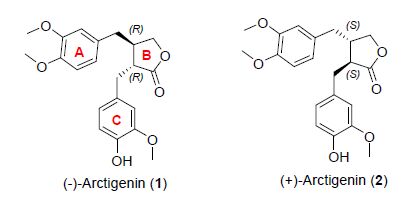
 图 1
(-)-牛蒡苷元及其对映异构体结构
Figure 1.
Structure of (-)-arctigenin and its enantiomer
图 1
(-)-牛蒡苷元及其对映异构体结构
Figure 1.
Structure of (-)-arctigenin and its enantiomer
引用本文:
吴平, 徐凯, 付莹, 康廷国, 窦德强, 翟延君. (-)-牛蒡苷元及其对映异构体的不对称合成新方法[J]. 有机化学,
2016, 36(5): 1111-1117.
doi:
10.6023/cjoc201511040

Citation: Wu Ping, Xu Kai, Fu Ying, Kang Tingguo, Dou Deqiang, Zhai Yanjun. A New Method for Asymmetric Synthesis of (-)-Arctigenin and Its Enantiomer[J]. Chinese Journal of Organic Chemistry, 2016, 36(5): 1111-1117. doi: 10.6023/cjoc201511040
Citation: Wu Ping, Xu Kai, Fu Ying, Kang Tingguo, Dou Deqiang, Zhai Yanjun. A New Method for Asymmetric Synthesis of (-)-Arctigenin and Its Enantiomer[J]. Chinese Journal of Organic Chemistry, 2016, 36(5): 1111-1117. doi: 10.6023/cjoc201511040
(-)-牛蒡苷元及其对映异构体的不对称合成新方法
摘要:
(-)-牛蒡苷元属于二苄基丁内酯型木脂素,是中药牛蒡子的主要活性成分.为了研究牛蒡苷元的构效关系,报道了(-)-牛蒡苷元及其对映异构体的不对称合成新方法.以苯丙酸衍生物为起始原料,首先利用噁唑烷酮类手性辅基构建丁内酯β位的手性中心,R构型和S构型β-苄基丁内酯的ee值分别为98%和96%.再利用空间位阻效应在α位构建第二个手性中心,最后脱除保护基得到目标产物.经6步反应,分别以58%、55%的总收率和97%、96%的ee值得到(-)-牛蒡苷元和(+)-牛蒡苷元.为接下来拟进行的结构优化奠定了技术基础.
English
A New Method for Asymmetric Synthesis of (-)-Arctigenin and Its Enantiomer
Abstract:
(-)-Arctigenin, the main active ingredient of traditional chinese medicine (TCM) arctii fructus, belongs to dibenzyl butyrolactone lignans. In order to study the structure-activity relationship of arctigenin, a new method for asymmetric synthesis of (-)-arctigenin and its enantiomer was developed. Phenylpropanoic acid derivate was used as starting material and the chiral center of beta butyrolactone was constructed by using oxazolidinone chiral auxiliary. The eesof R and S configurations are 98% and 96%, respectively. Then the second chiral center in the alpha position was constructed benefitting from the steric effect. After removal of protecting group, the target compounds were obtained in 58 % and 55% overall yield of (-)-arcti-genin and (+)-arctigenin with 97% and 96% ee,respectively. This work paved the way for further structural optimization of arctigenin.
-
Key words:
- arctigenin
- / oxazolidinone
- / Evans
- / lignan
- / butyrolactone
- / asymmetric synthesis
-
(-)-牛蒡苷元(1),(3R,4R)-3-(3-甲氧基-4-羟基苄基)-4-(3,4-二甲氧基苄基)-丁内酯,是中药牛蒡子(Arctii Fructus)的最主要活性成分(图 1). 研究表明,(-)-牛蒡苷元具有抗炎[1]、抗心律失常[2]、降血糖[3]、保护神经[4]、抗肿瘤[5]、缓解内质网应激[6]、抗老年痴呆[7]等多种生物活性,因此近年来受到了国内外越来越多的科研工作者的关注[8]. 本课题组长期致力于中药牛蒡子的开发研究,包括牛蒡子主要活性成分(-)-牛蒡苷元的衍生化[9]. 本课题组最近在先前发现牛蒡苷元具有较好神经保护作用的基础上又研究了其可能的作用机理[10]. 为了系统深入地研究牛蒡苷元的构效关系,利用化学方法对其进行不对称全合成具有重要的意义. 通过综合分析现有
图 1
(-)-牛蒡苷元及其对映异构体结构
Figure 1.
Structure of (-)-arctigenin and its enantiomer
(-)-牛蒡苷元的不对称合成路线[11],我们决定开发一条更适于大量制备及衍生物快速合成的新路线.
1 结果与讨论
(-)-牛蒡苷元不对称合成新方法如Scheme 1所示. 以3,4-二甲氧基苯丙酸(3)为起始原料,与手性辅基4-苄基噁唑烷酮4缩合得化合物5,随后在大位阻有机碱双(三甲基硅基)氨基钠(NaHMDS)的作用下发生Evans辅基介导的羰基α-烷基化反应,得到化合物6,还原消除手性辅基后得到醇7. 在对甲苯磺酸催化下发生酯交换反应得到ee值分别为98%的丁内酯8a(文献报道[11]利用其它路线合成8a,ee值为91%~96%不等)和96%的8b,随后再利用大位阻有机碱二异丙基胺基锂(LDA)在8的酯羰基α位引入衍生化的苄基得到10a/10b (de>99%),最后Pd/C催化氢解除去酚羟基保护基,分别以6步58%、55%的总收率和97%、96 %的ee值得到目标产物(-)-牛蒡苷元1和(+)-牛蒡苷元2.
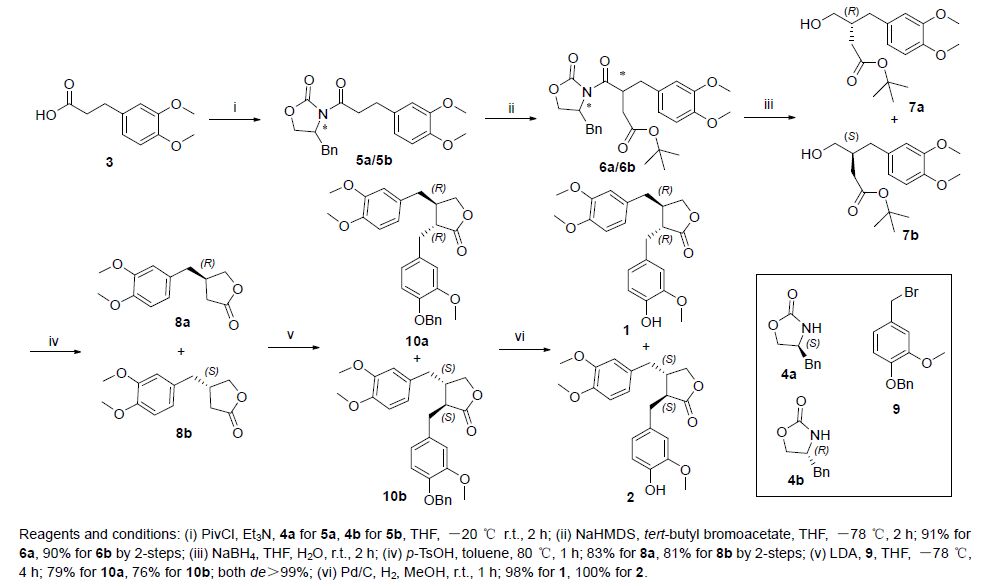 图 图式1
(-)-牛蒡苷元全合成路线
Figure 图式1.
Total synthetic route for (-)-arctigenin
图 图式1
(-)-牛蒡苷元全合成路线
Figure 图式1.
Total synthetic route for (-)-arctigenin
化合物5和8羰基α-烷基化立体选择性的模型如图 2所示. 当钠离子与化合物5a发生配位后,由于苄基的位阻作用,溴乙酸叔丁酯的羰基α碳主要从Re面进攻,主产物为6b,而从Si面进攻则为副产物. 根据化合物8b的ee值测定结果,Re面进攻产物与Si面进攻产物的比值为98:2. 同理,由于化合物5a中Evans辅基上苄基构型,溴乙酸叔丁酯的羰基α碳主要从Si面进攻,根据8a的ee值测定结果,Si面进攻产物与Re面进攻产物的比值为99:1.
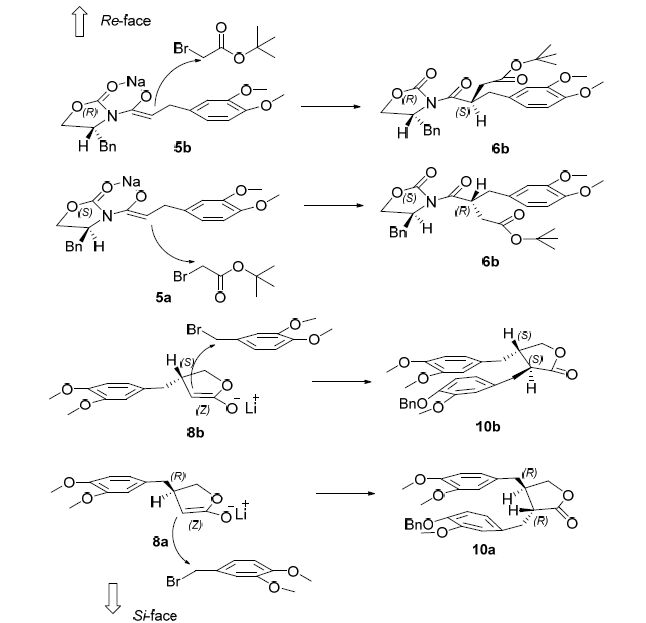 图 2
化合物5和8羰基α-烷基化立体选择性模型
Figure 2.
Proposed models for the stereochemical course of the alkylation of 5 and 8
图 2
化合物5和8羰基α-烷基化立体选择性模型
Figure 2.
Proposed models for the stereochemical course of the alkylation of 5 and 8
五元环内稀醇的非对映选择性烷基化反应中,anti-诱导的烷基化过渡态主要受到空间位阻的控制方式所支配[12]. 对于化合物8而言,由于丁内酯β位已经有了一个位阻较大的基团,所以同样具有大位阻作用的取代溴苄的亚甲基碳只能从相反的方向进攻生成相应的化合物10,同侧进攻则生成syn-异构体副产物. 由于Pd/C氢解不涉及到手性问题,根据化合物1/2的光学纯度测定结果,anti-诱导产物与syn-异构体的比例大于99:1.
Scheme 1所示路线适用于牛蒡苷元C环具有不同取代基的衍生物的快速制备. 随后,我们参考文献[13]尝试开发另一条合成路线(Scheme 2),希望通过这条路线制备(+)-牛蒡苷元的同时,可以以13或其对映异构体为中间体快速制备A环具有不同取代基的β-苄基丁内酯衍生物. 然而,当我们顺利得到中间体13后,发现随后的羰基α-位烷基化反应并没有区域选择性,13的酯羰基α位和β位均发生了反应. 我们调整有机碱的种类和原料的当量比均未能解决该问题.
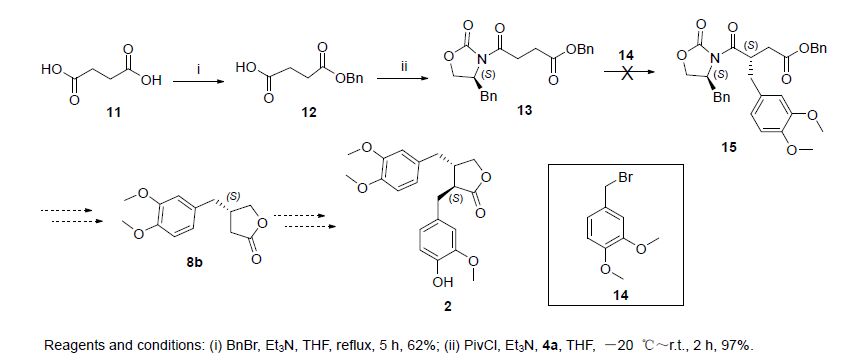 图 图式2
(+)-牛蒡子苷元全合成路线
Figure 图式2.
Attempted total synthetic route for (+)-arctigenin
图 图式2
(+)-牛蒡子苷元全合成路线
Figure 图式2.
Attempted total synthetic route for (+)-arctigenin
上述路线中涉及的溴苄衍生物,按照文献[14]所述方法制备(Scheme 3). 以香草醛为起始原料,经三步反应,分别以82%和79%的总收率得到9和14.
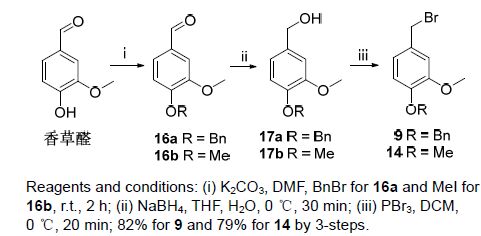 图 图式3
溴苄衍生物合成路线
Figure 图式3.
Synthetic route for benzyl bromide derivatives
图 图式3
溴苄衍生物合成路线
Figure 图式3.
Synthetic route for benzyl bromide derivatives
2 结论
本文研究了用于不对称合成天然产物(-)-牛蒡苷元及其对映异构体的新方法.
我们利用噁唑烷酮类手性辅基(Evans)成功制备了关键中间体(R)-4-(3,4-二甲氧基苄基)丁内酯(8a)和(S)-4-(3,4-二甲氧基苄基)丁内酯(8b),其ee值分别为98%、96%. 并在此基础上制备了(-)-牛蒡子苷元1及其对映异构体(+)-牛蒡子苷元2,其ee值分别为97%、96%. 为接下来拟进行的(-)-牛蒡苷元结构优化奠定了技术基础.
3 实验部分
3.1 仪器与试剂
Bruker AM 400型核磁共振仪(瑞士Bruker公司),以CDCl3为溶剂,TMS为内标; Agilent LCMSD质谱仪(美国Agilent公司); AB 5600+Q高分辨质谱仪(美国AB SCIEX公司); Rudolph Autopol Ⅳ-T型自动旋光仪(美国鲁道夫); X-4显微熔点仪(上海精密科学仪器有限公司). 薄层层析硅胶、柱层析硅胶购自烟台江友硅胶开发有限公司,所有试剂购自上海泰坦科技股份有限公司旗下探索平台.
3.2 化合物的合成
3.2.5 (R)-3-羟甲基-4-(3,4-二甲氧基苯基)-丁酸叔丁酯(7a)的合成
将上步产物6a (2.50 g,5.2 mmol)溶于80 mL四氢呋喃中,加入20 mL水,分批加入硼氢化钠(0.29 g,5.8 mmol),室温搅拌2 h. 减压浓缩除去大部分四氢呋喃,乙酸乙酯稀释,缓慢滴加1 mol/L盐酸至无气泡产生. 依次用5%硫酸氢钾和饱和氯化钠洗涤,无水硫酸钠干燥. 抽滤,滤液减压浓缩,得1.52 g无色透明液体7a. 不经纯化,可直接用于下步反应. -10.2 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 1.45 (s,9H),2.18 (br,1H),2.23~2.36 (m,3H),2.55 (dd,J=13.6,6.2 Hz,1H),2.65 (dd,J=13.6,6.2 Hz,1H),3.52 (dd,J=10.4,4.6 Hz,1H),3.62 (dd,J=10.3,3.8 Hz,1H),3.85 (s,3H),3.87 (s,3H),6.66~6.78 (m,2H),6.79 (d,J=8.6 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 172.4,148.2,146.8,131.6,120.6,111.7,110.5,80.1,64.4,55.3,55.2,39.2,36.8,36.3,27.5; ESI-MS m/z: 333.2 [M+Na]+; HRMS (ESI-TOF) calcd for C17H26O5Na [M+Na]+ 333.1672,found 333.1686.
3.2.14 (S)-4-苄基-3-(单苄酯基丁酰基)-2-噁唑烷酮(13)的合成
参照5a的合成方法,将3,4-二甲氧基苯丙酸变更为丁二酸单苄酯,得无色透明液体,产率97%. +54.3 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.82~2.71(m,3H),3.34~3.21 (m,3H),4.21~4.14 (m,2H),4.67~4.61 (m,1H),5.16 (s,2H),7.20~7.19 (m,2H),7.37~7.25 (m,8H); 13C NMR (100 MHz,CDCl3) δ: 172.2,171.8,153.5,135.8, 135.2,129.4,129.0,128.6,128.2,127.4,66.6,66.3,55.1,37.7,30.8,28.4; ESI-MS 368.2 [M+H]+,390.2 [M+Na]+; HRMS (ESI-TOF) calcd for C21H21NO5Na [M+Na]+ 390.1312,found 390.1312.
3.2.4 (R)-4-苄基-3-[(S)-2-乙酸叔丁酯基-3-(3,4-二甲氧基苯基)丙酰基]-2-噁唑烷酮(6b)的合成
同化合物6a合成. 白色固体,两步产率90%.m.p. 77~78 ℃; -117.4 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 1.41 (s,8.7H),1.46 (s,0.3H),2.38 (dd,J=16.9,3.0 Hz,1H),2.56 (dd,J=12.5,9.7 Hz,1H),2.75 (dd,J=12.7,10.6 Hz,1H),2.83 (dd,J=16.7,11.1 Hz,1H),2.97 (dd,J=12.9,5.8 Hz,1H),3.30 (d,J=13.3 Hz,1H),3.85 (s,3H),3.89 (s,3H),3.97 (t,J=8.2 Hz,1H),4.10 (d,J=8.7 Hz,1H),4.41~4.47 (m,1H),4.52~4.59 (m,1H),6.75~6.77 (m,2H),6.87 (s,1H),7.26~7.34 (m,5H); 13C NMR (100 MHz,CDCl3) δ: 175.5,171.4,153.2,149.0,147.9,135.8,130.7,129.6,129.0,127.4,121.4,112.3,111.1,80.9,66.0,56.01,55.97,55.7,41.5,38.0,37.6,36.8,28.2; ESI-MS m/z: 506.1 [M+Na]+.
3.2.12 (+)-牛蒡苷元(2)的合成
同化合物1合成. 白色固体,产率100%. m.p. 99~102 ℃; ee值96%; +30.3 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.42~2.74 (m,4H),2.77~3.08 (m,2H),3.82 (s,3H),3.83 (s,3H),3.85 (s,3H),3.88 (dd,J=8.9,7.4 Hz,1H),4.14 (dd,J=8.9,7.1 Hz,1H),5.57 (s,1H),6.46 (s,1H),6.55 (d,J=8.0 Hz,1H),6.59~6.68 (m,2H),6.75 (d,J=8.1 Hz,1H),6.83 (d,J=7.9 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 178.9,149.2,148.0,146.8,144.7,130.6,129.6,122.2,120.7,114.2,111.9,111.6,111.4,71.5,56.02,55.97,55.93,46.7,41.1,38.3,34.6; ESI-MS m/z: 373.2 [M+H]+,395.1 [M+Na]+.
3.2.1 (S)-4-苄基-3-(3-(3,4-二甲氧基苯基)丙酰基)-2-噁唑烷酮(5a)的合成
将3,4-二甲氧基苯丙酸(2.10 g,10.0 mmol)溶于60 mL无水THF中,于-20 ℃滴加特戊酰氯(1.20 mL,10.0 mmol)和三乙胺(4.20 mL,30.0 mmol),此温度下继续搅拌20 min. 滴加(S)-4-苄基-2-噁唑烷酮(1.60 g,9.0 mmol)的THF溶液(20 mL),一次性加入无水LiCl (0.42 g,10.0 mmol),继续搅拌20 min后升至室温,再继续搅拌2 h. 反应液减压浓缩至约20 mL,以100 mL乙酸乙酯稀释,依次用1 0%碳酸氢钠、5%硫酸氢钾、饱和氯化钠溶液洗涤,无水硫酸钠干燥. 抽滤,滤液减压浓缩,得3.27 g白色固体5a,不经纯化,可直接用于下步反应. m.p. 88~90 ℃(文献值[15] 91.5~92 ℃); +45.1 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.75 (dd,J=13.3,9.6 Hz,1H),3.91~3.04 (m,2H),3.16~3.35(m,3H),3.86 (s,3H),3.88 (s,3H),4.15~4.21 (m,2H),4.67 (ddt,J=9.9,6.6,3.2 Hz,1H),6.77~6.81 (m,3H),7.17 (d,J=6.7 Hz,2H),7.31~7.38 (m,3H); 13C NMR (100 MHz,CDCl3) δ: 172.6,153.6,149.0,147.6,135.3,133.2,129.6,129.2,129.1,127.5,120.6,112.0,111.4,66.3,56.1,56.0,55.3,38.0,37.4,30.1,26.6; ESI-MS m/z: 370.2 [M+H]+,392.1 [M+Na]+.
3.2.7 (R)-4-(3,4-二甲氧基苄基)-丁内酯(8a)的合成
将上步产物7a (1.50 g,4.8 mmol)溶于20 mL甲苯,加入对甲苯磺酸(41.6 mg,0.24 mmol),于80 ℃搅拌1 h. 减压浓缩除去甲苯. 残渣溶于乙酸乙酯,依次用10%碳酸氢钠和饱和氯化钠溶液洗涤,无水硫酸钠干燥,硅胶柱层析[V(石油醚):V(乙酸乙酯)=2:1],得1.14 g无色透明液体8a,两步产率83%,ee值98%[大赛璐CHIRALPAK®IF色谱柱,以V(正己烷):V(异丙醇):V(叔丁基甲醚)=80:10:10为流动相,流率0.8 mL/min,检测波长为UV 220 nm,温度25 ℃. 8a的保留时间为47.5 min,8b的保留时间为51.3 min]. +6.4 (c 1.0,CHCl3)[文献值[16] [α]D+4.3 (c 2.6,CHCl3)]; 1H NMR (400 MHz,CDCl3) δ: 2.30 (dd,J=17.5,6.8 Hz,1H),2.61 (dd,J=17.5,8.1 Hz,1H),2.66~2.78 (m,2H),2.79~2.90 (m,1H),3.87 (s,3H),3.88 (s,3H),4.05 (dd,J=9.0,6.1 Hz,1H),4.34 (dd,J=9.0,7.1 Hz,1H),6.67 (s,1H),6.70 (d,J=8.1 Hz,1H),6.82 (d,J=8.1 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 177.0,149.2,148.0,130.9,120.8,111.9,111.5,72.7,56.02,55.99,38.7,37.4,34.3; ESI-MS m/z: 237.1 [M+H]+,259.1 [M+Na]+.
3.2.3 (S)-4-苄基-3-[(R)-2-乙酸叔丁酯基-3-(3,4-二甲氧基苯基)丙酰基]-2-噁唑烷酮(6a)的合成
将上步产物5a (2.13 g,5.8 mmol)溶于100 mL无水THF,于-78 ℃滴加NaHMDS (4.3 mL,8.6 mmol,2 mol/L in THF). 搅拌1 h滴加溴乙酸叔丁酯(1.70 mL,11.5 mmol),继续在此温度下搅拌2 h. 饱和氯化铵溶液淬灭反应,减压浓缩除去大部分四氢呋喃,剩余物溶于100 mL乙酸乙酯,依次用5%硫酸氢钾和饱和氯化钠溶液洗涤,无水硫酸钠干燥. 抽滤,滤液减压浓缩,硅胶柱层析[V(石油醚):V(乙酸乙酯)=4:1],得2.58 g白色固体6a,两步产率91%. m.p. 77~79 ℃; +117.9 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 1.40 (s,8.45H),1.46 (s,0.55H),2.38 (dd,J=16.9,3.9 Hz,1H),2.56 (dd,J=13.1,9.4 Hz,1H),2.74 (dd,J=13.4,10.0 Hz,1H),2.83 (dd,J=17.0,11.0 Hz,1H),2.96 (dd,J=13.1,6.0 Hz,1H),3.31 (dd,J=13.4,2.7 Hz,1H),3.85 (s,3H),3.89 (s,3H),3.97 (t,J=8.3 Hz,1H),4.10 (dd,J=9.0,2.2 Hz,1H),4.41~4.49 (m,1H),4.49~4.62 (m,1H),6.77~6.79 (m,2H),6.87 (s,1H),7.26~7.29 (m,3H),7.31~7.38 (m,2H); 13C NMR (100 MHz,CDCl3) δ: 175.6,171.4,153.2,149.0,147.9,135.8,130.7,129.6,129.0,127.4,121.4,112.3,111.1,81.0,66.0,56.03,55.99,55.7,41.5,38.1,37.7,36.8,28.2; ESI-MS m/z: 506.1 [M+Na]+; HRMS (ESI-TOF) calcd for C27H33NNaO7 [M+ Na]+ 506.2149,found 506.2156.
3.2.2 (R)-4-苄基-3-[3-(3,4-二甲氧基苯基)丙酰基]-2-噁唑烷酮(5b)的合成
同化合物5a合成. 白色固体. m.p. 88~90 ℃; -45.5 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.75 (dd,J=13.3,9.6 Hz,1H),2.92~3.05 (m,2H),3.17~3.39 (m,3H),3.85 (s,3H),3.88 (s,3H),4.14~4.20 (m,2H),4.61~4.75 (m,1H),6.77~6.81 (m,3H),7.17 (d,J=6.7 Hz,2H),7.27~7.36 (m,3H); 13C NMR (100 MHz,CDCl3) δ: 172.6,153.6,149.0,147.6,135.3,133.2,129.5,129.2,129.1,127.5,120.6,112.0,111.4,66.3,56.1,56.0,55.2,37.9,37.4,30.1,26.6; ESI-MS m/z: 370.2 [M+H]+,392.1 [M+Na]+.
3.2.6 (S)-3-羟甲基-4-(3,4-二甲氧基苯基)-丁酸叔丁酯(7b)的合成
同化合物7a合成. 无色透明液体. +9.8 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 1.45 (s,9H),2.26~2.34 (m,3H),2.55 (dd,J=13.6,6.5 Hz,1H),2.65 (dd,J=13.6,6.6 Hz,1H),3.48~3.56 (m,1H),3.62~3.73 (m,1H),3.86 (s,3H),3.87 (s,3H),6.72~6.73 (m,2H),6.79 (d,J=8.6 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 172.3,148.2,146.8,131.6,120.6,111.7,110.5,80.1,64.4,55.3,55.2,39.2,36.8,36.3,27.5; ESI-MS m/z: 3 33.2 [M+Na]+.
3.2.9 (3R,4R)-3-(3-甲氧基-4-苄氧基苄基)-4-(3,4-二甲氧基苄基)-丁内酯(10a)的合成
将二异丙胺(360.0 μL,2.5 mmol)溶于20 mL干燥的四氢呋喃,冰浴滴加正丁基锂(1.6 mL,2.5 mmol,1.6 mol/L in THF),反应30 min后降至-78 ℃,滴加上步产物8a (400.0 mg,1.7 mmol)的1 mL四氢呋喃溶液,继续搅拌1 h,滴加化合物9 (519.0 mg,1.69 mmol)的1 mL四氢呋喃溶液,继续搅拌4 h,饱和氯化铵淬灭反应,减压浓缩除去大部分四氢呋喃,剩余物溶于乙酸乙酯,依次用5%硫酸氢钾和饱和氯化钠洗涤,无水硫酸钠干燥后,硅胶柱层析[V(石油醚):V(乙酸乙酯)=4:1],得614 mg无色粘稠液体10a,收率79%. -26.2 (c 1.3,CHCl3){文献值[17]-26.8 (c 1.0,CHCl3)}; 1H NMR (400 MHz,CDCl3) δ: 2.45~2.53 (m,2H),2.55~2.62 (m,2H),2.87~2.99 (m,2H),3.80 (s,3H),3.84 (s,3H),3.85 (s,3H),3.88~3.90 (m,1H),4.12 (t,J=7.7 Hz,1H),5.13 (s,2H),6.47 (s,1H),6.53 (d,J=8.0 Hz,1H),6.59 (d,J=7.5 Hz,1H),6.71 (s,1H),6.74 (d,J=8.2 Hz,1H),6.78 (d,J=7.9 Hz,1H),7.29~7.43 (m,5H); 13C NMR (100 MHz,CDCl3) δ: 178.9,149.9,149.2,148.0,147.2,137.3,131.0,130.6,128.7,128.0,127.8,127.4,127.1,121.5,120.7,114.2,113.0,112.0,111.5,71.4,71.2,65.5,56.12,56.05,55.96,46.7,41.3,38.3,34.7; ESI-MS m/z: 462.9 [M+H]+,484.9 [M+Na]+.
3.2.8 (S)-4-(3,4-二甲氧基苄基)-丁内酯(8b)的合成
同化合物8a合成. 无色透明液体,两步产率81%,ee值96%. -6.3 (c 1.0,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.29 (dd,J=17.5,6.9 Hz,1H),2.61 (dd,J=17.5,8.1 Hz,1H),2.70~2.77 (m,2H),2.80~2.88 (m,1H),3.86 (s,3H),3.87 (s,3H),4.04 (dd,J=8.9,6.2 Hz,1H),4.33 (dd,J=8.9,7.1 Hz,1H),6 .67 (s,1H),6.70 (d,J=8.2 Hz,1H),6.82 (d,J=8.0 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 177.0,149.2,148.0,130.9,120.8,111.9,111.5,72.8,56.02,56.00,38.7,37.4,34.3; ESI-MS m/z: 237.1 [M+H]+,259.1 [M+Na]+.
3.2.10 (3S,4S)-3-(3-甲氧基-4-苄氧基苄基)-4-(3,4-二甲氧基苄基)-丁内酯(10b)的合成
将二异丙胺(145 μL,1.02 mmol)溶于15 mL干燥的四氢呋喃,冰浴滴加正丁基锂(0.64 mL,1.02 mmol,1.6 mol/L in THF),反应30 min后降至-78 ℃,滴加上步产物8b (160.0 mg,0.68 mmol)的1 mL四氢呋喃溶液,反应1 h后,滴加化合物9 (208.9 mg,0.68 mmol)的1 mL四氢呋喃溶液,继续搅拌4 h至TLC显示反应完全,饱和氯化铵淬灭反应,减压浓缩除去大部分四氢呋喃,剩余物溶于乙酸乙酯,依次用5%硫酸氢钾和饱和氯化钠洗涤,无水硫酸钠干燥后,硅胶柱层析[V(石油醚):V(乙酸乙酯)=4:1]. 得239.0 mg无色粘稠液体10b,收率76%,de>99%. +26.5 (c 1.3,CHCl3); 1H NMR (400 MHz,CDCl3) δ: 2.54~2.43 (m,2H),2.64~2.54 (m,2H),2.93 (qd,J=14.1,6.0 Hz,2H),3.80 (s,3H),3.84 (s,3H),3.85 (s,3H),3.88 (dd,J=10.9,3.3 Hz,1H),4 .11 (dd,J=9.0,7.0 Hz,1H),5.13 (s,2H),6.47 (d,J=1.8 Hz,1H),6.52 (dd,J=8.1,1.8 Hz,1H),6.59 (dd,J=8.1,1.8 Hz,1H),6.71 (d,J=1.8 Hz,1H),6.74 (d,J=8.1 Hz,1H),6.78 (d,J=8.1 Hz,1H),7.42~7.29 (m,5H); 13C NMR (100 MHz,CDCl3) δ: 178.9,149.9,149.1,148.0,147.2,137.3,131.0,130.6,128.7,128.0,127.4,121.5,120.7,114.1,113.0,111.9,111.4,71.4,71.2,56.12,56.04,55.96,46.7,41.3,38.3,34.7; ESI-MS m/z: 462.9 [M+H]+,484.9 [M+Na]+.
3.2.11 (-)-牛蒡苷元(1)的合成
将上步产物10a (600.0 mg,1.3 mmol)溶于20 mL甲醇,加入10% Pd/C (60.0 mg),氢气氛下室温搅拌1 h. 抽滤,滤液减压浓缩得米色固体,乙醚洗涤数次,得474 mg白色粉末状固体1,产率98%,ee值97%[大赛璐CHIRALPAK® ID色谱柱,以V(正己烷):V(异丙醇):V(叔丁基甲醚)=80:10:10为流动相,流率0.8 mL/min,检测波长为UV 220 nm,温度25 ℃. 1的保留时间为40.9 min,2的保留时间为35.2 min]. m.p. 100~102 ℃; -30.7 (c 1.0,CHCl3)[文献值[18] m.p. 100~101 ℃,-31.5 (c 1.06,CHCl3),天然产物]; 1H NMR (400 MHz,CDCl3) δ: 2.42~2.74 (m,4H),2.77~3.08 (m,2H),2.82 (s,3H),2.83 (s,3H),3.85 (s,3H),3.88 (dd,J=8.9,7.4 Hz,1H),4.14 (dd,J=8.9,7.1 Hz,1H),5.57 (s,1H),6.46 (s,1H),6.55 (d,J=8.0 Hz,1H),6.59~6.68 (m,2H),6.75 (d,J=8.1 Hz,1H),6.83 (d,J=7.9 Hz,1H); 13C NMR (100 MHz,CDCl3) δ: 178.9,149.2,148.0,146.8,144.7,130.6,129.6,122.2,120.7,114.2,111.9,111.6,111.4,71.5,56.02,55.97,55.93,46.7,41.1,38.3,34.6; ESI-MSm/z: 373.2 [M+H]+,395.1 [M+ Na]+; HRMS (ESI-TOF) calcd for C21H25O6 [M+H]+ 373.1646,found 373.1642.
3.2.13 丁二酸单苄酯(12)的合成
参照文献[19]所述方法合成,产率62%. 化合物表征数据与文献一致.
致谢
感谢大赛璐药物手性技术(上海)有限公司对化合物光学纯度的测定.
辅助材料(Supporting Information) 中间体和目标产物的1H NMR、13C NMR图谱以及(-)-牛蒡子苷元1的HRMS图谱. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
Hyam, S. R.; Lee, I. A.; Gu, W.; Kim, K. A.; Jeong, J. J.; Jang, S. E.; Han, M.-J.; Kim, D. H. Eur. J. Pharmacol. 2013, 708, 21. doi: 10.1016/j.ejphar.2013.01.014
-
[2]
Zhao, Z.-Y.; Yin, Y.-Q.; Wu, H.; Jiang, M.; Lou, J.-S.; Bai, G.; Luo, G.-A. Cell. Physiol. Biochem. 2013, 32, 1342.
-
[3]
Miele, C.; Beguinot, F. Diabetologia 2012, 55, 1244.
-
[4]
Zhang, N.; Wen, Q.-P.; Ren, L.; Liang, W.-B.; Xia, Y.; Zhang, X.-D; Zhao, D.; Sun, D.; Hu, Y.; Hao, H.-G.; Yan, Y.-P.; Zhang, G.-X.; Yang, J.-X.; Kang, T.-G. Int. J. Mol. Sci. 2013, 14, 18657.
-
[5]
Gu, Y.; Scheuer, C.; Feng, D.-L; Menger, M. D.; Laschke, M. W. Anti-Cancer Drug 2013, 8, 781.
-
[6]
Gu, Y.; Qi, C.-T.; Sun, X.-X.; Ma, X.-Q.; Zhang, H.-H.; Hu, L.-H.; Yuan, J.-Y.; Yu, Q. Acta Pharmacol. Sin. 2012, 33, 941.
-
[7]
Zhu, Z.-Y.; Yan, J.-M.; Jiang, W.; Yao, X.-G.; Chen, J.; Chen, L.-L.; Li, C.-J.; Hu, L.-H.; Jiang, H.-L.; Shen, X. J. Neurosci. 2013, 33, 13138. doi: 10.1523/JNEUROSCI.4790-12.2013
-
[8]
Landete, J. M. Food Res. Int. 2012, 46, 410. doi: 10.1016/j.foodres.2011.12.023
-
[9]
Wang, H.-H.; Wu, P.; Kang, H.; Xu, L.; Zhu, R.-X.; Kang, T.-G. Chin. J. Org. Chem. 2012, 32, 1894 (in Chinese). (王欢欢, 吴平, 康宏, 许亮, 朱瑞新, 康廷国, 有机化学, 2012, 32, 1894.) (b) Xu, Y.-B.; Dou, D.-Q. J. Liaoning Univ. Tradit. Chin. Med. 2013, (7), 64 (in Chinese). (徐煜彬, 窦德强, 辽宁中医药大学学报, 2013, (7), 64.) (c) Chen, G.-R.; Li, H.-F.; Dou, D.-Q; Xu, Y.-B.; Jiang, H.-S.; Li, F.-R.; Kang, T.-G. Nat. Prod. Res. 2013, 23, 2251.
-
[10]
Li, D.-W.; Liu, Q.-P.; Jia, D.; Dou, D.-Q.; Wang, X.-F.; Kang, T.-G. Planta Med. 2014, 80, 48.
-
[11]
Amancha, P. K.; Liu, H.-J.; Ly, T. W.; Shia, K. S. Eur. J. Org. Chem. 2010, 18, 3473. (b) Eich, E.; Pertz, H.; Kaloga, M.; Schulz, J.; Fesen, M. R.; Mazumder, A.; Pommier, Y. J. Med. Chem. 1996, 39, 86. (c) Bode, J. W.; Doyle, M. P.; Protopopova, M. N.; Zhou, Q.-L. J. Org. Chem. 1996, 61, 9146. (d) Fischer, J.; Reynolds, A. J.; Sharp, L. A.; Sherburn, M. S. Org. Lett. 2004, 6, 1345. (e) Pohmakotr, M.; Soorukram, D.; Tuchinda, P.; Prabpai, S.; Kongsaeree, P.; Reutrakul, V. Tetrahedron Lett. 2004, 45, 4315.
-
[12]
Lin, G.-Q.; Chen, Y.-Q.; Chen, X.-Z.; Lin, Y.-M. Chiral Synthesis, Asymmetric Reaction and Its Application, Beijing Science Press, Beijing, 2000, p. 56 (in Chinese). (林国强, 陈耀全, 陈新滋, 李月明, 手性合成-不对称反应及其应用, 科学出版社, 北京, 2000, p. 56.)
-
[13]
Markus, N.; Christine, W.; Tobias, B.; Felix, M.; Jens, C.; Benedikt, S.; Ralf, P.; Soledad, R. G.; Thomas, P.; Norbert, S. J. Med. Chem. 2013, 56, 1853. doi: 10.1021/jm301346z
-
[14]
Woo, L. W. L.; Bubert, C.; Sutcliffe, O. B.; Smith, A.; Chander, S. K.; Mahon, M. F.; Purohit, A.; Reed, M. J.; Potter, B. V. L. J. Med. Chem. 2007, 50, 3540. doi: 10.1021/jm061462b
-
[15]
So, M.; Kotake, T.; Matsuura, K.; Inui, M.; Kamimura, A. J. Org. Chem. 2012, 77, 4017. doi: 10.1021/jo300380z
-
[16]
Hughes, G.; Kimura, M.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11253. doi: 10.1021/ja0351692
-
[17]
Sibi, M. P.; Liu, P.; Ji, J.; Hajra, S.; Chen, J. J.Org. Chem. 2002, 67, 1738 doi: 10.1021/jo015501x
-
[18]
Moritani, S.; Nomura, M.; Takeda, Y.; Miyamoto, K. Biol. Pharm. Bull. 1996, 19, 1515. doi: 10.1248/bpb.19.1515
-
[19]
Chen, X.-Y.; Zenger, K.; Lupp, A.; Kling, B.; Heilmann, J.; Fleck, C.; Kraus, B.; Decker, M. J. Med. Chem. 2012, 55, 5231. doi: 10.1021/jm300246n
-
[1]
-

图 2 化合物5和8羰基α-烷基化立体选择性模型
Figure 2 Proposed models for the stereochemical course of the alkylation of 5 and 8
Reagents and conditions: (i) BnBr,Et3N,THF,reflux,5 h,62%; (ii) PivCl,Et3N,4a,THF,-20 ℃~r.t.,2 h,97%.
-


 下载:
下载:




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1098
- HTML全文浏览量: 203
 下载:
下载:
