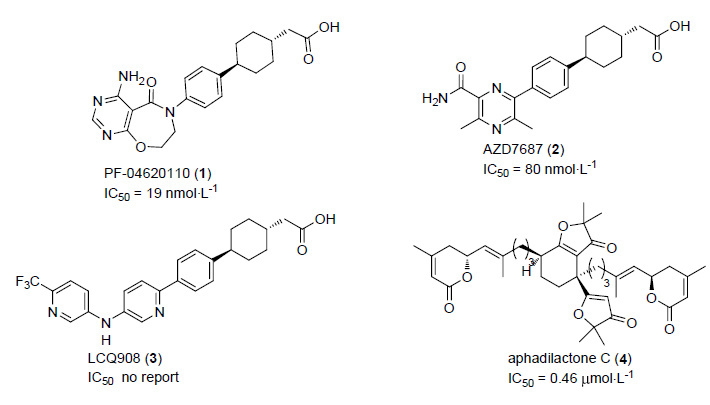
 图 1
已进入临床研究的DGAT1抑制剂和天然产物aphadilactone C
Figure 1.
DGAT1 inhibitors in clinical study and natural compound aphadilactone C
图 1
已进入临床研究的DGAT1抑制剂和天然产物aphadilactone C
Figure 1.
DGAT1 inhibitors in clinical study and natural compound aphadilactone C
引用本文:
李丹, 尹建朋, 李静雅, 南发俊. 基于天然产物Aphadilactone C结构关键片段的二酰基甘油酰基转移酶1抑制剂的设计与合成[J]. 有机化学,
2016, 36(6): 1359-1367.
doi:
10.6023/cjoc201511037

Citation: Li Dan, Yin Jianpeng, Li Jingya, Nan Fajun. Design and Synthesis of Diacylglycerol Acyltransferase 1 Inhibitors Based on Aphadilactone C[J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1359-1367. doi: 10.6023/cjoc201511037
Citation: Li Dan, Yin Jianpeng, Li Jingya, Nan Fajun. Design and Synthesis of Diacylglycerol Acyltransferase 1 Inhibitors Based on Aphadilactone C[J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1359-1367. doi: 10.6023/cjoc201511037
基于天然产物Aphadilactone C结构关键片段的二酰基甘油酰基转移酶1抑制剂的设计与合成
摘要:
二酰基甘油酰基转移酶(DGAT)作为甘油三酯合成的唯一限速酶, 成为治疗肥胖以及其他代谢综合征的重要靶标. 利用从具有DGAT1抑制活性的天然产物aphadilactone C分子中重要内酯环与进入临床研究的2-((1R,4R)-4-(4-(4-氨基-5-氧亚基-7,8-二氢嘧啶[5,4-f][1,4] 氧氮杂-6(5H)-基)苯基)环己基)乙酸(PF-04620110), 2-((1R,4R)-4-(4-(6-氨甲酰基-3,5-二甲基对二氮杂苯-2-基)苯基)环己基)乙酸(AZD-7687)分子中杂环单元的重新组合, 设计合成了化合物5~8, 以此来验证aphadilactone C中内酯环单元是否与已知DGAT1抑制剂中的环己基乙酸部分相等同. 最终的活性测试结果显示, 化合物5~8并不具有DGAT1的抑制作用. 天然产物aphadilactone C的DGAT抑制机制值得深入研究.
-
关键词:
- aphadilactone C
- / DGAT1
- / 选择性抑制剂
- / PF-04620110
- / AZD-7687
English
Design and Synthesis of Diacylglycerol Acyltransferase 1 Inhibitors Based on Aphadilactone C
Abstract:
Diacylglycerol acyltransferase (DGAT), the only limited enzyme in the synthesis of triacylglycerol (TAG), is regarded as an important therapeutic target for human obesity and other metabolic syndromes. Compounds 5~8 were designed and synthesized, in which the lactone group of aphadilactone C was introduced into the PF-04620110 and AZD-7687, which have entered into the clinical research, to verify whether the lactone in aphadilactone C played the same role as carboxylic group in PF-04620110 and AZD-7687. The final vitro assay showed that compounds 5~8 have not the inhibition activity to DGAT1. This might suggest that inhibition mechanism of aphadilactone C was not the same as PF-04620110 and AZD-7687.
-
Key words:
- aphadilactone C
- / DGAT1
- / selective inhibitor
- / PF-04620110
- / AZD-7687
-
甘油三酯(TG)是人体内含量最多的脂类,亦是体内能量的主要来源,大部分组织均可以利用甘油三酯的分解产物供给能量. 人体合成甘油三酯的途径主要有磷脂酸途径和甘油一酯途径. (1)甘油一酯(MG)途径: 以MG为起始物,与脂酰辅酶A (FA CoA)在单酰基甘油脂酰转移酶(MGAT)作用下酯化生成甘油二脂(DG),DG再与FA CoA在二酰基甘油酰基转移酶 (DGAT)作用下酯化生成TG. (2)磷脂酸(3-GP)途径: 3-磷酸甘油在脂酰转移酶(GPAT)作用下,与两分子FA CoA反应生成3-GP,3-GP在磷脂酸磷酸酶(PAP)作用下,水解释放出无机磷酸而转变为DG,在二酰基甘油酰基转移酶(DGAT)作用下酯化生成TG[1]. DGAT是TG合成最后一步的催化酶,也是TG合成的唯一限速酶. 它主要结合在内质网膜上,是微粒体酶,由酰基化的辅酶A和二酰基甘油以共价键的形式结合起来,可以被DG特异性激活[2]. 人体中DGAT有DGAT1和DGAT2两种类型,两者有54%的同源性,但是分属于不同的基因家族. DGAT1属于酰基辅酶A胆固醇酰基转移酶家族,而DGAT2则属于DGAT2超家族[3].
研究表明,抑制DGAT1的活性可以降低TG的合成,改善机体对胰岛素和瘦素敏感性,增加葡萄糖摄取率和能量消耗,抵抗高脂饮食导致的肥胖等,可作为治疗肥胖和其他代谢综合征的的靶标[4]; 而DGAT2抑制剂并不适用于肥胖或代谢疾病的药物治疗[5].
DGAT1作为治疗肥胖和糖尿病的新型靶点,目前还没有上市的药物,国内外各大医药公司及科研机构寻找DGAT1选择性抑制剂的活动如火如荼地展开着. 例如,辉瑞公司开发的化合物PF-04620110对人DGAT1具有较高的选择性抑制. 目前该化合物已完成I期临床的实验[6]; 阿斯利康公司的AZD-7687也在2011年4月完成了多剂量递增的I期临床研究; 诺华公司的LCQ-908已启动Ⅱ期临床研究.
aphadilactone C (4)是由中国科学院上海药物研究所岳建民研究员[7]在2013年从楝科植物大叶山楝中分离而来. 它是目前已发现的天然来源结构新颖的DGAT1抑制剂,IC50值为(0.46±0.09) μmol•L-1. 而且它对DGAT1选择性相对于DGAT2也是高达217倍. 鉴于aphadilactone C良好的活性和选择性,引起了我们对这个分子的兴趣. 我们课题组[8]首次完成了aphadilactone C天然产物的全合成,在此基础上,展开了对其构效关系的进一步探索.
对目前的已进入临床研究阶段的DGAT1抑制剂PF-04620110(辉瑞公司)、AZD-7687(阿斯利康公司)以及LCQ-908(诺华公司)分析可以发现,这三个化合物都是含有一个相同的、必须的药效团——苯基环己基乙酸基团,而另一端一般都是一个含有氨基的芳香杂环结构. 进一步的文献调研发现,绝大多数的DGAT1抑制剂都是在一端含有芳香杂环结构; 而另一端的乙酸基团也是必须的[9],将羧基转换成中性的电子等排体,或者将与羧基相连的linker做一些调整,但其活性仍然是一定程度上保持. 这就说明了在小分子对DGAT1活性的抑制过程中,除了芳香杂环结构在与蛋白结合中起到非常重要的作用,乙酸或其替代基团在对DGAT1抑制过程中也起到了非常重要的作用.
目前绝大多数DGAT1抑制剂中的乙酸基团可能发挥如下三种作用: 羧基与DGAT1特定的氢键供体氨基酸残基相互接近时,能够作为一个氢键的受体,与其形成氢键作用[10]; 末端的羧基基团也有可能模拟了DGAT1的脂肪酸底物[11],与油酸-辅酶A结合位点竞争性地结合(Scheme 1). 这一生化机制已经被辉瑞公司的研究小组以T863作为小分子抑制剂在小鼠体内实验深入地研究过,它除了能使小鼠的体重降低大约6.7%之外,还能够明显地降低小鼠体内甘油三酯以及胆固醇的含量[9a]; 最后,羧基还可以起到增加溶解度、改善其药代动力学、增强活性以及解决一些由PXR诱导和CYP抑制引起的一些潜在的脱靶效应等[9a].
图 1
已进入临床研究的DGAT1抑制剂和天然产物aphadilactone C
Figure 1.
DGAT1 inhibitors in clinical study and natural compound aphadilactone C
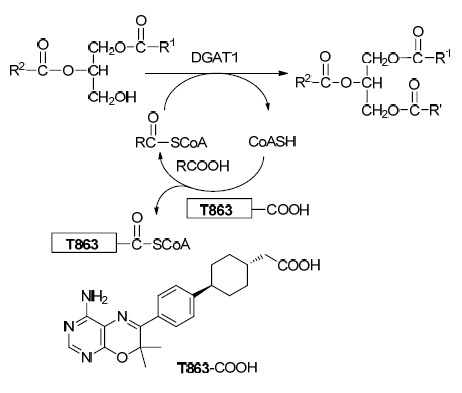 图 图式1
DGAT1小分子抑制剂与辅酶A竞争性结合示意图
Figure 图式1.
Schematic of inhibitors binding competitively at the oleoly-CoA binding site
图 图式1
DGAT1小分子抑制剂与辅酶A竞争性结合示意图
Figure 图式1.
Schematic of inhibitors binding competitively at the oleoly-CoA binding site
对aphadilactone C所表现出来的良好抑制活性进行分析,我们认为,尽管aphadilactone C与目前常见的DGAT1抑制剂有显著不同的结构骨架,但其与DGAT1的作用形式与已知的DGAT1抑制剂可能有一定的相似性. 天然产物aphadilactone C中存在的内酯环部分可能作为一个氢键的受体或者模拟了DGAT1的脂肪酸底物,而呋喃酮部分与PF-04620110,AZD-7687,LCQ-908等分子的芳香杂环部分起相类似的作用,含双键的长链则替代linker苯基环己基部分,内酯环可能作为一个氢键的受体,或者作为羧基的一个前药基团,当受到水解酶水解之后成为羧基发挥药效. 基于这一个猜测,首先我们选取了已知DGAT1抑制剂AZD7687及PF-04620110,保留其芳香杂环部分不变,而将其苯基环己基乙酸部分换成天然产物aphadilactone C中的内酯环,并以不同linker连接两部分,初步设计了4个化合物作为目标.
首先是以天然产物aphadilactone C的脂肪链结构作为linker,以内酯作为一个氢键的受体,我们设计并合成了化合物5. 在此基础上,又考虑是否为内酯环受水解酶作用开环成羧基的可能性,根据其开环后linker所需长度,我们将其开环之前的链长缩短,设计合成了化合物6. 除此之外,我们还选择了辉瑞公司PF-04620110 的芳香杂环部分作为亲水端,并且在其与内酯环中间引入直线性更好的炔基作为linker,我们设计了化合物7; 考虑到aphadilactones A~D四个化合物中,仅仅只有aphadilactone C具有良好的 DGAT1的抑制活性,提示了我们对化合物的特定的立体构型的考虑,因此我们又设计出了化合物8 (Scheme 2).
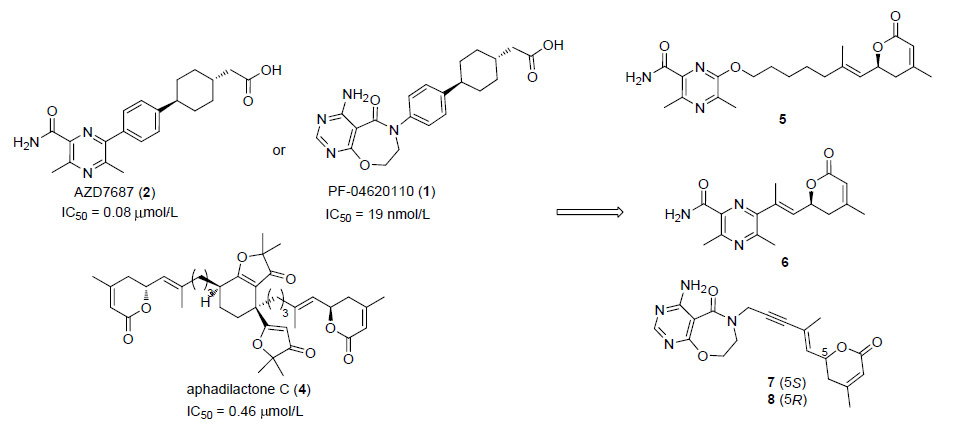 图 图式2
化合物5~8设计思路
Figure 图式2.
Designing of compounds 5~8
图 图式2
化合物5~8设计思路
Figure 图式2.
Designing of compounds 5~8
1 结果与讨论
1.1 合成部分
化合物5的合成是由两个主要的中间体15,16经亲核取代以及后续的酸性条件脱异丙基、氧化生成. 首先是对中间体15的合成. 以乙酰乙酸乙酯作为起始原料,在冰醋酸中经亚硝酸钠作用,很容易转化成中间体肟10. 中间体肟在氯化氢的乙醇溶液中,经Pd/C催化氢化,以很高的收率得到α-氨基-β-酮基丁酸乙酯盐酸盐(11). 化合物11与Boc保护的丙氨酸反应,生成酰胺12. 酰胺12经过三氟乙酸脱去Boc保护基,紧接着分子内的酰胺键的生成,顺利得到内酰胺13. 内酰胺13在微波反应的条件下,经三氯氧磷作用进行氯取代,得到氯代吡嗪14. 化合物14在氨水中,经过氨基的取代,最终得到氯代吡嗪甲酰胺15. 重要中间体16的合成参照文献[8] aphadilactone C (4)全合成中的步骤. 化合物16经氢化钠的作用生成氧负离子,然后氧负离子对氯代吡嗪酰胺上的氯原子亲核取代,得到化合物17. 化合物17首先在弱酸对甲苯磺酸吡啶盐(PPTS)的作用下脱去异丙基得到半缩醛,半缩醛不经纯化,直接采用MnO2氧化,就可以得到最终产物5 (Scheme 3).
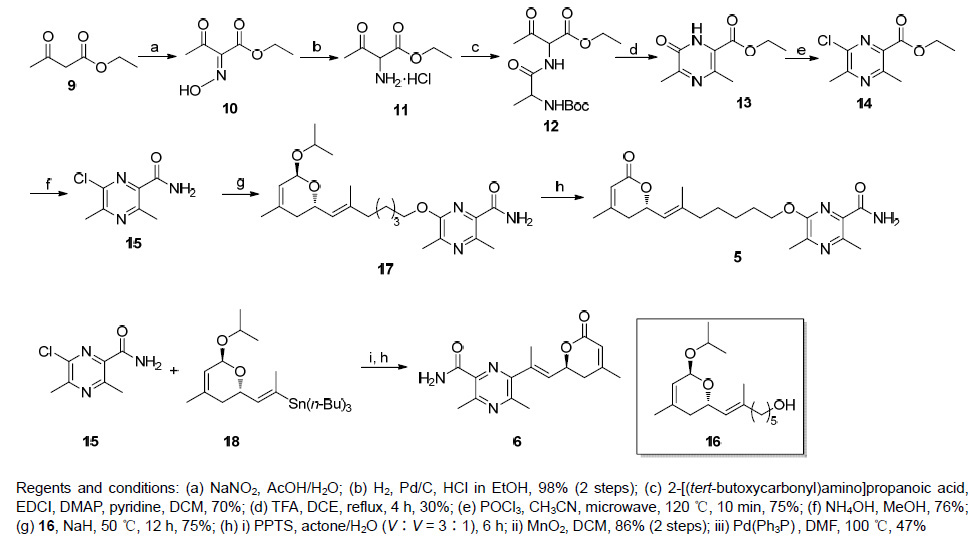 图 图式3
化合物5和6的合成
Figure 图式3.
Synthesis of compounds 5 and 6
图 图式3
化合物5和6的合成
Figure 图式3.
Synthesis of compounds 5 and 6
化合物6的合成,首先是经过中间体15与18经钯催化偶联,然后在酸性条件脱异丙基以及之后的MnO2氧化得到(此步骤同化合物5的合成步骤),中间体18的合成参照文献[8] aphadilactone C (4)合成中的步骤(Scheme 3).
化合物7的合成,首先是由中间体炔25与烯基碘26经过偶联,然后脱异丙基、氧化(此步骤同5的合成)、氨基取代得到. 炔25的合成首先从工业原料嘧啶醛20开始,在偶氮二异丁腈(AIBN)的催化下,与磺酰氯反应,得到嘧啶酰氯21. 嘧啶酰氯再与炔丙基胺衍生物22反应,得到酰胺23. 酰胺23经过氯化氢的甲醇溶液作用,脱去硅醚保护基,再经过分子内的亲核取代,生成七元环的内酰胺中间体25. 烯基碘化物26的合成参照文献aphadilactone C (4)全合成中的步骤9[8](Scheme 4).
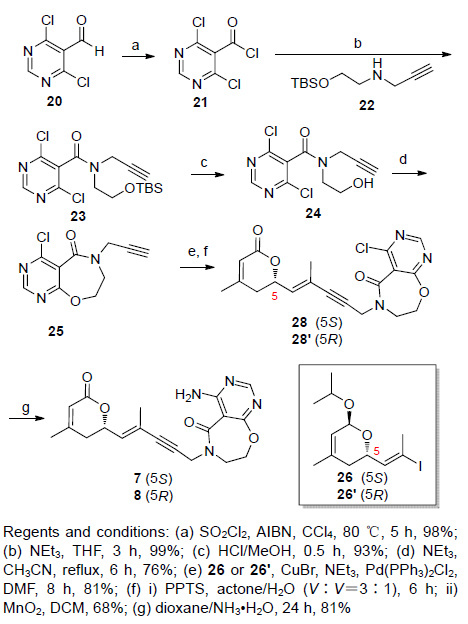 图 图式4
化合物7和8的合成
Figure 图式4.
Synthesis of compounds 7 and 8
图 图式4
化合物7和8的合成
Figure 图式4.
Synthesis of compounds 7 and 8
另外一种构型的化合物8,则是经过中间体25与26'偶联得到. 化合物26'的合成步骤与26类似,只是由化合物29和30发生的不对称杂Diels-Alder反应的催化剂S1构型与aphadilactone C全合成中不对称杂Diels-Alder反应用到的催化剂构型相反. 但此处所得到的化合物31的ee值也明显下降,仅有大约60%. 由化合物31到化合物26'的合成过程参照文献[8] aphadilactone C的全合成步骤(Scheme 5).
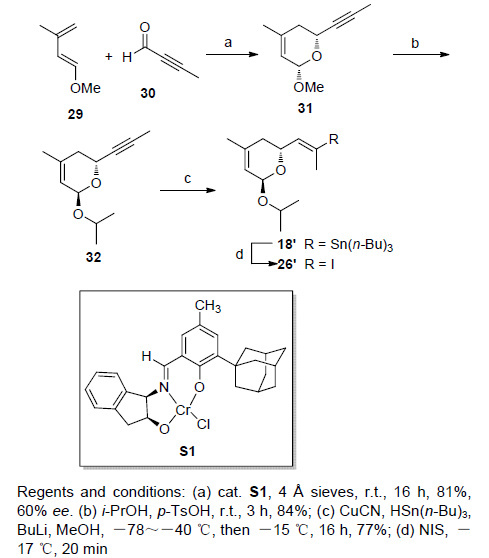 图 图式5
化合物26'的合成
Figure 图式5.
Synthesis of compound 26'
图 图式5
化合物26'的合成
Figure 图式5.
Synthesis of compound 26'
1.2 化合物5~8的DGAT1抑制活性测试
我们分离正常Sprague Dawley大鼠的肝脏微粒体, 并利用同位素法对上述合成的化合物进行体外分子活性测试. 各化合物对肝脏DGAT的抑制活性如表 1. 在化合物浓度均为1 μmol•L-1 时,AZD-7867和aphadilactone C都表现出较好的DGAT抑制活性,其抑制率分别为89.32%和89.23%,而改造后的化合物5~8对DGAT1的抑制率均小于10%. 故而我们所设计的4个化合物并不具有DGAT的抑制活性.
 表 1
化合物5~8对大鼠肝脏DGAT的抑制率
Table 1.
DGAT inhibitive rate of compounds 5~8
表 1
化合物5~8对大鼠肝脏DGAT的抑制率
Table 1.
DGAT inhibitive rate of compounds 5~8
编号 抑制率/% AZD-7867 89.32±2.06 Aphadilactone C 89.23±3.35 5 4.72±10.49 6 0±8.22 7 3.49±2.44 8 6.14±4.78 表 1 化合物5~8对大鼠肝脏DGAT的抑制率
Table 1. DGAT inhibitive rate of compounds 5~82 结论
PF-04620110 (1),AZD-7687 (2)等这些已经进入临床试验的DGAT1抑制剂,都具有着相似度非常高的结构骨架——芳香杂环结构和苯基环己基乙酸. 而抑制活性和选择性都较好的天然产物aphadilactone C跟已知抑制剂却有着明显不同的结构骨架. 因此,我们基于已知抑制剂PF-04620110,AZD-7687和天然产物aphadilactone C,将二者的优势结构相结合,采用aphadilactone C中的内酯环结构代替已知抑制剂中的苯基环己基乙酸部分,合成了化合物5,并根据链长不同进一步设计合成了化合物6. 除此之外我们还考察了内酯环的立体化学因素,分别合成了化合物7和8. 通过DGAT1的抑制实验发现,我们设计的化合物5~8都几乎失去对DGAT1的抑制作用. 因此我们推测,天然产物aphadilactone C对DGAT1的抑制作用与已知抑制剂有可能是不相同的作用模式. 此外,由于我们合成的化合物数目有限,也有可能在链长、立体化学等方面仍存在差异,进而影响其对DGAT1的抑制活性. 本工作的尝试也表明了aphadilactone C作为新型DGAT1抑制剂需要进行深入的构效关系研究.
3 实验部分
3.1 仪器与试剂
核磁氢谱(1H NMR)和碳谱(13C NMR)采用Bruker BioSpin AG (Ultrashield Plus AV-300,400,500)型核磁共振仪测定,CDCl3为溶剂,TMS 为内标; 质谱(ESI- MS)采用Agilent G6120单四级杆质谱仪测定; 高分辨质谱(HRMS)采用Q-Tof micro四级杆飞行时间串联质谱仪测定; 匀浆器、高速离心机(HITACHI)、超高速离心机(HITACHI)、Perkin Elmer 1450-023液闪计数仪(Perkin Elmer)、涡旋器; sn-二油酸甘油酯(sn-1,2-dioleoylgly- cerol) (sigma),牛白蛋白(不含游离脂肪酸)[BSA (FFA free)],其它试剂均采用市售化学纯或者分析纯.
3.2 实验方法
3.3 体外抑制DGAT1活性实验方法
提取大鼠肝脏微粒体[14, 15]: 将成年SD大鼠(约250 g)颈椎脱臼处死,迅速打开腹腔,剪开下腔静脉待血流尽后摘下肝脏,称湿重,剪碎,加入(即1 g肝脏至4 mL缓冲液)湿重的预冷缓冲液1 (0.25 mol•L-1蔗糖,10 mmol•L-1 Tris-HC1,pH 7.4,1.0 mmol•L-1乙二胺四乙酸二钠),用匀浆器将肝脏匀成均匀的悬浊液,将悬浊液12000 r/min,4 ℃离心2次,每次30 min,取上清. 将上清用超高速离心机105000 g,4 ℃,1 h离心,取沉淀,用缓冲液2 (0.25 mol•L-1 蔗糖,10 mmol•L-1 Tris-HC1,pH 7.4)重悬,-80 ℃保存备用.
活性测试[15]: 200 μL反应体系中含200 μmol•L-1 sn-1,2-二油酸甘油酯,30 μmol•L-1 [14C] 油酸辅酶A,8 mmol•L-1 MgCl2,200 μg BSA (FFA free),10 μg 肝脏微粒体蛋白和175 mmol•L-1 Tris-HCl buffer,pH 8.0,与化合物(溶于DMSO中)一起于96孔透明尖底板25 ℃反应30 min; 之后每孔加50 μL Stop buffer(异丙醇/正庚烷/水,V:V:V=80:20:2)停止反应,将全部液体转移到2 mL EP管中; 每管加入1.45 mL 终止反应液,并加400 μL水彻底涡旋,12000 r/min离心10 min; 取上层液体200 μL,烘干; 加1.7 mL闪烁液,读14C信号值.
辅助材料(Supporting Information) 所有化合物的1H NMR以及13C NMR原始谱图,所有新化合物的HR-MS原始谱图,已知化合物的LR-MS原始谱图. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载
3.2.5 (S,E)-3,5-二甲基-6-((6-甲基-7-(4-甲基-6-氧亚基-3,6-二氢吡喃-2-基)庚-6-烯-1-基)氧基)吡嗪-2-甲酰胺(5)的合成
将化合物17 (40 mg,0.093 mmol)溶于丙酮/水(V: V=3:1) (4 mL)的混合溶液中,室温下加入PPTS (28 mg,0.11 mmol),3 h后反应完全,加水稀释,乙酸乙酯萃取,饱和食盐水洗一次. 无水硫酸钠干燥,减压浓缩,得到粗产物半缩醛直接溶于10 mL干燥DCM中,室温下加入MnO2 (81 mg,0.93 mmol),5 h后原料反应完全. 反应液直接过滤,滤液减压浓缩,柱层析纯化,得到无色油状化合物5 (31 mg,86%). 1H NMR (300 MHz,CDCl3) δ: 1.44~1.51 (m,4H),1.71 (d,J=1.2 Hz,3H),1.82 (t,J=7.2 Hz,2H),1.98 (s,3H),2.07 (t,J=6.6 Hz,2H),2.16~2.23 (m,1H),2.32~2.43 (m,1H),2.49 (s,3H),2.83 (s,3H),4.29 (t,J=6.6 Hz,2H),5.07~5.14 (m,1H),5.32 (d,J=8.4 Hz,1H),5.74 (br s,1H),5.81 (s,1H),7.51 (br s,1H); 13C NMR (75 MHz,CDCl3) δ: 16.67,19.32,21.97,22.95,25.61,27.09,28.59,35.09,39.16,66.28,74.14,116.61,121.99,134.67,142.45,145.68,147.36,154.77,157.00,165.29,167.63; HRMS calcd for C21H30N3O4 [M+H]+ 388.2236,found 388.2212.
3.2.9 N-(2-((叔丁基二甲基硅基)氧基)乙基)丙-2-炔- 1-胺(22)的合成
参考文献[13]方法,将2-羟基乙基炔丙基胺(495 mg,5 mmol)溶于DCM中,加入咪唑(439 mg,6.5 mmol),5 min后加入叔丁基二甲基氯硅烷(TBSCl,982 mg,6.5 mmol),室温反应5 h,原料反应完全,加水稀释,DCM萃取. 减压浓缩,柱层析纯化,得无色液体化合物22 (1.02 g,96%). 1H NMR (CDCl3,300 MHz) δ: 0.05 (s,6H),0.88 (s,9H),2.20 (t,J=3.0 Hz,1H),2.78 (t,J=2.4 Hz,2H),3.44 (d,J=3.0 Hz,2H),3.73 (t,J=2.4 Hz,2H); 13C NMR (75 MHz,CDCl3) δ: 82.15,71.22,62.32,50.53,38.17,25.89 (3C),18.27,-5.36 (2C); EI-MS m/z: 214 (M+H)+.
3.2.3 3,5-二甲基-6-氯吡嗪-2-甲酰胺(15)的合成
参考文献[9b]方法,将化合物14 (530 mg,2.5 mmol)溶于10 mL甲醇中,加入4 mL氨水. 50 ℃反应过夜,直接将反应液减压浓缩,得浅黄色粉末. 经柱层析纯化得到浅黄色固体15 (350 mg,76%). 1H NMR (300 MHz,CDCl3) δ: 2.68 (s,3H),2.92 (s,3H),5.64 (br s,1H),7.55 (br s,1H); 13C NMR (75 MHz,CDCl3) δ: 22.27,22.71,138.79,143.72,153.06,154.99,165.90; ESI-MS m/z: 208 (M+Na)+.
3.2.11 4-氯-6-(丙-2-炔-1-基)-7,8-二氢嘧啶[5,4-f]- [1, 4]氧氮杂-5(6H)-酮(25)的合成
将化合物23 (400 mg,0.80 mmol)溶于5 mL甲醇中,室温下将HCl/MeOH (0.5 mL/5 mL)加入其中,室温反应0.5 h,原料反应完全. 加水稀释,乙酸乙酯萃取,碳酸氢钠溶液洗涤有机相,饱和食盐水洗一次,无水硫酸钠干燥,柱层析纯化,得油状化合物24 (290 mg,93%),一对非对映异构体(轴手性),二者比例约1:1.2. 1H NMR (300 MHz,CDCl3) δ: 2.35~2.37 (m,2.2H),3.44 (t,J= 2.4 Hz,2H),3.81 (t,J=2.4 Hz,2H),3.88 (t,J=2.4 Hz,2.4H),3.98 (t,J=2.4 Hz,2.4H),4.07 (d,J=1.2 Hz,2.4H),4.49 (d,J=1.2 Hz,2 H),8.82 (s,1H),8.84 (s,1.2H); 13C NMR (75 MHz,CDCl3) δ: 34.62,39.65,48.40,50.23,59.65,60.99,72.90,74.54,76.43,77.37,129.01,129.05,157.93,158.14,158.64,158.76,162.46,162.89; HRMS calcd for C10H9Cl2N3NaO2 [M+Na]+ 295.9970,found 295.9963.
将化合物24 (300 mg,1.1 mmol)溶于15 mL乙腈中,加入1 mL三乙胺,置于80 ℃下回流过夜,冷制室温,减压浓缩,直接柱层析纯化,得油状化合物25 (200 mg,76%). 1H NMR (300 MHz,CDCl3) δ: 2.38 (t,J=2.4 Hz,1H),3.81 (t,J=4.8 Hz,2H),4.50 (d,J=2.4 Hz,2H),4.75 (t,J=4.5 Hz,2H),8.71 (s,1H); 13C NMR (75 MHz,CDCl3) δ: 35.76,44.48,73.93,74.28,76.96,116.46,159.28,162.17,162.52,165.66; HRMS calcd for C10H9Cl- N3O2 [M+H]+ 238.0383,found 238.0372.
3.2.12 (R,E)-4-氯-6-(4-甲基-5-(4-甲基-6-氧亚基-3,6-二氢吡喃-2-基)戊-4-烯-2-炔-1-基)-7,8-二氢嘧啶[5,4-f][1, 4]氧氮杂-5(6H)-酮(28)的合成
将化合物25 (150 mg,0.63 mmol),化合物26 (305 mg,0.95 mmol),CuBr (20 mg),NEt3 (0.4 mL),溶于20 mL DMF中. 充分换Ar气氛围,然后再加入催化剂Pd(PPh3)2Cl2 (50 mg),室温反应过夜,原料反应完全,加水稀释,乙酸乙酯萃取,无水硫酸钠干燥,过滤,减压浓缩. 柱层析,得油状化合物27 (220 mg,81%). 1H NMR (300 MHz,CDCl3) δ: 1.16 (d,J=3.0 Hz,3H),1.21 (d,J=3.0 Hz,3H),1.74 (s,3H),1.80 (d,J=18 Hz,1H),1.87 (s,3H),2.04 (d,J=18 Hz,1H),3.80 (t,J=3.0 Hz,2H),3.96~4.00 (m,1H),4.61 (s,2H),4.70~4.73 (m,1H),4.74 (t,J=3.0 Hz,2H),5.07 (s,1H),5.45 (s,1H),5.86 (d,J=3.0 Hz,1H),8.71 (s,1H); 13C NMR (75 MHz,CDCl3) δ: 17.64,21.92,22.79,23.82,34.72,36.47,44.36,63.21,69.33,74.40,80.14,87.65,93.22,116.57,119.83,120.19,136.65,138.06,159.23,162.10,162.50,165.66; HRMS calcd for C22H26ClN3NaO4 [M+Na]+ 454.1510,found 454.1501. 得到的粗产物也可不经纯化直接用于下一步反应.
将以上粗产物27直接溶于丙酮/水(15 mL/5 mL)的混合溶液中,加入PPTS (173 mg),室温反应3 h后,原料反应完全,乙酸乙酯萃取,饱和食盐水洗一次,无水硫酸钠干燥,减压浓缩. 将浓缩物直接溶于20 mL无水DCM中,加入新制MnO2室温反应5 h,硅藻土过滤,柱层析纯化,得油状化合物28 (166 mg,0.43 mmol,68%). 1H NMR (300 MHz,CDCl3) δ: 1.89 (s,3H),2.0 (s,3H),2.23~2.46 (m,2H),3.80 (t,J=4.5 Hz,2H),4.61 (s,2H),4.74 (t,J=4.5 Hz,2H),5.10~5.18 (m,1H),5.84 (s,1H),5.93 (d,J=8.4 Hz,1H),8.71 (s,1H); 13C NMR (75 MHz,CDCl3) δ: 17.86,23.02,34.22,36.45,44.54,73.04,74.32,81.78,86.62,116.53,116.73,122.68,134.04,156.57,159.28,162.16,162.49,164.25,165.66; HRMS calcd for C19H18ClN3NaO4 [M+Na]+ 410.0884,found 410.0877.
3.2.13 (R,E)-4-氨基-6-(4-甲基-5-(4-甲基-6-氧亚基- 3,6-二氢吡喃-2-基)戊-4-烯-2-炔-1-基)-7,8-二氢嘧啶 [5,4-f][1, 4]氧氮杂-5(6H)-酮(7)的合成
将化合物28 (80 mg,0.21 mmol)溶于二氧六环与NH3•H2O的混合溶液中(15 mL/2 mL),室温反应24 h,原料反应完全,减压浓缩,直接柱层析纯化,得浅黄色粉末化合物7 (63 mg,81%). m.p. 95~99 ℃; 1H NMR (300 MHz,CDCl3) δ: 1.87 (s,3H),2.00 (s,3H),2.22~2.41 (m,2H),3.76 (t,J=4.2 Hz,2H),4.53 (s,2H),4.62 (t,J=4.2 Hz,2H),5.10~5.16 (m,1H),5.83 (d,J=1.5 Hz,1H),5.91 (dd,J=1.2,8.4 Hz,1H),8.24 (s,1H); 13C NMR (100 MHz,CDCl3) δ: 17.90,22.99,34.22,37.55,46.32,72.44,73.07,77.02,82.29,86.01,95.36,116.71,122.81,133.74,156.56,159.53,164.29,166.11,166.24; HRMS calcd for C19H21N4O4 [M+H]+ 369.1563,found 369.1559.
3.2.14 (S,E)-4-氯-6-(4-甲基-5-(4-甲基-6-氧亚基-3,6-二氢吡喃-2-基)戊-4-烯-2-炔-1-基)-7,8-二氢嘧啶[5,4-f][1, 4]氧氮杂-5(6H)-酮(28')的合成
将化合物25 (150 mg,0.63 mmol),26' (305 mg,0.95 mmol),CuBr (20 mg),NEt3 (0.4 mL),溶于20 mL DMF中. 充分换Ar气氛围,然后再加入催化剂Pd(PPh3)2Cl2 (50 mg),室温反应过夜,原料反应完全,加水稀释,乙酸乙酯萃取,无水硫酸钠干燥,过滤,减压浓缩. 柱层析,收无色油状化合物27' (210 mg,77%). 1H NMR (300 MHz,CDCl3) δ: 1.16 (d,J=3.0 Hz,3H),1.21 (d,J=3.0 Hz,3H),1.74 (s,3H),1.80 (d,J=18 Hz,1H),1.87 (s,3H),2.04 (d,J=18 Hz,1H),3.80 (t,J=3.0 Hz,2H),3.96~4.00 (m,1H),4.61 (s,2H),4.71~4.73 (m,1H),4.74 (t,J=3.0 Hz,2H),5.07 (s,1H),5.45 (s,1H),5.86 (d,J=3.0 Hz,1H),8.71 (s,1H); 13C NMR (100 MHz,CDCl3) δ: 17.64,21.92,22.79,23.82,34.72,36.47,44.36,63.21,69.33,74.40,80.14,87.65,93.22,116.57,119.83,120.19,136.65,138.06,159.23,162.10,162.50,165.66; HRMS calcd for C22H26ClN3NaO4 [M+Na]+ 454.1510,found 454.1500. 得到的粗产物也可不经纯化直接用于下一步反应.
将以上粗产物直接溶于丙酮/水(15 mL/5 mL)的混合溶液中,加入PPTS (173 mg),室温反应3 h后,原料反应完全,乙酸乙酯萃取,饱和食盐水洗一次,无水硫酸镁干燥,减压浓缩. 将浓缩物直接溶于20 mL无水DCM中,加入新制MnO2室温反应5 h,硅藻土过滤,柱层析纯化,收无色油状化合物28' (166 mg,68%). 1H NMR (300 MHz,CDCl3) δ: 1.89 (s,3H),2.0 (s,3H),2.23~2.46 (m,2H),3.80 (t,J=4.5 Hz,2H),4.61 (s,2H),4.74 (t,J=4.5 Hz,2H),5.10~5.18 (m,1H),5.84 (s,1H),5.93 (d,J=8.4 Hz,1H),8.71 (s,1H); 13C NMR (100 MHz,CDCl3) δ: 17.86,23.02,34.22,36.45,44.54,73.04,74.32,81.78,86.62,116.53,116.73,122.68,134.04,156.57,159.28,162.16,162.49,164.25,165.66; HRMS calcd for C19H18ClN3NaO4 [M+Na]+ 410.0884,found 410.0877.
3.2.15 (S,E)-4-氨基-6-(4-甲基-5-(4-甲基-6-氧亚基- 3,6-二氢吡喃-2-基)戊-4-烯-2-炔-1-基)-7,8-二氢嘧啶[5,4-f][1, 4]氧氮杂-5(6H)-酮(8)的合成
将化合物28' (80 mg,0.21 mmol)溶于二氧六环与NH3•H2O (15 mL/2 mL)的混合溶液中,室温反应24 h,原料反应完全,减压浓缩,直接柱层析纯化,得浅黄色粉末8 (63 mg,81%). m.p. 97~100 ℃; 1H NMR (300 MHz,CDCl3) δ: 1.87 (s,3H),2.00 (s,3H),2.22~2.41 (m,2H),3.76 (t,J=4.2 Hz,2H),4.53 (s,2H),4.62 (t,J=4.2 Hz,2H),5.10~5.16 (m,1H),5.83 (d,J=1.5 Hz,1H),5.91 (dd,J=1.2,8.4 Hz,1H),8.24 (s,1H); 13C NMR (100 MHz,CDCl3) δ: 17.90,22.9,34.22,37.55,46.32,72.44,73.07,77.02,82.29,86.01,95.36,116.71,122.81,133.74,156.56,159.53,164.29,166.11,166.24; HRMS calcd for C19H21N4O4 [M+H]+ 369.1563,found 369.1556.
3.2.2 3,5-二甲基-6-氯吡嗪-2-甲酸乙酯(14)的合成
参考文献[9b]方法,将化合物13 (700 mg,3.6 mmol)溶于12 mL新鲜处理好的乙腈中(悬浮液),换Ar,搅拌下逐滴加入三氯氧磷(830 μL,9 mmol),置微波反应器中120 ℃反应10 min,冷置室温后,直接短硅胶柱过滤一遍,然后再柱层析纯化,DCM洗脱,得浅黄色液体14 (580 mg,75%). 1H NMR (300 MHz,CDCl3) δ: 1.41 (t,J=7.2 Hz,3H),2.67 (s,3H),2.76 (s,3H),4.44 (q,J=7.2 Hz,2H); 13C NMR (75 MHz,CDCl3) δ: 14.18,22.27,22.36,62.19,139.52,145.09,152.50,154.92,164.37; ESI -MS m/z: 215 (M+H)+.
3.2.7 (S,E)-3,5-二甲基-6-(1-(4-甲基-6-氧亚基-3,6-二氢吡喃-2-基)丙-1-烯-2-基)吡嗪-2-甲酰胺(6)的合成
具体反应步骤参照化合物17→5的合成,得到最终产物油状化合物6 (8 mg,81%). 1H NMR (300 MHz,CDCl3) δ: 2.03 (s,3H),2.13 (s,3H),2.33~2.57 (m,2H),2.62 (s,3H),2.92 (s,3H),5.30~5.38 (m,1H),5.70 (br s,1H),5.74 (dd,J=1.2,8.1 Hz,1H),5.88 (s,1H),7.68 (br s,1H); 13C NMR (75 MHz,CDCl3) δ: 17.03,22.77,23.03,23.23,34.52,73.65,116.81,129.51,138.13,138.29,150.48,152.79,152.85,156.67,164.52,167.06; HRMS calcd for C16H19N3NaO3 [M+Na]+ 324.1324,found 324.1319.
3.2.1 3,5-二甲基-6-氧亚基-1,6-二氢吡嗪-2-甲酸乙酯(13)的合成
参考文献[9b,12]方法,将乙酰乙酸乙酯(3.9 g,30 mmol)溶于30 mL冰醋酸,冰水浴下,缓慢加入亚硝酸钠的水溶液17 mL (75 mmol),滴加过程中保持温度低于5 ℃,当棕色气体挥发完毕后,将反应液在0 ℃下反应2 h,再升至室温反应1 h,原料反应完全. 向反应混合物中加水稀释,乙酸乙酯萃取,有机相依次经过水洗、饱和碳酸氢钠水溶液洗、饱和食盐水洗. 无水硫酸钠干燥之后,减压浓缩. 得到无色油状粗产物10,不经纯化直接用于下一步反应. ESI-MS m/z: 182 (M+Na)+.
将化合物10 (30 mmol)直接溶于90 mL EtOH中,换Ar,加入Pd/C (10%,0.33 equiv.),换H2氛围,然后向反应液中加入HCl的EtOH溶液30 mL (3 mol•L-1,3 equiv.),置室温反应24 h,原料反应完全. 反应液直接通过硅藻土过滤. 滤液经减压浓缩,得到浅白色粉末11(5.3 g,98%,两步). ESI-MS m/z: 146 (M+H)+.
将化合物11 (29 mmol)溶于80 mL二氯甲烷(DCM)中,再加入Boc-Ala-NH2 (34.8 mmol),1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI,34.8 mmol),4-二甲氨基吡啶(DMAP,34.8 mmol),吡啶(58 mmol),室温反应过夜. 加入冰水稀释,乙酸乙酯萃取,柱层析分离提纯,收浅黄色液体12 (6.5 g,70%). ESI-MS m/z: 339 (M+Na)+.
将化合物12 (4.0 g,12.7 mmol),溶于干燥的1,2-二氯乙烷中,搅拌下缓慢加入三氟乙酸(10 equiv.,127 mmol),回流4 h原料反应完全. 直接减压浓缩除去溶剂,粗产物经柱层析纯化,得到黄色粉末13 (750 mg,30%). 1H NMR (300 MHz,CDCl3) δ: 1.42 (t,J=7.2 Hz,3H),2.52 (s,3H),2.63 (s,3H),4.43 (q,J=7.2 Hz,2H); 13C NMR (75 MHz,CDCl3) δ: 14.14,20.83,20.91,62.88,120.51,136.33,154.30,161.59,163.67; ESI-MS m/z: 197 (M+H)+.
3.2.10 N-(2-((叔丁基二甲基硅基)氧基)乙基)-4,6-二氯-N-(丙-2-炔-1-基)嘧啶-5-甲酰胺(23)的合成
将化合物22 (300 mg,0.90 mmol)溶于5 mL无水四氢呋喃中,Ar氛围下加入三乙胺(137 μL,0.99 mmol),室温搅拌下将酰氯21 (200 mg,0.95 mmol)的四氢呋喃溶液2 mL逐滴加入. 室温反应3 h,原料反应完全,减压浓缩,直接柱层析纯化,得油状化合物23 (450 mg,99%),一对非对映异构体(轴手性),二者比例约1:0.8. 1H NMR (300 MHz,CDCl3) δ: 0.03 (s,4.8H),0.08 (s,6H),0.86 (s,7.2H),0.90 (s,9H),2.30~2.31 (m,1.8H),3.39 (t,J=3.0 Hz,1.6H),3.76 (t,J=3.0 Hz,1.6H),3.83 (t,J=3.0 Hz,2H),3.94 (t,J=3.0 Hz,2H),4.12 (d,J=1.2 Hz,2H),4.54 (d,J=1.2 Hz,1.6H),8.82 (s,0.8H),8.82 (s,1H); 13C NMR (75 MHz,CDCl3) δ: -5.53,-5.52,18.07,18.21,25.80,34.93,39.93,47.64,49.71,61.35,61.58,72.67,73.96,129.26,129.34,157.91,157.95,158.68,161.90,162.10; HRMS calcd for C16H23Cl2N3NaO2Si [M+Na]+410.0834,found 410.0829.
3.2.6 6-((E)-1-((2S,6S)-6-异丙氧基-4-甲基-3,6-二氢吡喃-2-基)丙-1-烯-2-基)-3,5-二甲基吡嗪-2-甲酰胺 (19)的合成
将化合物15 (150 mg,0.31 mmol)和18 (57 mg,0.31 mmol)溶于3 mL四氢呋喃中,换Ar氛围,再加入催化剂Pd(PPh3)4 (36 mg,0.1 equiv.),110 ℃反应过夜. 冷却至室温,乙酸乙酯萃取,水洗两次,饱和食盐水洗一次,无水硫酸钠干燥,减压浓缩. 柱层析纯化,得无色油状化合物19 (50 mg,47%). 1H NMR (300 MHz,CDCl3) δ: 1.20 (d,J=6.0 Hz,3H),1.28 (d,J=6.0 Hz,3H),1.77 (s,3H),1.94 (dd,J=15,3.6 Hz,1H),2.11 (s,3H),2.12~2.16 (m,1H),2.64 (s,3H),2.93 (s,3H),4.02~4.07 (m,1H),4.89~4.94 (m,1H),5.13 (s,1H),5.50 (s,1H),5.67 (dd,J=1.5,8.4 Hz,1H),7.73 (br s,1H); 13C NMR (75 MHz,CDCl3) δ: 16.79,2.06,22.83,22.94,23.18,23.88,34.97,63.77,69.49,93.43,120.32,133.25,135.20,136.76,137.99,151.34,152.20,152.90,167.29; HRMS calcd for C19H27N3NaO3 [M+Na]+ 368.1950,found 368.1941.
3.2.4 6-(((E)-7-((2S,6S)-6-异丙氧基-4-甲基-3,6-二氢吡喃-2-基)-6-甲基-庚-6-烯-1-基)氧基)-3,5-二甲基吡嗪-2-甲酰胺(17)的合成
称取化合物16 (60 mg,0.21 mmol)溶于5 mL四氢呋喃中,换Ar,室温搅拌下加入NaH (50 mg,10 equiv.),5 min后将化合物15 (0.32 mmol,68 mg)的四氢呋喃溶液加入其中. 50 ℃反应12 h,原料反应完全. 加水淬灭反应,乙酸乙酯萃取,饱和食盐水洗一遍,无水硫酸钠干燥,减压浓缩. 经过柱层析分离提纯,得无色油状化合物17 (68 mg,75%). 1H NMR (300 MHz,CDCl3) δ: 1.15 (d,J=6.0 Hz,3H),1.21 (d,J=6.0 Hz,3H),1.43~1.52 (m,4H),1.69 (s,3H),1.71 (s,3H),1.73~1.86 (m,3H),1.95~2.47 (m,2H),2.49 (s,3H),2.83 (s,3H),3.96~4.01 (m,1H),4.28 (t,J=6.6 Hz,2H),4.60~4.72 (m,1H),5.07 (s,1H),5.21 (d,J=8.1 Hz,1H),5.42 (s,1H),5.89 (br s,1H),7.51 (br s,1H); 13C NMR (75 MHz,CDCl3) δ: 16.54,19.30,21.86,21.96,22.79,23.78,25.68,27.21,28.58,35.69,39.27,63.42,66.34,68.92,93.28,120.07,124.85,134.62,137.18,139.63,145.64,147.38,154.78,167.70; HRMS calcd for C24H37N3NaO4 [M+Na]+ 454.2682,found 454.2670.
3.2.8 4,6-二氯嘧啶-5-甲酰氯(21)的合成
将化合物20 (1.0 g,6.0 mmol)和AIBN (60 mg)溶于10 mL CCl4中,换Ar氛围,搅拌下滴加SO2Cl2 (1.0 mL),将反应液置80 ℃下回流5 h,原料反应完全. 直接减压浓缩,得到油状液体酰氯不经纯化,直接用于下一步反应.
-
-
[1]
Mu, H.; Høy, C. E. Prog. Lipid Res. 2004, 43, 105. doi: 10.1016/S0163-7827(03)00050-X
-
[2]
Cases, S.; Stone, S. J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K. D.; Voelker, T.; Farese, R. V. Jr. J. Biol. Chem. 2001, 276, 38870. doi: 10.1074/jbc.M106219200
-
[3]
Lardizabal, K. D.; Mai, J. T.; Wagner, N. W.; Wyrick, A.; Voelker, T.; Hawkins, D. J. J. Biol. Chem. 2001, 276, 38862. doi: 10.1074/jbc.M106168200
-
[4]
Smith, S. J.; Cases, S.; Jensen, D. R.; Chen, H. C.; Sande, E.; Tow, B.; Sanan, D. A.; Raber, J.; Eckel, R. H.; Farese, R. V. Jr. Nat. Genet. 2000, 25, 87. (b) Chen, H. C.; Smith, S. J.; Ladha, Z.; Jensen, D. R.; Ferreira, L. D.; Pulawa, L. K.; McGuire, J. G.; Pitas, R. E.; Eckel, R. H.; Farese, R. V. Jr. J. Clin. Invest. 2002, 109, 1049. (c) Chen, H. C.; Jensen, D. R.; Myers, H. M.; Eckel, R. H.; Farese, R. V. Jr. J. Clin. Invest. 2003, 111, 1715. (d) Chen, H. C.; Ladha, Z.; Smith, S. J.; Farese, R. V. Jr. Am. J. Physiol. Endocrinol. Metab. 2003, 284, 213. doi: 10.1038/75651
-
[5]
Stone, S. J.; Myers, H. M.; Watkins, S. M.; Brown, B. E.; Feingold, K. R.; Elias, P. M.; Farese, R. V. Jr. J. Biol. Chem. 2004, 279, 11767. (b) Chen, H. C.; Farese, R. V. Jr. Arterioscler., Thromb., Vasc. Biol. 2005, 25, 482. doi: 10.1074/jbc.M311000200
-
[6]
Dow, R. L.; Andrews, M. P.; Li, J. C.; Gibbs, E. M.; Guzman-Perez, A.; LaPerle, J. L.; Li, Q.; Mather, D.; Munchhof, M. J.; Niosi, M.; Patel, L.; Perreault, C.; Tapley, S.; Zavadosk, W. J. Bioorg. Med. Chem. 2013, 21, 5081. doi: 10.1016/j.bmc.2013.06.045
-
[7]
Liu, J.; He, X. F.; Wang, G. H.; Merino, E. F.; Yang, S. P.; Zhu, R. X.; Gan, L. S.; Zhang, H.; Cassera, M. B.; Wang, H. Y.; Kingston, D. G. I.; Yue, J. M. J. Org. Chem. 2013, 79, 599.
-
[8]
Yin, J. P.; Gu, M.; Li, Y.; Nan, F. J. J. Org. Chem. 2014, 79, 6294. doi: 10.1021/jo501117k
-
[9]
Devita, R. J.; Pinto, S. J. Med. Chem. 2013, 56, 9820. (b) Barlind, J. G.; Bauer, U. A.; Birch, A. M.; Birtles, S.; Buckett, L. K.; Butlin, R. J.; Davies, R. D. M.; Eriksson, J. W.; Hammond, C. D.; Hovland, R.; Johannesson, P.; Johansson, M. J.; Kemmitt, P. D.; Lindmark, B. T.; Gutierrez, P. M.; Noeske, T. A.; Nordin, A.; O'Donnell, C. J.; Petersson, A. U.; Redzic, A.; Turnbull, A. V.; Vinblad, J. J. Med. Chem. 2012, 55, 10610. doi: 10.1021/jm4007033
-
[10]
Dow, R. L.; Andrews, M.; Aspnes, G. E.; Balan, G.; Gibbs, E. M.; Perez, A. G.; Karki, K.; LaPerle, J. L.; Li, J. C.; Litchfield, J.; Munchhof, M. J.; Perreault, C.; Patel, L. Bioorg. Med. Chem. Lett. 2011, 21, 6122. doi: 10.1016/j.bmcl.2011.08.028
-
[11]
Zhou, G.; Ting, P. C.; Wishart, G.; Zorn, N.; Aslanian, R. G.; Lin, M.; Smith, M.; Walker, S. S.; Cook, J.; Heek, M. V.; Lachowicz, J. Bioorg. Med. Chem. Lett. 2014, 24, 1790. doi: 10.1016/j.bmcl.2014.02.028
-
[12]
Mordant, C.; Dunkelmann, P.; Ratovelomanana-Vidal, V.; Genet, J.-P. Eur. J. Org. Chem. 2004, 14, 3017.
-
[13]
Efthymiou, T. C.; Huynh, V.; Oentoro, J., Peel, B., Desaulniers, J. P. Bioorg. Med. Chem. Lett. 2012, 22, 1722. doi: 10.1016/j.bmcl.2011.12.104
-
[14]
Wu, W. N.; Mckown, L. A. Optimization in Drug Discovery, Vol. 11, Eds.: Yan, Z.-Y.; Caldwell, G. W., Humana Press Inc., Totowa, 2004, p. 163.
-
[15]
Coleman, R. A. Methods Enzymol. 1992, 209, 98. doi: 10.1016/0076-6879(92)09013-S
-
[1]
-

图 1 已进入临床研究的DGAT1抑制剂和天然产物aphadilactone C
Figure 1 DGAT1 inhibitors in clinical study and natural compound aphadilactone C

图式1 DGAT1小分子抑制剂与辅酶A竞争性结合示意图
Scheme 1 Schematic of inhibitors binding competitively at the oleoly-CoA binding site
表 1 化合物5~8对大鼠肝脏DGAT的抑制率
Table 1. DGAT inhibitive rate of compounds 5~8
编号 抑制率/% AZD-7867 89.32±2.06 Aphadilactone C 89.23±3.35 5 4.72±10.49 6 0±8.22 7 3.49±2.44 8 6.14±4.78  下载: 导出CSV
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1483
- HTML全文浏览量: 212
 下载:
下载:
