 图 1
常见的铁配合物催化剂类型
Figure 1.
Type of iron complexe catalysts
图 1
常见的铁配合物催化剂类型
Figure 1.
Type of iron complexe catalysts
引用本文:
何心伟, 胡小倩, 陶佳佳, 韩光, 商永嘉. 铁配合物催化的有机反应研究进展[J]. 有机化学,
2016, 36(7): 1465-1483.
doi:
10.6023/cjoc201601007

Citation: He Xinwei, Hu Xiaoqian, Tao Jiajia, Han Guang, Shang Yongjia. Progress in Iron Complexes-Catalyzed Organic Reactions[J]. Chinese Journal of Organic Chemistry, 2016, 36(7): 1465-1483. doi: 10.6023/cjoc201601007
Citation: He Xinwei, Hu Xiaoqian, Tao Jiajia, Han Guang, Shang Yongjia. Progress in Iron Complexes-Catalyzed Organic Reactions[J]. Chinese Journal of Organic Chemistry, 2016, 36(7): 1465-1483. doi: 10.6023/cjoc201601007
铁配合物催化的有机反应研究进展
English
Progress in Iron Complexes-Catalyzed Organic Reactions
Abstract:
In recent years, metal and their complexes-catalyzed organic reactions have received much attention in organic chemistry due to their rapidity and efficiency. Iron complexes have attracted much attention from chemical society due to the advantages of high catalytic activity and selectivity. The advances of iron complexes-catalyzed organic reactions are reviewed, such as polymerisation reactions, hydrosilylation/hydroboration, cycloaddition, redox reactions, cross-coupling reactions and 1,4-additions, and the prospects of its development are forecasted.
-
Key words:
- catalysis
- / iron complexe
- / organic reaction
- / research progress
-
铁是地球上储量仅次于铝的元素之一(约4.7 wt%),原子量为55.847,在元素周期表中位于第Ⅷ族,其电子排布是[Ar]3d64s2,易失去4s轨道上的两个电子和3d轨道上的一个电子,成为二价铁和半充满稳定结构的三价铁离子,由于铁具有良好的可变氧化态,易于与氮、氧、磷等元素配位形成稳定的配合物. 1949年,Reppe[1]将五羰基配位的铁催化剂应用于烯烃的胺甲基化反应而使得铁配合物催化剂成为有机化学研究工作者关注的焦点,随后出现了诸如二茂铁(1951年)[2]、Collman试剂Na2Fe(CO)4 (1959年)[3]等关于铁配合物的研究. 此后许多关于铁催化剂的设计合成及应用研究取得了系列重要研究成果,2004年,Bolm等[4]综述了铁催化剂的重要研究进展,随后有关铁催化的有机反应的综述文章[5]及专著[6]相继出版. 本文综述了2000~2015年期间发展的有关铁配合物催化剂类型及其催化的有机反应研究方面所取得的进展.
1 常见铁配合物催化剂类型
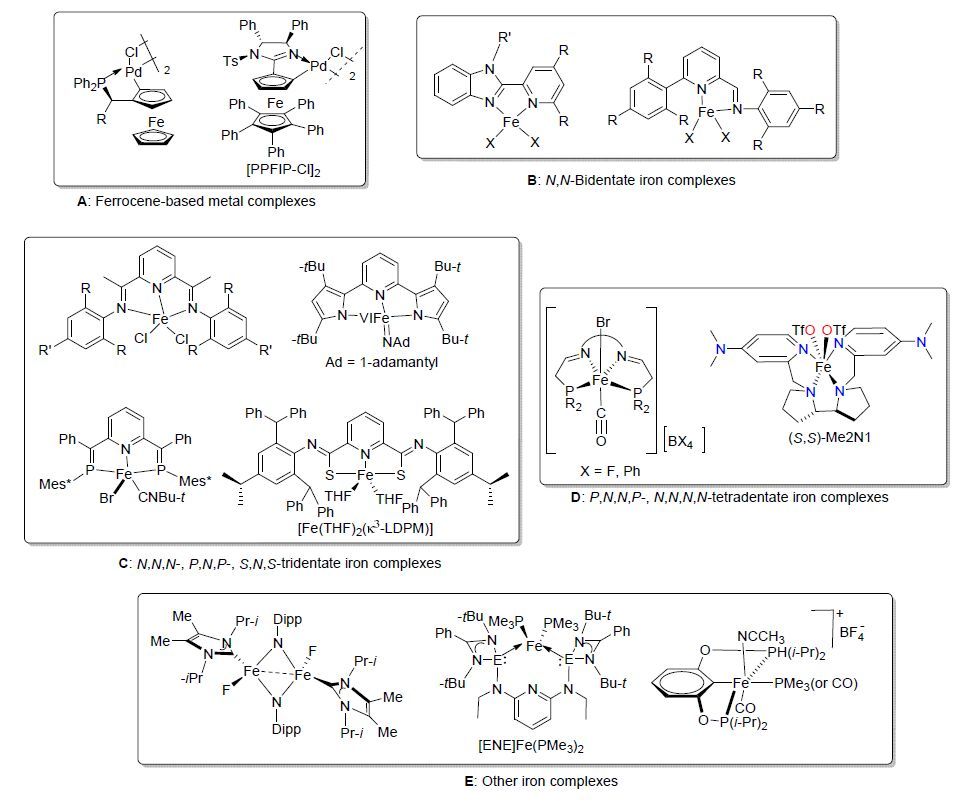
近年来,有关铁配合物的研究受到有机化学工作者的广泛关注,研究的热点集中在配体结构的设计合成及其催化的有机反应等方面. 因此,设计合成结构新颖、易配位的过渡金属配合物配体成为配位化学发展的热点,近年来发展的铁配合物类型主要有二茂铁及其金属配合物[7](图 1,A)、双齿配位铁配合物[8](图 1,B,如N,N-双齿)、三齿铁配合物[9](图 1,C,如吡啶双亚胺N,N,N-三齿、吡啶双吡咯N,N,N-三齿、吡啶双亚磷P,N,P-三齿以及S,N,S-三齿铁配合物等)、四齿配位铁配合物[10](图 1,D,如P,N,N,P-四齿、N,N,N,N-四齿)、N-杂环卡宾(NHC)铁配合物[11]及其他结构新颖的铁配合物(图 1,E).
图 1
常见的铁配合物催化剂类型
Figure 1.
Type of iron complexe catalysts
多数铁配合物由于合成简便,易与含O,N,P等配位原子的配体配位而形成稳定的配合物及具有高催化活性、高选择性等优点在有机合成中有着广泛的应用,如催化聚合反应、硅/硼氢化反应、环加成反应、氧化与还原反应、交叉偶联反应以及1,4-加成反应等.
2 聚合反应
近年来,聚乙烯、聚丙烯、聚苯乙烯等传统的烯烃聚合物及其与环烯烃、极性分子的共聚体在科学界和工业界备受关注. 一方面,大量聚烯烃已成为商品,深入到人们生活的方方面面; 另一方面,许多具有新型功能的聚合物不断涌现,并成为潜在的现有产品的替代品或全新的功能材料. 烯烃在催化剂的作用下,转变成聚烯烃,并随着催化剂结构的变化,聚烯烃产物的性能也不断改变,因此催化剂是研究聚烯烃材料的核心和原动力. 有机金属烯烃聚合物催化剂的出现不仅可以在温和的反应条件下得到用传统方法通常要在高温高压或自由基引发等苛刻条件下方可实现的烯烃聚合,而且这些新型催化剂可以剪裁聚合物的微结构,实现聚合物物理性质的调控,使得化学家、材料学家可以在分子水平上设计新型功能聚烯烃材料和改善已有聚合物的性能.
2.1 炔烃聚合
2011年,McGuinness等[12]首次将一种2,6-二芳亚氨基吡啶铁(Ⅱ)配合物1作为催化剂应用于乙炔聚合反应 (Eq. 1),研究结果表明,该类催化剂催化活性极高,催化剂用量较传统催化剂可降低三个数量级,且在较低的催化剂用量下催化乙炔聚合反应在催化剂表面可以得到乙炔聚合物膜. 但是该类铁配合物催化剂虽能很好地催化乙炔的聚合反应,却无法实现对聚合物链的控制合成,有望通过对配体结构的进一步修饰来完成.
2.2 烯烃聚合
2013年,Sun等[13]设计合成并表征了一种双吡啶双亚胺铁配合物催化剂2,并将该配合物催化剂应用于催化烯烃聚合反应(Scheme 1). 结果发现,该类催化剂具有较高的催化活性,在甲基铝氧烷(MAO)或改良的甲基铝氧烷(MMAO)共催化作用下,其催化活性可高达 1.3×107 g•mol-1(Fe)•h-1,与类似单金属和其他双核金属配合物催化剂相比,该双金属催化剂具有很高的热稳定性,在70 ℃高温下同样具有较高的催化活性,且催化剂寿命也得到相应提高 ,运用该系列催化剂催化乙烯聚合时,乙烯聚合度较高,聚合后的聚乙烯材料的分子量可达到383 kg/mol (Mw). 该类铁配合物催化剂合成方法简便,在工业上具有潜在的应用前景.
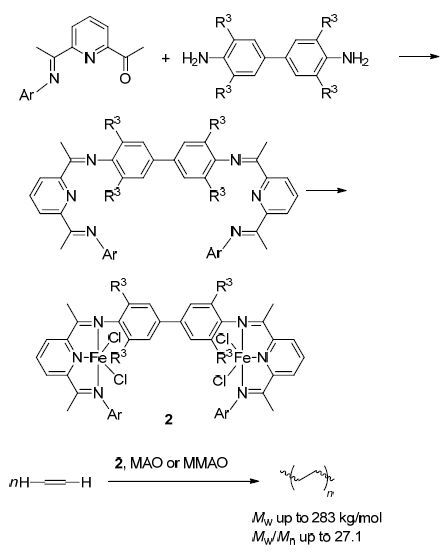 图 图式1
双吡啶双亚胺铁配合物的合成及其催化烯烃聚合反应
Figure 图式1.
Synthesis of bispyridine-bisimino iron complexes and catalysis of olefin polymerization
图 图式1
双吡啶双亚胺铁配合物的合成及其催化烯烃聚合反应
Figure 图式1.
Synthesis of bispyridine-bisimino iron complexes and catalysis of olefin polymerization
2014年,Sun等[14]设计合成了系列2-(1-芳亚胺基)-乙基-9-芳亚胺基-5,6,7,8-四氢环庚烷并吡啶配体(Scheme 2),并与氯化亚铁进行反应合成得到系列相应的Fe(Ⅱ)配合物3a~3e,配合物单晶结构和密度泛函(DFT)理论计算证实,该配合物中心原子铁周围的配体形状为扭曲的双锥体几何形状. 该类配合物作为催化剂在乙烯聚合反应中,显示了更高的催化活性(高达 1.56×107 g PE•mol-1(Fe)•h-1),在其催化活性及相关反应参数的研究中发现配体中环庚烷的环张力对其催化活性有较大影响. 利用该类铁配合物催化的乙烯聚合反应,可通过控制实验参数来得到不同分子量和分散性的聚乙烯产品,从而使得该配合物催化剂有望在工业上实现大规模应用.
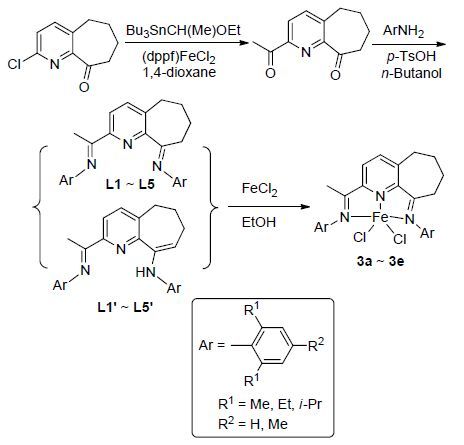 图 图式2
吡啶双亚胺铁配合物的设计合成
Figure 图式2.
Synthesis of bisnminopyridine iron complexes
图 图式2
吡啶双亚胺铁配合物的设计合成
Figure 图式2.
Synthesis of bisnminopyridine iron complexes
3 硅/硼氢化反应
不对称硅/硼氢化反应可用于合成手性醇、胺、硅烷等手性化合物,自从20世纪70年代初发现以来,有关前手性酮、烯烃、亚胺等底物催化不对称硅氢化反应的研究报道呈现出两个研究高峰. 第一个研究高潮出现于20世纪20年代中期至80年代初,研究重点侧重于各种反应底物,手性催化剂的开发研究报道不多,主要是采用其他催化不对称反应如不对称加氢、氢甲酰化反应中常用的手性催化剂. 第二个研究高潮出现于20世纪90年代初,至今方兴未艾,研究重点侧重于各种高效手性催化剂配体的合成,在酮、烯烃和亚胺等的不对称硅/硼氢化反应研究中均合成出了多种具有高活性、高手性诱导效应的手性配体.
3.1 烯烃的不对称硅/硼氢化反应
烯烃类化合物不对称硅/硼氢化反应是最早被研究的,一般是经硅/硼氢化反应以后,用双氧水进行氧化处理,可得到手性醇. 由于其反应比较复杂,立体选择性和位置选择性要兼顾,增加了研究的困难性,同样是到了20世纪90年代后,找到了高效的手性催化剂,尤其是含有手性轴的联萘类配体的发现和应用,才真正使烯烃的不对称硅/硼氢化反应研究取得了突破性进展,并重新受到重视. 近年来铁配合物催化剂的合成及应用得到快速发展,由于其易于合成,催化活性和选择性高,研究工作者逐渐将铁配合物催化剂应用于烯烃的不对称硅/硼氢化反应中, 并且取得了良好催化效果.
3.2 酮的硅氢化反应
酮类化合物的不对称硅氢化反应研究最早,也最为系统. 近年来不断有高效手性配体或催化剂的研究报道出现,有的已达到几乎定量的化学产率和光学纯度[17]. 2014年,Driess等[11b]报道了一种新颖的双金属烯基吡啶钳形(ENE)铁配合物6的合成及其催化苯乙酮的硅氢化反应研究. 首先从2,6-二乙胺基吡啶出发,分别与2 equiv.的正丁基锂和N,N′-二叔丁基苯甲脒基氯硅烯或N,N′-二叔丁基苯甲脒基氯锗烯反应得到相应的SiNSi型或GeNGe型配体,该配体室温下与1.1 equiv.的氯化亚铁作用四氢呋喃溶剂中反应3 h即可得到相应的钳形铁配合物[SiNSi]FeCl2,然后在PMe3作用下与KC8反应可以得到一种富电子的Fe(0)配合物[SiNSi]Fe(PMe3)2. 配合物[SiNSi]Fe(PMe3)2在苯乙酮的硅氢化还原反应中表现出良好的催化活性(Eq. 3),结果发现,底物取代基的电子效应对反应的影响不大,无论是含有强供电子基或强吸电子基的对位取代苯乙酮,均能以优异的产率获得相应的α-苯乙醇产物,但是底物的普适性和催化剂的催化作用机理仍需进一步探索,以找到底物取代基效应的一般规律和配合物在羰基的硅氢化反应过程中的作用.
2014年,Guan等[18]报道了一种POCOP钳形阳离子铁配合物{[2,6-(i-Pr2PO)2C6H3]Fe(CO)(PMe3)(CH3CN)}+[BF4]- (7,[Fe]+-BF4-,trans CO/CH3CN)在催化醛酮的硅氢化反应中的应用(Eq. 4),该配合物催化剂在1 mol%的用量下即可实现苯甲醛的定量转化,该工作首次实现了阳离子型铁配合物催化剂在醛酮的硅氢化反应中的应用.
2015年,Huang等[19]设计合成了一种新颖的手性噁唑啉吡啶亚胺配体(IPO ligands),该配体中含有两个二苯甲基和一个叔丁基等三个具有较大位阻的取代基,与FeBr2进行配位得到一种相应的铁(Ⅱ)配合物(S)-8 (Scheme 4),并将其应用于芳基酮的不对称硅氢化反应. 由于配体中的大位阻作用而使得该铁配合物具有较高的催化活性(>99%)和对映选择性(ee>93%). 结果发现,该催化剂对各种取代基的芳基乙酮或脂肪族酮均有良好的催化效果,均能以优异产率获得相应的醇产物,但是对脂肪族酮或二苯甲酮的催化对映选择性较差,因此需通过进一步修饰配体的空间位阻来提高其催化脂肪族酮硅氢化反应的对映选择性.
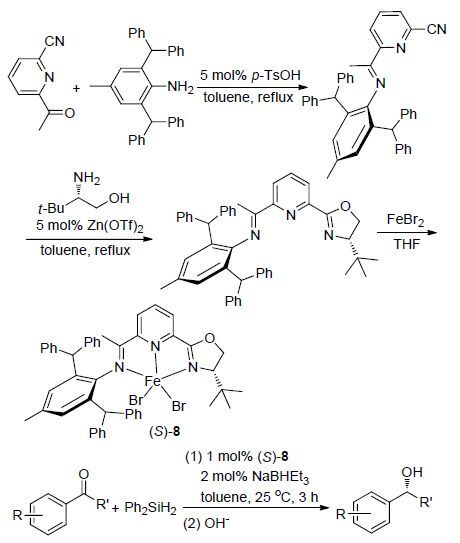 图 图式4
手性噁唑啉基吡啶亚胺铁配合物的设计合成及其催化活性
Figure 图式4.
Synehtsis of iminopyridine-oxazoline iron complexes and their catalytic activity
图 图式4
手性噁唑啉基吡啶亚胺铁配合物的设计合成及其催化活性
Figure 图式4.
Synehtsis of iminopyridine-oxazoline iron complexes and their catalytic activity
2015年,Gade等[20]设计合成了一种含双噁唑啉配体的铁烷基配合物9和铁烷氧基配合物10,将其应用于催化烷基酮和芳基酮的不对称硅氢化反应(Scheme 5). 结果发现,该类催化剂具有很高的催化活性和对映选择性(TOF=240 h-1,-40 ℃,ee>99%). 该配合物催化剂对底物的适用范围很广,即使底物酮中含有较大位阻的取代基时也可以高于95%的产率得到相应高ee值的手性产物,当底物酮中不含长链烷基时,催化的对映选择性更高(ee>99%). 该配合物中配体结构的进一步修饰及其催化机理的研究尚在继续中.
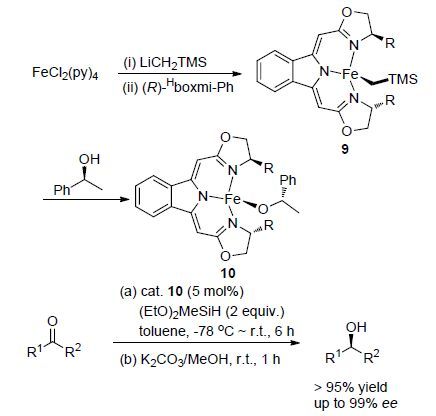 图 图式5
手性双噁唑啉基烷氧基铁配合物的设计合成及其催化活性
Figure 图式5.
Synthesis of bisoxazoline-alkoxido iron complexes and their catalytic activity
图 图式5
手性双噁唑啉基烷氧基铁配合物的设计合成及其催化活性
Figure 图式5.
Synthesis of bisoxazoline-alkoxido iron complexes and their catalytic activity
2015年,Nishiyama等[21]以手性和非手性的双噁唑啉三甲基硅基或锡基苯配体11 (abbreviated as phebox)与Fe(CO)5在光照条件下氧化加成反应得到系列NCN型钳形铁配合物[(phebox)Fe(CO)2(MMe3)] (12),其中M为Si或Sn,随后将该系列铁配合物应用于苯乙酮的不对称硅氢化反应中(Scheme 6),结果表明,系列配合物中M为Si的配合物的催化活性最高(产率>99%),遗憾的是该类配合物催化苯乙酮的硅氢化反应的对映选择性不高(ee>34%). 为进一步提高该类配合物的催化对映选择性,该课题组在对配体的结构作进一步的修饰研究.
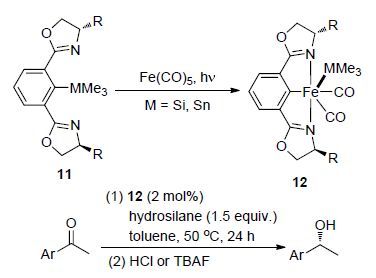 图 图式6
NCN 钳形铁配合物的设计合成及其催化活性
Figure 图式6.
Synthesis of NCN pincer iron complexes and their catalytic activity
图 图式6
NCN 钳形铁配合物的设计合成及其催化活性
Figure 图式6.
Synthesis of NCN pincer iron complexes and their catalytic activity
近年来,一类自身携带氢的铁配合物催化剂受到关注,此类催化剂将金属本身配位的氢作为硅氢化反应的氢源来完成还原反应. 2014年,Li等[22]利用二苯甲酮亚胺配体与Fe(PMe3)4或FeMe2(PMe3)4配位得到四种含氢铁的配合物13a~13d,随后将该铁配合物应用于催化醛酮的硅氢化反应. 结果表明,该类铁配合物催化剂对醛酮的硅氢化反应具有良好的催化活性,四种配合物催化剂相比较而言,配合物13a的催化效果最好,并对该类配合物的催化硅氢化反应的催化循环提出了一种可能的反应机理(Scheme 7). 提出的机理指出,含氢铁配合物催化剂13首先会脱去一个PMe3配体形成具有催化活性的不饱和中间体A,继而酮羰基与中心原子铁进行配位得到中间体B,进一步发生氢转移而转化为不饱和配合物中间体C,最后是硅烷还原酮生成硅醚产物并释放中间体A完成催化循环.
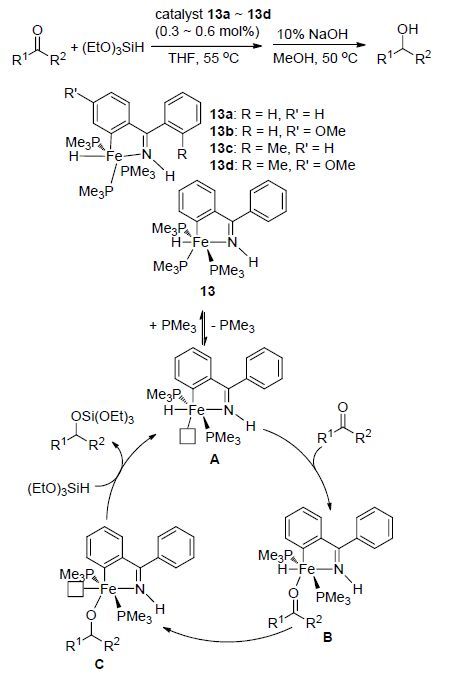 图 图式7
含氢铁配合物催化剂催化酮的硅氢化反应及其机理
Figure 图式7.
Mechanism of catalytic hydrosilylation with hydrido iron complexes
图 图式7
含氢铁配合物催化剂催化酮的硅氢化反应及其机理
Figure 图式7.
Mechanism of catalytic hydrosilylation with hydrido iron complexes
Li和Sun等[23]利用Csp3—H键和Si—H键活化设计合成了一类新颖的PXP (X=C,Si)钳形含氢铁配合物催化剂14和15,并将其应用于醛酮的不对称硅氢化反应中(Eq. 5),结果显示该类催化剂在醛酮的不对称硅氢化反应中显示了优异的催化活性,催化剂用量降低至0.3 mol%时仍有良好的催化活性,且催化底物的适用范围很广,各种类型的醛(芳醛、杂芳醛、苄基醛)和酮(芳基酮、杂芳基酮、二苯甲酮、环烷酮)均能很好地应用该类催化剂进行硅氢化还原得到相应的醇类化合物,但是当芳醛对位连有强供电子基(如甲氧基)时,仅能以中等左右收率得到目标产物,而使用二苯甲酮作底物时其反应性较差,产率仅有21%. 因该类催化剂的合成条件温和、实验操作简单使得其具有有潜在的应用价值和广泛的应用前景.
2015年,Sun等[24]又用类似的方法合成得到一种与上述配合物结构相似的POCOP钳形含氢铁配合物催化剂16 (Eq. 6),该类催化剂较上述催化剂的优点在于其催化活性更高,使用相同量的催化剂催化各种芳香醛或杂芳香醛时均能以优异收率得到相应的醇类化合物,基本不受芳环上取代基电子效应和空间效应的影响. 该催化剂虽也能很好地催化芳基酮的硅氢化反应,但其催化产率普遍偏低,仅有催化二苯甲酮和环己酮的硅氢化反应的分离产率有所提高. 由此可见,延长钳形铁配合物的钳形链长可提高其催化活性和催化选择性,但是合成该类铁配合物催化剂的时间较长(0 ℃下反应3 d),因此需要发展更为简单高效的合成方法来进一步扩大该类铁配合物催化剂的在有机反应中的实际应用.
随后,Li等[25]利用选择性C—F/C—H键活化的方法合成得到两种含氟二芳基甲亚胺的含氢铁配合物17,该配合物在醛酮的硅氢化还原反应中显示了更加优异的催化活性,不仅降低了反应的时间,而且大大提高了反应的转化率和产物的分离收率. 研究发现,底物适用范围很广,各种取代基的芳香醛和芳香酮都能获得优异的收率,即使脂肪族的环己酮和环戊酮也有中等以上收率. 研究中还发现,随着催化剂用量的减少,该种含氢铁配合物催化剂对芳香醛和芳香酮的硅氢化反应中表现出良好的化学选择性,当催化剂用量降低至0.3 mol%时可选择性还原芳香醛(Eq. 7),而芳香酮不被还原.
为进一步研究底物的适用范围,他们用该配合物测试了对α,β-不饱和醛的硅氢化还原反应(Eq. 8),在相同条件下也显示良好的催化活性,且当α,β-不饱和醛的α位连有取代基时,其反应效果较好,产率可高达93%. 即使使用3-苯基丙炔醇作为反应底物时也能以88%产率获得相应的产物.
近期,该课题组[26]设计合成了一种P,S-螯合的含氢铁(Ⅱ)配合物19,并将其应用于醛酮的硅氢化还原反应中(Eq. 9),结果发现,该类配合物具有良好的催化活性,对苯环上连有供电子的芳香醛时其反应性较差,但对二苯甲酮的硅氢化反应较之前的PC(Si)P钳形或POCOP钳形铁配合物催化剂催化活性有较大提高.
3.1.2 烯烃的不对称硅氢化反应
2015年,Lu等[16]将上述配合物的配体进行了结构修饰,合成了几种新型噁唑啉吡啶亚胺铁(Ⅱ)配合物5a~5e,将其应用于烯烃的反马氏加成硅氢化反应(Eq. 2),以良好区域选择性和对映选择性(ee>99%)合成得到系列手性硅烷的衍生物. 该类铁配合物催化剂消除了对底物取代基电子效应和空间效应的影响,即使α-位取代的芳基乙烯苯环上连有强吸电子的三氟甲基或α-位连有较大位阻的叔丁基时,产率仍能保持在95%以上,且产物的ee值也可达到99%. 同时通过偶联环化、氧化开环和分子内Mitsunobu反应等实现了烯烃硅氢化产物在(-)-thespesone和(S)-(+)-3-methyl dihydrobenzofuran等天然产物合成中的应用.
3.1.1 烯烃的不对称硼氢化反应
2014年,Lu等[15]设计合成了一种手性噁唑啉吡啶双亚胺铁(Ⅱ)配合物4,并将其应用于1,1-二取代烯烃的反马氏硼氢化反应(Scheme 3). 结果表明,该催化剂催化的烯烃硼氢化反应具有较高的区域选择性和对映选择性,配体中的吡啶亚胺基团可以稳定铁的配位,利用手性噁唑啉基团控制烯烃硼氢化反应的对映选择性. 该催化剂具有良好的反应性能和稳定性,可实现温和条件下系列手性含硼衍生物的高效合成,但是反应仍存在以下局限性,如只适用于α-位取代的苯乙烯衍生物,当苯环上连有强吸电子基或α-位连有位阻较大的基团时,反应的对映选择性虽能得到保持,但产率却出现明显下降等.
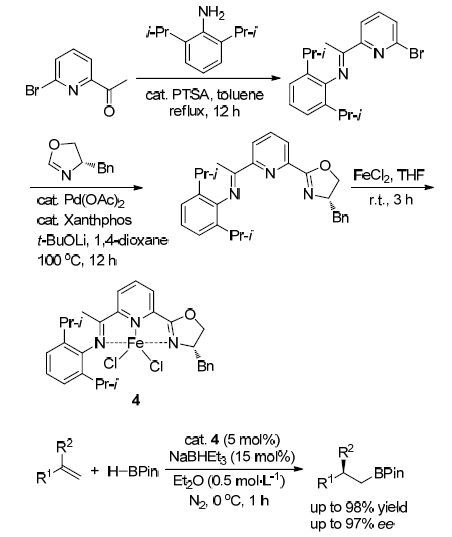 图 图式3
手性噁唑啉基吡啶双亚胺铁配合物的设计合成及其催化活性
Figure 图式3.
Synehtsis of chiral iminopyridine oxazoline iron complexes and catalytic activity
图 图式3
手性噁唑啉基吡啶双亚胺铁配合物的设计合成及其催化活性
Figure 图式3.
Synehtsis of chiral iminopyridine oxazoline iron complexes and catalytic activity
4 环加成反应
4.1 不对称[3+2]环加成反应
二茂铁是最早发现的铁的配合物,其衍生物此后一直作为配合物催化剂或金属配体使用,并得到快速发展. 近年来基于二茂铁衍生物为配体的不对称催化反应受到有机化学家的广泛关注. 2010年,Hu等[27]以N,N-二甲基- (R)-1-(S)-2-二苯基膦基二茂铁乙胺为原料合成得到系列二茂铁基P,S-配体20,随后将该配体与[Cu(CH3CN)4]- ClO4或AgOAc一起作为催化剂催化亚氨基酯和2-环戊烯酮或查尔酮的[3+2]环加成反应(Scheme 8). 结果表明,应用[Cu(CH3CN)4]ClO4/(Rc,SFc)-20f催化体系催化2-环戊烯酮与亚氨基酯的[3+2]环加成反应时,以近乎完美的对映选择性(>99% ee)得到唯一endo-型加成产物,而应用AgOAc/(Rc,SFc)-20f催化体系催化查尔酮与亚氨基酯的环加成反应时,显示了良好的endo/exo选择性(endo/exo=91/9~96/4)和较高的对映选择性(ee>98%). 这一成果在二茂铁衍生物作为配体的金属催化的五元杂环化合物的合成中具有重要意义.
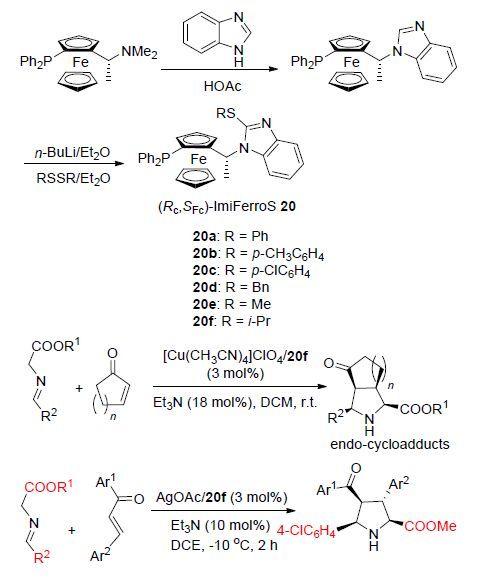 图 图式8
新型手性二茂铁基P,S-配体的合成及其催化不对称 [3+2]环加成反应
Figure 图式8.
Synthesis of new chiral ferrocenyl P,S-ligands and catalytic asymmetric [3+2] cycloaddition reactions
图 图式8
新型手性二茂铁基P,S-配体的合成及其催化不对称 [3+2]环加成反应
Figure 图式8.
Synthesis of new chiral ferrocenyl P,S-ligands and catalytic asymmetric [3+2] cycloaddition reactions
2015年,Scheidt等[28]设计合成了一种二茂铁基N-杂环卡宾(NHC)手性咔唑并吡啶盐催化剂(+)-21,并将其应用于催化苯甲酰甲酸酯和肉桂醛的环加成反应中(Eq. 10). 结果发现,与同类非二茂铁基催化剂相比,该二茂铁基N-杂环卡宾手性咔唑并吡啶盐催化剂具有良好的催化活性和对映选择性,主要是由于NHC中引
入二茂铁基团后有效增加了NHC的电子效应. 同时该二茂铁基NHC催化剂与CuCl进行配位可得到一种二茂铁基NHC-铜配合物,其对烯烃的不对称还原硼基化偶联反应具有良好的催化活性和对映选择性,进一步说明该二茂铁基N-杂环卡宾(NHC)手性咔唑并吡啶盐不仅可以作为Lewis碱催化剂使用,而且是一种良好的过渡金属配合物催化剂的配体.
4.2 不对称[3+3]环加成反应
2012年,Hu等[29]报道了一种三齿二茂铁基P,N,N-配体22与Cu(OAc)2•H2O共催化的炔丙基酯与环状烯胺的不对称[3+3]环加成反应(Eq. 11),该催化体系在温和条件下可以非常高的非对映选择性选择性(endo/exo>98/2)和对映选择性(up to 98% ee)得到endo-加成产物,该反应底物适用范围广,不仅具有较高的收率及较好的非对映选择性和对映选择性,通过该反应合成的产物在进一步衍生化的过程中其构型可以得到有效保持. 该反应发展了一种条件温和、底物适用范围广、高立体选择性和高对映选择性的合成具有重要光学活性价值的双环[n.3.1]骨架化合物的方法.
4.3 [2+2+2]环加成反应
环加成反应可以一步同时构建多个化学键,是目前国内外研究最为活跃的领域之一. 相比于传统方法,过渡金属催化的[2+2+2]环加成反应是合成多取代苯或吡啶衍生物的有效手段. 2015年,Fürstner等[30]报道了一种基于烷基化的二磷铁配合物23催化的炔烃或炔烃与氰基化合物的[2+2+2]环加成反应合成六取代苯或六取代吡啶的研究(Scheme 9). 该反应在二烷基溴化镁和二炔烃的作用下,[L2FeX2]型铁配合物23被还原得到二烯或二炔配位的[L2Fe(alkene)2]或[L2Fe(diene)]型零价铁配合物中间体,继而再与一分子炔烃或氰基化合物发生环加成反应得到目标产物,产率可高达97%. 其最大的意义在于首次使用铁配合物催化剂来催化炔烃的三聚反应,通过该反应不仅可以实现六取代苯的高效合成,而且可以高效合成五取代吡啶衍生物. 该反应条件相对较温和,原料容易获得,因此极具应用潜力.
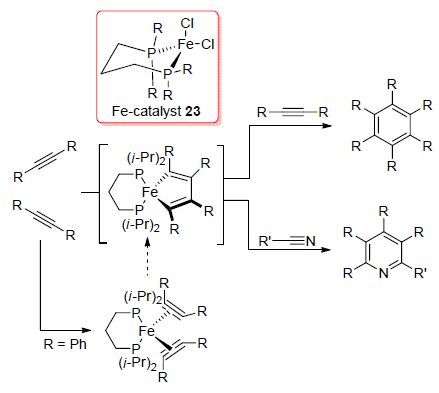 图 图式9
[L2FeX2]型铁配合物及其催化炔烃的[2+2+2]环三聚反应
Figure 图式9.
Iron complexes of type [L2FeX2] and their catalyzed the [2+2+2] cyclotrimerization of alkynes
图 图式9
[L2FeX2]型铁配合物及其催化炔烃的[2+2+2]环三聚反应
Figure 图式9.
Iron complexes of type [L2FeX2] and their catalyzed the [2+2+2] cyclotrimerization of alkynes
5 氧化与还原反应
5.1 氧化反应
5.2 不对称还原反应
5.1.1 烯烃不对称环氧化反应
光学活性环氧化合物是非常高效的有机合成中间体,其结构中的三元环具有特殊的张力,可通过选择性开环和官能团转化等反应合成许多有价值的手性化合物和天然产物. 环氧化合物也是一类具有良好粘结、耐腐蚀、高绝缘强度等性能的材料,已广泛应用于许多工业领域. 因此,探索制备环氧化合物特别是具有光学活性的环氧化合物一直是有机化学研究的重点. 2013年,Costas等[10b]设计合成了几种non-heme铁配合物催化剂24,在催化量的有机羧酸存在下将该配合物应用于烯烃与双氧水的不对称环氧化反应(Eq. 12). 结果表明,该配
合物催化剂具有较高的对映选择性,且作用于中心铁原子的配体的电子效应可与有机羧酸协同作用可有效促进O—O键断裂,在较短的时间内即可以较高产率得到相应的环氧化产物,该催化剂同时显示了较高的化学选择性,不足之处是该催化剂对末端烯烃的环氧化反应没有催化活性.
2015年,该课题组[31]将上述配合物配体结构进行了修饰,合成得到几种新颖的non-heme铁配合物催化剂25a~25c (PDP-Fe),在N-保护的氨基酸衍生物N-Npha-ILeu-OH作为共配体的作用下,将该配合物应用于α-取代苯乙烯的不对称环氧化反应中(Eq. 13). 结果表明,当以氨基酸作为共配体与PDP-Fe型铁配合物共同作用时,氨基酸可以与中心铁原子协同作用,能够有效提高双氧水的活性,催化一些难以环氧化的末端烯烃而得到相应的环氧化产物,并具有较高的对映选择性(up to 97% ee). 该催化剂显示了良好的催 化活性,具有底物适用范围广、实验条件温和、反应时间短等优点,发展了一种高效催化末端烯烃环氧化反应的配合物催化剂.
5.1.2 芳环的氧化开环反应
2015年,Paine等[32]报道了以一种三齿铁配合物为催化剂,O2存在下的邻氨基苯酚(2-Aminophenol,AP)氧化芳环开环合成吡啶-2-甲酸的研究(Scheme 10). 所使用的铁配合物为一种双吡啶-脲基三齿铁(Ⅱ)配合物26,该铁配合物首先与邻氨基苯酚发生配位反应分离得到一种2-氨基苯酚盐的铁(Ⅲ)配合物[(t-Bu-LMe)FeⅢ- (4,6-di-t-Bu-AP)](ClO4),该铁(Ⅲ)配合物继而与O2反应可使得4,6-二叔丁基-2-氨基苯酚中羟基邻位的C—C键区域选择性断裂,然后再发生羰基与氨基的缩合反应得到最终产物吡啶-2-甲酸衍生物. 研究结果表明,所使用的铁配合物中脲基基团是必须的,如果配体中不含脲基团,则配合物不具有使芳环C—C键断裂开环的催化活性,这将对邻氨基苯酚类化合物的氧化芳环开环反应配合物催化剂中配体结构的设计合成提供一种新的思路.
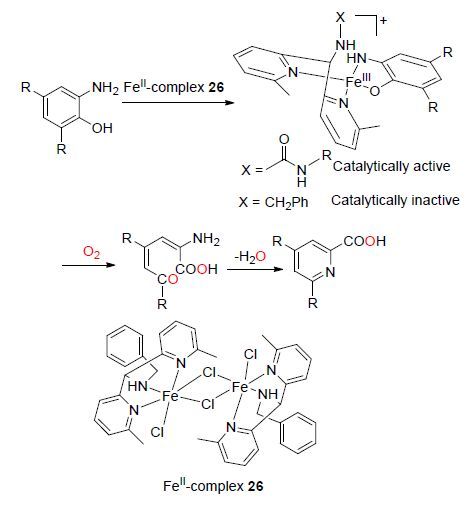 图 图式10
非血红素铁配合物催化的2-氨基苯酚与氧的芳环氧化开环反应
Figure 图式10.
Oxygenative aromatic ring cleavage of 2-amino- phenol with dioxygen catalyzed by a nonheme iron complex
图 图式10
非血红素铁配合物催化的2-氨基苯酚与氧的芳环氧化开环反应
Figure 图式10.
Oxygenative aromatic ring cleavage of 2-amino- phenol with dioxygen catalyzed by a nonheme iron complex
5.2.2 其他还原反应
2012年,Morris课题组[46]设计合成了几种不对称P,N,N,P-型四齿铁配合物44,以异丙醇作为氢源条件下,将该类配合物催化剂应用于催化亚胺的不对称氢化还原反应,取得了良好的结果(Eq. 17). 温和条件下,以较低的催化剂用量和94%~99%的ee值将亚胺还原为二级胺类化合物,但是该催化剂对底物的空间位阻较为敏感,而几乎不受底物电子效应的影响.
2015年,Hu等[47]报道了一种新型PONOP-型钳形铁(Ⅱ)配合物催化的醛的加氢还原反应(Eq. 18). 设计合成的三种铁配合物催化剂中均含有2,6-二膦氧基吡啶配体,且均能在室温下很好的活化H2用于醛的加氢还原反应,其中配合物46和47的催化选择性较高. 当芳醛中含有烯基或酮羰基时,以配合物46和47作为催化剂时,即可实现室温下醛基的加氢还原,而保留烯基或酮羰基不受影响. 研究结果表明,该类配合物催化剂对底物的普适性非常好,无论是芳香醛或是脂肪醛在最优化条件下均能以较高产率被还原为相应的伯醇衍生物; 对芳醛而言即使醛基邻位含有强的供电子基(如OMe)或强的吸电子基(如NO2)也能得到良好以上产率的催化效果. 且当以甲酸钠作为氢源时, 配合物46和47也能很好的催化醛的氢化还原反应得到相应的苯甲醇的衍生物.
Kuwano等[48]以手性二茂铁双磷(Josiphos ligand)为配体48,分别与碘、Yb(OTf)3、[IrCl(cod)]2组成催化体系(cod=1,5-cyclooctadiene),用于催化嘧啶类化合物的不对称加氢还原反应(Eq. 19). 结果表明,该催化体系可将各种4-取代嘧啶衍生物高产率地还原为1,4,5,6-四氢嘧啶化合物,其中三氟甲磺酸镧系金属对芳环的催化氢化具有很高的对映选择性(up to 99% ee),且具有较好的底物适用性.
5.2.1 苯乙酮的不对称还原反应
具有光学活性的α-苯乙醇作为一种基本的手性中间体,在手性化合物的合成中具有重要地位,α-苯乙醇在医药、农药、香料等化工行业有着极其广泛的应用[33]. 在不对称合成的研究中,手性α-苯乙醇的合成有着极为重要的作用,它可以被用来从一些非常简单的起始原料制备一些重要的手性中间体,也能够在一些复杂天然产物的不对称合成中用来创造最初的手性中心.
苯乙酮的不对称还原得到具有光学活性的α-苯乙醇的研究较早,也较为系统,Morris课题组在该领域坚持不懈地工作,并合成得到多种与P,N原子配位的三齿、四齿铁配合物,以异丙醇作为氢源,实现了苯乙酮的不对称氢化还原反应,以优异的对映选择性实现手性α-苯乙醇的不对称合成. 近年来该课题组[34]在六元和五元四齿P,N,N,P-型铁配合物的设计合成及其催化苯乙酮的不对称氢化反应领域开展了系统研究工作,取得了系列重要研究成果.
2011年,该课题组[35]报道了一种五元芳基取代的铁(Ⅱ)羰基P,N,N,P-型配合物26及其对苯乙酮的不对称催化氢化反应(asymmetric transfer hydrogenation,ATH)研究(Eq. 14). 首先合成了系列五元(S,S)-trans-[Fe(CO)- (Br)(PR2-CH2CHdNCH(Ph)CH(Ph)NdCHCH2-PR2)]X配合物(27a~27c,X=BPh4; 27d,27e,X=BF4),其中三种含有甲基取代的芳基基团[a: R=para-CH3C6H4; b: R=ortho-CH3C6H4; c: R=3,5-(CH3)2C6H3],另外两种含有三氟甲基取代的芳基基团(d: R=para-CF3C6H4; e: R=3,5-(CF3)2C6H3). 以异丙醇为氢源时,随后将这五种铁的配合物应用于催化苯乙酮的不对称氢化反应(ATH)研究,结果表明,配合物27b,27d,27e的对该反应几乎没有催化活性,而配合物27a却显示了非常高的催化活性,催化活性在28 ℃时高达30000 h-1,是已见报道的催化ATH反应的铁配合物催化剂中催化活性最高的催化剂. 配合物27c催化ATH反应时,显示了较高的对映选择性,ee值高达90%,是已见报道的选择性最高的铁配合物催化剂.
随后,该课题组[36]利用动力学及其研究方法又对上述铁配合物催化苯乙酮的氢化还原反应的催化机理进行了深入研究(Scheme 11),并撰文详细报道.
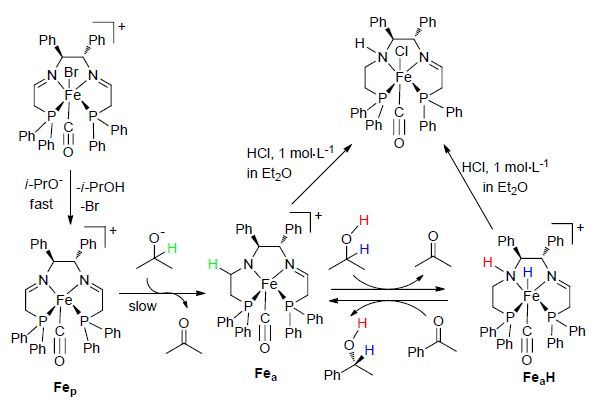 图 图式11
铁配合物催化苯乙酮的不对称氢转移反应及其机理
Figure 图式11.
Mechanism of asymmetric transfer hydrogenation of acetophenone using an iron(Ⅱ) complexes
图 图式11
铁配合物催化苯乙酮的不对称氢转移反应及其机理
Figure 图式11.
Mechanism of asymmetric transfer hydrogenation of acetophenone using an iron(Ⅱ) complexes
2012年,该课题组[37]用铁配合物trans-[Fe(CO)- (MeCN)(PPh2C6H4CH=NCH2)2-κ4P,N,N,P](BF4)2 (28) 在苯溶剂中与异丙醇钾作用得到一种结构不同寻常的铁配合物trans-[Fe(CO)(PPh2C6H4CH=NCH2CH2NH- CHC6H4PPh2)-κ5P,N,C,N,P][BF4] (29). 并将其应用于催化苯乙酮的氢化还原反应中(Scheme 12),进一步的研究发现,使用配合物28作为苯乙酮的氢化还原反应的催化剂时,在完成苯乙酮的氢化还原反应的同时配合物28也可以转化为配合物29,经密度泛函理论(DFT)计算及机理研究发现,反应体系中加入的异丙醇可作为苯乙酮氢化还原反应的氢源,首先异丙醇配位到中心铁原子上得到配合物30DFT,继而发生氢转移还原亚胺反应得到含活性氢的配合物31DFT,最后配合物31DFT完成苯乙酮的氢化还原反应而得到铁配合物29. 这一研究可以实现P,N,N,P-型铁配合物不同结构之间的相互转化,同时实现苯乙酮的催化氢化还原,这种“一箭双雕”的方法同时解决了配合物合成及其催化活性应用的问题.
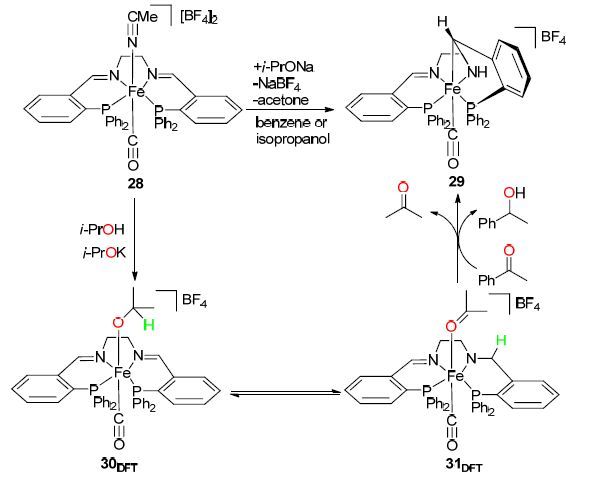 图 图式12
反式铁配合物的相互转化及催化苯乙酮的不对称氢转移反应
Figure 图式12.
Asymmetric transfer hydrogenation of acetophenone using the trans-iron(Ⅱ) complexes
图 图式12
反式铁配合物的相互转化及催化苯乙酮的不对称氢转移反应
Figure 图式12.
Asymmetric transfer hydrogenation of acetophenone using the trans-iron(Ⅱ) complexes
2013年,该课题组[38]从一种季磷盐出发,经多步反应合成得到一种部分饱和的胺/亚胺二磷配位的P,NH,N,P-型铁(Ⅱ)配合物33a~33c. 在异丙醇作氢源的条件下,将其用于还原酮或亚胺得到具有光学活性的醇或胺的衍生物(Scheme 13). 结果表明,该类催化剂在温和条件下,有很高的催化活性,28 ℃时其催化转化效率可高达200 s-1,同时对该催化反应提出了一种催化循环机理,并通过光谱学手段检测到了其中的铁的氢化物及酰胺的中间体.
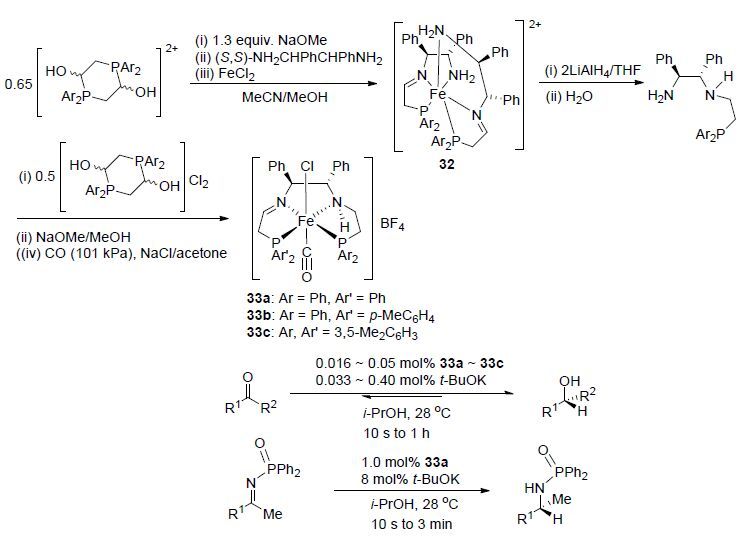 图 图式13
胺-亚胺双膦铁配合物的设计合成及其在酮和亚胺的不对称氢化反应中的应用
Figure 图式13.
Synthesis of amine(imine)diphosphine iron complexes and their application of the asymmetric transfer hydrogenation of ketones and imines
图 图式13
胺-亚胺双膦铁配合物的设计合成及其在酮和亚胺的不对称氢化反应中的应用
Figure 图式13.
Synthesis of amine(imine)diphosphine iron complexes and their application of the asymmetric transfer hydrogenation of ketones and imines
2012年,该课题组[39]发展了一种内球活化-外球催化的P,N,N,P-型铁配合物催化剂34,并利用DFT量子化学计算方法研究了其催化苯乙酮还原为α-苯乙醇的催化机理(Scheme 14),研究发现,在最初的活化阶段需要1 equiv.的异丙醇还原双胺配合物,继而逐步完成酮的氢化还原反应. 该配合物催化剂被认为在酮的加氢反应中具有较好的催化活性,同时在后续的氢负离子转移阶段具有良好的阻隔能力和较高的活化能,有望为其他更有效的铁催化剂的设计合成提供研究思路和理论依据.
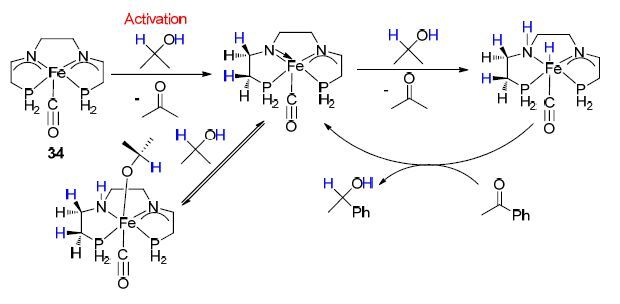 图 图式14
PNNP型铁配合物催化酮的氢转移反应机理
Figure 图式14.
Mechanism of transfer hydrogenation of ketones using iron(Ⅱ) PNNP eneamido complexes
图 图式14
PNNP型铁配合物催化酮的氢转移反应机理
Figure 图式14.
Mechanism of transfer hydrogenation of ketones using iron(Ⅱ) PNNP eneamido complexes
2014年,Morris课题组[40]设计合成了一种新型P,N,N,P-型铁配合物(S,S)-trans-[FeCl(CO)(PAr2-NH- N-PAr′2)]BF4 (35a: Ar,Ar′=Ph; 35b: Ar=Ph,Ar′=4-MeC6H4; 35c: Ar,Ar′=3,5-Me2C6H3),并应用于催化苯乙酮的不对称加氢还原反应(Scheme 15). 研究结果表明,在t-BuOK/2-propanol体系中,该催化剂对酮的不对称加氢还原反应具有良好的催化活性,当配体中含有较大的双二甲苯基膦基基团时,催化活性明显降低,但有良好的对映选择性. 同时结合DFT量子化学理论计算,研究了该类配合物催化剂的催化循环机理.
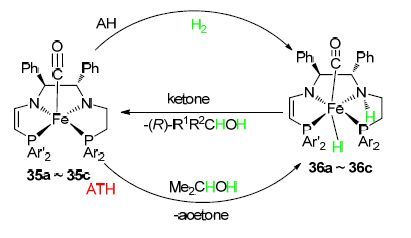 图 图式15
含胺-亚胺双膦配体的P-NH-N-P型铁配合物催化酮的不对称氢化和不对称转移氢化反应
Figure 图式15.
Iron catalysts containing amine(imine)diphosphine P-NH-N-P ligands catalyze both the asymmetric hydrogenation (AH) and asymmetric transfer hydrogenation (ATH) of ketones
图 图式15
含胺-亚胺双膦配体的P-NH-N-P型铁配合物催化酮的不对称氢化和不对称转移氢化反应
Figure 图式15.
Iron catalysts containing amine(imine)diphosphine P-NH-N-P ligands catalyze both the asymmetric hydrogenation (AH) and asymmetric transfer hydrogenation (ATH) of ketones
2014年,Morris课题组[41]合成了一类P,N,P′-型和P,NH,P′-型钳形手性铁配合物mer,trans-[Fe(P-N-P′)- (CO)2Br]BF4 (37),并将其应用于酮的加氢还原反应(Eq. 15). 首先合成了R取代基分别为苯基、苯甲基和异丙基的三种[Fe(PCy2CH2CH=NCH(R)CH2PPh2)(CO)2Br]BF4配合物,并用于苯乙酮加氢还原反应中,显示了较好的催化效果. 在对配体结构进行修饰时发现,当R基团换为含氮的基团时,所获得的铁配合物的催化活性大为降低. 为进一步拓展配体结构,该课题组又合成了系列六元P,N,P′-型和P,NH,P′-型铁配合物催化剂[Fe(P-N-P′)-(NCMe)3][BF4]2和[Fe(P-NH-P′)(NCMe)3][BF4]2,但是催化结果表明这两类催化剂均没有催化活性. 最后该课题组合成了一种非手性的mer,cis-Fe(PPh2-(o-C6H4)- CHNCH2CH2PPh2)(CO)Br2催化剂,催化苯乙酮的直接加氢还原反应. 结果表明,该类催化剂具有良好的催化活性和对映选择性(TOF up to 1980 h-1,ee up to 80%).
同年,该课题组[42]以配合物38为底物,经LiAlH4和醇处理后得到一种新颖的P,N,P'-型羰基含氢铁配合物催化剂39 (mer-trans-[Fe(Br)-(CO)2(P-CH=N-P')]- [BF4]) (P-CH=N-P'=R2PCH2CH=NCH2CH2PPh2,R=Cy,i-Pr和P-CH=N-P'=(S,S)-Cy2PCH2CH=NCH(Me)- CH(Ph)PPh2). 并将该系列配合物催化剂应用于催化酮和亚胺的不对称加氢还原反应(Scheme 16),结果表明,当催化剂配体中R=Ph基团时,配合物无催化活性. 其余手性铁配合物催化剂催化酮的不对称氢化还原反应中可以高达85%的ee值得到系列(S)-醇,催化剂的TOF达到2000 h-1,TON达到5000. 对亚胺的不对称催化氢化时ee值可达到90%,但TOF和TON则分别只有20 h-1和99. 同时该课题组利用NMR和DFT等手段对该配合物不对称催化的催化机理进行了研究.
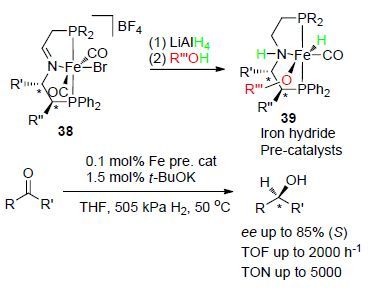 图 图式16
非对称PNP'钳形铁配合物催化剂及其催化酮的不对称氢化反应
Figure 图式16.
Iron(Ⅱ) complexes containing unsymmetrical PNP' pincer ligands and the catalytic asymmetric hydrogenation of ketones
图 图式16
非对称PNP'钳形铁配合物催化剂及其催化酮的不对称氢化反应
Figure 图式16.
Iron(Ⅱ) complexes containing unsymmetrical PNP' pincer ligands and the catalytic asymmetric hydrogenation of ketones
2015年,该课题组[43]又报道了一种从简单易得原料Ph2PH出发经多步反应合成得到铁配合物四氟硼酸化反式胺-亚胺二膦基一氯羰基铁(Ⅱ)配合物(40)的研究,以异丙醇作为氢源将其应用与芳乙酮和亚胺的不对称氢化还原反应(Scheme 17),并与传统的钌配合物催化剂的催化性能进行了比较. 结果表明,该类铁配合物催化剂催化还原芳乙酮的反应时催化效果较好,以良好至优秀产率和ee值得到系列手性(R)-醇的衍生物. 在催化含有二苯基膦酰基[P(O)Ph2]基团的亚胺时,该类配合物催化具有很高的催化性能和对映选择性,以更高的产率和ee值得到系列手性仲胺衍生物. 与传统的钌配合物催化剂相比,虽然钌配合物催化能够得到更高的ee值,但钌配合物催化往往需要更长的反应时间和较大的催化剂用量.
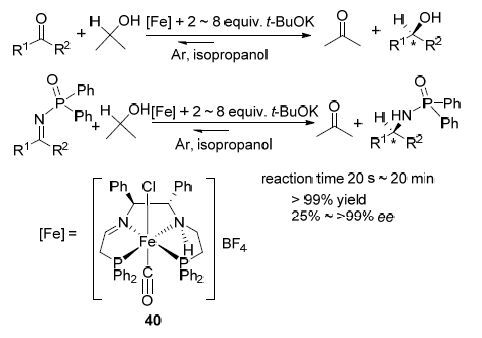 图 图式17
胺-亚胺双膦铁配合物催化酮和亚胺的不对称转移氢化反应
Figure 图式17.
Amine(imine)diphosphine iron catalyzed the asymmetric transfer hydrogenation of ketones and imines
图 图式17
胺-亚胺双膦铁配合物催化酮和亚胺的不对称转移氢化反应
Figure 图式17.
Amine(imine)diphosphine iron catalyzed the asymmetric transfer hydrogenation of ketones and imines
近期,该课题组[44]又利用上述方法合成得到上述配合物的一种衍生物41,以异丙醇作为氢源的条件下将其应用于催化芳乙酮的不对称还原反应,仅需0.02~0.2 mol%的催化剂用量,便可以40%~99%的转化率和高达99%的ee值得到系列手性α-苯乙醇衍生物(Eq. 16).
在Morris课题组研究工作的带动和鼓舞下,近年来不断有高效手性铁配合物催化剂应用于苯乙酮的不对称氢化反应的研究报道出现. 2014年,Mezzetti等[45]报道了一种含对称N2P2大环配体的双异腈铁(Ⅱ)配合物的合成,该配合物可通过简单易得的双异腈类似物合成得到(Scheme 18). 该类配合物催化剂的合成由相应的大环配体底物出发,先得到相应的乙腈铁(Ⅱ)配合物[Fe(MeCN)2(N2P2)](BF4)2 (42),继而与4 equiv. 的取代异腈反应即可得到双异腈铁(Ⅱ)配合物催化剂43,该类配合物催化剂应用于催化酮的氢化还原反应,结果表明,使用双叔丁基异腈铁(Ⅱ)配合物催化剂43a时,可以优秀产率得到相应的还原产物,且具有良好的对映选择性. 同时相对于已见报道的铁配合物催化剂来说,该催化剂对底物的容忍性更好,即使催化剂的用量降低至1 mol%时,其催化活性仍未见明显降低.
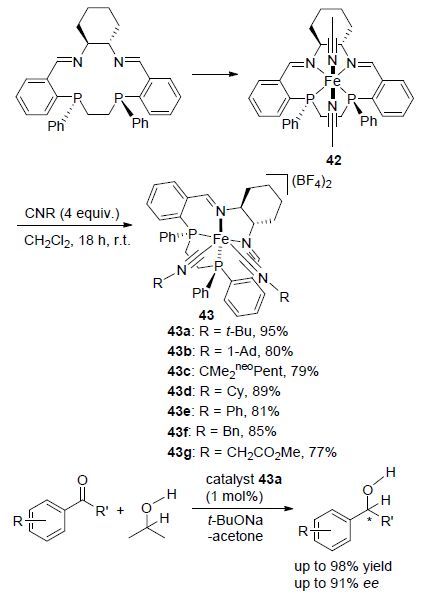 图 图式18
含手性N2P2大环配体的双异腈铁配合物的设计合成及其催化酮的对映选择性转移氢化反应
Figure 图式18.
Synthesis of diisonitile iron(Ⅱ) complexes with chiral N2P2 macrocycles and the catalytic of enantioselective transfer hydrogenation of ketones
图 图式18
含手性N2P2大环配体的双异腈铁配合物的设计合成及其催化酮的对映选择性转移氢化反应
Figure 图式18.
Synthesis of diisonitile iron(Ⅱ) complexes with chiral N2P2 macrocycles and the catalytic of enantioselective transfer hydrogenation of ketones
6 其他反应
6.1 1,4-加成反应
有机金属试剂对α,β-不饱和羰基化合物的不对称共轭加成是形成C—C键的重要反应之一,其加成产物可用于制备天然产物和具有生理活性的药物或中间体. 2014年,Pullarkat等[49]设计合成了一种二茂铁基磷配体,继而合成得到一种二茂铁基磷环钯配合物49,随后将其应用于环烯酮与芳基苯硼酸的不对称1,4-加成反应中(Eq. 20). 结果表明,该二茂铁配合物催化剂具有良好的催化活性和较高对映选择性,遗憾的是作者并未进一步拓展底物以研究该催化剂在不对称1,4-加成反应中的应用,因此,关于该类配合物在其他不对称有机合成中的应用及其作用机理尚需投入进行大量研究工作.
2015年,Peters等[50]设计合成了几种二茂铁基钯配合物50,并将其应用于催化异噁唑啉酮和甲基乙烯基酮的不对称1,4-加成反应(Eq. 21). 研究结果表明,不对称加成的对映选择性来自于二茂铁的空间作用. 这是第一例成功的将二茂铁基钯配合物应用于异噁唑啉酮的C-直接烷基化反应的例子.
6.2 芳基卤代烃/芳香烃的氨基化反应
CAr—N键普遍存在于生物活性物及药物中,芳胺类化合物广泛用作药物、染料、杀虫剂,因此含有 CAr—N化合物的合成引起了研究者的兴趣. 经典的合成方法有硝化还原法、Ulmann合成法以及SNAr合成法. 但是这些方法存在通用性差,合成步骤多,化学选择不确定,需要苛刻的反应条件等缺点. 在过去几年里,因为选择性和官能团兼容性的提高,钯催化卤代芳烃的胺化已广泛应用于合成芳胺,并且逐渐发展成一个普遍、可靠和实用的方法. 近年来随着铁配合物催化剂的广泛应用,研究者将目光指向了铁及其配合物催化合成芳基胺的研究,并取得了初步成果.
2014年,Baran等[51]以二茂铁为催化剂,在较温和的实验条件下,实现了芳香环C—H键的直接氨基化反 应(Scheme 19). 该反应中利用自制的N-琥珀酰亚胺过酸酯(NSP,N-succinimidyl perester)为底物与各种取代的芳香烃进行反应,可以中等左右的产率得到系列琥珀酰亚胺基取代的氨基化产物. 机理研究证实,二茂铁在反应过程中扮演电子穿梭的角色,通过单电子转移过程促使过酸酯O—O键的断裂而形成自由基,继而实现琥珀酰亚胺自由基对芳香环的加成反应而得到最终产物. 该方法除具有实验条件温和、底物适用范围广等优点外,关键是首次实现了二茂铁催化的单电子转移过程,为其他类型有机反应的设计研究提供了一种新思路.
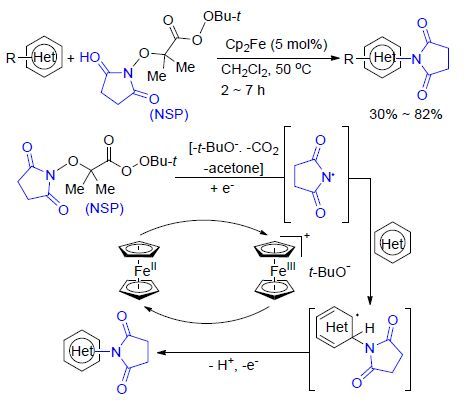 图 图式19
二茂铁催化(杂)芳香烃C—H键的氨基化反应及其可能的机理
Figure 图式19.
Ferrocene-catalyzed C—H imidation of (hetero)- arenes and its proposed mechanism
图 图式19
二茂铁催化(杂)芳香烃C—H键的氨基化反应及其可能的机理
Figure 图式19.
Ferrocene-catalyzed C—H imidation of (hetero)- arenes and its proposed mechanism
2015年,Hartwig等[52]以Josiphos配体和η2-bound 苯甲腈配体与[Ni(cod)]2进行配位得到一种二茂铁基镍配合物51,该配合物作为催化剂可以很好地催化芳基亲电试剂(如芳基卤)的氨基化反应(Scheme 20). 研究结果表明,在该催化剂作用下,芳基卤试剂可以与多种胺化试剂(如氨气、甲基氯化铵、乙基氯化铵等)进行偶联得到伯胺、仲胺的衍生物. 在该催化剂作用下,硫酸铵等多种铵盐也可代替氨气作为胺化试剂与芳基卤发生氨基化反应得到相应的伯胺或仲胺衍生物,扩展了该类反应底物的适用范围,同时发展了一些难以用常规方法制备的复杂有机胺类化合物的合成方法,有望在工业上得到广泛应用.
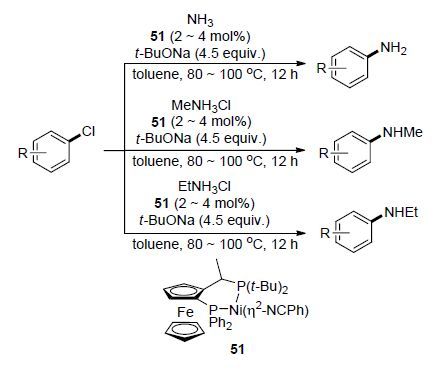 图 图式20
二茂铁基镍配合物催化的芳基氯与氨或铵盐的氨基化反应
Figure 图式20.
Ferrocenyl-nickel complexes-catalyzed amination of aryl chlorides with ammonia or ammonium salts
图 图式20
二茂铁基镍配合物催化的芳基氯与氨或铵盐的氨基化反应
Figure 图式20.
Ferrocenyl-nickel complexes-catalyzed amination of aryl chlorides with ammonia or ammonium salts
Stradiotto等[53]报道了JosiPhos (52)/[Ni(cod)2]催化体系催化的芳基卤或芳基甲苯磺酸盐的氨基化反应(Eq. 22). 研究结果表明,该催化体系对底物的适用范围非常广,无论是芳基或杂芳基的氯化物、溴化物,还是芳基或杂芳基的对甲苯磺酰化合物均适用于该反应,且芳基上取代基的适用范围也很广,即使芳环上连有强供电子基(如甲氧基)、强吸电子基(如氰基、三氟甲基等)或连有杂芳基(如吡咯基、吡啶基等)均能以较高产率得到相应的氨基化产物. 该反应具有底物适用范围广、配体价廉易得、实验操作简单等特点.
6.3 偶联反应
交叉偶联反应是一类用于碳碳键形成的重要化学反应,在有机合成中应用十分广泛[54]. 近年来,我们课题组研究发现二茂铁异噁唑(啉)配体与钯催化剂在催化Heck及卤代烃和炔的交叉偶联反应中显示了较高的催化活性及选择性(Eq. 23). 由芳香二醛出发设计合成了几种双二茂铁双异噁唑(啉)钯配合物53,并将其应用于Heck及卤代烃和炔的交叉偶联反应中[55~57]. 结果表明,这种双二茂铁双异噁唑(啉)钯配合物稳定性高,催化活性及选择性均较好,催化Heck及卤代烃和炔的交叉偶联反应时可在水溶剂中进行,无需惰性气体保护和添加其他含磷配体或铜催化剂,且催化Heck偶联反应中得到的烯烃以E-式构型为主. 该催化体系的优点是可使用无毒无害的水作溶剂,而且催化剂可通过过滤的方式进行回收和循环使用.
2012年,Deng等[58]报道了一种双N-杂环卡宾双核铁配合物[(IPr2Me2)Fe(μ2-NDipp)2Fe(IPr2Me2)] (54)催化的非活性的烷基氟化物与芳基格氏试剂的交叉偶联反应(Eq. 24). 这是首例报道的铁配合物催化的有机氟化物作为亲电体的交叉偶联反应,该催化剂催化的 C(sp3)—F的烷基化反应对底物的普适性很好,适用于各种取代基的芳基格氏试剂和末端取代的烷基氟化物. 该反应经历自由基历程,这是目前首次报道的铁配合物催化的自由基型交叉偶联反应,进一步拓展了铁配合物催化的有机反应类型.
2015年,Hu等[59]设计合成了一种双噁唑啉苯胺(Bopa)手性铁配合物催化剂57,该配合物中双噁唑啉配体可以稳定具有催化活性中心的铁原子,同时将该配合物催化剂应用于烷基卤代烃与芳基格氏试剂的烷基-芳基Kumada交叉偶联反应(Scheme 21),并对其催化机理进行了研究. 研究结果表明,该配合物催化剂的催化活性较高,反应在很短的时间内即可完成,且该催化剂对卤代烃的适用范围较广,一级或二级的溴代烷烃或碘代烷烃均能很好的发生反应. 机理研究表明,配合物中[Fe(Bopa-Ph)(Ph)2]-作为氧化试剂与卤代烷先发生氧化加成,继而与格式试剂发生反应完成芳基-烷基偶联,该机理经自由基捕捉剂和DFT理论计算证实. 该工作对深入理解铁配合物催化的烷基卤代烃的交叉偶联反应提供了有力的理论依据和实验支撑.
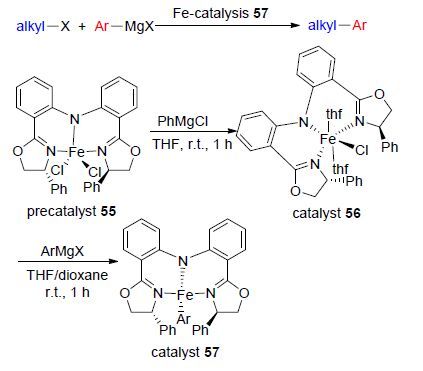 图 图式21
双噁唑啉苯胺基钳形铁配合物催化的烷基-芳基Kumada偶联反应
Figure 图式21.
Bis(oxazolinylphenyl)amido pincer iron complexes catalyzed the alkyl-aryl Kumada coupling reactions
图 图式21
双噁唑啉苯胺基钳形铁配合物催化的烷基-芳基Kumada偶联反应
Figure 图式21.
Bis(oxazolinylphenyl)amido pincer iron complexes catalyzed the alkyl-aryl Kumada coupling reactions
以廉价易得的有机酸作为反应底物的脱羧偶联反应作为一种新的形成碳碳键的方法受到研究者的青睐,成为近年来的研究热点. 2013年,Mao等[60]以二茂铁为催化剂,肉桂酸和非活化的苄基化合物为原料,在无任何配体存在的条件下,以二叔丁基过氧化物(DTBP)为引发剂实现了C(sp2)—C(sp3)键间的脱羧偶联反应(Eq. 25),该反应的显著特点使用价廉易得且性质稳定的非活化的苯甲基化合物作为底物可有效避免使用对金属偶联反应敏感的底物,且该反应兼具底物适用范围广,官能团兼容性好等优点.
6.4 N-甲基化反应
碳酸二甲酯(DMC)作为一种绿色化学试剂,困其分子中含有多种活泼的官能团,可取代卤代甲烷、硫酸二甲酯等甲基化试剂被广泛应用于有机合成中. 近年来碳酸二甲酯(DMC)作为甲基化试剂与相应化合物发生甲基化反应备受关注,成为有机合成领域研究的热点之一. 2014年,Sortais等[61]报道了N-杂环卡宾铁配合物催化仲胺与碳酸二甲酯或碳酸二乙酯的N-甲基化反应(Eq. 26). 该反应以配合物[CpFe(CO)2(IMes)]I (58)为催化剂,苯基硅烷(PhSiH3)为还原剂,光照条件下可高效实现仲胺类化合物N-甲基化合成多种叔胺衍生物. 与传统方法相比,使用该类铁配合物催化剂催化仲胺的N-甲基化反应实验条件更温和,且产率有明显提高.
7 结论与展望
综上所述,新颖配体结构的铁配合物的设计合成及其催化的有机反应是近年来配位化学研究的热点领域之一. 从报道的许多成功例子来看,与钌、铑、钯等贵金属配合物催化剂以及其它具有毒性的过渡金属催化剂相比,铁配合物催化的有机反应的研究已经取得了很大的突破. 大量的基础研究和应用研究正在不断深入进行,铁配合物催化中心的本质和催化机理逐渐得到阐明. 因此,随着科研工作者对铁配合物催化剂结构及性能的构效关系认识的不断加深,新颖铁配合物催化剂的设计合成及其在催化有机反应中的应用将得到持久和深入的发展,对有机金属化学、配位化学等学科的发展也将起到至关重要的作用.
尽管铁配合物的合成及其催化性能研究已经取得了一定进展,但是相对于一些钌、钯等贵金属配合物催化剂的研究而言,新颖铁配合物催化剂的开发仍然是配合物化学研究的一个新兴的研究领域. 铁配合物催化的有机反应仍然存在反应条件苛刻、底物普适性差等缺点. 多数铁配合物在催化苯乙酮或亚胺的氢化还原反应中具有良好的催化活性或对映选择性,而在其他类型的反应中应用较少,且个别催化剂还存在配体合成困难、价格昂贵等问题,使得其应用受到限制. 因而设计并合成一些制备简单、价格低廉、结构稳定、底物普适性好的手性铁配合物催化剂是铁配合物研究领域的核心内容之一.
未来对铁配合物催化的有机反应研究中还存在诸多挑战,一方面烯烃不对称氢化反应及不对称交叉偶联反应构建C—C键的研究将是铁配合物催化的有机反应面临的一大挑战. 另一方面,认识和理解手性铁配合物催化剂中结构及性能的关系以及催化体系中添加物对催化反应机理的作用等方面将成为未来研究铁配合物的另一重要研究方向. 因此,我们相信未来铁配合物在配体的设计开发、催化的有机反应,尤其是不对称催化等方面都将有着更加广阔的发展前景.
-
-
[1]
Reppe, W. Experientia 1949, 5, 93.
-
[2]
Kealy, T. J.; Pauson, P. L. Nature 1951, 168, 1039.
-
[3]
Hieber, V. W.; Braun, G. Z. Naturforsch. 1959, 146, 132.
-
[4]
Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Chem. Rev. 2004, 104, 6217.
-
[5]
Rose, E.; Andrioletti, B.; Zrig, S.; Quelquejeu-Ethève, M. Chem. Soc. Rev. 2005, 34, 573. (b) Correa, A.; García Mancheño, O.; Bolm, C. Chem. Soc. Rev. 2008, 37, 1108. (c) Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293. (d) Gopalaiah, K. Chem. Rev. 2013, 113, 3248. 10.1039/b405679p
-
[6]
Plietker, B. Iron Catalysis in Organic Chemistry: Reactions and Applications, Wiley-VCH, Weinheim, Germany, 2008. (b) Plietker, B. Topics in Organometallic Chemsitry, Iron Catalysis: Fundamentals and Applications, Springer-VBH, Berlin, 2011, Vol. 33.
-
[7]
Gan, K.; Sadeer, A.; Xu, C.; Li, Y.; Pullarkat, S. A. Organometallics 2014, 33, 5074. 10.1021/om5007215
-
[8]
Champouret, Y. D. M.; Fawcett, J.; Nodes, W. J.; Singh, K.; Solan, G. A. Inorg. Chem. 2006, 45, 9890. (b) Sun, W.-H.; Hao, P.; Zhang, S.; Shi, Q.; Zuo, W.; Tang, X.; Lu, X. Organometallics 2007, 26, 2720. 10.1021/ic061286x
-
[9]
Ma, J.; Feng, C.; Wang, S.; Zhao, K.-Q.; Sun, W.-H.; Redshaw, C.; Solan, G. A. Inorg. Chem. Front. 2014, 1, 14. (b) Searles, K.; Fortier, S.; Khusniyarov, M. M.; Carroll, P. J.; Sutter, J.; Meyer, K.; Mindiola, D. J.; Caulton, K. G. Angew. Chem., Int. Ed. 2014, 53, 14139. (c) Lin, Y.-F.; Ichihara, N.; Nakajima, Y.; Ozawa, F. Organometallics 2014, 33, 6700. (d) Suzuki, T.; Matsumoto, J.; Kajita, Y.; Inomata, T.; Ozawaa, T.; Masuda, H. Dalton Trans. 2015, 44, 1017. 10.1039/C3QI00028A
-
[10]
Zuo, W.; Tauer, S.; Prokopchuk, D. E.; Morris, R. H. Organometallics 2014, 33, 5791. (b) Cussó, O.; Garcia-Bosch, I.; Ribas, X.; Lloret-Fillol, J.; Costas, M. J. Am. Chem. Soc. 2013, 135, 14871. 10.1021/om500479q
-
[11]
Zhang, Q.; Xiang, L.; Deng, L. Organometallics 2012, 31, 4537. (b) Gallego, D.; Inoue, S.; Blom, B.; Driess, M. Organometallics 2014, 33, 6885. (c) Bhattacharya, P.; Krause, J. A.; Guan, H. Organometallics 2014, 33, 6113.
-
[12]
Karpiniec, S. S.; McGuinness, D. S.; Britovsek, G. J. P.; Wierengaa, T. S.; Patel, J. Chem. Commun. 2011, 47, 6945.
-
[13]
Xing, Q.; Zhao, T.; Qiao, Y.; Wang, L.; Redshaw, C.; Sun, W.-H. RSC Adv. 2013, 3, 26184.
-
[14]
Huang, F.; Xing, Q.; Liang, T.; Flisak, Z.; Ye, B.; Hu, X.; Yang, W.; Sun, W.-H. Dalton Trans. 2014, 43, 16818.
-
[15]
Chen, J.; Xi, T.; Lu, Z. Org. Lett. 2014, 16, 6452.
-
[16]
Chen, J.; Cheng, B.; Cao, M.; Lu, Z. Angew. Chem., Int. Ed. 2015, 54, 4661. 10.1002/anie.201411884
-
[17]
Morris, R. H. Chem. Soc. Rev. 2009, 38, 2282. 10.1039/b806837m
-
[18]
Bhattacharya, P.; Krause, J. A.; Guan, H. Organometallics 2014, 33, 6113. 10.1021/om500758j
-
[19]
Zuo, Z.; Zhang, L.; Leng, X.; Huang, Z. Chem. Commun. 2015, 51, 5073.
-
[20]
Bleith, T.; Wadepohl, H.; Gade, L. H. J. Am. Chem. Soc. 2015, 137, 2456. 10.1021/ja512986m
-
[21]
Ito, J.; Hosokawa, S.; Khalid, H. B.; Nishiyama, H. Organometallics 2015, 34, 1377. 10.1021/acs.organomet.5b00082
-
[22]
Zuo, Z.; Sun, H.; Wang, L.; Li, X. Dalton Trans. 2014, 43, 11716.
-
[23]
Wu, S.; Li, X.; Xiong, Z.; Xu, W.; Lu, Y.; Sun, H. Organometallics 2013, 32, 3227. (b) Zhao, H.; Sun, H.; Li, X. Organometallics 2014, 33, 3535.
-
[24]
Huang, S.; Zhao, H.; Li, X.; Wang, L.; Sun, H. RSC Adv. 2015, 5, 15660.
-
[25]
Wang, L.; Sun, H.; Li, X. Eur. J. Inorg. Chem. 2015, 2732.
-
[26]
Xue, B.; Sun, H.; Li, X. RSC Adv. 2015, 5, 52000.
-
[27]
Zhang, C.; Yu, S.-B.; Hu, X.-P.; Wang, D.-Y.; Zheng, Z. Org. Lett. 2010, 12, 5542.
-
[28]
Check, C. T.; Jang, K. P.; Schwamb, C. B.; Wong, A. S.; Wang, M. H.; Scheidt, K. A. Angew. Chem., Int. Ed. 2015, 54, 4264. 10.1002/anie.201410118
-
[29]
Zhang, C.; Hu, X.-H.; Wang, Y.-H.; Zheng, Z.; Xu, J.; Hu, X.-P. J. Am. Chem. Soc. 2012, 134, 9585.
-
[30]
Casitas, A.; Krause, H.; Goddard, R.; Fürstner, A. Angew. Chem., Int. Ed. 2015, 54, 1521. 10.1002/anie.201410069
-
[31]
Cussó, O.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Angew. Chem., Int. Ed. 2015, 54, 2729. 10.1002/anie.201410557
-
[32]
Chatterjee, S.; Paine, T. K. Inorg. Chem. 2015, 54, 1720. 10.1021/ic502658p
-
[33]
Li, Y.; Yu, S.; Wu, X.; Xiao, J.; Shen, W.; Dong, Z.; Gao, J. J. Am. Chem. Soc. 2014, 136, 4031. (b) Li, Y.; Yu, S.; Shen, W.; Gao. J. Acc. Chem. Res. 2015, 48, 2587. (c) Yoshimura, M.; Tanaka, S.; Kitamura, M. Tetrahedron Lett. 2014, 55, 3635. (d) Foubelo, F.; Nájera, C.; Yus, M. Tetrahedron: Asymmetry 2015, 26, 769. 10.1021/ja5003636
-
[34]
Mikhailine, A. A.; Morris, R. H. Inorg. Chem. 2010, 49, 11039. (b) Lagaditis, P. O.; Lough, A. J.; Morris, R. H. Inorg. Chem. 2010, 49, 10057. (c) Meyer, N.; Lough, A. J.; Morris, R. H. Chem. Eur. J. 2009, 15, 5605. (d) Mikhailine, A.; Lough, A. J.; Morris, R. J. Am. Chem. Soc. 2009, 131, 1394. (e) Sui-Seng, C.; Freutel, F.; Lough, A. J.; Morris, R. Angew. Chem., Int. Ed. 2008, 47, 940. 10.1021/ic101548j
-
[35]
Sues, P. E.; Lough, A. J.; Morris, R. H. Organometallics 2011, 30, 4418. 10.1021/om2005172
-
[36]
Mikhailine, A. A.; Maishan, M. I.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2012, 134, 12266. 10.1021/ja304814s
-
[37]
Prokopchuk, D. E.; Sonnenberg, J. F.; Meyer, N.; Iuliis, M. Z.-D.; Lough, A. J.; Morris, R. H. Organometallics 2012, 31, 3056. 10.1021/om201170f
-
[38]
Zuo, W.; Lough, A. J.; Li, Y. F.; Morris, R. H. Science 2013, 342, 1080. 10.1126/science.1244466
-
[39]
Prokopchuk, D. E.; Morris, R. H. Organometallics 2012, 31, 7375. 10.1021/om300572v
-
[40]
Zuo, W.; Tauer, S.; Prokopchuk, D. E.; Morris, R. H. Organometallics 2014, 33, 5791. 10.1021/om500479q
-
[41]
Sonnenberg, J. F.; Lough, A. J.; Morris, R. H. Organometallics 2014, 33, 6452. 10.1021/om5008083
-
[42]
Lagaditis, P. O.; Sues, P. E.; Sonnenberg, J. F.; Wan, K. Y.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2014, 136, 1367. 10.1021/ja4082233
-
[43]
Zuo, W.; Morris, R. H. Nat. Protoc. 2015, 10, 241. 10.1038/nprot.2015.012
-
[44]
Smith, S. A. M.; Morris, R. H. Sythesis 2015, 47, 1775.
-
[45]
Bigler, R.; Mezzetti, A. Org. Lett. 2014, 16, 6460.
-
[46]
Mikhailine, A. A.; Maishan, M. I.; Morris, R. H. Org. Lett. 2012, 14, 4638. 10.1021/ol302079q
-
[47]
Mazza, S.; Scopelliti, R.; Hu, X. Organometallics 2015, 34, 1538.
-
[48]
Kuwano, R.; Hashiguchi, Y.; Ikeda, R.; Ishizuka, K. Angew. Chem., Int. Ed. 2015, 54, 2393. 10.1002/anie.201410607
-
[49]
Gan, K.; Sadeer, A.; Xu, C.; Li, Y.; Pullarkat, S. A. Organometallics 2014, 33, 5074. 10.1021/om5007215
-
[50]
Hellmuth, T.; Frey, W.; Peters, R. Angew. Chem., Int. Ed. 2015, 54, 2788. 10.1002/anie.201410933
-
[51]
Foo, K.; Sella, E.; Thomé, I.; Eastgate, M. D.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 5279. 10.1021/ja501879c
-
[52]
Green, R. A.; Hartwig, J. F. Angew. Chem., Int. Ed. 2015, 54, 3768. 10.1002/anie.201500404
-
[53]
Borzenko, A.; Rotta-Loria, N. L.; MacQueen, P. M.; Lavoie, C. M.; McDonald, R.; Stradiotto, M. Angew. Chem., Int. Ed. 2015, 54, 3773. 10.1002/anie.201410875
-
[54]
Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215. (b) Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417. (c) Cherney, A. H.; Kadunce, N. T.; Reisman, S. E. Chem. Rev. 2015, 115, 9587. (d) Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Chem. Rev. 2015, 115, 12138. 10.1021/cr100280d
-
[55]
Shang, Y.-J.; Wu, J.-W.; Fan, C.-L.; Hu, J.-S.; Lu, B.-Y. J. Organomet. Chem. 2008, 693, 2963.
-
[56]
Feng, Z.-J.; Yu, S.-Y.; Shang, Y.-J. Appl. Organomet. Chem. 2008, 22, 577.
-
[57]
Yu, S.-Y.; Zhang, Z.-Q.; Yu, Z.-Y.; Shang, Y.-J. Appl. Organomet. Chem. 2014, 28, 657.
-
[58]
Mo, Z.; Zhang, Q.; Deng, L. Organometallics 2012, 31, 6518.
-
[59]
Bauer, G.; Wodrich, M. D.; Scopelliti, R.; Hu, X. Organometallics 2015, 34, 289.
-
[60]
Yang, H.; Yan, H.; Sun, P.; Zhu, Y.; Lu, L.; Liu, D.; Rong, G.; Mao, J. Green Chem. 2013, 15, 976.
-
[61]
Zheng, J.; Darcel, C.; Sortais, J.-B. Chem. Commun. 2014, 50, 14229.
-
[1]
-

图式1 双吡啶双亚胺铁配合物的合成及其催化烯烃聚合反应
Scheme 1 Synthesis of bispyridine-bisimino iron complexes and catalysis of olefin polymerization

图式3 手性噁唑啉基吡啶双亚胺铁配合物的设计合成及其催化活性
Scheme 3 Synehtsis of chiral iminopyridine oxazoline iron complexes and catalytic activity

图式4 手性噁唑啉基吡啶亚胺铁配合物的设计合成及其催化活性
Scheme 4 Synehtsis of iminopyridine-oxazoline iron complexes and their catalytic activity

图式5 手性双噁唑啉基烷氧基铁配合物的设计合成及其催化活性
Scheme 5 Synthesis of bisoxazoline-alkoxido iron complexes and their catalytic activity

图式6 NCN 钳形铁配合物的设计合成及其催化活性
Scheme 6 Synthesis of NCN pincer iron complexes and their catalytic activity

图式7 含氢铁配合物催化剂催化酮的硅氢化反应及其机理
Scheme 7 Mechanism of catalytic hydrosilylation with hydrido iron complexes

图式8 新型手性二茂铁基P,S-配体的合成及其催化不对称 [3+2]环加成反应
Scheme 8 Synthesis of new chiral ferrocenyl P,S-ligands and catalytic asymmetric [3+2] cycloaddition reactions

图式9 [L2FeX2]型铁配合物及其催化炔烃的[2+2+2]环三聚反应
Scheme 9 Iron complexes of type [L2FeX2] and their catalyzed the [2+2+2] cyclotrimerization of alkynes

图式10 非血红素铁配合物催化的2-氨基苯酚与氧的芳环氧化开环反应
Scheme 10 Oxygenative aromatic ring cleavage of 2-amino- phenol with dioxygen catalyzed by a nonheme iron complex

图式11 铁配合物催化苯乙酮的不对称氢转移反应及其机理
Scheme 11 Mechanism of asymmetric transfer hydrogenation of acetophenone using an iron(Ⅱ) complexes

图式12 反式铁配合物的相互转化及催化苯乙酮的不对称氢转移反应
Scheme 12 Asymmetric transfer hydrogenation of acetophenone using the trans-iron(Ⅱ) complexes

图式13 胺-亚胺双膦铁配合物的设计合成及其在酮和亚胺的不对称氢化反应中的应用
Scheme 13 Synthesis of amine(imine)diphosphine iron complexes and their application of the asymmetric transfer hydrogenation of ketones and imines

图式14 PNNP型铁配合物催化酮的氢转移反应机理
Scheme 14 Mechanism of transfer hydrogenation of ketones using iron(Ⅱ) PNNP eneamido complexes

图式15 含胺-亚胺双膦配体的P-NH-N-P型铁配合物催化酮的不对称氢化和不对称转移氢化反应
Scheme 15 Iron catalysts containing amine(imine)diphosphine P-NH-N-P ligands catalyze both the asymmetric hydrogenation (AH) and asymmetric transfer hydrogenation (ATH) of ketones

图式16 非对称PNP'钳形铁配合物催化剂及其催化酮的不对称氢化反应
Scheme 16 Iron(Ⅱ) complexes containing unsymmetrical PNP' pincer ligands and the catalytic asymmetric hydrogenation of ketones

图式17 胺-亚胺双膦铁配合物催化酮和亚胺的不对称转移氢化反应
Scheme 17 Amine(imine)diphosphine iron catalyzed the asymmetric transfer hydrogenation of ketones and imines

图式18 含手性N2P2大环配体的双异腈铁配合物的设计合成及其催化酮的对映选择性转移氢化反应
Scheme 18 Synthesis of diisonitile iron(Ⅱ) complexes with chiral N2P2 macrocycles and the catalytic of enantioselective transfer hydrogenation of ketones

图式19 二茂铁催化(杂)芳香烃C—H键的氨基化反应及其可能的机理
Scheme 19 Ferrocene-catalyzed C—H imidation of (hetero)- arenes and its proposed mechanism

图式20 二茂铁基镍配合物催化的芳基氯与氨或铵盐的氨基化反应
Scheme 20 Ferrocenyl-nickel complexes-catalyzed amination of aryl chlorides with ammonia or ammonium salts
-


 下载:
下载:





















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 7147
- HTML全文浏览量: 3069
 下载:
下载:
