应. 当底物羟基的对位未被取代时,该反应只得到对位CF3S取代的产物; 当对位存在取代基时,得到临位取代产物. 同时由于PhNHSCF3与烯烃可以发生反应,该课题 组[34]也进行了2-烯丙基苯酚与PhNHSCF3的反应结果得到CF3S连接在烯烃的端位CH2上的苯并呋喃类产物(Scheme 6).
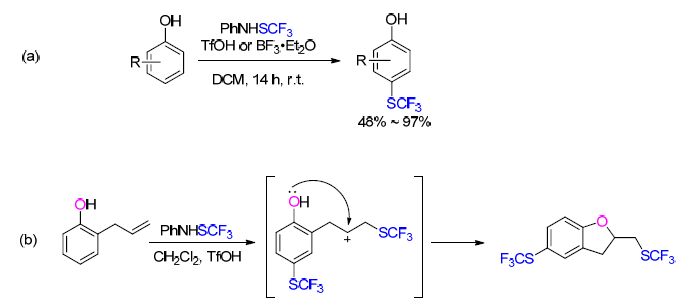 图 图式 6
酚的三氟甲硫基化和2-烯丙基苯酚的双官能团化
Figure 图式 6.
Trifluoromethylthiolation of phenols and the double functionalization of 2-allylphenol
图 图式 6
酚的三氟甲硫基化和2-烯丙基苯酚的双官能团化
Figure 图式 6.
Trifluoromethylthiolation of phenols and the double functionalization of 2-allylphenol
曹松课题组[35]也报道了以PhNHSCF3作为三氟甲硫基化试剂,在乙酰氯(CH3COCl)的存在下,对芳基酮、二烷基酮以及环酮直接三氟甲硫基化的方案(Scheme 7). 当芳基酮作底物时,芳环上存在吸电子基或给电子基时都可以得到良好的收率,但是连接吸电子基的底物比连接给电子基的底物得到的产物收率稍微低一些; 当二烷基铜做底物时,反应拥有较高的区域选择性,CF3S主要取代仲碳上的氢;当环酮做底物时,六元环酮可以得到单-三氟甲硫基化产物且有良好的收率,而五元环酮主要得到双-三氟甲硫基化产物.
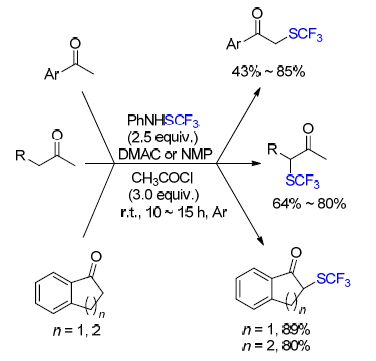 图 图式 7
各种酮类的三氟甲硫基化反应.
Figure 图式 7.
Trifluoromethylthiolation of various ketones
图 图式 7
各种酮类的三氟甲硫基化反应.
Figure 图式 7.
Trifluoromethylthiolation of various ketones
在天然产物和药物中,二氢呋喃作为一个重要的结构是由于其拥有卓越的生物活性[36]. 最近,刘雪原等[37]报道了一个合成4-三氟甲硫基-2,3-二氢呋喃类化合物的便利方法. 该反应方法是以高炔丙醇作为底物,与PhNHSCF3一起被对甲苯磺酸(p-TsOH)和氯化铋共活化后,通过进一步反应得到目标产物. 反应拥有高的区域选择性(Scheme 8). 该方法是底物中含有未被保护的羟基,且作为亲核体与分子内的叁键一起参与三氟甲硫基化/环化反应的首例报道.
2014年,Billard课题组[38]发现在与苯乙酮反应时,N-甲基-N-三氟甲硫基对甲苯磺酰胺[TsN(Me)SCF3]表现出的活性比N-甲基-N-三氟甲硫基苯胺[PhN(Me)- SCF3]更强. 并且TsN(Me)SCF3可以在20 g的规模下制备,拥有80%的收率. 于是该课题组试验了该试剂与一些结构简单的酮类在碱性条件下的反应活性. 虽然有好的反应活性,但是由于碱性条件,出现了一些 双-三氟甲硫基化的副产物.
随后该课题组[39]进一步优化反应条件,得出了酸性条件下酮的三氟甲硫基化反应(Scheme 9). 在该反应条件下,即使是更加复杂的酮类也有不错的收率. 与之前的报道[38]相比,该反应只得到单-三氟甲硫基化产物. 例外的是,3-乙酰吲哚只得到双-三氟甲硫基取代的化合物. 可能是其烯醇式存在较大的共轭体系大大增强了反应活性,即使是已经添加上一个三氟甲硫基,也可以进行进一步反应. 该反应的一个缺点是,底物中存在羟基会使反应效率下降,需要加入质子源(NH4Cl)来增强反应效率. 值得注意的是,之前报道[38]的碱性条件下不易被三氟甲硫基化的醛类,通过调节具体反应条件也可以得到不错的收率(Scheme 9). 但是该反应与羟醛缩合反应相互之间存在竞争.
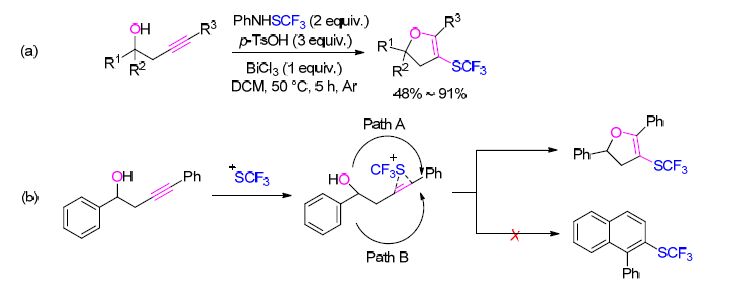 图 图式 8
4-三氟甲硫基-2,3-二氢呋喃类的合成和反应途径
Figure 图式 8.
Synthesis of 4-(trifluoromethyl)thio-2,3-dihydrofurans and its reaction pathway
图 图式 8
4-三氟甲硫基-2,3-二氢呋喃类的合成和反应途径
Figure 图式 8.
Synthesis of 4-(trifluoromethyl)thio-2,3-dihydrofurans and its reaction pathway
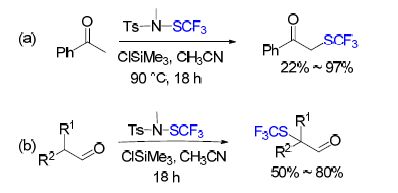 图 图式 9
酮和醛的α-三氟甲硫基化反应
Figure 图式 9.
α-Trifluoromethylthiolation of ketones and aldehydes
图 图式 9
酮和醛的α-三氟甲硫基化反应
Figure 图式 9.
α-Trifluoromethylthiolation of ketones and aldehydes
该课题组[40]又使用Knochel-Hauser碱对杂环化合物进行选择性去质子化,完成了对多种芳杂环化合物的选择性三氟甲硫基化(Scheme 10). 有趣的是,该反应允许合成的三氟甲硫代杂环化合物上带有溴原子. 然而,当使用Turbo格氏试剂(i-PrMgCl•LiCl)代替Knochel-Hauser碱时,3-溴吡啶环上的溴取代基被三氟甲硫基取代(Scheme 10).
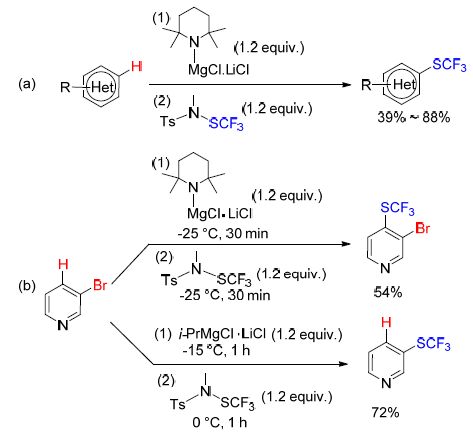 图 图式 10
芳杂环的选择性三氟甲硫基化反应
Figure 图式 10.
Selective trifluoromethylthiolation of heteroarenes
图 图式 10
芳杂环的选择性三氟甲硫基化反应
Figure 图式 10.
Selective trifluoromethylthiolation of heteroarenes
最近,Billard课题组[41]通过实验对比,认证了第二代三氟甲基次磺酰胺试剂[TsN(Me)SCF3]比第一代[PhN(R)SCF3]具有更强的亲电能力,但是PhN(R)SCF3存在一些特异性,在一些特定情况下不能被TsN(Me)SCF3轻易代替.
N-三氟甲硫基邻苯二甲酰亚胺结构上与N-溴代丁二酰亚胺(NBS)类似,是一个亲电子的三氟甲硫基化试剂. 首先由Munavalli等[42]在0 ℃条件下,用CF3SCl对邻苯二甲酰亚胺钾盐进行亲核取代制得. 同时该课题组试验了该试剂与烯胺以及活泼亚甲基化合物的反应. 该试剂与烯胺反应可以得到相应的α-三氟甲硫基酮; 而与活泼亚甲基化合物(乙酰乙酸乙酯、丙二酸二乙酯)反应时,没有观察到相应的三氟甲硫基化产物. 由于CF3SCl具有较高的毒性和腐蚀性,需要特殊的仪器和技术,因此限制了N-三氟甲硫基邻苯二甲酰亚胺的应用.
2014年,沈其龙课题组[43]报道了一个高收率的,制备N-三氟甲硫基邻苯二甲酰亚胺的新方法(Scheme 11). 同时报道了用该试剂作为三氟甲硫基化试剂,在铜催化条件下芳基/乙烯基硼酸的三氟甲硫基化反应(Scheme 11). 该方法可以在空气条件下进行,但是相对的收率会有所下降.
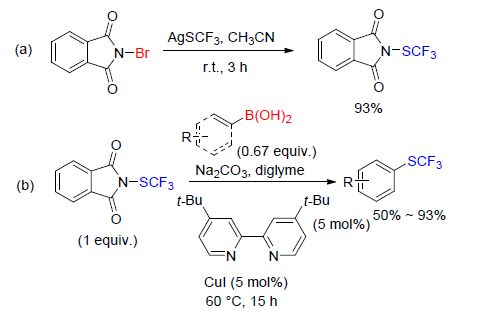 图 图式 11
N-三氟甲硫基邻苯二甲酰亚胺的合成以及芳基/乙烯基硼酸的三氟甲硫基化反应
Figure 图式 11.
Synthesis of N-(trifluoromethylthio)phthalimide and trifluoromethylthiolation of aryl/vinyl boronic acids
图 图式 11
N-三氟甲硫基邻苯二甲酰亚胺的合成以及芳基/乙烯基硼酸的三氟甲硫基化反应
Figure 图式 11.
Synthesis of N-(trifluoromethylthio)phthalimide and trifluoromethylthiolation of aryl/vinyl boronic acids
最近,Glorius等[44]报道了没有过渡金属参与的,直接三氟甲硫基化生物相关的N-杂环化合物(吡咯、吲哚)的反应(Scheme 12). 该反应以常见的NaCl作为催化剂,并且不需要保护基团的参与. 在该反应条件下,其它一些生物相关的N-杂环化合物,如7-氮杂吲哚、中氮茚衍生物和咪唑并[1,2-a]吡啶,也被成功地三氟甲硫基化,产率分别为94%、96%、86%. 该反应转化过程简单有力,有高的普适性. 因此该反应表现出了其在医药与农药研究开发领域内的应用潜能.
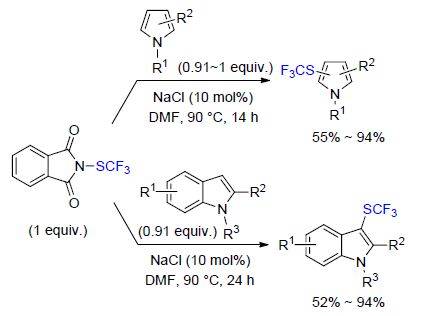 图 图式 12
N-杂环化合物的三氟甲硫基化反应
Figure 图式 12.
Trifluoromethylthiolation of N-heteroarenes
图 图式 12
N-杂环化合物的三氟甲硫基化反应
Figure 图式 12.
Trifluoromethylthiolation of N-heteroarenes
Besset课题组[45]报道了温和条件下钯催化的 C(sp3)—H键的官能团化反应,该反应在烷基酰胺的β-位形成C(sp3)—SCF3键(Scheme 13). 反应起初,PdⅡ-催化剂与双配位基的螯合物发生螯合作用得到配位中间体,随后该中间体通过协同的金属化-去质子化反应形成关键的环钯配合物. 然后经过氧化加成还原消除的途径得到相应产物(Scheme 13). 同时,该方法被用于合成含CF3S的生物活性分子类似物,如布洛芬、萘普生,收率分别为33%、23%,展示了该方法在药物合成中的应用潜能.
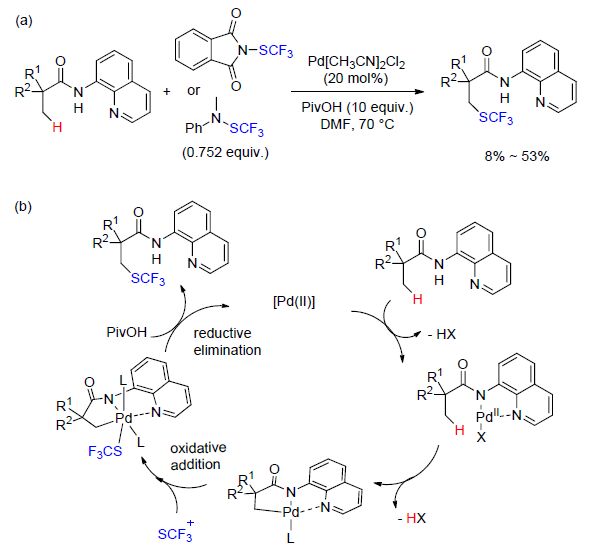 图 图式 13
PdⅡ催化的三氟甲硫基化反应和假设的反应机理
Figure 图式 13.
PdⅡ-catalyzed trifluoromethylthiolation and its proposed mechanism
图 图式 13
PdⅡ催化的三氟甲硫基化反应和假设的反应机理
Figure 图式 13.
PdⅡ-catalyzed trifluoromethylthiolation and its proposed mechanism
2013年,Shibata课题组[46]首次报道了三氟甲磺酰基高价碘叶立德试剂在催化剂CuCl存在下,可以与亲核体如烯胺、吲哚和
-酮酯反应得到三氟甲基硫醚化合物. 最近,该课题组[47]又报道了在CuF2催化下,用该试剂对烯丙基三甲基硅烷和三甲基硅基烯醚的三氟甲硫基化反应(Eq. 6). 值得注意的是,当三甲基硅基烯醚作为底物时,总是选择性的得到单-三氟甲硫基化产物. 而Billard课题组[38]之前报道的以TsN(Me)SCF3作为三氟甲硫基化试剂时,主要得到双-三氟甲硫基化产物. 最近,Zhang和Yi等[48]报道了基于Langlois试剂CF3SO2Na的C(sp2)—H键的直接三氟甲硫基化反应(Scheme 14). 该反应体系包含有一个还原剂(EtO)2P(O)H以及一个氧化剂二甲基亚砜(DMSO). CF3SO2Na可以在反应体系中生成CF3SSCF3,后者在CuCl催化下产生CF3S阳离子,直接用来对吲哚、吡咯以及烯胺类化合物进行亲电的三氟甲硫基化反应. 该反应方案经济适用并且操作简单,可用于最后一步进行三氟甲硫基化反应的方案. 同时,与该反应过程相似的有Vicic课题组[49]最近所报道的反应. 在后者的报道中,CF3SO2Na可以在反应体系中生成CuSCF3,从而发生芳基碘化物的亲核三氟甲硫基化反应.
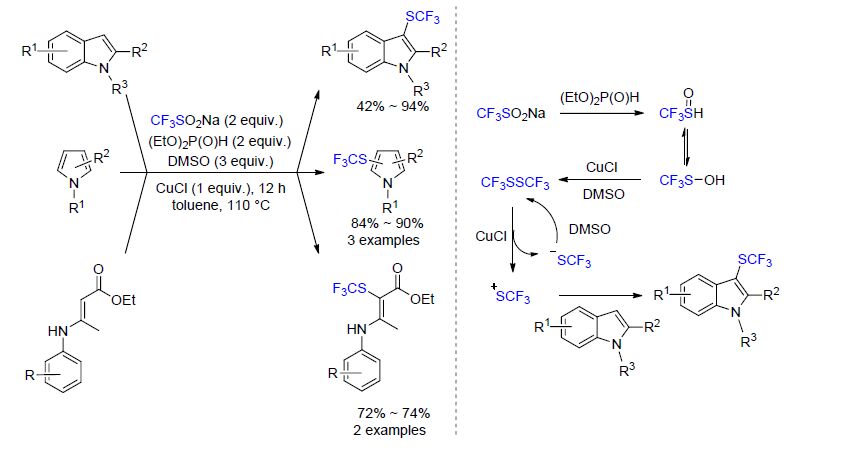 图 图式 14
CF3SO2Na与C(sp2)—H键之间的三氟甲硫基化反应及其可能的机理
Figure 图式 14.
Trifluoromethylthiolation of C(sp2)—H bonds with CF3SO2Na and its proposed mechanism
图 图式 14
CF3SO2Na与C(sp2)—H键之间的三氟甲硫基化反应及其可能的机理
Figure 图式 14.
Trifluoromethylthiolation of C(sp2)—H bonds with CF3SO2Na and its proposed mechanism
几年前,沈其龙和吕龙等[50]受到Togni试剂与芳基/乙烯基硼酸之间发生的三氟甲基化反应的启发,经过多次的尝试制得了一种新型的三氟甲硫基化试剂A[51],并且最终由Buchwald等[52]通过光谱等技术手段确认了该试剂为三氟甲基取代的硫代过氧化物(OSCF3),SCF3连接在氧原子上而不是碘原子上. 随后通过实验探究[53],沈其龙等[54]发现A苯环上的碘原子并不是必不可少的部分,从而得到了简化的三氟甲硫基化试剂B. 与A相比,B虽然活性降低了一些,但是更加便宜. 最近该课题组对先前工作进行了总结,概述了A和B分别与吲哚、芳基/乙烯基/烷基硼酸、烯烃、脂肪族羧酸、β-酮酸酯、氧化吲哚、炔烃之间的反应(Scheme 15). 值得注意的是,烷基羧酸的脱羧三氟甲硫基化反应以及烯烃的三氟甲硫基化反应可能涉及到烷基自由基历程.
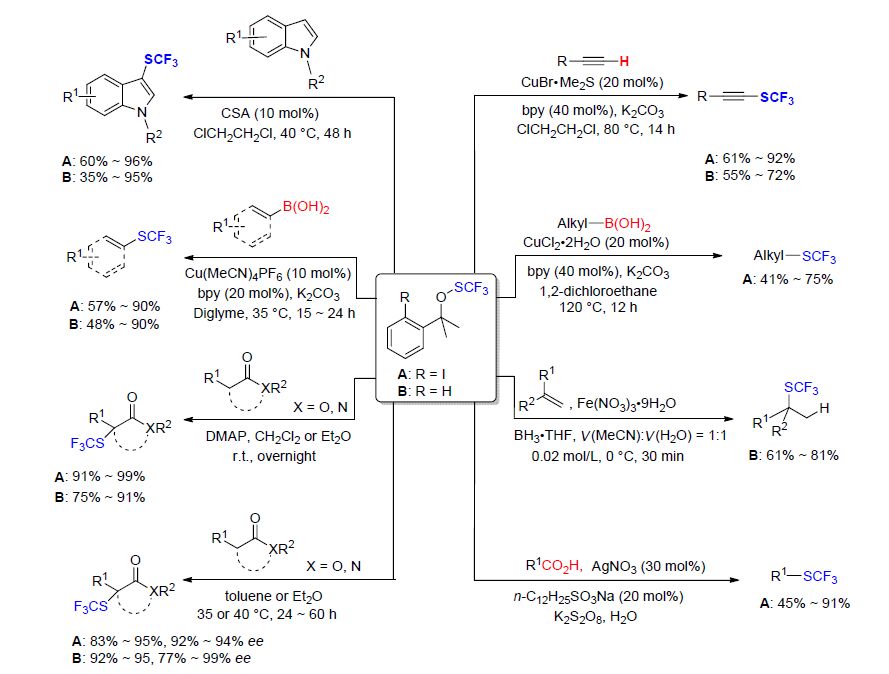 图 图式 15
三氟甲基取代的硫代过氧化物A/B与吲哚衍生物、硼酸、烯烃、脂肪族羧酸、β-酮酸酯以及炔烃之间的三氟甲硫基化反应
Figure 图式 15.
Trifluoromethylthiolation of indoles,boronic acids,alkenes,aliphatic carboxylic acids,β-keto esters and alkynes with the trifluoromethyl-substituted thioperoxide reagent A/B
图 图式 15
三氟甲基取代的硫代过氧化物A/B与吲哚衍生物、硼酸、烯烃、脂肪族羧酸、β-酮酸酯以及炔烃之间的三氟甲硫基化反应
Figure 图式 15.
Trifluoromethylthiolation of indoles,boronic acids,alkenes,aliphatic carboxylic acids,β-keto esters and alkynes with the trifluoromethyl-substituted thioperoxide reagent A/B
Dai课题组[55]又使用A对环丙醇进行了开环的亲电三氟甲硫基化反应,得到了β-三氟甲硫基的羰基化合物(Eq. 7). 由于羰基的存在,该反应产物可以容易地转化为其他含CF3S化合物. 虽然该反应机理尚不明确,但是该催化反应模式为探索C(sp3)—杂原子键的形成模式提出了新方向.
在沈其龙等的总结[54]中,同时涉及了基于N-溴代丁二酰亚胺(NBS)和糖精分子设计的新型三氟甲硫基化试剂——N-三氟甲硫基邻苯甲酰磺酰亚胺与各类化合物的反应. 该试剂可以在温和条件下与醇、胺、硫醇、末端炔烃、羰基化合物以及富电子的芳烃发生亲电的三氟甲硫基化反应,并且拥有良好的收率[56](Scheme 16).
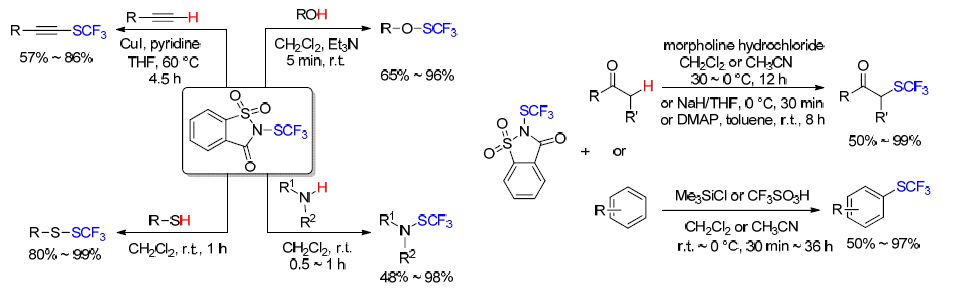 图 图式 16
N-三氟甲硫基糖精与各种亲核体之间发生的三氟甲硫基化反应
Figure 图式 16.
Trifluoromethylthiolation of various nucleophilic substrates with N-trifluoromethylthiosaccharin
图 图式 16
N-三氟甲硫基糖精与各种亲核体之间发生的三氟甲硫基化反应
Figure 图式 16.
Trifluoromethylthiolation of various nucleophilic substrates with N-trifluoromethylthiosaccharin
随后沈其龙课题组[57]又使用该试剂在路易斯酸催化下进行了烯烃的三氟甲硫内酯化/内酰胺化反应. 该反应过程经过一个Thiiranium离子中间体,该中间体随后被羧酸或者酰胺进攻生成目标产物(Eq. 8).
N-三氟甲硫基糖精良好的反应活性同样引起了其他课题组的研究兴趣. 例如Li课题组[58]最近报道了该试剂在铑Ⅲ催化下与吲哚发生的反应,C—S键选择性的出现在2-位上(Eq. 9). Cahard课题组[59]也报道了用烯丙基醇和该试剂的反应(Eq. 10). 该反应首先得到的是三氟甲基取代的硫代过氧化物中间体OSCF3,随后该中间体经过[2, 3]-σ迁移重排得到三氟甲基亚砜类化合物.
1.3 CF3S阴离子参与的三氟甲硫基化反应
最近,杨春皓和邓廉夫等[60]成功地使用AgSCF3作为三氟甲硫基化试剂,完成了α-卤代酮和缺电子的苄基卤化物的三氟甲硫基化反应(Scheme 17). AgSCF3和KI在反应过程中生成活泼但对水分和空气不敏感的CF3S阴离子,后者可以作为亲核体直接进行下一步反应. 另外,该反应条件温和,并且在一些情况下拥有近乎定量的收率(>99%). 值得注意的是,富电子的苄基卤化物如三甲基苄溴无法提供相应的产物.
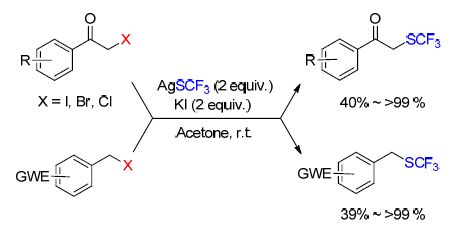 图 图式 17
AgSCF3与α-卤代酮以及缺电子的苄基卤化物之间的三氟甲硫基化反应
Figure 图式 17.
Trifluoromethylthiolation of α-haloketones and electron-deficient benzyl halides with AgSCF3
图 图式 17
AgSCF3与α-卤代酮以及缺电子的苄基卤化物之间的三氟甲硫基化反应
Figure 图式 17.
Trifluoromethylthiolation of α-haloketones and electron-deficient benzyl halides with AgSCF3
催化剂在定义上是反应过程中没有消耗,但是实际操作中的障碍依然会造成损失. 最近,Schoenebeck课题组[61]报道了一个直接三氟甲硫基化芳基碘化物和芳基溴化物的反应方法,拥有高效率并且操作简单. 该方法以双核PdⅠ作为催化剂,易获取的[Me4N]SCF3作为三氟甲硫基化试剂(Scheme 18). PdⅠ催化剂对空气、水分以及热表现出好的稳定性,使其可以在实验室的开放环境下用普通的柱色谱法进行回收,因此该方法与其他使用敏感的Pd0和Ni0作催化剂的方法相比,拥有相当大的实践优势.
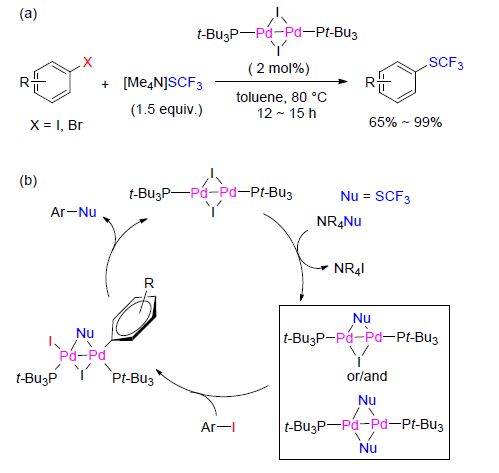 图 图式 18
PdⅠ催化的芳基碘化物/溴化物与[Me4N]SCF3之间的三氟甲硫基化反应以及其可能的机理
Figure 图式 18.
PdⅠ-catalyzed trifluoromethylthiolation of aryl iodides/bromides with [Me4N]SCF3 and its proposed mechanism
图 图式 18
PdⅠ催化的芳基碘化物/溴化物与[Me4N]SCF3之间的三氟甲硫基化反应以及其可能的机理
Figure 图式 18.
PdⅠ-catalyzed trifluoromethylthiolation of aryl iodides/bromides with [Me4N]SCF3 and its proposed mechanism
2014年钟伟课题组[62]报道了使用易获取的试剂Na2S2O3和CF3SiMe3,在铜和邻二氮菲催化下,对芳基碘化物或烷基碘化物进行三氟甲硫基化反应(Eq. 11). 该反应分两步进行,S-芳基或S-烷基硫代硫酸酯是关键中间体. 大的空间位阻会影响反应效率,如邻叔丁基碘苯,不发生该反应.
铜-SCF3复合物[49]在很多情况下是一种引人注目的试剂,可以将芳基卤化物转化成相应的芳基三氟甲基硫醚(AgSCF3). CuSCF3是一个典型的三氟甲硫基化试剂,能与多种亲电试剂反应[14c]. 但是传统制备CuSCF3的方法存在一些缺点,如缺乏效率或者使用昂贵的AgSCF3和具有毒性的CF3SSCF3作为起始底物[49].
近日,Vicic课题组[49]报道了一个合成CuSCF3的实用方法. 该方法通过一个三苯基膦作为还原剂参与的脱氧还原过程,使用Langlois试剂CF3SO2Na生成CuSCF3. 后者可以与一系列的配体(L)发生反应,得到一些在空气中稳定的[LCu(SCF3)]复合物,这些复合物可作为有价值的三氟甲硫基化试剂. 另外,反应中生成的CuSCF3可以与芳基碘化物在1,3-二甲基-2-咪唑啉酮(DMI)溶剂中直接反应,得到一系列芳基三氟甲基硫醚. 该反应高效(74%~93%)且操作简单,多种官能团可以适用于该反应条件(Scheme 19).
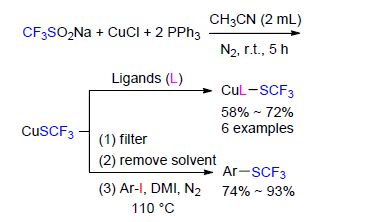 图 图式 19
CuSCF3的合成以及其应用
Figure 图式 19.
Synthesis of CuSCF3 and its application
图 图式 19
CuSCF3的合成以及其应用
Figure 图式 19.
Synthesis of CuSCF3 and its application
2013年,翁志强课题组[63]用CuF2和MeSiCF3以及S8与2,2'-联吡啶配位,成功制备了2,2'-联吡啶三氟甲硫铜金属络合物[(bpy)Cu(SCF3)]. 该络合物以固态存在于空气中可以在几天内保持稳定,以液态存在于空气中也可以在几个小时内没有变化. 该络合物成功被用于芳香(杂)环卤化物[63]、苄基溴[64]、卤代烷[65]的三氟甲硫基化反应. 最近,通过对前期工作[30] 的进一步研究,该课题组[66]报道了使用[(bpy)Cu(SCF3)]合成α-三氟甲硫基酮的反应,在该反应条件下,即使底物芳环上连有硝基或氰基,反应仍然能够得到良好的收率(77%~83%)(Scheme 20).
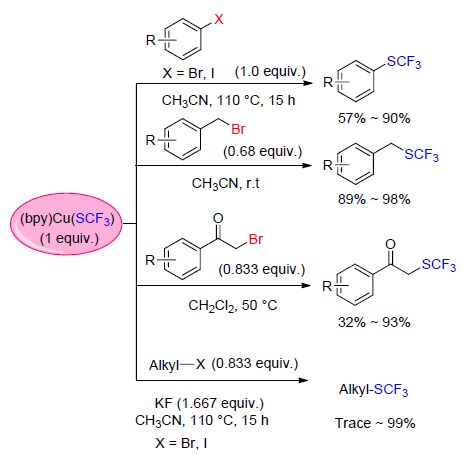 图 图式 20
(bpy)Cu(SCF3)与芳基碘化物/溴化物、苄基溴化物、酮以及卤代烷之间发生的三氟甲硫基化反应
Figure 图式 20.
Trifluoromethylthiolation of aryl bromides/ iodides,benzyl bromides,ketones and haloalkanes with (bpy)Cu(SCF3)
图 图式 20
(bpy)Cu(SCF3)与芳基碘化物/溴化物、苄基溴化物、酮以及卤代烷之间发生的三氟甲硫基化反应
Figure 图式 20.
Trifluoromethylthiolation of aryl bromides/ iodides,benzyl bromides,ketones and haloalkanes with (bpy)Cu(SCF3)
随后,α-三氟甲硫基-α,β-不饱和羰基化合物的合 成[67]、β-三氟甲硫基-α,β-不饱和羰基化合物的合成[68],以及乙烯基三氟甲基硫酯的合成[69]也相继被报道(Scheme 21). 值得注意的是,α-三氟甲硫基-α,β-不饱和羰基化合物的合成以及乙烯基三氟甲基硫酯的合成都经历了氧化加成-还原消除的反应过程,而β-三氟甲硫基-α,β-不饱和羰基化合物的合成则是经历了1,4-加成-消除的反应过程.
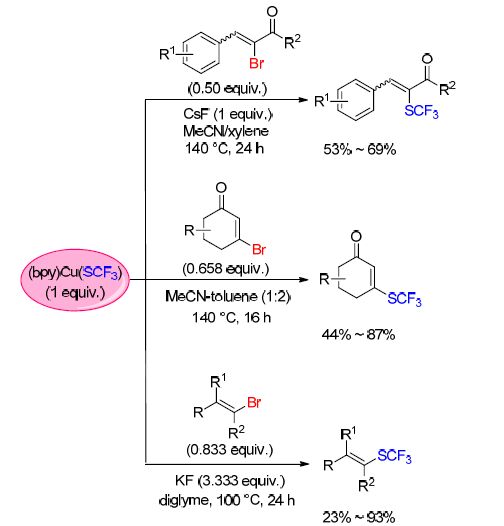 图 图式 21
α,β-不饱和羰基化合物以及乙烯基三氟甲基硫酯的合成
Figure 图式 21.
Synthesis of α,β-unsaturated carbonyl compounds and vinyl trifluoromethyl thioethers
图 图式 21
α,β-不饱和羰基化合物以及乙烯基三氟甲基硫酯的合成
Figure 图式 21.
Synthesis of α,β-unsaturated carbonyl compounds and vinyl trifluoromethyl thioethers
基于之前的研究,翁志强课题组[70]考虑到膦配体作为强力的σ-供体以及在金属参与反应中的广泛适用性,用CuF2和MeSiCF3以及S8在三苯基膦的存在下成功地合成了三苯基膦三氟甲硫铜络合物[(PPh3)2Cu- (SCF3)]. 同时,用该络合物与烯丙基溴化物反应,得到了烯丙位CF3S取代的产物,并且拥有良好的收率(Eq. 12).
最近,曾步兵等[71]报道了在铜参与的情况下,使用氟化钾、单质硫和三氟甲基三甲基硅烷(TMSCF3),在无水二甲基甲酰胺(DMF)中对α-溴代酮进行三氟甲硫基化反应. 反应起初可能是TMSCF3在KF,S8和DMF存在下生成CF3S负离子,随后CF3S负离子与铜盐反应得到一个铜Ⅰ-复合物中间体. 该中间体与底物反应得到一个铜Ⅲ-复合物中间体,随后进一步转化得到目标产物(Scheme 22). 该反应条件温和且不需要任何配体参与,反应所需的试剂都是便宜且无毒的.
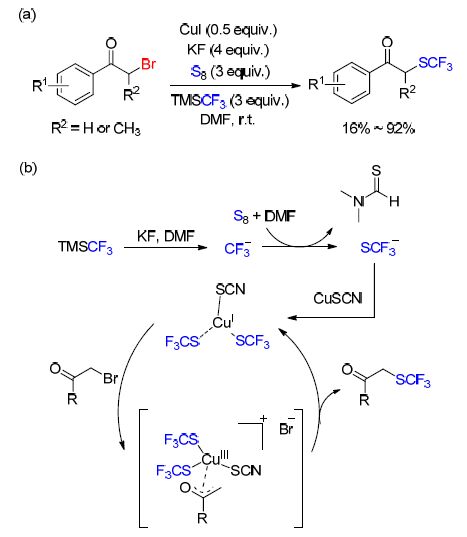 图 图式 22
α-溴代酮的三氟甲硫基化反应及其可能的机理
Figure 图式 22.
Trifluoromethylthiolation of α-bromoketones and its proposed mechanism
图 图式 22
α-溴代酮的三氟甲硫基化反应及其可能的机理
Figure 图式 22.
Trifluoromethylthiolation of α-bromoketones and its proposed mechanism
Liang课题组[72]首次报道了在单质硫和其他氟离子存在下,用反应过程中生成的二氟卡宾与烷基亲电体进行的三氟甲硫基化反应(Scheme 23). 该反应用到二氟亚甲基磷酸酯甜菜碱(Ph3P+CF2CO2-,PDFA)作为卡宾试剂. PDFA可以在没有其他添加剂的中性条件下脱羧产生Ph3P+CF2-,随后生成二氟卡宾. 后者直接与氟离子以及单质硫反应产生CF3S负离子,与烷基亲电体反应得到目标产物[73]. 该反应拥有良好的普适性,并且过渡态金属对于该反应不是必不可少的.
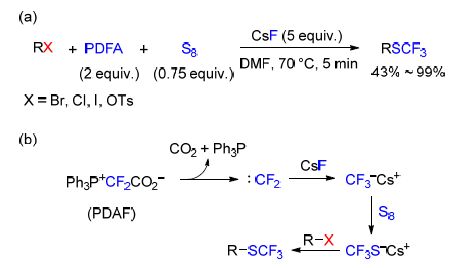 图 图式 23
烷基亲电体的三氟甲硫基化反应及其可能的机理
Figure 图式 23.
Trifluoromethylthiolation of alkyl electrophiles and its proposed mechanism
图 图式 23
烷基亲电体的三氟甲硫基化反应及其可能的机理
Figure 图式 23.
Trifluoromethylthiolation of alkyl electrophiles and its proposed mechanism
2013年,Zard和Li[74]首次提出了O-十八烷基-S-三氟甲基碳酸酯作为三氟甲硫基化试剂,在氟化钾和吡咯的存在下,对芦竹碱和α-溴取代的酮类化合物表现出很好的活性. 最近,施敏和魏音等[75]报道了在胺催化条件下,Morita-Baylis-Hillman (MBH)碳酸酯和O-十八烷 基-S-三氟甲基碳酸酯之间可以发生亲核三氟甲硫基化反应(Scheme 24). 该反应的区域选择性可以通过选择不同的试剂进行控制,当四氢呋喃(THF)作为溶剂时,伯碳型的 烯丙位上连有CF3S基团的化合物为主要产物; 当CHCl3为溶剂时,仲碳型的烯丙位上连有CF3S基团的化合物为主要产物. 在反应过程中,1,4-二氮杂二环[2.2.2]辛烷(DABCO)起到一个双重角色,既活化O-十八烷基-S-三氟甲基碳酸酯,又活化MBH碳酸酯.
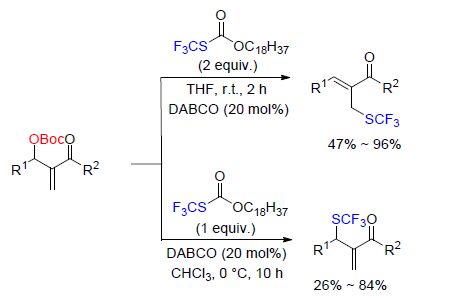 图 图式 24
溶剂控制的MBH碳酸酯与O-十八烷基-S-三氟甲基碳酸酯之间发生的三氟甲硫基化反应
Figure 图式 24.
Trifluoromethylthiolation of MBH carbonates with O-octadecyl-S-trifluoromethylcarbonate controlled by solvent
图 图式 24
溶剂控制的MBH碳酸酯与O-十八烷基-S-三氟甲基碳酸酯之间发生的三氟甲硫基化反应
Figure 图式 24.
Trifluoromethylthiolation of MBH carbonates with O-octadecyl-S-trifluoromethylcarbonate controlled by solvent
2 直接三氟甲硒基化反应
在农药和医药分子研究开发中,引入三氟甲硒基团(CF3Se)是一个有前途的研究领域. CF3Se与三氟甲硫基(CF3S)拥有相似的性质(Hammet和Taft参数,亲核性),可以控制分子的膜通透性和生物利用度[76]. 虽然过量的硒对人体具有毒性[77],但是低剂量的硒对人类和其他的生命系统是一种必需营养素[76]. 另外,许多有机硒衍生物表现出了显著的抗氧化、抗癌、抗菌以及抗病毒性能[78],体现出了三氟甲硒基化反应的研究价值,
最近,Schoenebeck课题组[76]报道了一个高效的钯催化的三氟甲硒基化反应. 该反应以催化手段对芳基碘化物进行三氟甲硒基化,得到相应的芳香族三氟甲硒基化产物(Eq. 13). 所需的催化剂是热稳定的双核PdⅠ,同时该催化剂对空气,水也表现出较好的稳定性. 该反应操作方法简单,有较好的普适性,但是所需的芳基碘化物较为昂贵.
Goossen课题组[29]在报道芳环以及芳杂环重氮盐的三氟甲硫基化反应的同时,也报道了同种底物的三氟甲硒基化反应(Eq. 14). 该反应在催化量的硫氰酸亚铜存在下,[Me4N]SeCF3作为三氟甲硒基化试剂,在室温下进行. 该反应适用于一系列功能化的分子并且可以与重氮化反应结合以“一锅煮”的方法进行试验.
最近,Rueping课题组[79]以[Me4N]SeCF3为三氟甲硒基化试剂,进行了硼酸、硼酸频哪醇酯以及末端炔烃的氧化三氟甲硒基化反应(Scheme 25). 该方法反应条件温和,可以在室温下空气中进行,同时该反应拥有较为灵活的溶剂选择,并且拥有较好的普适性. [Me4N]SeCF3可以由红硒和TMSCF3合成. 该反应是第一例炔类的三氟甲硒基化反应,不仅适用于芳香族的炔烃,脂肪族的炔烃也同样适用. 硼酸和硼酸频哪醇酯与[Me4N]SeCF3反应时,得到的反应结果相似,且富电子和缺电子的芳香族硼酸及硼酸频哪醇酯都有较好的收率.
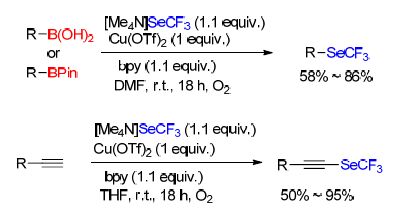 图 图式 25
[Me4N]SeCF3与硼酸、硼酸频哪醇酯以及末端炔烃之间发生的三氟甲硒基化反应
Figure 图式 25.
Trifluoromethylselenolation of boronic acids,boronic pinacol esters and terminal alkynes with [Me4N]SeCF3
图 图式 25
[Me4N]SeCF3与硼酸、硼酸频哪醇酯以及末端炔烃之间发生的三氟甲硒基化反应
Figure 图式 25.
Trifluoromethylselenolation of boronic acids,boronic pinacol esters and terminal alkynes with [Me4N]SeCF3
2014年,翁志强课题组[80]报道了在铜Ⅰ催化下,芳基和烷基卤化物的三氟甲硒基化反应(Scheme 26). 该反应经过一个关键的中间体[(phen)Cu(SeCF3)]2, 并且该中间体被成功分离出来. 该反应体系中,银可以将CF3Se基团转移至铜,促进了中间体[(phen)Cu(SeCF3)]2的生成,从而对反应的进行起到了重要的推动作用. 另外,该催化体系可以与大范围的官能团相适应.
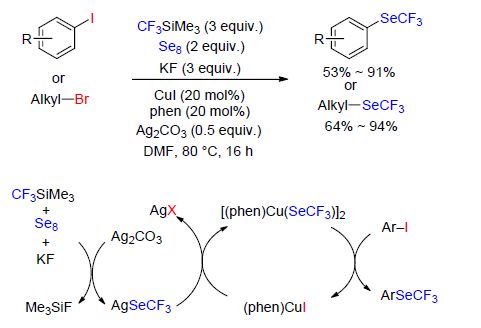 图 图式 26
芳基碘化物和烷基溴化物的三氟甲硒基化反应及其可能的机理
Figure 图式 26.
Trifluoromethylselenolation of aryl iodides,alkyl bromides and its proposed reaction pathway
图 图式 26
芳基碘化物和烷基溴化物的三氟甲硒基化反应及其可能的机理
Figure 图式 26.
Trifluoromethylselenolation of aryl iodides,alkyl bromides and its proposed reaction pathway
同时,翁志强课题组[81]又成功合成了上述中间体[(phen)Cu(SeCF3)]2。该中间体是一种高效率的亲核三氟甲硒基化试剂,可以由CuI、Me3SiCF3、单质硒和KF以及联二吡啶在室温下合成,并且可以在空气中稳定数月. 但是在四甲基哌啶氮氧化物(TEMPO)存在下易于分解[82]. 该课题组[81]试验了[(bpy)Cu(SeCF3)]2与卤代芳烃和卤代烷烃之间的反应(Scheme 27). 结果显示,大范围官能团化的卤代芳烃和卤代烷烃可以与该试剂反应,并且得到中等到优越收率的相应三氟甲硒基化产物.
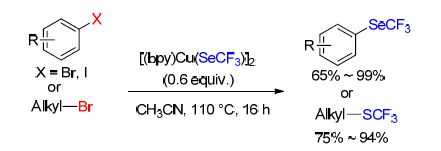 图 图式 27
[(bpy)Cu(SeCF3)]2与卤代芳烃和卤代烷烃之间发生的三氟甲硒基化反应
Figure 图式 27.
Trifluoromethylselenolation of aryl halides and haloalkanes with [(bpy)Cu(SeCF3)]2
图 图式 27
[(bpy)Cu(SeCF3)]2与卤代芳烃和卤代烷烃之间发生的三氟甲硒基化反应
Figure 图式 27.
Trifluoromethylselenolation of aryl halides and haloalkanes with [(bpy)Cu(SeCF3)]2
同年,炔丙基三氟甲基硒醚以及烯丙基三氟甲基硒醚的合成也被报道[82a](Scheme 28),反应可能经过氧化加成-还原消除历程,并且拥有良好的普适性以及良好的收率. 试验表明,该反应方法可以扩展至克级. 随后,该课题组[67, 68]报道了α,β-不饱和羰基化合物的三氟甲硒基化反应(Scheme 28). 该反应同样经历了氧化加成-还原消除历程,且产物主要为E-同分异构体. 反应产物收率比相应的α,β-不饱和羰基化合物的三氟甲硫基化反应收率高,可能是因为CF3Se——拥有更高的亲核能力.
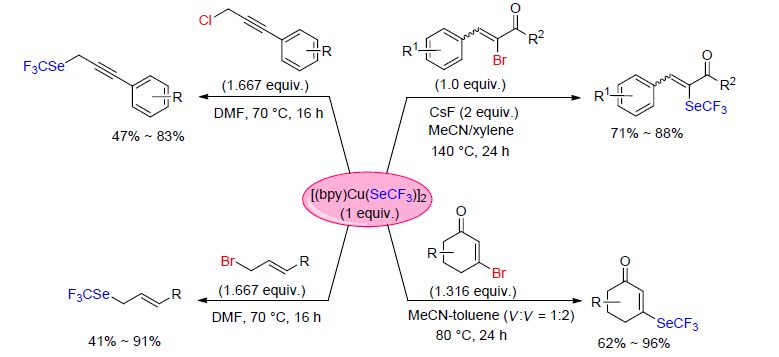 图 图式 28
炔丙基/烯丙基三氟甲基硒醚以及三氟甲硒基取代的α,β-不饱和羰基化合物的合成
Figure 图式 28.
Synthesis of propargylic/allylic trifluoromethyl selenoethers and trifluoromethylseleno-substituted α,β-unsaturated carbonyl compounds
图 图式 28
炔丙基/烯丙基三氟甲基硒醚以及三氟甲硒基取代的α,β-不饱和羰基化合物的合成
Figure 图式 28.
Synthesis of propargylic/allylic trifluoromethyl selenoethers and trifluoromethylseleno-substituted α,β-unsaturated carbonyl compounds
最近,翁志强课题组[83]又报道了在铜参与的情况下,端基炔的氧化三氟甲硒基化反应. 反应需要加入氧化剂戴斯-马丁试剂(DMP),在KF的存在下,DMP与[(bpy)Cu(SCF3)]2反应形成中间体. 随后该中间体与端基炔反应得到相应的产物(Scheme 29). 反应底物芳环上的电子效应对反应的结果没有显著的影响,并且底物的芳环上可以存在卤素,如氟原子、氯原子以及溴原子. 该反应不仅适用于芳香族炔烃,脂肪族炔烃同样适用. 乙烯基三氟甲基硒醚的合成也同样被报道[84]. 在铜参与的温和条件下,乙烯基卤化物发生三氟甲硒基化反应(Scheme 29). 该反应经过氧化加成-还原消除过程. 值得注意的是,该方法同时适用于富电子和缺电子的乙烯基卤化物. 并且产物乙烯基三氟甲基硒醚的E/Z立体结构与起始底物相对应.
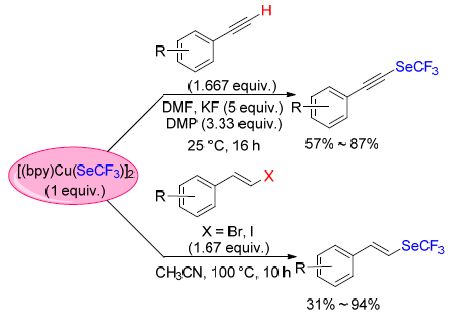 图 图式 29
[(bpy)Cu(SCF3)]2与末端炔烃和乙烯基卤化物之间发生的三氟甲硒基化反应
Figure 图式 29.
Trifluoromethylselenolation of terminal alkynes and vinyl halide with [(bpy)Cu(SCF3)]2
图 图式 29
[(bpy)Cu(SCF3)]2与末端炔烃和乙烯基卤化物之间发生的三氟甲硒基化反应
Figure 图式 29.
Trifluoromethylselenolation of terminal alkynes and vinyl halide with [(bpy)Cu(SCF3)]2
3 直接三氟甲氧基化反应
三氟甲氧基(CF3O)拥有与三氟甲硫基(CF3S)的相近的亲脂性(πx=1.04)[85],高的亲脂性增强了含CF3O化合物穿越脂质细胞膜的能力,提高了其生物利用度. 因此含CF3O化合物在农药和医药研发领域也有很高应用价值.
最近,翁志强课题组报道[85]了含银配合物(Aryl-BIAN)Ag(OCF3)•THF的制备过程(Scheme 30). 该配合物由AgOCF3的乙腈溶液与1 equiv. Aryl-BIAN在THF中合成,是一个对空气轻微敏感的固体. 当在室温条件下,该配合物再次溶解于THF中时开始分解. 同时,翁志强等试验了该配合物与有机卤化物之间的反应,结果显示,该配合物可以与苄基溴平稳的反应,并且可以得到较好收率的苄基三氟甲基醚(Scheme 30). 一系列的官能团如烷基、甲氧基、硝基卤素等可以适用于该反应条件,苄基氟是主要的副产物. 另外,苄基氯在该反应条件下没有相应的产物被观察到.
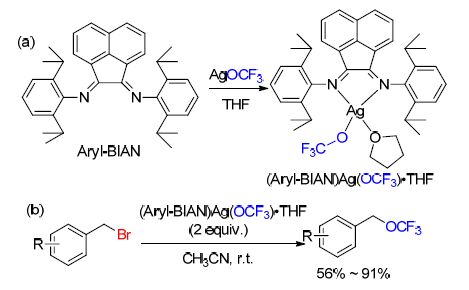 图 图式 30
(Aryl-BIAN)Ag(OCF3)•THF的制备及其与苄基溴的反应
Figure 图式 30.
Preparation of the (Aryl-BIAN)Ag(OCF3)•THF and its reaction with benzyl bromides
图 图式 30
(Aryl-BIAN)Ag(OCF3)•THF的制备及其与苄基溴的反应
Figure 图式 30.
Preparation of the (Aryl-BIAN)Ag(OCF3)•THF and its reaction with benzyl bromides
2014年,Ngai课题组[86]首次报道了N-芳基-N-三氟甲氧基胺类化合物的合成,以及其在合成邻三氟甲氧基苯胺中的应用(Scheme 31). 首先,被保护的N-芳基-N-羟胺与Togni试剂反应,生成N—O—CF3结构; 其次,在MeNO2溶液中,CF3O通过N—O键异裂迁移到被 保护的氨基的邻位,从而得到邻三氟甲氧基苯胺. 该反应能够扩展至克级并且有好的普适性. 同时富电子的底物能够得到较高的收率,缺电子的底物则多数可以得到较好的收率,个别存在例外. 随后,该课题组[86]通过进一步研究,将两步反应整合成“一锅煮”的反应方法,从而简化了反应方案.
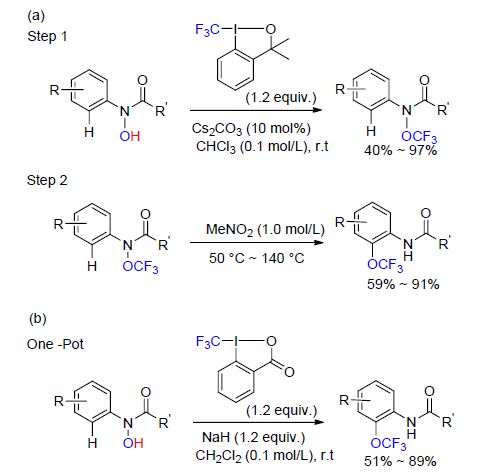 图 图式 31
邻三氟甲氧基苯胺衍生物的合成
Figure 图式 31.
Synthesis of ortho-Trifluoromethoxylated aniline derivatives
图 图式 31
邻三氟甲氧基苯胺衍生物的合成
Figure 图式 31.
Synthesis of ortho-Trifluoromethoxylated aniline derivatives
最近,Ngai课题组[87]又报道了用Togni试剂对官能团化的吡啶和嘧啶进行区域选择性三氟甲氧基化的反应(Scheme 32). 该反应首先进行的是氨羟基的三氟甲基化形成O—CF3键,随后OCF3迁移到吡啶或嘧啶的α'位. 该反应方法操作简单反应条件温和,适用于多种官能团的存在. 当吡啶或嘧啶环上羟胺基的对位有给电子基团时有利于反应进行.
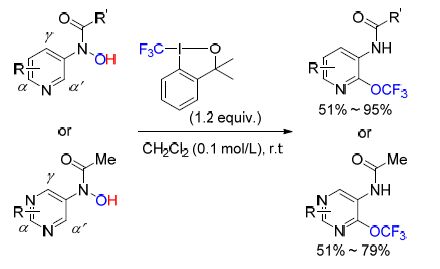 图 图式 32
官能团化的吡啶和嘧啶的区域选择性三氟甲氧基化反应
Figure 图式 32.
Regioselective trifluoromethoxylation of functionalized pyridines and pyrimidines
图 图式 32
官能团化的吡啶和嘧啶的区域选择性三氟甲氧基化反应
Figure 图式 32.
Regioselective trifluoromethoxylation of functionalized pyridines and pyrimidines
4 直接三氟乙氧基化反应
三氟乙氧基(CF3CH2O)拥有极好的稳定性、高的电负性以及亲脂性[88]. 这些特性使其如上述的含氟基团一样,可以用来提高分子的生物利用度和代谢稳定性,体现出了该基团的研究价值.
2013年,赵康和杜云飞等[89]提出一系列的烯胺酮以及烯胺羧酸酯与PhIO在三氟乙醇中反应时,可以转换成三氟乙氧代的2H-氮丙啶类化合物(Scheme 33). 该串联反应可能首先是PhIO参与的氧化三氟乙氧基化反应,随后发生α-三氟乙氧基烯胺中间体的氮丙啶化反应.
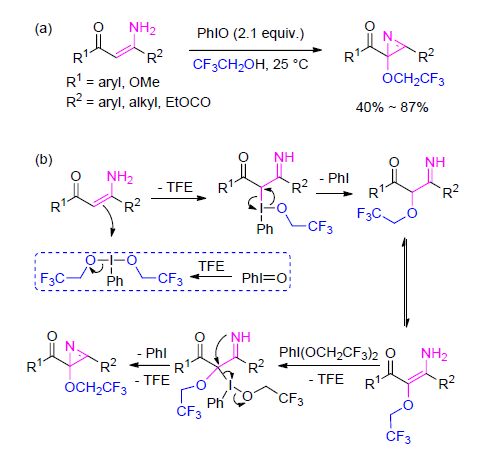 图 图式 33
2H-氮丙啶的合成以及其可能的机理
Figure 图式 33.
Synthesis of 2H-azirines and its proposed mechanism
图 图式 33
2H-氮丙啶的合成以及其可能的机理
Figure 图式 33.
Synthesis of 2H-azirines and its proposed mechanism
最近,翁志强课题组在之前[(bpy)Cu(SCF3)][63]以及[(bpy)Cu(SeCF3)]2[81]的研究基础上,继续探索并报 道[88, 90]了铜Ⅰ-氟化醇盐复合物[(phen)2Cu][OCH2CF3]的合成,同时展示了该试剂在芳基和杂芳基溴化物的三氟乙氧基化反应中的应用(Scheme 34). 该试剂由CuOBu-t和邻二氮菲在THF中反应,随后加入三氟乙醇而制得. 底物芳环上连接有吸电子基或给电子基与该试剂反应皆可得到较好的收率. 通过调节所使用的含氟醇可以得到另外两个氟化醇盐复合物[(phen)2Cu][OCH2CF2CF3]和[(phen)2Cu][OCH2CF2CF2H],这两个试剂分别可以用于芳基和杂芳基溴化物的五氟丙氧基化反应以及四氟丙氧基化反应.
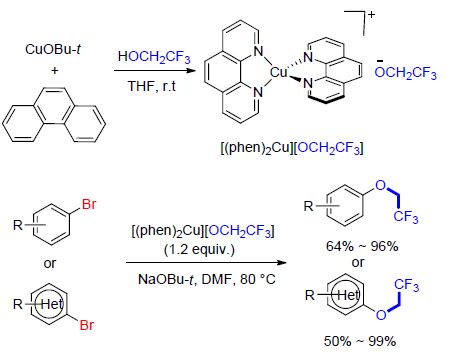 图 图式 34
[(phen)2Cu][OCH2CF3]的合成及其与芳基和杂芳基溴化物的反应
Figure 图式 34.
Synthesis of [(phen)2Cu][OCH2CF3] and its reaction with aryl and heteroaryl bromides
图 图式 34
[(phen)2Cu][OCH2CF3]的合成及其与芳基和杂芳基溴化物的反应
Figure 图式 34.
Synthesis of [(phen)2Cu][OCH2CF3] and its reaction with aryl and heteroaryl bromides
随后,该课题组[91]又报道了三氟乙氧基化α-溴代酮的反应(Scheme 35). 该反应以三氟乙醇为三氟乙氧基化试剂,在Cs2CO3参与下直接引入三氟乙氧基. 当反应底物为缺电子体系时,反应效率降低并且伴随有未识别的副产物. 值得注意的是,该反应可以扩展至克级,并且反应产物α-三氟乙氧基酮可以进一步合成三氟乙氧代醇和三氟乙氧代吡唑(Scheme 35),从而扩大了该反应的应用前景.
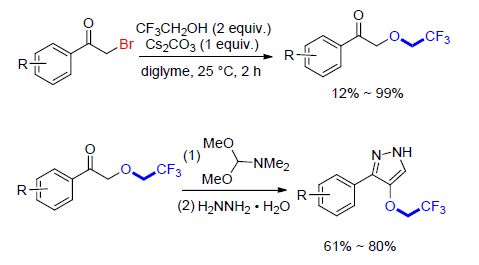 图 图式 35
α-溴代酮的三氟乙氧基化反应以及三氟乙氧基取代的吡唑的合成
Figure 图式 35.
Trifluoroethoxylation of α-bromoketones and the synthesis of trifluoroethoxy-substituted pyrazole
图 图式 35
α-溴代酮的三氟乙氧基化反应以及三氟乙氧基取代的吡唑的合成
Figure 图式 35.
Trifluoroethoxylation of α-bromoketones and the synthesis of trifluoroethoxy-substituted pyrazole
在钯催化下,芳基溴代物以及溴代查尔酮可以与三氟乙醇进行三氟乙氧基化反应得到CF3CF2O基团连接在芳环上的产物[92](Scheme 36). 该反应用到的Pd/t-BuXPhos或者Pd/JohnPhos配体催化体系可以促进三氟乙醇与芳基溴化物以及溴代查尔酮的交叉偶合反应,但是对富电子的芳香体系没有作用.
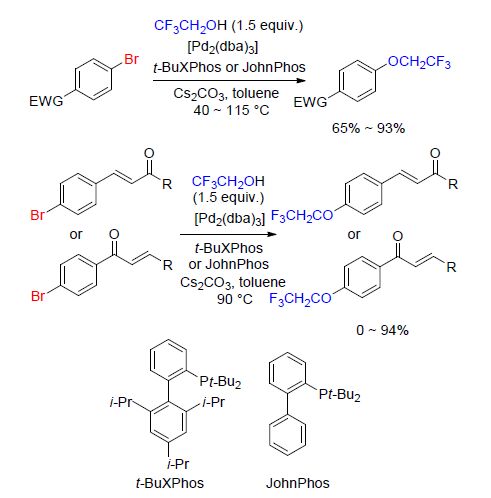 图 图式 36
钯催化的三氟乙醇与芳基溴代物以及溴代查尔酮之间的三氟乙氧基化反应
Figure 图式 36.
Palladium-catalyzed trifluoroethoxylation of aryl bromides and bromo-chalcones with trifluoroethanol
图 图式 36
钯催化的三氟乙醇与芳基溴代物以及溴代查尔酮之间的三氟乙氧基化反应
Figure 图式 36.
Palladium-catalyzed trifluoroethoxylation of aryl bromides and bromo-chalcones with trifluoroethanol
5 18F参与的放射性标记
正电子发射断层显像(PET)是一种核医学成像技术,用来提供放射示踪剂在体内的生物分布图像[93]. 18F拥有几乎理想的原子核性能,包括高的正电子衰变率(β+,97%)、低的正电子能量(0.635 MeV)所导致的有限的正电子迁移距离(<2 mm)以及适当的半衰期(109.7 min),因此18F是临床成像使用最广泛的PET放射性核素[94]. 鉴于此,新型18F标记的放射示踪剂的研发成为了很多工作者的研究热点,其中的关键在于用18F对生物活性分子进行标记. 之前有几篇综述概述了18F-标记反应的研究进展[95],本文将主要介绍最近的一些研究成果.
点击反应是最频繁用于对新的生物活性化合物进行18F-标记的方法之一[96]. 在2002年,Rostovtsec课题 组[97]以及Torne课题组[98]分别独立报道了铜Ⅰ催化的叠氮-炔基环加成反应(CuAAC). 该方法的优势是拥有出色的反应效率、区域选择性以及特别适合用于18F-标记敏感的生物分子的特点,即可以在室温下快速形成1,4-二取代-1,2,3-三唑类化合物.
2006年,Marik和Sutcliffe等[99]首次报道了CuAAC在18F-标记策略中的应用. 他们放射性标记了3个不同的炔烃前体,随后将这3个炔烃前体与叠氮基-功能化的肽类反应得到放射化学收率(RCY)为54%~99%的18F-标记的生物分子. 2014年,Ross课题组[96]、Lee课题组[100]以及Maschauer和Prante等[101]针对点击反应在放射性标记方面做了不同程度的总结性报道,随后Kim通过一篇微综述[102]报道了没有铜催化的点击反应SPAAC (力引发的叠氮-炔基环加成反应)在18F-标记生物分子方面的应用,同时还报道了在活体内肿瘤部位18F-标记介孔二氧化硅纳米颗粒(MSNs)的一些应用.
最近,Perrin等[103]报道了一种新型的两性放射性化学合成子N-烷基胺甲基三氟硼酸盐(AMBF3). AMBF3容易由胺与碘代甲基硼酸频哪醇酯(Iodomethylboronyl pinacolate)发生烷基化反应,随后与KHF2反应制得(Scheme 37). 该合成子容易与生物分子发生点击反应,环加成得到生物偶联复合物. 生物偶联复合物可以通过18F-19F同位素交换(IEX)[104]在较短的时间内完成18F标记,在高比活度以及高纯度(>98%)下得到较好收率的放射性示踪剂,并且不用HPLC进行提纯.
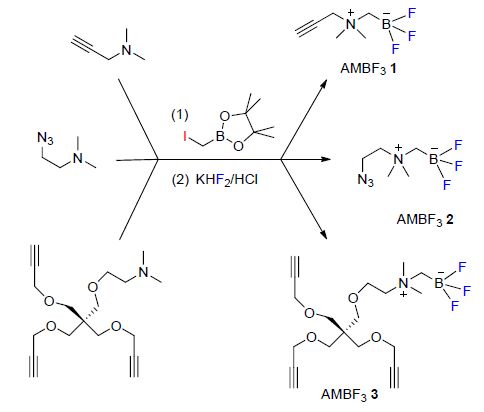 图 图式 37
放射性合成子AMBF3 1~3的合成
Figure 图式 37.
Synthesis of radiosynthons AMBF3 1~3
图 图式 37
放射性合成子AMBF3 1~3的合成
Figure 图式 37.
Synthesis of radiosynthons AMBF3 1~3
该课题组通过将AMBF3 2与炔烃-罗丹明偶联,随后用18F进行标记,得到典型的荧光PET追踪剂[18F]AMBF3-罗丹明. Octreotate是一种生长抑素2型受体(sstr2)激动剂,被放射性标记后在临床上用来成像sstr2-阳性神经内分泌瘤. 将AMBF3 1和叠氮-octreotate偶联并标记后得到[18F]AMBF3-octreotate,在AR42J胰腺肿瘤异种移植以及阻塞控制有高的吸收(>10% ID/g). 将AMBF3 1 与1个octreotate类似物偶联并且标记得到[18F]AMBF3-LM3,LM3是一种生长抑素受体拮抗剂. 该试剂在小鼠的AR42J异体移植胰腺肿瘤也有很高的吸收. 同时该课题组也尝试并首次制备了对含碳的脱水酶-IX (CA-IX) 靶点成像的18F放射示踪剂,该试剂由AMBF3 3与磺胺类衍生物偶联并且用18F进行标记得到.
通过进一步研究,Perrin和Bénard等[105]又通过使用放射性合成子AMBF3 1制备了新颖的18F-标记的蛙皮素衍生物18F-AMBF3-MJ9 (Scheme 38). 该试剂以胃泌素释放肽受体(GRPR)为目标可用于前列腺癌的成像.
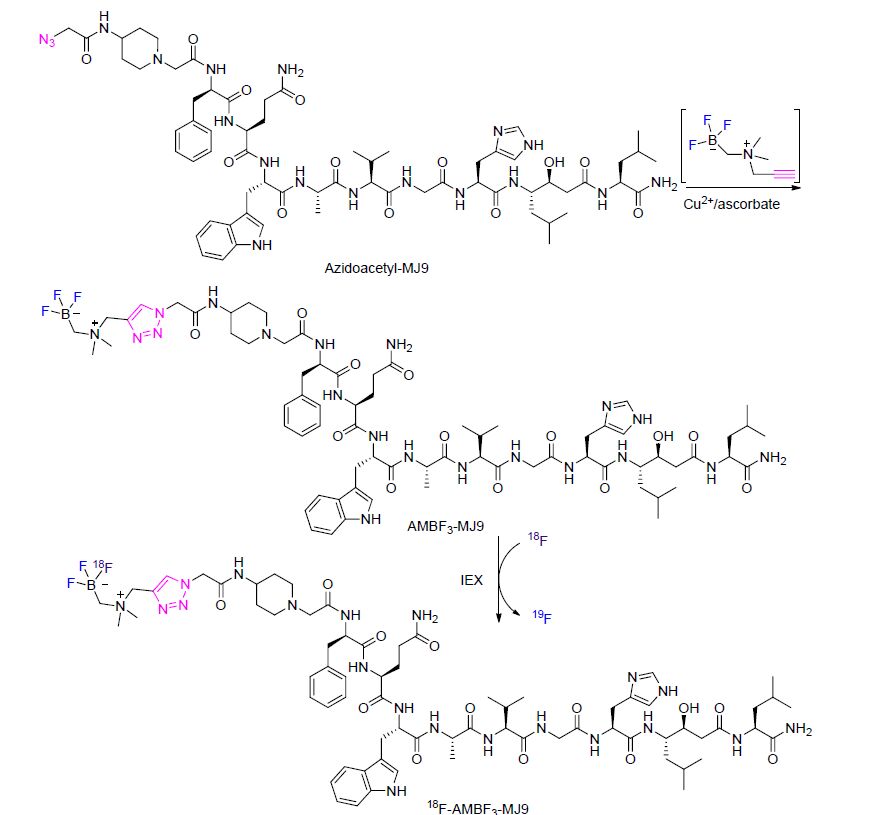 图 图式 38
18F-AMBF3-MJ9的合成
Figure 图式 38.
Synthesis of 18F-AMBF3-MJ9
图 图式 38
18F-AMBF3-MJ9的合成
Figure 图式 38.
Synthesis of 18F-AMBF3-MJ9
同样是运用点击反应,O'Hagan等[106]表述了一个通过两步反应直接放射性标记与恶性肿瘤相关的cRGD肽的方法(Scheme 39). 通常的18F-标记方法需要[18F]氟化物存在于干燥无水溶剂中,而该方法在37 ℃下发生在缓冲溶液中(pH 7.8). Fluorinase L-硒代蛋氨酸(L-SeMet)被用来催化反式卤化反应得到[18F]FDEA,随后[18F]FDEA与叠氮-cRGD肽发生点击反应得到放射性标记的生物相关的肽. 该放射性标记的肽在小鼠体内代谢稳定并且与αvβ3整合蛋白保留有较高的亲和力.
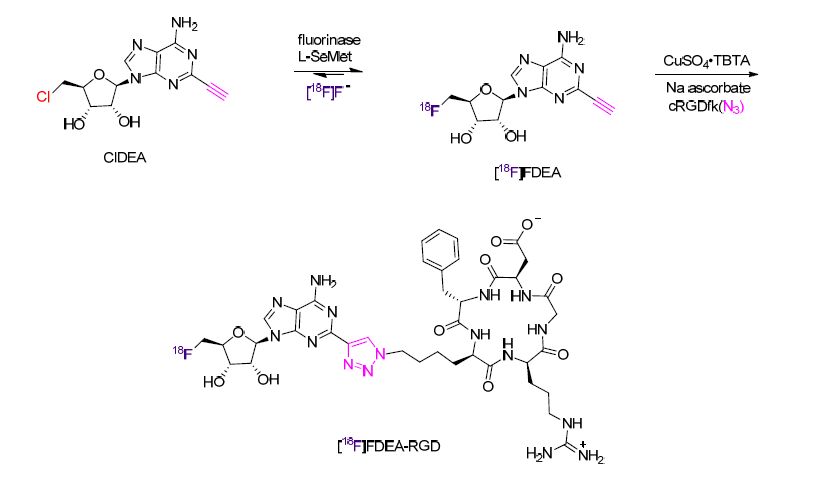 图 图式 39
[18F]FDEA-RGD的合成
Figure 图式 39.
Synthesis of [18F]FDEA-RGD
图 图式 39
[18F]FDEA-RGD的合成
Figure 图式 39.
Synthesis of [18F]FDEA-RGD
Liang和Xiao等[72]首次报道了在单质硫以及放射性氟离子(18F)存在下,烷基亲电体的[18F]三氟甲硫基化反应(Scheme 40). 反应过程中生成的二氟卡宾可以直接参与下一步反应转化为[18F]CF3S负离子. 该反应可以得到相应的18F-标记的三氟甲硫基取代的烷烃,且拥有不错的RCY. 同时,该反应也拥有良好的普适性.
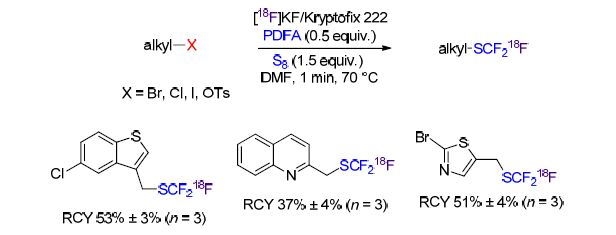 图 图式 40
二氟卡宾参与的脂肪族卤化物[18F]三氟甲硫基化反应
Figure 图式 40.
Difluorocarbene-mediated [18F]trifluoromethylthiolation of aliphatic halides
图 图式 40
二氟卡宾参与的脂肪族卤化物[18F]三氟甲硫基化反应
Figure 图式 40.
Difluorocarbene-mediated [18F]trifluoromethylthiolation of aliphatic halides
在刊登Liang和Xiao等[72]文章的同一期杂志中,Gouverneur等[107]介绍了亲卤的三氟甲磺酸银盐[AgⅠOTf]存在下,温和条件下前体aryl-OCHFCl、OCF2Br以及SCF2Br的亲核卤交换18F-氟化反应(Scheme 41). 该AgⅠOTf参与的反应使首次得到一系列18F标记的aryl-OCHF2、OCF3以及SCF3衍生物. 当前体aryl-SCF2Br为反应底物时,芳基上连接的多数基团都能适用于反应条件. 而前体aryl-OCF2Br为底物时,单-或双-保护的NBoc前体都不利于反应. 至于前体aryl-OCHFCl,芳基上连有吸电子基或给电子基都适用于该反应. 同时,该课题组通过竞争实验表明,前体与[18F]氟化物反应的活性顺序为ArOCHFCl>ArCF2Br≈ArCHFCl>ArSCF2Br>ArOCF2Br. 适用于治疗肌萎缩侧索硬化(ALS)的钠通道调节药利鲁唑通过该方法得到18F标记产物,并且拥有29%的RCY.
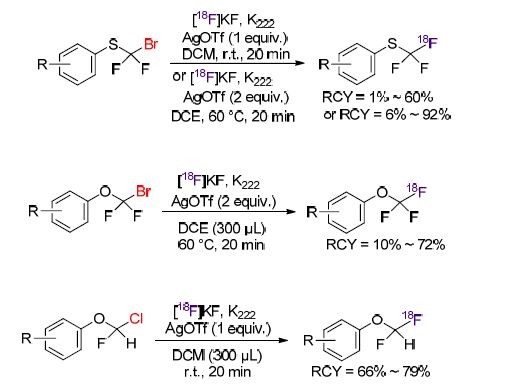 图 图式 41
银Ⅰ参与的卤交换[18F]氟化反应得到[18F]芳基-SCF3、OCF3以及OCHF2
Figure 图式 41.
AgⅠ-mediated halex 18F-fluorination towards [18F]aryl-SCF3,OCF3 and OCHF2
图 图式 41
银Ⅰ参与的卤交换[18F]氟化反应得到[18F]芳基-SCF3、OCF3以及OCHF2
Figure 图式 41.
AgⅠ-mediated halex 18F-fluorination towards [18F]aryl-SCF3,OCF3 and OCHF2
Groves和Hooker等[108]首次描述了在没有载体加入的情况下,用[18F]氟化物在最后一步对脂肪族苄基C—H键进行18F标记的化学过程. 该方法用Mn(salen)OTs作为F-传递催化剂,可以在10 min内标记多种生物活性分子以及合成模块,并且不需要对前体化合物进行预活化. 八种类药物分子包括布洛芬甲酯、N-三氟乙酰基雷沙吉兰、纳布美通、萨利麝香、塞来昔布类似物、受保护的依那普利拉、芬戈莫德以及西那卡塞通过该方法得到了相应的18F标记物(Scheme 42).
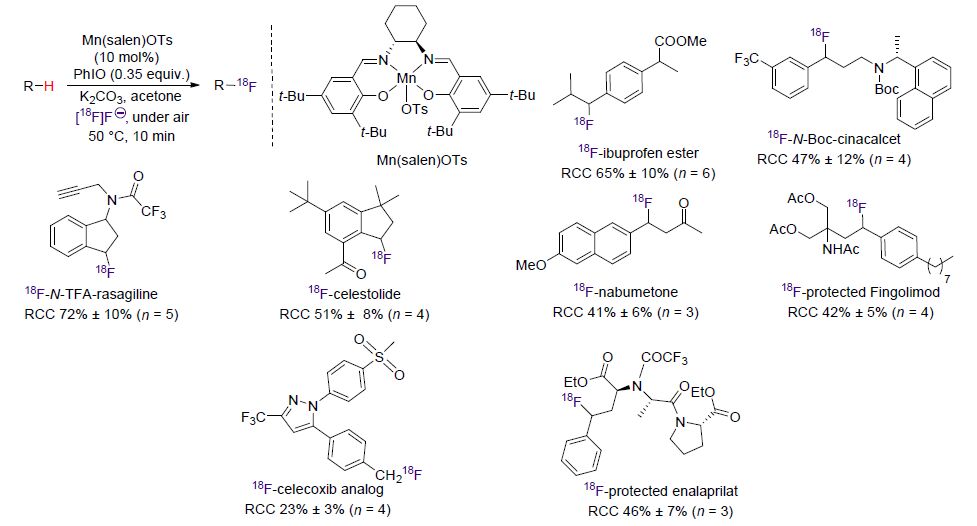 图 图式 42
直接18F-标记脂肪族C—H键
Figure 图式 42.
Direct 18F-labeling of aliphatic C—H bonds
图 图式 42
直接18F-标记脂肪族C—H键
Figure 图式 42.
Direct 18F-labeling of aliphatic C—H bonds
最近,陈小元课题组[109]报道了一个新颖的辅基18F-六氟苯(18F-HFB),该辅基可以用来制备放射性标记的二聚体生物分子或小分子(Scheme 43). 18F-HFB在室温下用K18F/K2,2,2通过氟原子交换反应制得. 18F-HFB通过蒸馏分离,随后在基础以及还原性条件下与硫醇化的肽类或小分子反应得到18F标记的二聚体. 该反应在加热条件下可以一步进行,也可以在室温条件下分两步进行. 由此法制得的18F-RGD-TFB-RGD表现出与整联蛋白受体存有特异性结合、细胞吸收以及活体内肿瘤部位出现聚集现象(Scheme 43).
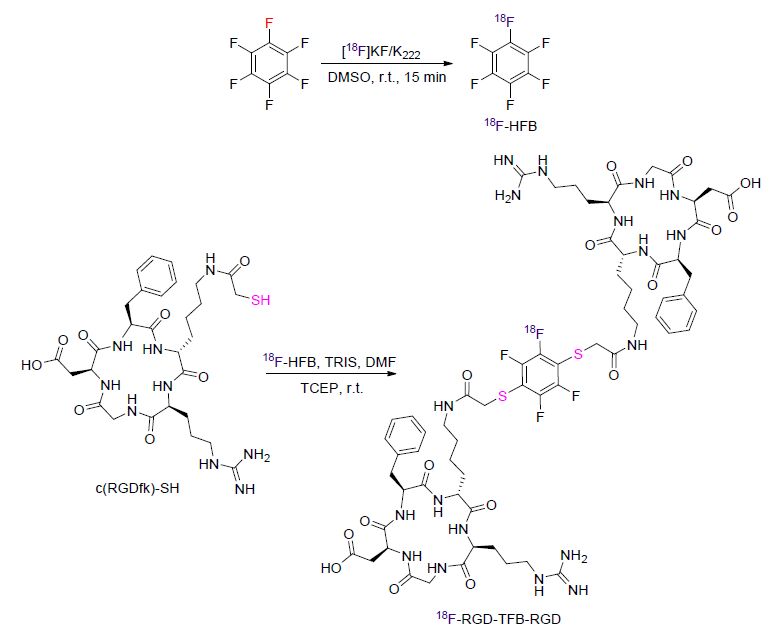 图 图式 43
18F-六氟苯以及18F-RGD-TFB-RGD的放射性合成
Figure 图式 43.
Radiosynthesis of 18F-hexafluorobenzene and 18F-RGD-TFB-RGD
图 图式 43
18F-六氟苯以及18F-RGD-TFB-RGD的放射性合成
Figure 图式 43.
Radiosynthesis of 18F-hexafluorobenzene and 18F-RGD-TFB-RGD
载药纳米载体以及用于药物释放的纳米药物体系需要非常仔细的对体内物理化学及药代动力学性质进行评估. Roig和Borrós等[110]报道了18F-放射性标记基于聚酯的纳米颗粒(NPs)的新方法. 该方法需要制备放射性标记的活性剂4-[18F]氟苄基-2-溴乙酰胺([18F]FBBA),该活性剂在温和条件下可以与聚合物结合生成[18F]-标记的聚合物(Scheme 44). 当[18F]FBBA与嵌段共聚物结合,最后被标记的嵌段共聚物作为构成要素通过一个改进的纳米共沉淀方法合并成NPs. 放射性标记过程在统计学上对NPs的物理性质没有显著的改变,通过PET/CT技术可以测定NPs在小鼠体内的生物分布模式. 目前该方法已经被应用于制备在给药方面有潜在应用的肽-功能化的NPs.
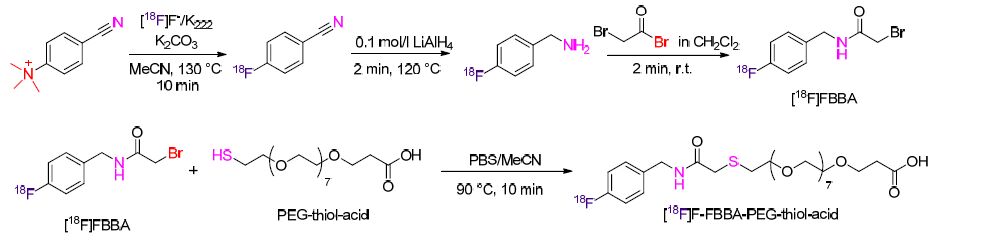 图 图式 44
[18F]FBBA的制备以及其与聚合物PEG-thiol-acid的反应
Figure 图式 44.
Preparation of [18F]FBBA and its reaction with polymer PEG-thiol-acid
图 图式 44
[18F]FBBA的制备以及其与聚合物PEG-thiol-acid的反应
Figure 图式 44.
Preparation of [18F]FBBA and its reaction with polymer PEG-thiol-acid
2009年McBride等[111]首次报道了通过铝缀合物进行18F标记的方法. 该方法使用[18F]氟化铝(Al18F)的组合形式以及它与双官能团配体NOTA衍生物——p-SCN-NOTA(图 2)的络合能力,通过“一锅煮”得到Al18F-螯合物-肽复合体. 最近杨敏等[112]将新型双官能团螯合剂p-SCN-NODA[113](图 2)与作用于胃泌素释放肽受体(GRPR)的肽——MATBBN结合探索了其应用能力(Scheme 45). p-SCN-NODA可以提高RCY至68.3%±1.8%. 18F-Al-NODA-MA-TBBN可以在25 min内制造出来并且拥有超过98%的放射化学纯度. 18F-Al-NODA- MATBBN在肿瘤部位的吸收值为3.23%±0.23% ID/g并且主要通过肾脏排出. 因此18F-Al-NODA-MATBBN可能成为监控前列腺癌的潜在的PET追踪剂候选物.
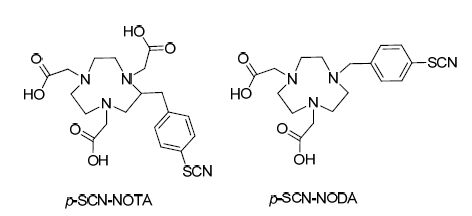 图 2
p-SCN-NOTA和p-SCN-NODA的化学结构
Figure 2.
Chemical structures of p-SCN-NOTA and p-SCN-NODA
图 2
p-SCN-NOTA和p-SCN-NODA的化学结构
Figure 2.
Chemical structures of p-SCN-NOTA and p-SCN-NODA
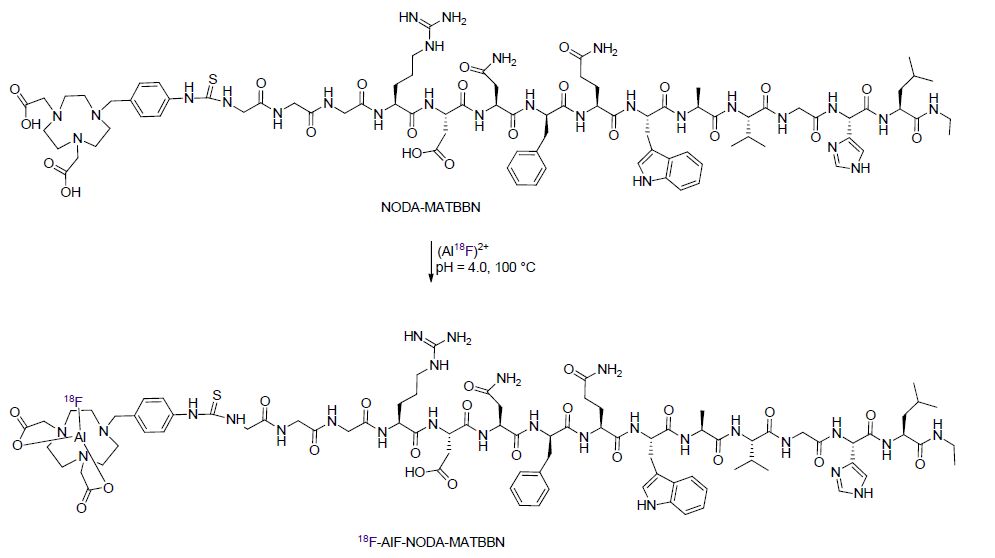 图 图式 45
18F 标记NODA-MATBBN得到18F-AlF-NODA-MATBBN
Figure 图式 45.
18F labeling of NODA-MATBBN to 18F-AlF-NODA-MATBBN
图 图式 45
18F 标记NODA-MATBBN得到18F-AlF-NODA-MATBBN
Figure 图式 45.
18F labeling of NODA-MATBBN to 18F-AlF-NODA-MATBBN
体内的生理过程对外源性外消旋体化合物表现出高度的手性识别,这是由于一对对映体与手性靶标如受体、酶以及离子通道之间拥有不同相互作用[114]. 在药物化学中,单一对映体的应用有利于减少对患者的用药剂量,最小化由于无活性的对映体造成的副作用[115]. 对于PET技术,手性放射性示踪剂拥有同样的重要性.
最近Gouverneur等[116]报道了首例有机催化的不对称18F-氟化作用. 该反应使用手性咪唑烷酮作为催化剂,[18F]N-氟代双苯磺酰胺([18F]NFSI)作为氟来源[117],得到18F标记的α-氟代醛,[18F]α-氟代醛是一种用于合成放射性示踪剂的拥有广泛用途的合成子. 虽然[18F]α-氟代醛无法被单独分离出来,但是可以通过“一锅煮”的方法在MeOH中与苯酰肼衍生化而得到相应的腙(>90% ee) (Schenme 46). 同时[18F]α-氟代醛因为醛基的存在可以进一步衍生为其他拥有高价值的18F标记的分子,如[18F]α-氟代羧酸、[18F]α-氟代酰胺等[116]. 该课题组[118]通过合成PET示踪剂(2S,4S)-4-[18F]氟代谷氨酸证明了该方法的可行性(Scheme 46). 谷氨酸在肿瘤的代谢过程中起到关键作用.
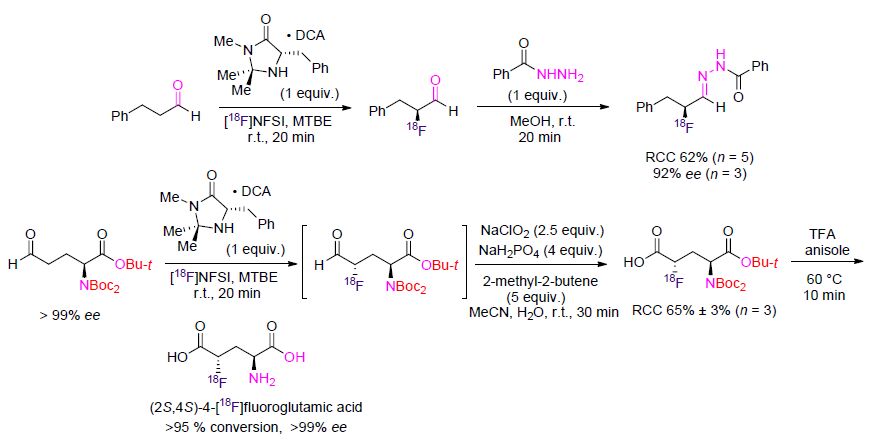 图 图式 46
放射性合成[18F]α-氟代醛及其应用
Figure 图式 46.
Radiosynthesis of [18F]α-fluoroaldehydes and its application
图 图式 46
放射性合成[18F]α-氟代醛及其应用
Figure 图式 46.
Radiosynthesis of [18F]α-fluoroaldehydes and its application
6 总结与展望
含氟基团拥有独特的性质,可以显著影响分子的性质和功能. 最近几年来,该领域的研究呈现出爆发式增长. 同时由于18F近乎理想的原子核性质,掀起了18F标记的放射性示踪剂的研究热潮. 本文主要总结了直接三氟甲硫基化反应、三氟甲硒基化反应、三氟甲氧基化反应和三氟乙氧基化反应最近几年的研究进展,同时介绍了最近一些18F-标记方法的研究成果. 关于该研究领域,作者认为还存在一些局限性: 例如上述的一些反应的机理以及反应的选择性研究尚不完全,一些反应需要使用较为昂贵的底物或特殊催化剂. 另外,只有部分反应可以用于大规模生产. 但是,正因为存在局限性,所以该研究领域还存在有巨大的发展空间.
未来该领域的发展应该着重于以下几个方面: (1)反应方法的便利性,即操作简单、反应条件温和、可以适用于普通的实验室环境. (2)反应的选择性研究是一个需要重点关注的方向,高的选择性可以很大地提高反应效率,以至于提高经济效益. (3)许多反应需要催化剂的存在,使用易获取的催化剂有利于反应方法的推广. (4)反应所需试剂的价格也应该引起科研工作者的注意,低额的成本结合高的反应选择性可以使经济效益最大化.
由于含氟基团和18F所特有的性质,分子内引入含氟基团以及18F-标记方法的研究成果将会大力推动医药和农药以及其他行业的发展,从而为人类社会的进步作出贡献.
-
-
[1]
Harper, D. B.; O'Hagan, D. Nat. Prod. Rep. 1994, 11, 123. doi: 10.1039/np9941100123
-
[2]
O'Hagan, D.; Harper, D. B. J. Fluorine Chem. 1999, 100, 127.(b) Zhang, W.; Cai, Z.; Zhou, W. Chin. J. Pharm. 2014, 45, 174 (in Chinese).(张文强, 蔡正艳, 周伟澄, 中国医药工业杂志, 2014, 45, 174.)(c) Zhu, Y.; Zhou, W. Chin. J. Pharm. 2015, 46, 74 (in Chinese).(朱怡君, 周伟澄, 中国医药工业杂志, 2015, 46, 74.)(d) Ma, S.; Wen, Y.; Zhou, W. Chin. J. Pharm. 2016, 47, 79 (in Chinese).(马帅, 温颖玲, 周伟澄, 中国医药工业杂志, 2016, 47, 79.) doi: 10.1016/S0022-1139(99)00201-8
-
[3]
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315.(b) Wang, J.; Liu, H. Chin. J. Org. Chem. 2011, 31, 1785 (in Chinese)(王江, 柳红, 有机化学, 2011, 31, 1785.) doi: 10.1021/acs.jmedchem.5b00258
-
[4]
Keating, G. M. Drugs 2014, 74, 207. doi: 10.1007/s40265-013-0170-8
-
[5]
Infante, J. R.; Fecher, L. A.; Falchook, G. S.; Nallapareddy, S.; Gordon, M. S.; Becerra, C.; DeMarini, D. J.; Cox, D. S.; Xu, Y.; Morris, S. R.; Peddareddigari, V. G. R.; Le, N. T.; Hart, L.; Bendell, J. C.; Eckhardt, G.; Kurzrock, R.; Flaherty, K.; Burris, H. A.; Messersmith, W. A. Lancet Oncol. 2012, 13, 773. doi: 10.1016/S1470-2045(12)70270-X
-
[6]
Rheault, T. R.; Stellwagen, J. C.; Adjabeng, G. M.; Hornberger, K. R.; Petrov, K. G.; Waterson, A. G.; Dickerson, S. H.; Mook, R. A.; Laquerre, S. G.; King, A. J.; Rossanese, O. W.; Arnone, M. R.; Smitheman, K. N.; Kane-Carson, L. S.; Han, C.; Moorthy, G. S.; Moss, K. G.; Uehling, D. E. ACS Med. Chem. Lett. 2013, 4, 358. doi: 10.1021/ml4000063
-
[7]
Fruman, D. A.; Cantley, L. C. N. Engl. J. Med. 2014, 370, 1061. doi: 10.1056/NEJMe1400055
-
[8]
Rice, K. D.; Aay, N.; Anand, N. K.; Blazey, C. M.; Bowles, O. J.; Bussenius, J.; Costanzo, S.; Curtis, J. K.; Defina, S. C.; Dubenko, L.; Engst, S.; Joshi, A. A.; Kennedy, A. R.; Kim, A. I.; Koltun, E. S.; Lougheed, J. C.; Manalo, J. C.; Martini, J. F.; Nuss, J. M.; Peto, C. J.; Tsang, T. H.; Yu, P.; Johnston, S. ACS Med. Chem. Lett. 2012, 3, 416.
-
[9]
Menear, K. A.; Adcock, C.; Boulter, R.; Cockcroft, X.-l.; Copsey, L.; Cranston, A.; Dillon, K. J.; Drzewiecki, J.; Garman, S.; Gomez, S.; Javaid, H.; Kerrigan, F.; Knights, C.; Lau, A.; Loh, V. M., Jr.; Matthews, I. T. W.; Moore, S.; O'Connor, M. J.; Smith, G. C. M.; Martin, N. M. B. J. Med. Chem. 2008, 51, 6581. doi: 10.1021/jm8001263
-
[10]
Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N. P.; Wang, Y.; Peukert, S.; Miller-Moslin, K.; Yuan, J.; Guo, R.; Matsumoto, M.; Vattay, A.; Jiang, Y.; Tsao, J.; Sun, F.; Pferdekamper, A. C.; Dodd, S.; Tuntland, T.; Maniara, W.; Kelleher, J. F., III; Yao, Y.-m.; Warmuth, M.; Williams, J.; Dorsch, M. ACS Med. Chem. Lett. 2010, 1, 130.
-
[11]
Wu, J. Tetrahedron Lett. 2014, 55, 4289.(b) Campbell, M. G.; Ritter, T. Org. Process Res. Dev. 2014, 18, 474.(c) Lin, J.-H.; Xiao, J.-C. Tetrahedron Lett. 2014, 55, 6147.(d) Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765.(e) Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826. doi: 10.1016/j.tetlet.2014.06.006
-
[12]
Lantaño, B.; Torviso, M. R.; Bonesi, S. M.; Barata-Vallejo, S.; Postigo, A. Coord. Chem. Rev. 2015, 285, 76.(b) Barata-Vallejo, S.; Torviso, M. R.; Lantaño, B.; Bonesi, S. M.; Postigo, A. J. Fluorine Chem. 2014, 161, 134.(c) Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J. L.; Izawa, K.; Liu, H.; Soloshonok, V. A. J. Fluorine Chem. 2014, 167, 37.(d) Xu, X. H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731.(e) Ni, C.; Zhu, L.; Hu, J. Acta Chim. Sinica 2015, 73, 90 (in Chinese)(倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90.)(f) Qing, F.-L. Chin. J. Org. Chem 2012, 32, 815 (in Chinese).(卿凤翎, 有机化学, 2012, 32, 815.)(g) Zhang, J.; Jin, C.; Zhang, Y. Chin. J. Org. Chem 2014, 34, 662 (in Chinese).(张霁, 金传飞, 张英俊, 有机化学, 2014, 34, 662.)
-
[13]
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165. doi: 10.1021/cr00002a004
-
[14]
Toulgoat, F.; Alazet, S.; Billard, T. Eur. J. Org. Chem. 2014, 2014, 2415.(b) Lin, J.-H.; Ji, Y.-L.; Xiao, J.-C. Curr. Org. Chem. 2015, 19, 1541.(c) Zhang, K.; Xu, X.; Qing, F. Chin. J. Org. Chem 2015, 35, 556 (in Chinese).(张柯, 徐修华, 卿凤翎, 有机化学, 2015, 35, 556.)
-
[15]
Zhang, K.; Liu, J. B.; Qing, F. L. Chem. Commun. 2014, 50, 14157. doi: 10.1039/C4CC07062C
-
[16]
Li, C.; Zhang, K.; Xu, X.-H.; Qing, F.-L. Tetrahedron Lett. 2015, 56, 6273.
-
[17]
Wu, H.; Xiao, Z.; Wu, J.; Guo, Y.; Xiao, J. C.; Liu, C.; Chen, Q. Y. Angew. Chem. 2015, 127, 4142. doi: 10.1002/ange.201411953
-
[18]
Guo, S.; Zhang, X.; Tang, P. Angew. Chem., Int. Ed. Engl. 2015, 54, 4065. doi: 10.1002/anie.201411807
-
[19]
Somei, M.; Yamada, F. Nat. Prod. Rep. 2005, 22, 73.(b) Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875. doi: 10.1039/b316241a
-
[20]
Fuentes, N.; Kong, W.; Fernandez-Sanchez, L.; Merino, E.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 964. doi: 10.1021/ja5115858
-
[21]
Huo, Y.; Fang, X.; Huang, B.; Zhang, K.; Nie, X.; Zeng, H. Chin. J. Org. Chem 2012, 32, 1169 (in Chinese).(霍延平, 方小明, 黄宝华, 张 焜, 聂晓李, 曾和平, 有机化学2012, 32, 1169.)
-
[22]
Qiu, Y. F.; Zhu, X. Y.; Li, Y. X.; He, Y. T.; Yang, F.; Wang, J.; Hua, H. L.; Zheng, L.; Wang, L. C.; Liu, X. Y.; Liang, Y. M. Org. Lett. 2015, 17, 3694. doi: 10.1021/acs.orglett.5b01657
-
[23]
Ong, E. B. B.; Watanabe, N.; Saito, A.; Futamura, Y.; Abd El Galil, K. H.; Koito, A.; Najimudin, N.; Osada, H. J. Biol. Chem. 2011, 286, 14049. doi: 10.1074/jbc.M110.185397
-
[24]
Taechowisan, T.; Lu, C.; Shen, Y.; Lumyong, S. Microbiology 2005, 151, 1691.
-
[25]
Taechowisan, T.; Lu, C.; Shen, Y.; Lumyong, S. Food Agric. Immunol. 2007, 18, 203.
-
[26]
Bailly, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.-P.; Hildebrand, M.-P.; Peyrot, V.; Wattez, N. J. Med. Chem. 2003, 46, 5437.(b) Rappl, C.; Barbier, P.; Bourgarel-Rey, V.; Gregoire, C.; Gilli, R.; Carre, M.; Combes, S.; Finet, J.-P.; Peyrot, V. Biochemistry 2006, 45, 9210. doi: 10.1021/jm030903d
-
[27]
Argotte-Ramos, R.; Ramirez-Avila, G.; Rodriguez-Gutierrez, M. d. C.; Ovilla-Munoz, M.; Lanz-Mendoza, H.; Rodriguez, M. H.; Gonzalez-Cortazar, M.; Alvarez, L. J. Nat. Prod. 2006, 69, 1442. doi: 10.1021/np060233p
-
[28]
Zeng, Y.-F.; Tan, D.-H.; Chen, Y.; Lv, W.-X.; Liu, X.-G.; Li, Q.; Wang, H. Org. Chem. Front. 2015, 2, 1511. doi: 10.1039/C5QO00271K
-
[29]
Matheis, C.; Wagner, V.; Goossen, L. J. Chem. Eur. J. 2016, 22, 79. doi: 10.1002/chem.201503524
-
[30]
Huang, Y.; He, X.; Lin, X.; Rong, M.; Weng, Z. Org. Lett. 2014, 16, 3284.
-
[31]
Sheng, J.; Wu, J. Org. Biomol. Chem. 2014, 12, 7629. doi: 10.1039/C4OB01451K
-
[32]
Xiao, Q.; Zhu, H.; Li, G.; Chen, Z. Adv. Synth. Catal. 2014, 356, 3809. doi: 10.1002/adsc.v356.18
-
[33]
Korte, A.; Legros, J.; Bolm, C. Synlett 2005, 36, 2397.
-
[34]
Jereb, M.; Gosak, K. Org. Biomol. Chem. 2015, 13, 3103. doi: 10.1039/C4OB02633K
-
[35]
Wu, W.; Zhang, X.; Liang, F.; Cao, S. Org. Biomol. Chem. 2015, 13, 6992. doi: 10.1039/C5OB00960J
-
[36]
Mortensen, D. S.; Rodriguez, A. L.; Carlson, K. E.; Sun, J.; Katzenellenbogen, B. S.; Katzenellenbogen, J. A. J. Med. Chem. 2001, 44, 3838.(b) Faul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407. doi: 10.1021/jm010211u
-
[37]
Chen, D. Q.; Gao, P.; Zhou, P. X.; Song, X. R.; Qiu, Y. F.; Liu, X. Y.; Liang, Y. M. Chem. Commun. 2015, 51, 6637. doi: 10.1039/C5CC00669D
-
[38]
Alazet, S.; Zimmer, L.; Billard, T. Chem. Eur. J. 2014, 20, 8589. doi: 10.1002/chem.201403409
-
[39]
Alazet, S.; Ismalaj, E.; Glenadel, Q.; Le Bars, D.; Billard, T. Eur. J. Org. Chem. 2015, 2015, 4607.
-
[40]
Alazet, S.; Zimmer, L.; Billard, T. J. Fluorine Chem. 2015, 171, 78. doi: 10.1016/j.jfluchem.2014.09.009
-
[41]
Glenadel, Q.; Alazet, S.; Billard, T. J. Fluorine Chem. 2015, 179, 89. doi: 10.1016/j.jfluchem.2015.06.007
-
[42]
Munavalli, S.; Rohrbaugh, D. K.; Rossman, D. I.; Berg, F. J.; Wagner, G. W.; Durst, H. D. Synth. Commun. 2000, 30, 2847. doi: 10.1080/00397910008087435
-
[43]
Kang, K.; Xu, C.; Shen, Q. Org. Chem. Front. 2014, 1, 294. doi: 10.1039/c3qo00068k
-
[44]
Honeker, R.; Ernst, J. B.; Glorius, F. Chem. Eur. J. 2015, 21, 8047. doi: 10.1002/chem.201500957
-
[45]
Xiong, H. Y.; Besset, T.; Cahard, D.; Pannecoucke, X. J. Org. Chem. 2015, 80, 4204. doi: 10.1021/acs.joc.5b00505
-
[46]
Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782. doi: 10.1021/ja402455f
-
[47]
Arimori, S.; Takada, M.; Shibata, N. Org. Lett. 2015, 17, 1063.
-
[48]
Jiang, L.; Qian, J.; Yi, W.; Lu, G.; Cai, C.; Zhang, W. Angew. Chem., Int. Ed. 2015, 54, 14965. doi: 10.1002/anie.201508495
-
[49]
Yang, Y.; Xu, L.; Yu, S.; Liu, X.; Zhang, Y.; Vicic, D. A. Chem. Eur. J. 2016, 22, 858. doi: 10.1002/chem.v22.3
-
[50]
Liu, T.; Shen, Q. Org. Lett. 2011, 13, 2342.
-
[51]
Shao, X.; Wang, X.; Yang, T.; Lu, L.; Shen, Q. Angew. Chem., Int. Ed. 2013, 52, 3457. doi: 10.1002/anie.v52.12
-
[52]
Vinogradova, E. V.; Mueller, P.; Buchwald, S. L. Angew. Chem., Int. Ed. 2014, 53, 3125. doi: 10.1002/anie.201310897
-
[53]
Shao, X.; Xu, C.; Lu, L.; Shen, Q. J. Org. Chem. 2015, 80, 3012. doi: 10.1021/jo502645m
-
[54]
Shao, X.; Xu, C.; Lu, L.; Shen, Q. Acc. Chem. Res. 2015, 48, 1227. doi: 10.1021/acs.accounts.5b00047
-
[55]
Li, Y.; Ye, Z.; Bellman, T. M.; Chi, T.; Dai, M. Org. Lett. 2015, 17, 2186.
-
[56]
Xu, C.; Ma, B.; Shen, Q. Angew. Chem., Int. Ed. 2014, 53, 9316. doi: 10.1002/anie.201403983
-
[57]
Xu, C.; Shen, Q. Org. Lett. 2015, 17, 4561.
-
[58]
Wang, Q.; Xie, F.; Li, X. J. Org. Chem. 2015, 80, 8361. doi: 10.1021/acs.joc.5b00940
-
[59]
Maeno, M.; Shibata, N.; Cahard, D. Org. Lett. 2015, 17, 1990.
-
[60]
Jiang, M.; Zhu, F.; Xiang, H.; Xu, X.; Deng, L.; Yang, C. Org. Biomol. Chem. 2015, 13, 6935. doi: 10.1039/C5OB00858A
-
[61]
Yin, G.; Kalvet, I.; Schoenebeck, F. Angew. Chem. 2015, 127, 6913.
-
[62]
Zhong, W.; Liu, X. Tetrahedron Lett. 2014, 55, 4909.
-
[63]
Weng, Z.; He, W.; Chen, C.; Lee, R.; Tan, D.; Lai, Z.; Kong, D.; Yuan, Y.; Huang, K. W. Angew. Chem., Int. Ed. 2013, 52, 1548. doi: 10.1002/anie.201208432
-
[64]
Kong, D.; Jiang, Z.; Xin, S.; Bai, Z.; Yuan, Y.; Weng, Z. Tetrahedron 2013, 69, 6046.
-
[65]
Lin, Q.; Chen, L.; Huang, Y.; Rong, M.; Yuan, Y.; Weng, Z. Org. Biomol. Chem. 2014, 12, 5500. doi: 10.1039/c4ob00403e
-
[66]
Huang, Y.; He, X.; Li, H.; Weng, Z. Eur. J. Org. Chem. 2014, 2014, 7324.
-
[67]
Zhu, P.; He, X.; Chen, X.; You, Y.; Yuan, Y.; Weng, Z. Tetrahedron 2014, 70, 672.
-
[68]
Hou, C.; Lin, X.; Huang, Y.; Chen, Z.; Weng, Z. Synthesis 2015, 47, 969.
-
[69]
Huang, Y.; Ding, J.; Wu, C.; Zheng, H.; Weng, Z. J. Org. Chem. 2015, 80, 2912. doi: 10.1021/acs.joc.5b00144
-
[70]
Wang, Z.; Tu, Q.; Weng, Z. J. Organomet. Chem. 2014, 751, 830. doi: 10.1016/j.jorganchem.2013.08.008
-
[71]
Li, J.; Xie, F.-F.; Wang, P.; Wu, Q.-Y.; Chen, W.-D.; Ren, J.; Zeng, B.-B. Tetrahedron 2015, 71, 5520.
-
[72]
Zheng, J.; Wang, L.; Lin, J. H.; Xiao, J. C.; Liang, S. H. Angew. Chem. 2015, 127, 13434. doi: 10.1002/ange.201505446
-
[73]
Zheng, J.; Cai, J.; Lin, J.-H.; Guo, Y.; Xiao, J.-C. Chem. Commun. 2013, 49, 7513.(b) Qiao, Y.; Si, T.; Yang, M.-H.; Altman, R. A. J. Org. Chem. 2014, 79, 7122.(c) Levin, V. V.; Trifonov, A. L.; Zemtsov, A. A.; Struchkova, M. I.; Arkhipov, D. E.; Dilman, A. D. Org. Lett. 2014, 16, 6256.
-
[74]
Li, S.-G.; Zard, S. Z. Org. Lett. 2013, 15, 5898. doi: 10.1021/ol403038f
-
[75]
Yang, H.-B.; Fan, X.; Wei, Y.; Shi, M. Org. Chem. Front. 2015, 2, 1088. doi: 10.1039/C5QO00198F
-
[76]
Aufiero, M.; Sperger, T.; Tsang, A. S.; Schoenebeck, F. Angew. Chem., Int. Ed. 2015, 54, 10322. doi: 10.1002/anie.201503388
-
[77]
Papp, L. V.; Holmgren, A.; Khanna, K. K. Antioxid. Redox Signaling 2010, 12, 793. doi: 10.1089/ars.2009.2973
-
[78]
Nogueira, C. W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255. doi: 10.1021/cr0406559
-
[79]
Lefebvre, Q.; Pluta, R.; Rueping, M. Chem. Commun. 2015, 51, 4394.
-
[80]
Chen, C.; Hou, C.; Wang, Y.; Hor, T. S.; Weng, Z. Org. Lett. 2014, 16, 524.
-
[81]
Chen, C.; Ouyang, L.; Lin, Q.; Liu, Y.; Hou, C.; Yuan, Y.; Weng, Z. Chem. Eur. J. 2014, 20, 657. doi: 10.1002/chem.v20.3
-
[82]
Rong, M.; Huang, R.; You, Y.; Weng, Z. Tetrahedron 2014, 70, 8872.(b) Feng, Z.; Chen, F.; Zhang, X. G. Org. Lett. 2012, 14, 1938.(c) Zhao, T. S. N.; Szabo, K. J. Org. Lett. 2012, 14, 3966.
-
[83]
Wang, Y.; You, Y.; Weng, Z. Org. Chem. Front. 2015, 2, 574. doi: 10.1039/C5QO00045A
-
[84]
Wu, C.; Huang, Y.; Chen, Z.; Weng, Z. Tetrahedron Lett. 2015, 56, 3838.
-
[85]
Chen, S.; Huang, Y.; Fang, X.; Li, H.; Zhang, Z.; Hor, T. S.; Weng, Z. Dalton Trans 2015, 44, 19682. doi: 10.1039/C5DT02078F
-
[86]
Hojczyk, K. N.; Feng, P.; Zhan, C.; Ngai, M. Y. Angew. Chem., Int. Ed. 2014, 53, 14559. doi: 10.1002/anie.201409375
-
[87]
Feng, P.; Lee, K. N.; Lee, J. W.; Zhan, C.; Ngai, M.-Y. Chem. Sci. 2016, 7, 424.
-
[88]
Huang, Y.; Huang, R.; Weng, Z. Synlett 2015, 2327.
-
[89]
Sun, X. Q.; Lyu, Y. R.; Zhang-Negrerie, D.; Du, Y. F.; Zhao, K. Org. Lett. 2013, 15, 6222.
-
[90]
Huang, R.; Huang, Y.; Lin, X.; Rong, M.; Weng, Z. Angew. Chem., Int. Ed. 2015, 54, 5736. doi: 10.1002/anie.201501257
-
[91]
Wang, Y.; You, Y.; Weng, Z. J. Fluorine Chem. 2015, 175, 51. doi: 10.1016/j.jfluchem.2015.03.009
-
[92]
Rangarajan, T. M.; Devi, K.; Ayushee; Prasad, A. K.; Singh, R. P. Tetrahedron 2015, 71, 8307. doi: 10.1016/j.tet.2015.08.071
-
[93]
Hargreaves, R. J.; Rabiner, E. A. Neurobiol. Dis. 2014, 61, 32. doi: 10.1016/j.nbd.2013.08.017
-
[94]
Cai, L.; Lu, S.; Pike, V. W. Eur. J. Org. Chem. 2008, 2853.(b) Miller, P. W.; Long, N. J.; Vilar, R.; Gee, A. D. Angew. Chem., Int. Ed. 2008, 47, 8998.(c) Jacobson, O.; Chen, X. Curr. Top. Med. Chem. 2010, 10, 1048.(d) Pimlott, S. L.; Sutherland, A. Chem. Soc. Rev. 2011, 40, 149.(e) Smith, G. E.; Sladen, H. L.; Biagini, S. C. G.; Blower, P. J. Dalton Trans. 2011, 40, 6196.
-
[95]
Zeng, J. L.; Wang, J.; Ma, J. A. Bioconjugate Chem. 2015, 26, 1000.(b) Jacobson, O.; Kiesewetter, D. O.; Chen, X. Bioconjugate Chem. 2015, 26, 1. doi: 10.1021/acs.bioconjchem.5b00180
-
[96]
Kettenbach, K.; Schieferstein, H.; Ross, T. L. BioMed. Res. Int. 2014, 2014, 361329.
-
[97]
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. doi: 10.1002/(ISSN)1521-3773
-
[98]
Tornoe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057. doi: 10.1021/jo011148j
-
[99]
Marik, J.; Sutcliffe, J. L. Tetrahedron Lett. 2006, 47, 6681. doi: 10.1016/j.tetlet.2006.06.176
-
[100]
Choi, J. Y.; Lee, B. C. Nucl. Med. Mol. Imaging 2015, 49, 258. doi: 10.1007/s13139-015-0377-6
-
[101]
Maschauer, S.; Prante, O. BioMed. Res. Int. 2014, 214748/1.
-
[102]
Kim, D. W. J. Fluorine Chem. 2015, 174, 142. doi: 10.1016/j.jfluchem.2014.11.009
-
[103]
Liu, Z.; Pourghiasian, M.; Radtke, M. A.; Lau, J.; Pan, J.; Dias, G. M.; Yapp, D.; Lin, K. S.; Bénard, F.; Perrin, D. M. Angew. Chem., Int. Ed. 2014, 53, 11876.(b) Liu, Z.; Lin, K. S.; Bénard, F.; Pourghiasian, M.; Kiesewetter, D. O.; Perrin, D. M.; Chen, X. Nat. Protoc. 2015, 10, 1423. doi: 10.1002/anie.201406258
-
[104]
Schirrmacher, R.; Bradmoeller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Bucholz, H. G.; Bartenstein, P.; Waengler, B.; Niemeyer, C. M.; Jurkschat, K. Angew. Chem., Int. Ed. 2006, 45, 6047.(b) Li, Z. B.; Chansaenpak, K.; Liu, S. L.; Wade, C. R.; Conti, P. S.; Gabbai, F. P. MedChemComm 2012, 3, 1305. doi: 10.1002/(ISSN)1521-3773
-
[105]
Pourghiasian, M.; Liu, Z.; Pan, J.; Zhang, Z.; Colpo, N.; Lin, K. S.; Perrin, D. M.; Bénard, F. Bioorg. Med. Chem. 2015, 23, 1500. doi: 10.1016/j.bmc.2015.02.009
-
[106]
Thompson, S.; Onega, M.; Ashworth, S.; Fleming, I. N.; Passchier, J.; O'Hagan, D. Chem. Commun. 2015, 51, 13542.
-
[107]
Khotavivattana, T.; Verhoog, S.; Tredwell, M.; Pfeifer, L.; Calderwood, S.; Wheelhouse, K.; Lee Collier, T.; Gouverneur, V. Angew. Chem., Int. Ed. 2015, 54, 9991. doi: 10.1002/anie.201504665
-
[108]
Huang, X.; Liu, W.; Ren, H.; Neelamegam, R.; Hooker, J. M.; Groves, J. T. J. Am. Chem. Soc. 2014, 136, 6842.(b) Liu, W.; Groves, J. T. Acc. Chem. Res. 2015, 48, 1727. doi: 10.1021/ja5039819
-
[109]
Jacobson, O.; Yan, X.; Ma, Y.; Niu, G.; Kiesewetter, D. O.; Chen, X. Bioconjugate Chem. 2015, 26, 2016. doi: 10.1021/acs.bioconjchem.5b00278
-
[110]
Di Mauro, P. P.; Gomez-Vallejo, V.; Baz Maldonado, Z.; Llop Roig, J.; Borrós, S. Bioconjugate Chem. 2015, 26, 582.
-
[111]
McBride, W. J.; Sharkey, R. M.; Karacay, H.; D'Souza, C. A.; Rossi, E. A.; Laverman, P.; Chang, C. H.; Boerman, O. C.; Goldenberg, D. M. J. Nucl. Med. 2009, 50, 991. doi: 10.2967/jnumed.108.060418
-
[112]
Chen, F.; Zhu, B.; Pan, D.; Xu, Y.; Lin, X.; Yang, R.; Wang, L.; Yang, M. J. Radioanal. Nucl. Chem. 2016, 308, 905. doi: 10.1007/s10967-015-4577-4
-
[113]
Shetty, D.; Jeong, J. M.; Kim, Y. J.; Lee, J. Y.; Hoigebazar, L.; Lee, Y. S.; Lee, D. S.; Chung, J. K. Bioorg. Med. Chem. 2012, 20, 5941. doi: 10.1016/j.bmc.2012.07.050
-
[114]
Agranat, I.; Caner, H.; Caldwell, J. Nat. Rev. Drug Discovery 2002, 1, 753. doi: 10.1038/nrd915
-
[115]
Brooks, W. H.; Guida, W. C.; Daniel, K. G. Curr. Top. Med. Chem. 2011, 11, 760. doi: 10.2174/156802611795165098
-
[116]
Buckingham, F.; Gouverneur, V.; Kirjavainen Anna, K.; Forsback, S.; Krzyczmonik, A.; Keller, T.; Solin, O.; Newington Ian, M.; Glaser, M.; Luthra Sajinder, K. Angew. Chem., Int. Ed. 2015, 54, 13366. doi: 10.1002/anie.201506035
-
[117]
Teare, H.; Robins, E. G.; Aarstad, E.; Luthra, S. K.; Gouverneur, V. Chem. Commun. 2007, 2330.
-
[118]
Hensley, C. T.; Wasti, A. T.; DeBerardinis, R. J. J. Clin. Invest. 2013, 123, 3678. doi: 10.1172/JCI69600
-
[1]
-

图式 1 醌的三氟甲硫基化反应以及其可能的机理
Scheme 1 Trifluoromethylthiolation of quinines and its proposed mechanism

图式 2 未活化的C(sp3)—H键的直接三氟甲硫基化反应以及其可能的机理
Scheme 2 Direct trifluoromethylthiolation of unactivated C(sp3)—H bond and its proposed mechanism

图式 3 吲哚[2,1-a]异喹啉类化合物的合成以及其可能的机理
Scheme 3 Synthesis of indolo[2,1-a]isoquinolines and its proposed mechanism

图式 4 1,6-烯炔的三氟甲硫基化反应以及其可能的机理
Scheme 4 Trifluoromethylthiolation of 1,6-enynes and its proposed mechanism

图式 5 1,2-二炔基苯与PhNHSCF3以及BiCl3或TsOH之间的反应
Scheme 5 Reaction of 1,2-bis(alkynyl)benzene and PhNHSCF3 with BiCl3 or TsOH

图式 6 酚的三氟甲硫基化和2-烯丙基苯酚的双官能团化
Scheme 6 Trifluoromethylthiolation of phenols and the double functionalization of 2-allylphenol

图式 8 4-三氟甲硫基-2,3-二氢呋喃类的合成和反应途径
Scheme 8 Synthesis of 4-(trifluoromethyl)thio-2,3-dihydrofurans and its reaction pathway

图式 11 N-三氟甲硫基邻苯二甲酰亚胺的合成以及芳基/乙烯基硼酸的三氟甲硫基化反应
Scheme 11 Synthesis of N-(trifluoromethylthio)phthalimide and trifluoromethylthiolation of aryl/vinyl boronic acids

图式 13 PdⅡ催化的三氟甲硫基化反应和假设的反应机理
Scheme 13 PdⅡ-catalyzed trifluoromethylthiolation and its proposed mechanism

图式 14 CF3SO2Na与C(sp2)—H键之间的三氟甲硫基化反应及其可能的机理
Scheme 14 Trifluoromethylthiolation of C(sp2)—H bonds with CF3SO2Na and its proposed mechanism

图式 15 三氟甲基取代的硫代过氧化物A/B与吲哚衍生物、硼酸、烯烃、脂肪族羧酸、β-酮酸酯以及炔烃之间的三氟甲硫基化反应
Scheme 15 Trifluoromethylthiolation of indoles,boronic acids,alkenes,aliphatic carboxylic acids,β-keto esters and alkynes with the trifluoromethyl-substituted thioperoxide reagent A/B

图式 16 N-三氟甲硫基糖精与各种亲核体之间发生的三氟甲硫基化反应
Scheme 16 Trifluoromethylthiolation of various nucleophilic substrates with N-trifluoromethylthiosaccharin

图式 17 AgSCF3与α-卤代酮以及缺电子的苄基卤化物之间的三氟甲硫基化反应
Scheme 17 Trifluoromethylthiolation of α-haloketones and electron-deficient benzyl halides with AgSCF3

图式 18 PdⅠ催化的芳基碘化物/溴化物与[Me4N]SCF3之间的三氟甲硫基化反应以及其可能的机理
Scheme 18 PdⅠ-catalyzed trifluoromethylthiolation of aryl iodides/bromides with [Me4N]SCF3 and its proposed mechanism

图式 20 (bpy)Cu(SCF3)与芳基碘化物/溴化物、苄基溴化物、酮以及卤代烷之间发生的三氟甲硫基化反应
Scheme 20 Trifluoromethylthiolation of aryl bromides/ iodides,benzyl bromides,ketones and haloalkanes with (bpy)Cu(SCF3)

图式 21 α,β-不饱和羰基化合物以及乙烯基三氟甲基硫酯的合成
Scheme 21 Synthesis of α,β-unsaturated carbonyl compounds and vinyl trifluoromethyl thioethers

图式 22 α-溴代酮的三氟甲硫基化反应及其可能的机理
Scheme 22 Trifluoromethylthiolation of α-bromoketones and its proposed mechanism

图式 23 烷基亲电体的三氟甲硫基化反应及其可能的机理
Scheme 23 Trifluoromethylthiolation of alkyl electrophiles and its proposed mechanism

图式 24 溶剂控制的MBH碳酸酯与O-十八烷基-S-三氟甲基碳酸酯之间发生的三氟甲硫基化反应
Scheme 24 Trifluoromethylthiolation of MBH carbonates with O-octadecyl-S-trifluoromethylcarbonate controlled by solvent

图式 25 [Me4N]SeCF3与硼酸、硼酸频哪醇酯以及末端炔烃之间发生的三氟甲硒基化反应
Scheme 25 Trifluoromethylselenolation of boronic acids,boronic pinacol esters and terminal alkynes with [Me4N]SeCF3

图式 26 芳基碘化物和烷基溴化物的三氟甲硒基化反应及其可能的机理
Scheme 26 Trifluoromethylselenolation of aryl iodides,alkyl bromides and its proposed reaction pathway

图式 27 [(bpy)Cu(SeCF3)]2与卤代芳烃和卤代烷烃之间发生的三氟甲硒基化反应
Scheme 27 Trifluoromethylselenolation of aryl halides and haloalkanes with [(bpy)Cu(SeCF3)]2

图式 28 炔丙基/烯丙基三氟甲基硒醚以及三氟甲硒基取代的α,β-不饱和羰基化合物的合成
Scheme 28 Synthesis of propargylic/allylic trifluoromethyl selenoethers and trifluoromethylseleno-substituted α,β-unsaturated carbonyl compounds

图式 29 [(bpy)Cu(SCF3)]2与末端炔烃和乙烯基卤化物之间发生的三氟甲硒基化反应
Scheme 29 Trifluoromethylselenolation of terminal alkynes and vinyl halide with [(bpy)Cu(SCF3)]2

图式 30 (Aryl-BIAN)Ag(OCF3)•THF的制备及其与苄基溴的反应
Scheme 30 Preparation of the (Aryl-BIAN)Ag(OCF3)•THF and its reaction with benzyl bromides

图式 31 邻三氟甲氧基苯胺衍生物的合成
Scheme 31 Synthesis of ortho-Trifluoromethoxylated aniline derivatives

图式 32 官能团化的吡啶和嘧啶的区域选择性三氟甲氧基化反应
Scheme 32 Regioselective trifluoromethoxylation of functionalized pyridines and pyrimidines

图式 34 [(phen)2Cu][OCH2CF3]的合成及其与芳基和杂芳基溴化物的反应
Scheme 34 Synthesis of [(phen)2Cu][OCH2CF3] and its reaction with aryl and heteroaryl bromides

图式 35 α-溴代酮的三氟乙氧基化反应以及三氟乙氧基取代的吡唑的合成
Scheme 35 Trifluoroethoxylation of α-bromoketones and the synthesis of trifluoroethoxy-substituted pyrazole

图式 36 钯催化的三氟乙醇与芳基溴代物以及溴代查尔酮之间的三氟乙氧基化反应
Scheme 36 Palladium-catalyzed trifluoroethoxylation of aryl bromides and bromo-chalcones with trifluoroethanol

图式 40 二氟卡宾参与的脂肪族卤化物[18F]三氟甲硫基化反应
Scheme 40 Difluorocarbene-mediated [18F]trifluoromethylthiolation of aliphatic halides

图式 41 银Ⅰ参与的卤交换[18F]氟化反应得到[18F]芳基-SCF3、OCF3以及OCHF2
Scheme 41 AgⅠ-mediated halex 18F-fluorination towards [18F]aryl-SCF3,OCF3 and OCHF2

图式 43 18F-六氟苯以及18F-RGD-TFB-RGD的放射性合成
Scheme 43 Radiosynthesis of 18F-hexafluorobenzene and 18F-RGD-TFB-RGD

图式 44 [18F]FBBA的制备以及其与聚合物PEG-thiol-acid的反应
Scheme 44 Preparation of [18F]FBBA and its reaction with polymer PEG-thiol-acid

图 2 p-SCN-NOTA和p-SCN-NODA的化学结构
Figure 2 Chemical structures of p-SCN-NOTA and p-SCN-NODA

图式 45 18F 标记NODA-MATBBN得到18F-AlF-NODA-MATBBN
Scheme 45 18F labeling of NODA-MATBBN to 18F-AlF-NODA-MATBBN
-


 下载:
下载:















































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 5352
- HTML全文浏览量: 1441
 下载:
下载:
