 图 1
几种prezizaanes型萜类化合物
Figure 1.
Examples of prezizaane-type terpenoids
图 1
几种prezizaanes型萜类化合物
Figure 1.
Examples of prezizaane-type terpenoids
引用本文:
李林斌, 沈洋, 张延东. Jiadifenolide的合成研究进展[J]. 有机化学,
2016, 36(3): 439-446.
doi:
10.6023/cjoc201511054

Citation: Li Linbin, Shen Yang, Zhang Yandong. Synthetic Progress of Jiadifenolide[J]. Chinese Journal of Organic Chemistry, 2016, 36(3): 439-446. doi: 10.6023/cjoc201511054
Citation: Li Linbin, Shen Yang, Zhang Yandong. Synthetic Progress of Jiadifenolide[J]. Chinese Journal of Organic Chemistry, 2016, 36(3): 439-446. doi: 10.6023/cjoc201511054
Jiadifenolide的合成研究进展
摘要:
Jiadifenolide是一个从我国特有药物假地枫皮中分离得到的结构新颖的seco-prezizaanes型倍半萜类化合物, 具有一个高度氧化的五环笼状结构, 7个连续手性中心.它在较低浓度下有效促进神经元的生长, 被认为是一种小分子营养剂, 具有治疗神经退行性疾病的潜力.总结了过去五年间关于此分子的合成研究进展.
-
关键词:
- jiadifenolide
- / seco-prezizaanes型倍半萜
- / 神经营养剂
- / 神经退行性疾病
- / 全合成
English
Synthetic Progress of Jiadifenolide
Abstract:
Jiadifenolide, a new seco-prezizaane-type sesquiterpenoid, was isolated from Chinese unique medicine Illicium jiadifengpi. It contains a highly oxygenated cagelike architecture with seven contiguous stereocenters. It displays potent neurotrophic activity at very low concentration and is regarded as a promising lead compound for developing drugs for neurodegenerative diseases. This review summarizes the synthetic efforts towards jiadifenolide in the last five years.
-
八角科(Illiciaceae)植物为一单属科, 仅八角属(Illicium)一属.全世界八角科植物约有50种, 仅分布于北半球, 其中大多数分布在东亚及东南亚地区, 少数分布在北美洲南部.中国独有30余种, 主要分布于西南至华东的广大南方地区.中草药假地枫皮主要源自我国特有八角科八角属植物假地枫皮(Illicium jiadifengpi)的干燥根皮和茎皮[1].
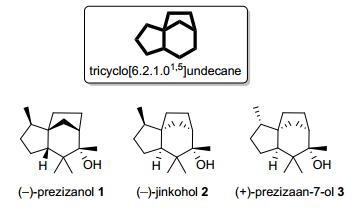
Prezizaanes[2]是一类广泛分布于植物中的具有三环[6.2.1.01, 5]十一烷骨架结构的有芳香气味的倍半萜天然产物(如图 1所示). seco-prezizaanes则是一类被看作由prezizaanes骨架发生键断裂衍生的化合物.自1952年由Lane等[3]从八角科植物Illicium anisatum果实中首次分离得到(-)-anisatin (4)开始, 在过去60多年的时间里, 科学家们从各种八角科植物中陆续分离出众多seco-prezizaanes型复杂的倍半萜, 其中具有代表性的有: jiadifenin, neomajucin和jiadifenolide等.这些化合物多具有强烈的神经毒性或促进神经元生长的生理活性.这些具有复杂结构和良好生物活性的天然产物吸引了众多合成化学家的关注, 近年来Yamada、Fukuyama等[4]完成了(-)-anisatin (4)的全合成, Danishefsky、翟宏斌、Theodorakis等[5]完成了jiadifenine (6)的全合成.
图 1
几种prezizaanes型萜类化合物
Figure 1.
Examples of prezizaane-type terpenoids
1 Jiadifenolide的结构与生物活性
2009年Fukuyama和黄建梅等[6]报道了从采自我国西南地区的假地枫皮(Illicium jiadifengpi)中分离得到的三个具有高度氧化态的seco-prezizaanes型倍半萜类化合物: (-)-jiadifenoxolane A (7), (-)-jiadifenoxolane B (8)和(-)-jiadifenolide (9)(如图 2所示), 它们都具有五环笼状结构.
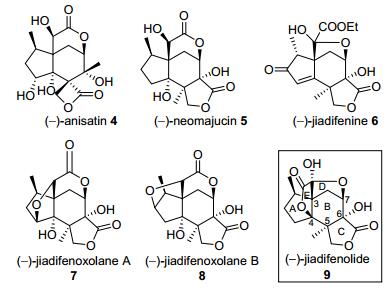 图 2
几种seco-prezizaanes型萜类化合物
Figure 2.
Examples of seco-prezizaane-type terpenoids
图 2
几种seco-prezizaanes型萜类化合物
Figure 2.
Examples of seco-prezizaane-type terpenoids
Jiadifenolide的结构通过高分辨质谱、红外光谱、NMR、比旋光、X射线单晶衍射分析及后续的全合成研究得到确认, 其化学结构如图 2所示.其结构具有以下特点: (1)拥有高度氧化的五环笼状结构, 包含连续的并环、桥环等结构特征; (2)含有7个连续手性中心, 其中核心六元B环包含5个手性碳, 且4个为四取代碳, 故十分拥挤; (3)包含多样的含氧官能团, 包含α-羟基内酯、α-半缩酮内酯等.由于jiadifenolide拥有一个高氧化的刚性骨架结构, 因此分子的全合成, 特别是无保护基的全合成对于合成化学家无疑是一个巨大的挑战.
此外, 初步的生理活性测试表明:在10 nmol/L至10 μmol/L的浓度范围内, (-)-jiadifenolide具有显著的促进原代培养大鼠皮质神经元生长的生理活性, 因而其可作为一种小分子神经营养剂, 并有望充当先导化合物用来开发治疗阿尔兹海默症、帕金森症、亨廷顿跳舞症等神经退行性疾病的药物.
Jiadifenolide复杂的结构、良好的促进神经细胞生长活性, 以及极低的天然分离产率(9×10-6)都引起了合成化学界的浓厚的研究兴趣.截至目前, 已有五个研究小组完成了这一分子的全合成工作, 其中四个小组完成了该分子的不对称全合成.本文将总结评述这五个小组的研究工作.
2 Jiadifenolide的合成研究
2009年, 在天然产物分离文献中[6]提及Fukuyama等通过借鉴Danishefsky等[5a]提出的jiadifenine (6)的相似合成转化, 提出了(-)-jiadifenolide (9)和其家族天然产物(-)-neomajucin (5)之间可能存在的生源转化关系, 并在实验室中使用Dess-Martin氧化剂以34%的产率实现了后者到前者的化学转化, 从而在一定程度上证明了上述假设的可靠性(图 3).
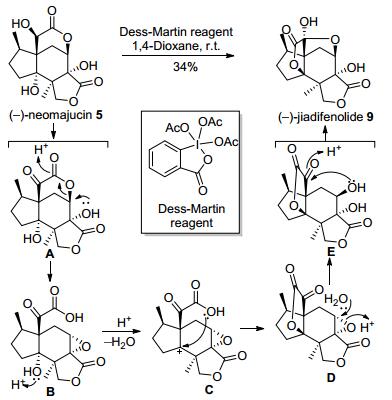 图 3
5到9的氧化转化机理
Figure 3.
Plausible mechanism for oxidation of 5 to 9
图 3
5到9的氧化转化机理
Figure 3.
Plausible mechanism for oxidation of 5 to 9
2011年3月, Theodorakis小组[7]在《德国应用化学》上以封面文章的形式报道了(-)-jiadifenolide (9)的首次不对称全合成, 并通过对比NMR数据和比旋光值, 证实了天然产物的绝对立体构型. 2013年3月, 该小组[8]又报道了从此前合成路线中的中间体18出发, 实现家族天然产物(-)-jiadifenine (6)的全合成的工作.此外, Theodorakis等对合成的天然产物及多个中间体进行了神经营养活性评测, 提出了初步的构效关系, 进一步确认了天然产物优秀的生理活性. 2014年4月Sorensen小组[9]报道了(-)-jiadifenolide (9)的第二条不对称全合成路线. 2014年5月Paterson小组[10]报道了一条jiadi-fenolide的消旋全合成路线. 2015年6月Shenvi小组[11]报道了第一条无保护8步克级规模合成(-)-jiadifeno-lide (9)的全合成路线.紧接着2015年10月张延东小组[12]报道了又一条无保护合成(-)-jiadifenolide (9)的全合成路线, 并报道了在全合成研究中发现的以β-羟基醛或酮为底物具有相当广谱适用性的形式[4+1]环化反应.此外鉴于之前天然产物分离者受限于天然产物分离量, 未能对该天然产物作出全面生物学评测, 张延东小组还对它的抗癌活性进行了初步评价.
2.1 Theodorakis小组对(-)-jiadifenolide (9)的全合成
Theodorakis小组[7]从商业原料1, 3-环戊二酮(10)出发以25步1.5%的总产率实现了对9的首次不对称全合成, 其关键步骤包括: (1) D-脯氨酰胺催化的不对称羟醛缩合反应; (2)钯催化的羰基化反应构建C环; (3)酸介导的串联反应构建E环.
如图 4所示, 起始原料10通过两步已知的烷基化反应制得不对称羟醛缩合反应前体11.在D-脯氨酰胺的催化下, 以>90% ee值引入了不对称元素并构建了A-B环系, 然后通过对酮12选择性的立体化学专一还原反应、并对还原得到的醇羟基进行保护得到了烯酮13.通过对不饱和酮13先后两次烯醇化并依次与镁碳酸甲酯(MMC)和Meerwein盐、碘甲烷作用, 先后引入乙酯基团和甲基, 从而构建了C-5季碳手性中心, 得到14.从化合物14出发, 经过四步官能团转化得到烯基三氟甲磺酸酯中间体, 其在Pd(PPh3)4作催化剂、CO气氛下, 发生插羰内酯化反应从而构建了天然产物的C环, 得到化合物16.后者经选择性地环氧化、双羟化氧化切断、Jones氧化、脱除硅保护基等一系列操作, 转化为具有[3.3.1]桥环体系的二醇18.将不饱和醇18环氧化后便得到了酸催化串联反应的前体, 这一具有α-羟基内酯的环氧化合物在Dess-Martin试剂的作用下, 发生类似Fukuyama等提出的生源转化假设中的酸介导串联反应, 生成含有E环的中间体19. 19经氢化、选择性保护转化为酮20.随后使用Comins试剂将这一酮转化为烯基三氟甲磺酸酯, 从而制得了钯催化的甲基化反应的底物.后者在三甲基铝和Pd(PPh3)4的作用下, 生成四环化合物21.最后将21进行立体专一的氢化、Davis氧化、Jones氧化后, 便完成了D环的构建, 并最终获得了5.1 mg的(-)-jiadifenolide, 实现了其首次全合成.
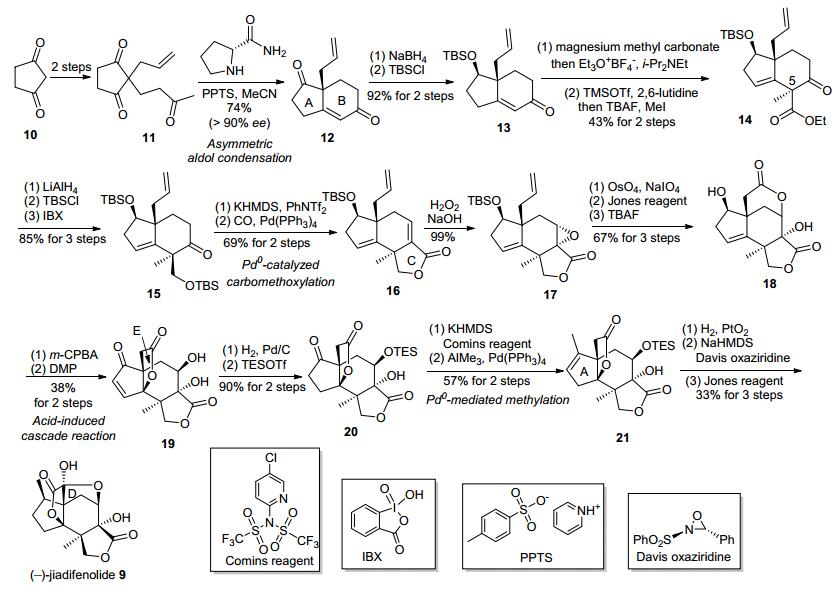 图 4
Theodorakis小组的合成路线
Figure 4.
Theodorakis' synthetic route
图 4
Theodorakis小组的合成路线
Figure 4.
Theodorakis' synthetic route
2.2 Sorensen小组对(-)-jiadifenolide (9)的全合成
Sorensen小组[9]从β-酮酯23出发以18步0.7%的产率实现了对9的不对称全合成.该路线的成环反应全部为常见的成环反应, 如Robinson环化反应、内酯化反应、碱条件下对环氧开环生成的醇与酮发生的分子内半缩酮化反应.整条路线最大的亮点是其首次在天然产物全合成中使用了以肟作为导向基团对非活化的甲基实现了C—H活化氧化反应.
如图 5所示, 通过文献报道的方法, 起始原料(R)-胡薄荷酮经三步转化为已知β-酮酯23, 后者与甲基乙烯基酮发生Robinson环化反应生成含有A-B环系的化合物24.然后通过对烯酮24进行甲基化、酮羰基保护、还原以及氧化等官能团转化, 制备了醛26.在对甲苯磺酰甲基异腈的作用下, 26经延伸碳链转化为多一个碳的腈27.后者在浓硫酸和含水甲醇的处理下, 发生氰基水解继而内酯化的串联反应, 构建出内酯E环, 与此同时, 缩酮保护基也在酸性条件下发生脱除, 制得酮28.酮28与盐酸羟氨发生缩合生成肟, 即获得甲基C—H活化反应的底物, 后者在醋酸钯和醋酸碘苯的参与下, 在乙酸酐/乙酸中100 ℃下反应12 h, 以总产率44%和1:1比例生成了甲基氧化产物29和它在C-5的差向异构体的混合物.随后在酸性条件下使用铁粉将肟29转化酮, 后者在Comins试剂的作用下转化为烯基三氟甲磺酸酯30.随后在醋酸钯的作用下, 30发生插羰内酯化反应, 以两步49%的产率生成不饱和甲酯31. 31通过酯交换形成内酯C环, 在环氧化、碘代、氧化等多步官能团化后转化为α-酮酯33.最后, 33在氢氧化锂的作用下, 发生串联关环反应形成D环, 由此Sorensen等获得了9.1 mg的(-)-jiadifenolide, 从而实现了天然产物的不对称全合成.
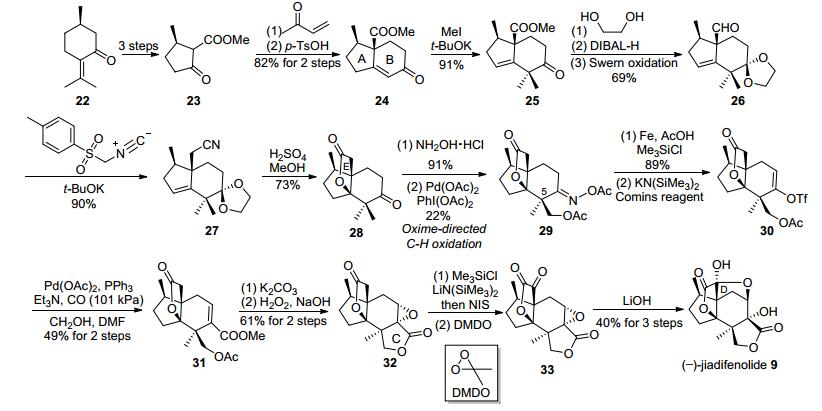 图 5
Sorensen小组的合成路线
Figure 5.
Sorensen's synthetic route
图 5
Sorensen小组的合成路线
Figure 5.
Sorensen's synthetic route
2.3 Paterson小组对(±)-jiadifenolide的全合成
Paterson小组[10]从商业原料3-甲基环戊烯酮(34)出发经过23步, 最终以2.3%的总产率实现了(±)-jiadi-fenolide的全合成.其关键反应是硼酸酯参与的选择性羟醛加成反应及后续的二碘化钐介导的还原环化反应, 这一转化实现了核心A-B-C三环骨架的构建.
如图 6所示, 从环戊烯酮出发, 通过Luche还原、羟基诱导的环氧化反应、对醇羟基的保护、路易斯酸介导的环氧异构化反应制备了取代环戊酮36.随后36发生Horner-Wadsworth-Emmons反应, 以E:Z>19:1, 73%的产率生成了α, β-不饱和酯37.后者经过还原、乙酰化转化为乙酸烯丙酯38.化合物38在二异丙基氨基锂和叔丁基二甲基氯硅烷、苯回流的条件下, 发生Ireland-Claisen重排反应, 以10:1非对映选择性地生成重排产物, 后者经氢化铝锂还原转化为伯醇39.化合物39经过两步官能团转化, 便可转化为γ-羰基醛40.然后醛40经过一步Bu2BOTf介导的羟醛加成反应, 可以近乎定量地以2:1的比例生成一对难分离的差向异构体混合物41.通过对醇41羟基的TES保护后, 便可分离这两个差向异构体, 从而获得二碘化钐介导的还原环化反应的底物42.在6 equiv.的二碘化钐存在的情况下, 在65 ℃的四氢呋喃里, 化合物42发生立体专一的还原环化反应, 以51%的产率生成具有A-B-C三环骨架的环化产物, 随后其在酸条件下脱除硅保护基生成二醇化合物44.最后通过多步氧化、还原、保护、脱保护转化, Paterson等最终获得了5.0 mg的(±)-jiadifenolide, 从而实现了该天然产物消旋体的全合成.
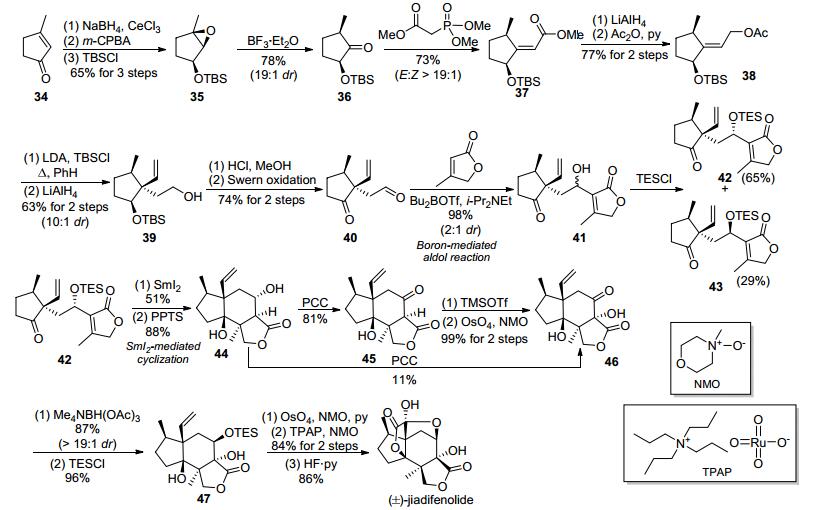 图 6
Paterson小组的合成路线
Figure 6.
Paterson's synthetic route
图 6
Paterson小组的合成路线
Figure 6.
Paterson's synthetic route
2.4 Shenvi小组对(-)-jiadifenolide (9)的全合成
Shenvi小组[11]的合成路线是一条典型的汇聚式合成路线, 两片段分别来源于商业可得的(+)-香茅醛(49)和2, 2, 6-三甲基-4H-1, 3-二噁英-4-酮(51).以(+)-香茅醛为起始原料计算, 以八步实现了(-)-jiadifenolide的克级制备.其关键步骤是一个[4+2]双Michael加成反应.
(+)-香茅醛(49)在(叔丁基亚氨基)三(吡咯烷)膦(BTPP)和全氟丁基磺酰氟(NfF)的作用下发生脱水反应制得端炔中间体, 后者再经过选择性的臭氧化反应后生成炔醛中间体, 然后在六羰基合钼的作用下发生分子内的氧杂Pauson-Khand反应, 从而以三步中等产率克级规模获得具有A-E环系的手性不饱和内酯50.另一方面, 原料51可以与羟基丙酮在甲苯回流条件下生成另一片段52.随后将不饱和内酯50用二异丙基氨基锂在-78 ℃进行去质子化形成烯醇负离子中间体, 再在-100 ℃加入另一片段52, 反应一段时间后, 在0 ℃加入四异丙醇钛和过量的二异丙基氨基锂后, 发生立体选择性的双Michael加成反应, 以20:1非对映选择性、70%的产率实现了克级规模制备四环体系53.最后对化合物53进行过酸参与的α-羟基化、立体选择地还原反应、α-溴代反应、Davis试剂参与的α-羟基化串联关环反应等一系列官能化操作, Shenvi等得到1.03 g的(-)-jiadifenolide, 实现了该天然产物的首次无保护基的克级规模全合成.
2.5 张延东小组对(-)-jiadifenolide (9)的全合成
张延东小组[12]的无保护基合成路线(如图 8所示)也从已知化合物β-酮酯23出发, 该化合物可由该小组改进的两步法从(R)-(+)-胡薄荷酮大量制备而来.首先化合物23通过烯丙基烷基化、臭氧化等反应高产率地转化为醛55.在Bu2BOTf的作用下, γ-内酯48选择性地与化合物55中的醛基发生羟醛加成反应, 生成Baylis-Hillman型产物56.接着后者在乙酸酐、三乙胺的作用下实现酯化-消除-双键异构串联反应, 生成了具有E式双键的不饱和化合物57. 57在二异丙基氨基锂的作用下, 分子内的酮羰基和内酯官能团发生烯醇化, 随后通过加入二异丁基氢化铝, 可以实现对乙酯基团选择性地还原, 最终生成伯醇58.在101 kPa的氢气氛下, 使用二氧化铂催化剂, 可以实现羟基导向的选择性氢化58中的二取代双键, 从而制备二碘化钐还原环化反应的底物59.该小组发现使用传统的SmI2还原条件, 均无法实现所期望的还原环化反应.随后, 在大剂量(7000 equiv.)水作为添加剂, 在四氢呋喃中则可以高效地实现高立体选择性的还原环化反应, 成功地以70%的产率获得具有A-B-C三环体系的二醇60.然后伯醇60经Swern氧化后, 以定量产率生成β-羟基醛61.该小组原本计划利用化合物61与三甲基硅烷重氮甲基锂的反应制备端炔中间体, 然而却意外发现了一个新颖的[4+1]环化反应, 并以较高的产率获得含有E环的内酯化合物62.这一发现大大推动了整个全合成的进程.在连续地三步氧化反应后, 实现了Sorensen合成路线中间体33的合成, 最后使用氢氧化锂处理这一中间体, 成功实现D环的合成, 获得了319 mg的天然产物, 从已知物23出发, 至此历经13步, 以总产率7.9%实现了(-)-jiadifenolide (9)的无保护基全合成.
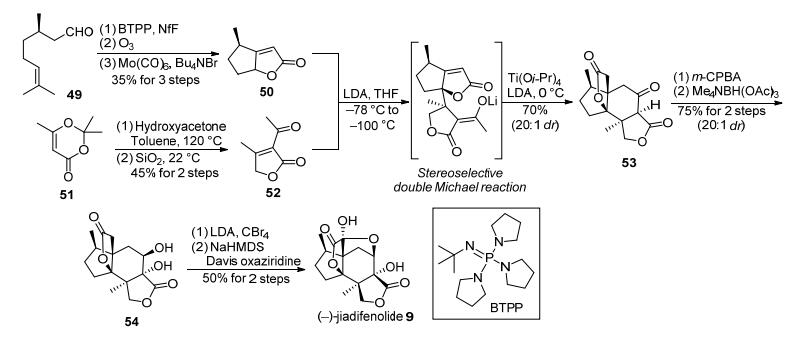 图 7
Shenvi小组的合成路线
Figure 7.
Shenvi's synthetic route
图 7
Shenvi小组的合成路线
Figure 7.
Shenvi's synthetic route
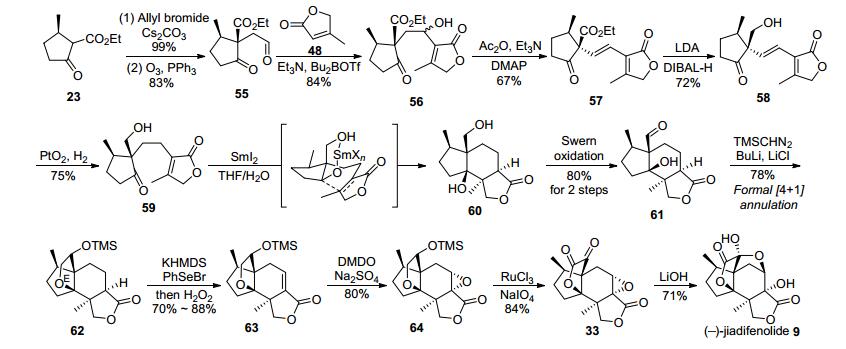 图 8
张延东小组的合成路线
Figure 8.
Zhang's synthetic route
图 8
张延东小组的合成路线
Figure 8.
Zhang's synthetic route
在完成了天然产物全合成后, 张延东小组也对新发现的[4+1]反应进行了相关的合成方法学研究.如图 9所示, 在对底物普适性的研究中发现这一方法对多种β-羟基醛和β-羟基酮均具有良好的效果, 可以实现多取代单环、螺环体系、并环体系的构筑.尤其令人惊喜的是, 利用该方法, 可以实现罕见的5-5反式并环体系(66f)的构建, 这在合成上具有非常重要的意义.在反应机理的研究方面, 该小组通过相关的氘代实验结果结合参考文献, 提出了如图 10所示的一个反应历程:首先, 三甲基硅烷重氮甲基锂对醛酮的羰基进行加成, 生成氧负离子中间体Ⅱ, 随后发生Brook重排、1, 5-质子转移, 产生中间体Ⅲ.此时存在两种可能的反应历程来生成最终产物:一是重氮化合物Ⅲ发生分解, 生成卡宾中间体, 之后发生氧负离子对卡宾的加成环化反应及后续质子化, 形成四氢呋喃环; 二是先发生质子化, 后发生氧-氢插入反应形成四氢呋喃环.
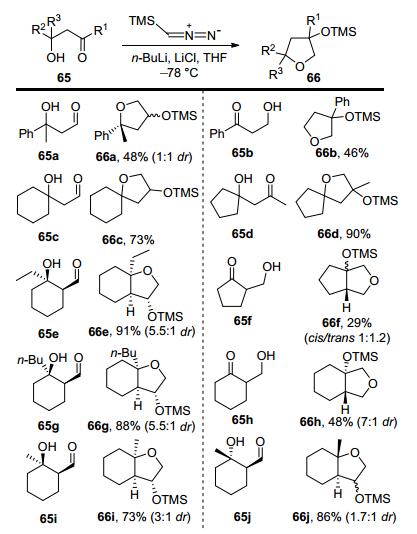 图 9
[4+1]反应底物的普适性考察
Figure 9.
Substrate scope of the [4+1] reaction
图 9
[4+1]反应底物的普适性考察
Figure 9.
Substrate scope of the [4+1] reaction
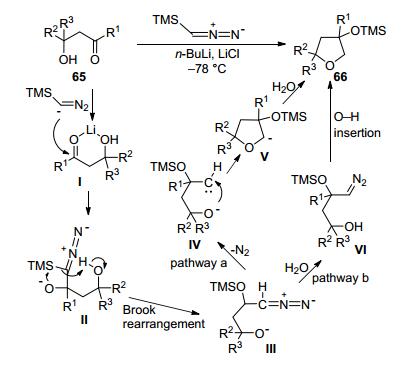 图 10
[4+1]环化反应的机理假设
Figure 10.
Possible mechanism of the [4+1] annulation
图 10
[4+1]环化反应的机理假设
Figure 10.
Possible mechanism of the [4+1] annulation
在获取了相当数量的天然产物后, 张延东小组对天然产物的抗癌活性进行了初步研究, 发现(-)-jiadifeno-lide (9)仅具有较弱的抗癌活性(IC50>100 μmol/L).
3 结语
目前, 已有五个研究小组完成了jiadifenolide (9)的全合成工作(如表 1所示), 其中四个小组实现了不对称全合成, 两个小组实现了无保护不对称全合成, 并制备了相当量的天然产物和相当种类的合成中间体, 但目前仅有Theodorakis小组[8]报道了天然产物和一些中间体的简单构效关系研究和张延东小组报道了抗癌活性的评测, 天然产物的全生理活性测试仍然是一个悬而未决的课题.因此我们相信还将会有更多的科研小组从开发新药物的角度出发, 加入到这一具有优秀生理活性的天然产物生物学和药学研究之中来.
 表 1
Jiadifenolide (9)的全合成总结
Table 1.
Summary of total syntheses of jiadifenolide
表 1
Jiadifenolide (9)的全合成总结
Table 1.
Summary of total syntheses of jiadifenolide
小组名 线性最长步骤数 总产率/% Theodorakis小组 25 1.5 Sorensen小组 18 0.7 Paterson小组a 23 2.3 Shenvi小组 8 8.3 张延东小组 13 7.9 a小组进行的是消旋体全合成. 表 1 Jiadifenolide (9)的全合成总结
Table 1. Summary of total syntheses of jiadifenolide从已报道的(-)-jiadifenolide (9)合成路线, 我们可以看出天然产物全合成研究不仅仅在天然产物结构确定和大量提供自然来源稀缺天然产物方面发挥重要作用, 同时对新反应的发现和发展也具有重要的启发意义.此外, 以天然产物全合成为基础的天然产物化学生物学研究将会成为未来天然产物化学领域的一个研究热点.
-
-
[1]
吴征镒, 路安民, 汤彦承, 陈之端, 李德铢, 中国被子植物科属综论, 科学出版社, 北京, 2003.Wu, Z.; Lu, A.; Tang, Y.; Chen, Z.; Li, D. The Families and Genera of Angiosperms in China, Science Press, Beijing, 2003 (in Chinese).
-
[2]
Goeke, A.; Mertl, D.; Brunner, G. Chem. Biodiversity 2004, 1, 1949. doi: 10.1002/(ISSN)1612-1880
-
[3]
Lane, J. F.; Koch, W. T.; Leeds, N. S.; Gorin, G. J. Am. Chem. Soc. 1952, 74, 3211. doi: 10.1021/ja01133a002
-
[4]
Niwa, H.; Nisiwaki, M.; Tsukada, I.; Ishigaki, T.; Ito, S.; Wakamatsu, K.; Mori, T.; Ikagawa, M.; Yamada, K. J. Am. Chem. Soc. 1990, 112, 9001.
(b) Ogura, A.; Yamada, K.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2012, 14, 1632. -
[5]
Cho, Y. S.; Carcache, D. A.; Tian, Y.; Li, Y.-M.; Danishefsky, S. J. J. Am. Chem. Soc. 2004, 126, 14358.
(b) Carcache, D. A.; Cho, Y. S.; Hua, Z.; Tian, Y.; Li, Y.-M.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 1016.
(c) Yang, Y.; Fu, X.; Chen, J.; Zhai, H. Angew. Chem., Int. Ed. 2012, 51, 9825.
(d) Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Org. Lett. 2011, 13, 4554. -
[6]
Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett. 2009, 11, 5190. doi: 10.1021/ol9021029
-
[7]
Xu, J.; Trzoss, L.; Chang, W. K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2011, 50, 3672. doi: 10.1002/anie.v50.16
-
[8]
Trzoss, L.; Xu, J.; Lacoske, M. H.; Mobley, W. C.; Theodorakis, E. A. Chem. Eur. J. 2013, 19, 6398. doi: 10.1002/chem.201300198
-
[9]
Siler, D. A.; Mighion, J. D.; Sorensen, E. J. Angew. Chem., Int. Ed. 2014, 53, 5332. doi: 10.1002/anie.v53.21
-
[10]
Paterson, I.; Xuan, M.; Dalby, S. M. Angew. Chem., Int. Ed. 2014, 53, 7286. doi: 10.1002/anie.201404224
-
[11]
Lu, H.-H.; Martinez, M. D.; Shenvi, R. A. Nat. Chem. 2015, 7, 604. doi: 10.1038/nchem.2283
-
[12]
Shen, Y.; Li, L.; Pan, Z.; Wang, Y.; Li, J.; Wang, K.; Wang, X.; Zhang, Y.; Hu, T.; Zhang, Y. Org. Lett. 2015, 17, 5480. doi: 10.1021/acs.orglett.5b02845
-
[1]
-
表 1 Jiadifenolide (9)的全合成总结
Table 1. Summary of total syntheses of jiadifenolide
小组名 线性最长步骤数 总产率/% Theodorakis小组 25 1.5 Sorensen小组 18 0.7 Paterson小组a 23 2.3 Shenvi小组 8 8.3 张延东小组 13 7.9 a小组进行的是消旋体全合成.  下载: 导出CSV
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2400
- HTML全文浏览量: 452
 下载:
下载:
